

1. Introduction
Diabetes mellitus is a worldwide epidemic characterized by chronic hyperglycemia that results from either a deficiency or tolerance in insulin.1 In the United States, 8.3% of the population currently has diabetes and that number is projected to increase to 1 in 3 adults by 2050 if current trends continue.2 As a consequence, diabetes is the seventh leading cause of death with an annual cost burden of $174 billion in the United States, including $116 billion in direct medical expenses.2 Blood glucose levels in diabetics fluctuate significantly throughout the day, resulting in serious complications including heart attacks, strokes, high blood pressure, kidney failure, blindness and limb amputation.1–2 Portable glucose sensors give patients the ability to monitor blood glucose levels, manage insulin levels, and reduce the morbidity and mortality of diabetes mellitus. Traditional glucose monitoring techniques are primarily based on the use of electrochemical amperometric glucose sensors. In 1987, Medisense Inc. launched the first personal glucose testing device consisting of a test strip and reader. Over 40 different commercial pocket-sized monitors have been introduced since then.3 To date, the U.S. Food and Drug Administration (FDA) has approved >25 glucose monitors with the majority employing test strips consisting of either glucose dehydrogenase (GDH) or glucose oxidase (GOx) immobilized on a screen-printed electrode.4 The analysis is based on obtaining a small blood sample (<1 μL) through a finger prick that is subsequently introduced into the test strip via capillary action.3–4 While these monitors have augmented the health outcomes for people with diabetes by improving blood glucose management, such monitoring only provides instantaneous blood glucose concentrations that are unable to warn of hyperglycemic or hypoglycemic events in advance. Additionally, the sample collection (i.e., finger prick) method is inconvenient resulting in poor patient compliance. Analytical methods that enable continuous monitoring of blood glucose have thus been sought.5 Continuous glucose monitoring (CGM) provides real-time information on trends (i.e., whether the glucose levels are increasing or decreasing), magnitude, duration, and frequency of glucose fluctuations during the day.5–6 Ideally, analytically functional continuous glucose monitoring devices could be linked to an insulin delivery pump, creating an artificial pancreas.5–6 In this review, we describe progress in the development of continuous glucose monitoring technologies, specifically focusing on subcutaneous implantable electrochemical glucose sensors, which are widely studied and commercially available. We discuss the challenges associated with the development of biocompatible coatings for electrochemical glucose sensors. Borrowing from the ideas of David Williams, we consider sensor coatings to be “biocompatible” if they optimize the clinical relevance of the sensor, avoid any negative local and systemic effects, and elicit the most appropriate local tissue response adjacent to the implant.7
2. Continuous glucose monitoring technologies
2.1. Electrochemical continuous glucose monitoring biosensors
Clark and Lyons were the first to publish on enzyme-modified electrodes with analyte selectivity.8 The first dedicated glucose analyzer for direct measurement of glucose in whole blood samples was launched by Yellow Spring Instruments in 1975.8 The most successful technologies for measuring glucose concentrations to date continue to be based on enzyme-modified electrodes and electrochemical detection (Scheme 1) with a number of useful designs. Updike and Hicks developed a sensor that detects glucose by monitoring the consumption of oxygen using two oxygen electrodes (one covered with the enzyme and one for reference). The differential current between these electrodes are compared so as to correct for background variations in oxygen.9 Alternatively, hydrogen peroxide produced enzymatically by glucose oxidase can be quantified amperometrically as first demonstrated by Guilbault and Lubrano in 1973.10 Amperometric enzyme electrodes vary greatly in electrode design, electrode material, enzyme immobilization method, and polymeric membrane compositions. The first continuous in vivo monitoring of blood glucose was proposed and demonstrated by Shichiri et al. in 1982 with the first FDA premarket approval of a CGM system occurring in 1999.11–12
Scheme 1.
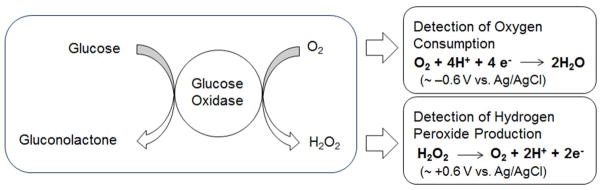
The enzymatic oxidation of glucose to gluconolactone by glucose oxidase (GOx) with subsequent electrochemical detection of oxygen depletion and/or hydrogen peroxide formation.
Continuous glucose monitoring devices are categorized according to the analytical method by which they quantify glucose and the level of their invasiveness, as summarized in Table 1. Due to their ability to be miniaturized and give high selectivity and sensitivity governed by the specific biocatalytic reactions of an immobilized enzyme, electrochemical biosensors are among the most widely studied devices.4 Non-enzymatic electrochemical glucose sensors have been reported whereby glucose is measured directly via direct electro-oxidation at high-surface area (i.e., porous) platinum electrodes,13–15 or through potentiometric detection dependent on pKa changes in a conducting polymer,16–17. However, poor selectivity has hindered their further development as clinical CGM devices. Enzyme-based sensors measure the rate of glucose oxidation through a change in oxygen or hydrogen peroxide concentrations upon reaction of glucose with a glucose-specific enzyme (e.g., GOx or GDH). First generation enzyme-based electrochemical biosensors were fabricated by immobilizing the enzyme on the surface of the electrode. The enzyme is reduced upon converting glucose to gluconolactone. Ambient oxygen facilitates the conversion of the reduced enzyme back to its oxidized form with concomitant production of hydrogen peroxide.18–19 As shown Scheme 1, the glucose concentration correlates with the amperometric signal obtained from either the electrochemical oxidation of produced hydrogen peroxide or the reduction of consumed oxygen. Although enzyme-based electrochemical glucose biosensors are characterized with high selectivity and sensitivity due their enzymatic nature, the dynamic range of such sensors is limited by co-substrate (i.e., oxygen) availability. An outer diffusion-limiting membrane is thus employed to control for this and eliminate oxygen deficiencies, albeit with a slightly delayed sensor response. Additionally, the working electrode potential required to monitor hydrogen peroxide (i.e., ~+0.6 V vs. Ag/AgCl) will also oxidize electroactive endogenous species (e.g., ascorbic acid and acetaminophen) and create high current densities.19 To address these shortcomings, second-generation electrochemical glucose biosensors employ electron mediators (e.g., [Os(4,4’-dimethoxy-2,2’-bipyridine)2Cl]+/2+) “wired” to the enzyme on a hydrophilic polymer matrix (e.g., poly(vinylpyridine) or poly(vinylimidazole)). Such mediators are capable of shuttling electrons from the redox center of the enzyme to the surface of the electrode, thus allowing for a lower applied electrode potential.20–21 In turn, the sensor response is independent of the co-substrate and interferences. Unfortunately, most mediators are toxic and competition between the mediator and oxygen still exists.19,22
Table 1.
Summary of continuous glucose monitoring technologies.
| Analytical technologies | Invasiveness | Species | Duration | Tissue-contacting membrane | Percent point in zone A and B in Clarke Error Grid | Ref | ||
|---|---|---|---|---|---|---|---|---|
| Electrochemical | Hydrogen peroxide detection | Invasive and minimal invasive (subcutaneous, percutaneous, and intravenous) | Human, pig, dog and rat | 3–7 d | Polyurethane or vinyl pyridine-styrene copolymer, | 97–98% | 241–242 | |
| Oxygen detection | Pig | 1 year | Polydimethylsiloxane (PDMS) and titanium housing | 96.2% | 31 | |||
| Redox mediated | Chimpanzee, dog and rat | <1 d | Redox hydrogel (Poly(vinylpridne) complex of osmium bipyridne) and polyethylene glycol | Linear regression R2=0.94 (rms=17%) | 123,143,243 | |||
| Iontophoresis | Minimal invasive (transcutaneous) | Human | 5 d | Hydrogel | 98.9% | 244–245 | ||
| Microdialysis | Invasive (percutaneous) | Human | 48 h | Polycarbonate/polyether | 94–98% | 23,246 | ||
| Optical | Fluorophore-based | FRET intensity | Invasive (Subcutaneous) Minimal invasive (sonophoresis and transdermal) | Human | 3 d | VEGF-loaded hydrogel | n/a | 247–248 |
| FRET lifetime | In vitro | n/a | n/a | n/a | 249 | |||
| Fluorescence intensity | Mice | 140 d | Hydrogel | n/a | 39 | |||
| Ocular spectroscopy | Noninvasive (body fluid) | Human | 1 month | Monosaccharide fluorescent signaling boronic acid doped contact lens | n/a | 37 | ||
| Non-fluorophore based | Optical coherence tomography (OCT) | Noninvasive (transdermal) | Human | 3 h | n/a | Linear regression (R2=0.95) | 250 | |
| Photoacoustic | Noninvasive (transdermal and body fluid) | Human | 6 h | n/a | 94.6% | 251 | ||
| Thermal infrared spectroscopy | Human | n/a | n/a | 100% | 252 | |||
| Polarimetry | Human | Ex vivo | n/a | >0.99 | 253–254 | |||
| Raman spectroscopy | Noninvasive (body fluid) | Human | 3 h | n/a | Linear regression (R2=0.83) | 255 | ||
| Combinational/Other | Piezoelectric | Noninvasive (transdermal) | In vitro | n/a | Photonic crystal doped polyacrylamide- poly(ethylene glycol) hydrogel | n/a | 256 | |
| Electromagnetic | In vitro | n/a | n/s | n/a | 257 | |||
| Impedance | Human | n/a | n/a | 78.4% | 258 | |||
An alternative method for evaluating glucose concentrations involves the use of implantable microdialysis probes. Glucose in the interstitial fluid is measured by collecting dialysate.23 Microdialysis avoids direct implantation of a sensor but the glucose measurement (i.e., recovery) remains erratic in vivo due to the foreign body response.19,24–25 Even more problematic, the analysis time is significantly increased by sampling and the devices have poor time resolution.24
Iontophoresis has also been used to detect glucose in a configuration similar to microdialysis of interstitial fluid in that it is measured ex vivo. A low electrical current is applied across the skin by two adjacent electrodes.26 The resulting current causes charged species to move across the dermis through pores in the skin where minute volumes of interstitial fluid are directed to an externally located sensor that measures glucose concentrations.27–28 Iontophoretic electrochemical sensors are not invasive and experience less fouling because of the skin’s ability to filter large biomolecules. However, the continuous applied current causes skin erythema and irritation. Additionally, sweating, often associated with hypoglycemia, affects the rate of extraction of glucose and therefore causes erroneous results when readings must be most accurate.29–30 Such side effects are undesirable with the latter resulting in inaccurate measurements.
2.2. Short-term and long-term electrochemical implantable glucose sensors (fully implantable versus percutaneous)
Enzyme-immobilized amperometric biosensors have been implanted fully subcutaneously as CGM devices for extended periods (months to years) or percutaneously for <1 month. Subcutaneous glucose sensors generally consist of a disk-type sensor with a titanium housing and measure oxygen consumption (Figure 1A). Gough et al. recently described a subcutaneous sensor design based on differential electrochemical detection of oxygen via a two-step chemical reaction catalyzed by GOx and catalase.31 Accurate glucose measurements were carried out for >1 year by taking into account the difference in oxygen reduction at an electrode producing a glucose-modulated current and a reference electrode producing an oxygen-dependent current.31 Sensor response changes due to collagen encapsulation, variations in local microvascular perfusion, and limitations in oxygen availability were thus minimized, reducing the demand of device calibrations. The subcutaneous nature of the sensors likely minimized any micromotion effects associated with the foreign body response.32 Of note, implantation and subsequent replacement of the sensors required surgery, with each instance followed by a long (~2–3 week) stabilization period. Faulty devices for whatever reason would be costly to remove and/or replace. The size of the sensor is larger than percutaneous CGM systems (~3 cm versus 350 microns) due to power (i.e., battery) requirements to support longer use.
Figure 1.
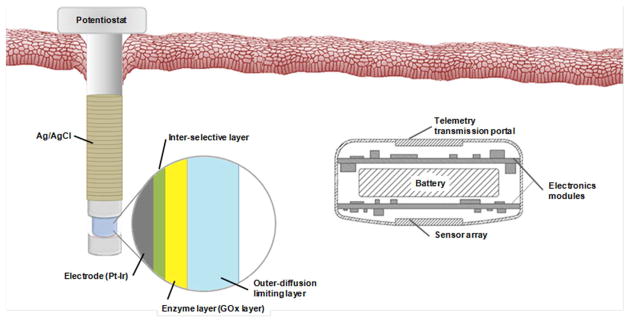
Sensor design of percutaneous (left) and fully implantable subcutaneous (right) electrochemical glucose biosensors.
Percutaneous needle-type microsensors monitor hydrogen peroxide production amperometrically as a measure of the glucose concentration (Figure 1B).22,33–34 The sensing cavity generally consists of a Pt-Ir wire working electrode coated with three functional layers: the inner selective layer, an enzyme layer, and the outer membrane. A silver/silver chloride (Ag/AgCl) wire is wrapped around the working electrode and serves as both a pseudo-reference and counter electrode. Though such sensors typically are characterized as having a shorter stabilization period (e.g., 2–4 h) compared to subcutaneous glucose sensors, the device penetrates through an opening in the dermis with concomitant infection risk. Frequent calibration (e.g., 2 times per day) is required even after the stabilization period due to changes in sensor response. Furthermore, the percutaneous nature of the device creates additional forces on the sensor that can lead to even greater inflammation.32
2.3. Glucose detection based on optical approaches or combination technologies
Almost all invasive sensing devices are characterized by infection risks, discomfort, and analytical response issues related to the foreign body response (FBR).29–30 Research has thus also focused on the development of minimally invasive and non-invasive spectroscopic methods to assay externally-accessible physiology (e.g., skin, saliva, tears).29 Glucose detection based on optical approaches are categorized as fluorophore- and non-fluorophore.19 Fluorophore-based approaches employ a spectroscopic affinity sensor wherein glucose and a fluorophore-labeled molecule bind competitively with a receptor (e.g., concanavalin A) specific to both ligands.35–36 Alternatively, only glucose binds to a recognition site (e.g., boronic acid derivatives, fluorescent moieties composite on a hydrogel) inhibiting photo-induced electron transfer (PET).37–38 The fluorescence emission intensity and/or lifetime of Förster Resonance Energy Transfer (FRET) are related to the glucose concentration.19 In this manner, Heo et al. used fluorescent hydrogel fibers (~1000 Vm dia.) to monitor interstitial glucose concentrations in a rodent model for 140 d.39 The hydrogel fibers allowed transdermal optical detection of the fluorescence intensity and were easy to remove.39 While optical detection provides sensitive detection without damaging the host, skin pigmentation and epidermal thickness can negatively impact the analytical response among hosts.35–36 Photobleaching of the fluorophore and scattering in tissue are significant drawbacks.19,35–36 Moreover, miniaturization of the instrumentation is not currently possible thus precluding personalized devices for continuous monitoring.19
Non-invasive optical techniques that avoid the use of fluorophores include optical coherence tomography, polarimetry, thermal infrared spectroscopy, photoacoustic spectroscopy, and Raman spectroscopy.19,29 Optical detection of glucose via near infrared (NIR) spectroscopy enables 90–95% of the light to pass through the human stratum corneum and epidermis into subcutaneous space with minimal tissue adsorption independent of skin pigmentation.19,40–41 The dielectric strength, polarizability and permittivity of the subcutaneous tissue adapt as a result of changes in glucose concentrations ultimately influencing the NIR absorbance, reflection, and refraction.19 Due to both a predominant absorption band in the NIR by water and light scattering in the tissue, the broader absorptivity of glucose negatively influences the sensitivity and selectivity of the measurement.42 Similarly, Raman spectroscopy has been used to determine glucose concentrations non-invasively through the measurement of inelastically scattered photons.19,30,43–44 The changes in energy from these photons is proportional to the vibrational or rotational energy of chemical bonds in the system (e.g., those associated with glucose). While Raman spectroscopy gives narrow and distinct peaks (in contrast to NIR absorbance), long spectral acquisition and stabilization times are required.30 Recent advances have demonstrated that surface enhanced Raman spectroscopy (SERS) dramatically decreases acquisition times with concomitant improvements in both sensitivity and limit of detection.45–46 Indeed, Van Duyne and coworkers demonstrated improved selectivity using SERS to measure glucose at a silver film on nanospheres (AgFON) coated with a (1-mercaptoundeca-11-yl)tri(ethylene glycol) (EG3) partitioning layer to pre-concentrate glucose within a zone of electromagnetic field enhancement.46 Clinical implementation of a SERS biosensor would require implantation of a device that could lead to photothermal damage of nearby tissue due to constant irradiation by the laser source.30 Other challenges associated with non-fluorophore-based optical detection include response effects due to motion, tissue heterogeneity, pH, and temperature resulting in poor blood glucose selectivity and accuracy.19,41,47
Impedance and electromagnetic spectroscopies represent methods other than traditional electrochemical or optical approaches that have been used to measure glucose. Impedance spectroscopy can be used to detect changes in plasma conductivity as a function of glucose concentration via the transport of alternating current through the tissue.48–49 An increase in the local glucose concentration results in decreased sodium and increased potassium concentrations in the plasma thus changing the dielectric strength, permittivity, and conductivity of the erythrocyte cell membrane.48 Unfortunately, the changes in blood dielectric properties are not specific to glucose, resulting in poor accuracy. Additionally, the temperature and disease state of the body have been shown to negatively impact such measurements.49–50
2.4. Commercially available glucose sensors
The global market for biosensors in medical diagnostics is estimated to reach $8.5 billion in 2012 and >$16 billion by 2017.51 More than 85% of the global biosensor market is captured by glucose biosensors, including CGM systems.4 The CGM device market is currently estimated at $92.2 million and forecasted to grow to a value of ~$200 million by 2017.52 Examples of commercial CGM systems are provided in Table 2. Since 1999, CGM systems have been approved by the FDA but only percutaneously implanted electrochemical glucose sensors are currently for sale in the United States.53 However, these sensors are not intended as warning or prognostic devices but rather as tools to inform users and physicians of trends in glucose fluctuations. Additionally, frequent calibration using intermittent glucose monitoring (via finger pricks) is still required due to erratic analytical performance. Due to finite FDA-regulated sensor lifetimes (5–7 d), the biosensor must be replaced on a regular basis with significant financial burden on the individual patient. In this regard, compliance regarding the use of such devices remains poor.
Table 2.
Specifications of example commercially available continuous glucose monitoring devices.
| Product | Technology and invasiveness | Implant location | Require to sensor warm up (h) | Calibration per day | Sensor life-time in vivo |
|---|---|---|---|---|---|
| iPro® Evaluation, Minimed Paradigm® Real-time Revel™ system and Guardian® Real-time CGM system (Medtronic)a | Electrochemical enzyme-based sensor (minimal invasive) | Subcutaneous abdomen | 2 | 4 | 3 d |
| Seven Plus (Dexcom)a | 2 | 2 | 7 d | ||
| Freestyle Navigator (Abott) | 10 | 4 | 5 d | ||
| GlucoDay® S (Menarini Diagnostics) | Microdialysis (minimal invasive) | 2 | 1 | 2 d | |
| GlucoWatch® Biographer (Animas) | Reverse iontophoresis (minimal invasive) | External on arm or wrist | 2 | 1 | 13 h |
| Pendra® (Pendragon Medical) | Impedance spectroscopy (noninvasive) | External on wrist | 1 | 1b | 3 d |
FDA approved and commercially available in USA
Calibration procedure required for 3 d by temperature and glucose challenge.
3. Issues in bioanalytical performance
3.1. Interferences
Although sensor accuracy is critical for compliance, many reports in the literature often neglect selectivity for glucose over interfering species during characterization.54 Indeed, a number of interferences (e.g., acetaminophen, ascorbic acid, and uric acid) can affect sensor response as they are electroactive at the electrode potential used to oxidize hydrogen peroxide. Selectivity evaluation for electrochemical CGM devices is clearly described by the Clinical and Laboratory Standards Institute (Guideline EP7-P, Interference Testing in Clinical Chemistry).54–55 As provided in Table 3, both the interfering species and its concentration are critical for full characterization.55–56 The response (i.e., signal increment) due to the interfering should be monitored in the presence of glucose at low and high glucose concentrations (4.4 and 6.7 mM, respectively).54–55
Table 3.
List of potential electrochemical interfering species problematic to amperometric enzyme-based glucose biosensors.a
| interfering species | test concentration recommended (μmol/L) | therapeutic (or biological) concentration (μmol/L)b | |
|---|---|---|---|
| exogenous drugs or drug metabolites | acetaminophen | 1324 | 66 – 200 |
| dopamine | 5.87 | 1.96 | |
| ibuprofen | 2425 | 48.5 – 340 | |
| methyldopa | 71 | 4.73 – 35.5 | |
| salicylic acid | 4340 | 720 – 2170 | |
| tetracycline | 34 | 4.5 – 11.3 | |
| tolbutamide | 2370 | 200 – 400 | |
| endogenous species | L-ascorbic acid | 170 | 23 – 85 |
| bilirubin (unconjugated) | 342 | 5 – 21 | |
| cholesterol | 13000 | 2950 – 5200 | |
| creatinine | 442 | 53 – 115 | |
| galactose | <280 | 840 | |
| triglycerides | 37000 | 340 – 3700 | |
| urea | 42900 | 1100 – 14300 | |
| uric acid | 1400 | 150 – 476 | |
Permselective membranes via size exclusion and/or electrostatic repulsion are often used to improve selectivity.4 The composition of such membranes must be accounted for when considering biocompatibility as the polymer contacts tissue and may ultimately dictate the foreign body response (FBR). The range of polymeric materials that have been evaluated as effective permselective films include cellulose acetate, Nafion, electropolymerized films (e.g., polyphenol), and multilayer hybrids of these polymers.57–58 Polyphenol permselective membranes are able to electropolymerize within an enzyme layer in a controllable manner, yielding a film with a thickness that is self-limiting (10–100 nm). As such, this simple approach is very attractive for reducing interferences.59–60 In some cases, such membranes also exclude surface-active macromolecules (i.e., proteins and platelets), protecting the surface from biofouling.61
As described earlier, the use of mediators to shuttle electrons between the enzyme and the electrode can also minimize the impact of interfering species by lowering the working potential required to oxidize hydrogen peroxide.56,58,62 Several redox mediators have been evaluated including ferrocene and osmium complexes, quinone compounds, metal phthalocyanines, carbon nanotubes, and conducting polymers.61
3.2. Oxygen dependence
The electrochemical detection of hydrogen peroxide requires oxygen, a cofactor in the GOx enzymatic reaction. Oxygen concentration in interstitial fluid is approximately ten times lower than the concentration of glucose in interstitial fluid resulting in an “oxygen deficit” state that is addressed by an outer diffusion-controlled membrane. Indeed, low concentrations of oxygen lead to problems with the biosensor response to glucose (particularly dynamic range) due to stoichiometric imbalance between the two cofactors.19,63 Oxygen deficiency is mitigated by using polymeric membranes that reduce glucose diffusion64 or employ alternative electron mediators (as noted above).5,56,65 Often, membranes similar to those that exclude polar interferences are employed to increase the ratio of oxygen/glucose permeability.4. Example polymers include polyurethane, Nafion, silicone elastomer, polycarbonate, and layer-by-layer assembled polyelectrolytes.66–69
Subcutaneous oxygen tension for humans ranges from 40 mm Hg (~5 kPa) to 130 mm Hg (~17kPa) with partial pressures affected by anesthetics and hypoxia at the implantation site.70 Sensor performance issues due to changing oxygen levels are exacerbated due to the foreign body response that results in local consumption of oxygen and glucose by inflammatory cells at the vicinity of the sensor. Of note, the oxygen diffusion to the sensor decays exponentially after sensor implantation due to changes in tissue permeability.31
3.3. Stability and degradation of sensor components
The failure of sensor components in vivo may be categorized as follows: 1) enzyme instability and leaching; 2) membrane degradation and delamination; and, 3) electrode passivation. Enzyme activity begins to decrease immediately both due to polymer entrapment and exposure to reactive oxidative species from sensor operation and the FBR (exposure to hydrogen peroxide and other reactive radicals).71–72 Considerable effort has been devoted to developing effective immobilization strategies to ensure enzyme stability. Examples of such strategies include crosslinking of the enzyme with bovine serum albumin (BSA) with glutaraldehyde, enzyme entrapment with or without covalent tethering within polymeric matrixes (e.g., hydrogels and sol-gel-derived materials), incorporation of the enzyme into electropolymerized conducting polymers such as polypyrrole, and fixation of the enzyme onto the electrode surface by electrostatic interactions generated by polyelectrolytes.56 Nevertheless, even properly immobilized enzymes inherently lose activity over time due primarily to loss of non-covalently bonded FAD cofactor. However, deactivation by endogenously produced hydrogen peroxide from oxidase reactions also contributes to the loss of activity.71 Large glucose concentrations and requirements for adequate sensor signals imply high rates of peroxide production and concomitant enzyme deactivation.
Almost all sensors to date consist of films or membranes used as sensing layers, barrier membranes, and/or biocompatible layers. These materials are prone to degradation from oxidative challenges (i.e., foreign body response), calcification, and delamination.73 When a film becomes detached or degrades, sensor instability or failure automatically results. Electrode fouling (often called electrode passivation) is another cause of sensor instability, and occurs when diffusible small molecules come into contact with the surface of the electrode after penetration of the sensor membrane.74
3.4. In vivo calibration
Since the analytical performance of CGM sensors changes drastically upon implantation, methods for defining and assessing sensor accuracy are critical for clinical use. Traditionally, the in vivo accuracy of such devices is evaluated using numerical point or rate accuracy.75 Current numerical and clinical accuracy criteria for CGM includes linear regressions and correlation coefficients; mean (or median) absolute and relative absolute difference (MAD and MARD); Clarke Error Grid Analysis (Clarke EGA); and, International Standard Organization (ISO) criteria.76 Because CGM systems also provide information on glucose fluctuations, the continuous glucose-error grid (CG-EGA) has been introduced comprising of (1) a point-error grid analysis (P-EGA) that evaluates the sensor accuracy in terms of accurate blood glucose measurements; and, (2) a rate-error grid analysis (R-EGA) that assesses the prediction capability of the sensor.77 These in vivo sensor evaluation methods require true blood glucose concentrations as determined using an external glucose measuring device (i.e., finger-prick glucose sensor). Reliable and reproducible procedures for calibration during in vivo monitoring are crucial to achieving accurate measurements. Of importance, CGM systems inherently estimate the blood glucose concentration by assuming the concentration of glucose in interstitial fluids will be similar enough. This assumption is problematic because the ratio of blood/tissue glucose is not constant, but rather depends on the metabolic rates related to glucose and insulin physiology including glucose uptake by cells or from blood vessels, blood flow, and permeability of capillaries.78 The glucose concentration discrepancies between blood and interstitial fluid are typically complex and vary based on time and concentration according to the physical state of the patient, including resting, hyperventilation, exercise, anoxia, and hypoxia.19,79 The lag time between blood and subcutaneous tissue glucose concentrations cause further inaccuracies for CGM devices.79–80 Under normal conditions (i.e., conditions in which glucose levels are not rapidly changing from activities such as exercising or eating), the physiological lag time between blood and interstitial fluid glucose ranges between 5 and 10 min.78,81 Longer, or unpredictable, lag times are created by physiological differences between individuals, intrinsic sensor lag time (typically on the order of seconds to a few minutes), and noise filtering. Lag is also created by tissue responses to the sensor such as electrode fouling, biofouling, and the foreign body encapsulation that impedes glucose diffusion to the sensor.5–6 Again, frequent calibrations using external glucose measuring devices are required to ensure CGM sensor accuracy.
Both “one-point” and “two-point” calibration procedures with blood glucose strips have been used to calibrate CGM sensors.80 The calibration process involves the conversion of the time-dependent current signal (i(t)) into an estimation of blood glucose concentration at a given time (CG(t)). Using the one-point calibration procedure, sensor sensitivity (S) is determined as the ratio between the current signal and the blood glucose concentration from a single blood glucose determination. This approach is useful for highly selective sensors with near-zero output current at zero glucose concentration. A two-point calibration procedure is preferred when the sensor output observed in the absence of glucose (i0) is not negligible. Two-point calibrations involve an estimate of two parameters, S and i0, by determining blood glucose concentration and concomitant sensor current at two different time points. The glucose concentration is then estimated from the response current according to eq. 1. The two-point calibration curve is
| (1) |
and is actually less accurate due to error associated with electronic noise and the “true” finger prick blood glucose measurement (accepted as ±10% error on commercial glucose meters) that results in significant positive or negative measurement artifacts.80,82 A one-point calibration is thus considered more appropriate. Even with an accurate calibration, repeated calibration is required as the sensor sensitivity changes over time due to physiological fluctuations and the foreign body response to the sensor.
3.5. Physiological fluctuations and sensor performance
In addition to the inherent sensor fabrication and analytical measurement challenges, patient variability is an equally important factor for CGM device utility. Since percutaneous sensors are implanted through the epidermis, dermis, and subcutaneous layers, the effects of tissue heterogeneity are relevant to the analytical performance of the sensor. Temporal and spatial glucose dynamics are both influenced by tissue composition, distribution, and thickness.47,83–84 Temperature fluctuations in subcutaneous tissue will also impact sensor performance by altering glucose oxidase activity.61
External pressure on the CGM sensor has also become a valid threat to analytical performance. For example, Gilligan et al. found that long-term implanted glucose sensors in humans provided accurate sensor readings while immobile, but posture changes (i.e., standing and/or moving) led to erratic outputs.85 Such behavior was attributed in part to blood occlusion due to pressures applied to tissue adjacent to the sensor.86–87 This performance limitation may be more pronounced in diabetic patients due to differences in physiology. Pressures as low as 2 kPa have been shown to diminish blood flow near the skin while only 7 kPa completely occluded blood circulation in diabetic patients whereas non-diabetics withstood pressures up to 22 kPa before total blood occlusion.88 These fluctuations and their possible impact on interstitial fluid physiology demand careful attention when characterizing in vivo sensor performance.
4. Consequences of the foreign body response on in vivo glucose sensor performance
While the above design, fabrication, and use criteria are of importance to CGM device use, none impacts the bioanalytical and clinical utility as much as the foreign body response (FBR). Most long-term medical implants to date are considered biocompatible or inert once the FBR resolves and the device is encapsulated by a collagen layer.89 Although CGM devices have a similar fate, the combination of the FBR, oxygen and glucose availability, and need for immediate calibration and use (e.g., in the physician’s office), have significantly impeded their utility and implementation as effective devices for diabetes management.
As shown in Figure 2, the FBR initiates upon the insertion of almost any material into subcutaneous tissue, starting with the creation of a wound and the wound healing cascade.90–91 Instantaneously, proteins adhere to the biomaterial surface in a process referred to as biofouling.91–92 The initial protein adsorption is an integral part of the overall FBR as the ensuing interface promotes the adhesion of inflammatory cells that subsequently stimulate blood clotting and the development of a provisional matrix.92 As part of the FBR, macrophages, monocytes, mast cells, and fibroblasts are recruited to the implant site to initiate clearance of the foreign body by releasing chemokines and cytokines.90–91 The concentrations and types of mediators released elicit further cell recruitment and ultimately phagocytosis93 as the body attempts to digest the implant. This process can result in a local pH’s dropping as low as 3.6 and disrupting biosensor performance as the activity of GOx is pH dependent.94 While preventing all macrophage migration and subsequent phagocytosis at a wound (glucose sensor) is unlikely, the activation state (i.e., M1 or M2) of the macrophage may influence the overall FBR. Indeed, macrophages serve three primary functions in the body: host defense, wound healing, and immune regulation.95 Since macrophages are vital for the wound-healing process that results from an injury, the phenotype of the cells present at an implant, rather than their concentration, is now believed to be a better indicator of tissue response.96 As the FBR progresses, frustrated phagocytosis from activated macrophages will lead to the fusion of macrophages into foreign body giant cells (FBGCs) that attempt to further breakdown the implant.73,94,97–98 For example, FBGC formation on polyurethanes has been shown to promote cracking of the underlying biomaterial.73 After one to two weeks, inflammatory cells deposit a collagen matrix that sequesters the implant from the native tissue. This collagen encapsulation lacks the microvasculature of native tissue.99 As blood vessels are the primary source of glucose, such encapsulation hinders accurate measurements of blood glucose. The extent of capsule development is dependent on all other preceding components of the FBR, including protein adhesion, cell activation, and cytokine signaling. The collagen encapsulation will persist for the lifetime of the device, negatively impacting sensor performance with respect to sensitivity and response (e.g., lag times).
Figure 2.
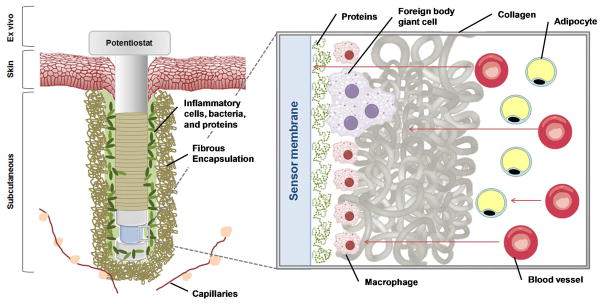
The foreign body response to a percutaneous glucose sensor upon tissue implantation. Arrows in the magnified area show diffusion of glucose from blood vessels towards the sensor through native tissue (e.g., adipocytes), the collagen capsule, localized inflammatory cells, and biofouling layer near the sensor surface. As illustrated, glucose may be consumed by native tissue or inflammatory cells prior to reaching the sensor.
While the individual effects of each step in the FBR on glucose sensor performance have been postulated, actual outcomes are more difficult to determine. Researchers have long sought to untangle the complexities that connect various events in the FBR with tissue integration and glucose sensor performance, as this knowledge could lead to the development of materials that address the specific tissue responses that most severely inhibit sensor performance.
The synthesis of antifouling materials has evolved to be a common strategy for improving glucose sensor functionality as the initial adhesion of proteins and cells onto a glucose sensor dampens sensor performance.100–101 Among the earliest reports describing reduced analytical performance, Thomé-Duret et al. implanted polyurethane-coated glucose biosensors to quantify changes in analytical sensitivity.100 Soon after implantation, the sensors were explanted and tested ex vivo.100 While the immediately explanted sensors had glucose sensitivities similar to those analyzed in vivo, response to glucose improved after rinsing, albeit not to pre-implantation levels.100 Nevertheless, this reversibility indicated that the process was passive and likely caused by biofouling on the sensor.100 Proteomic analysis revealed that the majority of the biofouling proteins on the sensor membrane were fragments <15 kDa.102–103 In contrast, Wisniewski et al. evaluated the impact of collagen encapsulation versus biofouling using microdialysis probes and found biofouling effects to actually be minimal.104 Specifically, the resistance to mass transfer of analyte (i.e., glucose) caused by the tissue over both short- (3 h) and long-term (8 d) implantation periods were typically 3 to 5 times greater than that caused by biofouling of the probes, regardless of implantation period.104 The observed biofouling was found to have only relatively small direct effects on the overall resistance and glucose extraction efficiency.104 It is important to note however that biofouling of proteins and cells at the sensor-tissue interface will also affect the tissue response to a biomaterial as will the composition of the polymers used to fabricate the sensors and dialysis probes. Furthermore, microdialysis probes consume significantly more glucose than electrochemical glucose sensors and therefore may underestimate the effects of biofouling in vivo.105
Following protein adhesion/biofouling, the FBR proceed with inflammatory cells responding to the injury, initiating a more profound immune response to the device. Klueh et al. reported on the effects of mast cells, regulators of inflammation.106 Both mast cell-sufficient and -deficient mice were implanted with subcutaneous glucose sensors for 28 days. During this period, glucose sensor performance in mast cell-sufficient mice was erratic with temporary response loss occurring within the first 3 weeks. Sensor performance in mast cell-deficient mice was markedly better with reliable sensor function throughout the 28-day period.106 Histology samples from both experimental groups confirmed that the mast cell-deficient mice exhibited reduced fibrosis and inflammation at the implantation site. To further confirm the effect of mast cells on glucose sensor performance, 104–105 mast cells were injected at the implant site.106 While glucose sensor performance recovered soon after injection (~15 min), the sensor response to glucose decreased after 1–2 days, further indicating a link between mast cell action and erratic glucose sensor performance.106
The most characteristic outcome of the FBR is collagen encapsulation around the foreign device. Early investigations of capsules formed around sensors focused on the influence of the capsule on glucose diffusion from native tissue. For example, Sharkawy and coworkers implanted non-porous polyvinyl alcohol (PVA) and stainless steel cages into the subcutaneous tissue of rats.107 Upon careful explantation of the collagen capsules, the diffusion of sodium fluorescein (376 g/mol) through the capsule was quantified and used to model small analyte transport.107 Diffusion of the fluorescein through the explanted capsules was ~50% that of normal (i.e., subcutaneous) tissue.107 Sensor lag times for native tissue was estimated at ~20 min, but tripled when modeled with decreased diffusion due to a capsule.107 Interestingly, empirical data suggests that lag times range from 10–15 min in vivo, indicating the integral function of angiogenesis in early granulation tissue. In subsequent experiments, Dungel et al. examined the effects of encapsulation on sensitivity by evaluation the response of glucose sensors inserted into polyvinyl alcohol sponges implanted in rats.108 The glucose sensitivity in vivo peaked at day 7, but then decreased for the duration of the study.108 Glucose sensitivity correlated well with the collagen encapsulation of the sponges with thicker collagen resulting in greater sensitivity loss.108 Koschwanez et al. investigated the effects of vascularity on glucose sensor performance in real-time by using an implanted optical window over the sensor along with microscopy, and laser Doppler flowmetry.109 The vessel length and perfusion of the vasculature increased during the implantation period (i.e., 14 d). Despite such increases in vasculature, the sensitivity of the sensor did not increase indicating that angiogenesis is not the only factor in assessing the biocompatibility of CGM sensors.109
Mathematical models and simulations of implantable glucose sensors have further helped understand how the processes of the FBR may affect glucose sensor performance. Simulations of glucose concentration oscillations by Jablecki and Gough concluded that increases in mass transfer would increase lag and could potentially decrease the magnitude and differences when fluctuating between high and low glucose sensor signals.110 While this conclusion is important for sensor design, the increases to mass transfer were not attributed to any specific part of the FBR and could originate from collagen capsule thickness, blood vessel density, or other unanticipated factors. To consider major tissue reactions individually, Novak and coworkers used a mathematical model to examine FBR effects on glucose sensor performance.111 Using previous histology data, five parameters (i.e., angiogenesis, cellular glucose consumption, capsule thickness, capsule diffusion coefficient, and capsule porosity) were used to design a mathematical model that mimicked glucose diffusion from capillaries to a glucose sensor in order to simulate the effect on sensor lag time and attenuation.111 The mathematical model treated vessels as sources of glucose and inflammatory cells as glucose sinks, while the encapsulation properties acted to impede the diffusion of glucose.111 Changes in cellular glucose uptake and the capsule diffusion coefficient of glucose had little effect on simulated sensor performance.111 The model was ultimately used to conclude that collagen capsule thickness, a common histological parameter, was the primary source of sensor lag time with little impact on sensor response attenuation.111 The positive correlation between lag time and capsule thickness supported the models described above.107,110–111 The two greatest factors in reducing sensor attenuation were a low capsule density and high degree of angiogenesis.111 While these are only mathematical models, the results do identify the histological parameters of key interest when assessing the biocompatibility of materials for glucose sensors.
5. Mitigating the foreign body response to improve analytical performance of in vivo glucose sensors
As the events in the FBR directly impact the utility of CGM devices, researchers have focused on improving the biocompatibility of the device as a strategy to improve sensor performance. These strategies range from chemical alteration at the tissue-sensor interface, changing physical properties of the device, and the release of biologically active molecules to influence the tissue reaction.
5.1. Models for assessing biocompatibility
Both in vitro and in vivo models are employed when developing implantable sensors. Although in vitro tests are less expensive and simpler to interpret, particularly for preliminary testing, their outcomes are not an accurate predictor of biocompatibility or glucose sensor performance in vivo.70,112 Indeed, testing in animal models is necessary to evaluate the intricate tissue reactions that confront a proposed biomaterial and fully represent FBR complexity. Most commonly, the tissue adjacent to the implanted biomaterial is excised and processed to analyze the collagen capsule, inflammatory response (i.e., cell amounts and types), and presence of capillaries. Histological analysis offers a snapshot of the relevant tissue reactions but requires an animal for each time point. As such, most initial testing occurs with small rodent models.
Methods have also been implemented for temporal monitoring of a single implant. First described by Marchant et al., the cage implant allows for the examination of the inflammatory response to a substrate of interest over time.113 The technique involves the implantation of a potential biomaterial inside a stainless steel cylinder (approximately 1 cm diameter by 3.5–4 cm when implanted into rats). Over time, exudates are removed from the cage and analyzed for inflammatory cells and cytokine expression.113–114 At the end of the experiment, the material is explanted and examined for the formation of FBGCs. Though this method is useful for temporal studies of the inflammatory response, it does not offer a complete analysis of the FBR as no information is gained with respect to collagen capsule formation, capillary density in the surrounding tissue, or sensor performance. Furthermore, as the materials are not directly contacting the native surrounding tissue, the specific inflammatory reactions observed are dependent only on cells in the exudates. In addition, the physical properties of the implant (e.g., size) impact the FBR, and the large size of the stainless steel cage will influence the overall immune response to the materials.115
Optical tissue windows have been implemented with implantable sensors as a method to visualize the FBR in real-time.109,116–118 The optical tissue window enables tissue monitoring and correlation to the sensor performance.116–118 Anesthesia is not required to observe the tissue in real-time, an important consideration for glucose sensor performance correlation as anesthesia can alter microvasculature circulation.116 The real-time tissue observation allows for a relation of angiogenesis and perfusion of the surrounding vasculature to the glucose sensor function. However, this technique is highly invasive involving implantation of additional materials (i.e., an optical window) that inherently elicits an additional tissue reaction with influence on the overall FBR to the closely placed biomaterial. As such, it is likely not an accurate characterization of sensor performance or the FBR.
5.1.1 Animal and diabetic models
When implanting in vivo, researchers often use rodent models (e.g., mouse119 or rat120–121) models to assess in vivo biocompatibility and/or glucose sensor functionality, though others (e.g., pigs,31 dogs,122 and chimpanzees123) are used as well. Such animal models are time and labor intensive while requiring surgical skills. Although an avian chorioallantoic membrane has been proposed as an alternative model for testing biomaterials with obvious expense benefits, long-term studies (i.e., >2 weeks) are not feasible.124–125
Unfortunately, the degree to which animal models accurately predict the FBR and analytical performance of glucose sensors in humans has come under scrutiny. For example, Wisniewski et al. compared the dialysate concentrations of glucose, pyruvate, lactate, glycerol, and urea at a microdialysis probe-tissue interface implanted in the subcutaneous space of both rats and humans to evaluate the validity of the cross-species relationship.126 The dialysate concentrations were significantly different for all metabolites in rats and humans throughout the first 6 days of the experiment, with the exception of glycerol on day 0.126 Additionally, the ratio of the glucose concentration in the dialysate to that in blood were significantly different between rats and humans indicating differences in the relative glucose availability in the tissue surrounding an implanted device.126 Histology samplings from human subcutaneous tissue showed a high concentration of adipose cells while rat tissue was characterized with larger amounts of collagen.126 Taken together, these results necessitate careful consideration and caution when extrapolating results from animal models to humans.
In addition to differences between species, one parameter that is not often considered is the differential healing response in diabetic versus healthy (i.e., non-diabetic) patients. Diabetics typically suffer from delayed and diminished wound healing.127 It is currently not known how such physiology affects the FBR and subsequent sensor performance. In a key study, Gerrtisen et al. investigated the differences in the FBR between diabetic and non-diabetic rabbits to percutaneous and subcutaneous materials.128 Histological analysis revealed delayed neovascularization and less matrix production in the diabetic rabbits that clearly could influence glucose sensor performance.128 Due to such important histological differences that likely impact glucose sensor performance, diabetic animals should be used to better simulate tissue response and characteristics of diabetic patients. However, the wide range of available diabetic models in animals (e.g., genetic or chemically induced) may further complicate comparisons between studies.129
5.2. Effects of sensor geometry and physical stress
Classically, the physiological response to a material as a function of mechanical and chemical properties has been the most studied parameter for understanding and improving biocompatibility. However, even more inherent variables influence the FBR. Glucose sensors have traditionally employed microsensors due to many practical concerns (e.g., facile implantation), but also a less significant FBR as a result of their smaller size. In an important study, Ward et al. implanted polyurethane substrates that were either 300 or 2000 Vm thick into the subcutaneous tissue of rats for 7 weeks.115 Subsequent histological analysis revealed that the capsule thickness was ~20% thinner surrounding the smaller implant. Sensor geometry plays an equally important role with respect to biocompatibility. To highlight these effects, Li and associates implanted rectangular substrates into rat subcutaneous tissue and examined the capsule formation at periods up to 20 months.130 At 20 months, thinner capsules were observed at the implant’s corners while the face of the implant closest to the skin had a thicker capsule than the opposite side.130 Although sensors are typically not rectangular, this study demonstrates that sensor geometry may be an important factor in determining biocompatibility.
Physical stress imposed by the CGM device represents another factor that contributes to the FBR. A recent two-part review by Helton and coworkers surveyed reports about physical stress and related implications on the FBR and glucose sensor performance.32,131 Picha et al. found that implant location within the subcutaneous tissue affects the FBR.132 Textured and non-textured surfaces were implanted at six different positions in the dorsal rat subcutaneous space.132 While textured surfaces elicited a more favorable FBR, much of the response that did occur was attributed to differential sheer stress caused by the interaction between the textured surface of the device and the surrounding fatty tissue.132 However, other factors such as localized tissue differences (e.g., adjacent joints that may create physical stress), may have affected the overall healing response. Another specific concern in examining physical forces is the difference between percutaneous and purely subcutaneous implants. Most preliminary testing of materials is done subcutaneously for simplicity, but these devices are intended to be implanted percutaneously and as such additional external forces may cause differential healing. A study by Koschwanez et al., in which the histology adjacent to glucose sensor membrane materials implanted either subcutaneously or percutaneously were examined supports this theory.121 Subcutaneous histology of a porous poly-L-lactic acid (PLLA) coating showed a threefold increase in vascularization and a ~65% reduction in collagen compared to bare glucose sensors at 3 weeks.121 However, when the same materials were implanted percutaneously, histological differences were less apparent, with porous coatings giving a twofold increase in vascularization and a twofold increase, rather than decrease, in collagen content compared to bare sensors at 2 weeks.121 While examination of the subcutaneous implant noted no blood vessels or collagen within the interior of the porous material, the percutaneous experiments revealed significant levels of collagen and vascularization, indicating a differential FBR dependent on the nature (i.e., purely subcutaneous vs. percutaneous) of the implant.121 In a follow-up study, Koschwanez et al. compared capillary density adjacent to subcutaneous and percutaneous materials using an optical window model in rat tissue.109 At 10 and 14 d post-implantation, the vessel lengths and blood perfusion of the porous materials was significantly greater than bare (i.e., non-porous) sensor coatings when implanted subcutaneously. Despite these observations, the same materials implanted percutaneously resulted in no observable differences. Furthermore, the capillary density near the bare (i.e., non-porous) materials was twofold greater for the percutaneous compared to subcutaneous devices indicating that motion effects may stimulate further tissue responses to implanted materials.109 It was hypothesized that the observed differences were caused by the additional physical forces imposed by micromotion at the percutaneous devices.
In addition to the FBR, other biological events may negatively influence sensor performance, including injuries caused by movement of the implanted material. For example, localized bleeding from damaged capillaries is a proposed source of seemingly random and short-term failures of in vivo glucose sensors. To examine this hypothesis, Klueh et al. injected whole and heparanized blood at the site of a glucose sensor and examined the subsequent analytical performance.133 As anticipated, the injection of blood, but not saline or plasma, led to a temporary (i.e., several hours) signal reduction that eventually returned to normal.133 This reduction was explained by the clot formation around the sensing portion of the CGM device that formed a glucose sink until clearance.133 Therefore, temporary signal reductions may actually be the result of local tissue damage caused by forces exerted on the sensor and may be solved by using more flexible materials.
While practical considerations regarding the size, shape, and implant site location may have a profound impact on the FBR, these universal concerns should not preclude the development of more biocompatible sensor coatings at the interfacial level. Rather, these factors must be explored alongside the development of improved coatings. Below, the design and testing of the newest materials that may mitigate the host immune response and improve the utility of the next generation CGM devices are discussed.
5.3. Polymer coatings
It is well known that certain materials elicit a more favorable FBR than others, and such materials are often chosen for use in implant design and fabrication. Polymer coatings are necessary for glucose sensors as they reduce the diffusion of interferences to the sensor while simultaneously balancing glucose and oxygen diffusion to enable an adequate glucose response, (i.e., dynamic range). These materials are also relevant as biomaterials as they are durable, inert, and capable of sustaining harsh environments produced by the FBR. Of the polymers commonly evaluated, Nafion, polyurethane, polyethylene glycol (PEG), and hydrogels have been implemented successfully as glucose sensor membranes for subcutaneous tissue use.
Nafion is a perfluorosulfonic acid-based polymer that has been implemented as a biocompatible coating.134 With respect to glucose sensing, Moussy et al. implanted Nafion-coated CGM probes into canines that proved functional for up 10 days, but ultimately the membranes were deemed unstable with significant cracking and eventual failure.122 Later studies by Mercado and Moussy demonstrated that this breakdown was likely caused by mineralization of the membrane.135
Polyurethane (PU) has been used extensively as an outer membrane to act as a biocompatible interface with the surrounding host tissue. Early studies by Zhang and Wilson evaluated the in vitro and in vivo performance of PU as an outer membrane, exhibiting its potential as a material for glucose sensor fabrication and use.136 Biocompatibility aside, PU allows for sufficient oxygen transport while limiting glucose diffusion to the sensor.136 Ward et al. examined the in vivo performance of Tecoflex SG-85A PU membranes manually loop coated onto needle-type glucose sensors in rats after 30 h of implantation.137 The Tecoflex SG-85A membrane provided the biocompatibility often observed with polyurethane while also ensuring ideal sensor performance by limiting glucose diffusion.34 The sensors were found to accurately track the glucose with relatively short lag times (~4.5 min). Several groups have found that copolymer blends with PU leads to both enhanced stability and biocompatibility. Anderson and coworkers investigated the host response to PU using the cage implant system.138–139 In long-term studies (i.e., 10 weeks), biodegradation of PU was slower for polycarbonate PUs, containing a soft segment of polyhexamethylene carbonate, compared to polyether Pus that have a soft segment composed of polytetramethylene oxide.138 However, no significant differences in cell adhesion or FBGC formation were noted. To investigate the response in situ, Ward and coworkers implanted PU and PU with silicone and polyethylene oxide (PU-S-PEO) into the subcutaneous tissue of a rat for periods up to 7 weeks.115 The collagen encapsulation surrounding the PU-S-PEO substrates was ~45% thinner than those around the PU substrates though no differences in capillary density were observed.115 In vitro examination of silicone-modified PU was also found to significantly inhibit adhesion and FBGC formation, possibly explaining the in vivo benefits observed.140 Yu et al. used differential polyurethane chemistry to create epoxy-polyurethane membranes in an effort to enhance the biocompatibility of implantable glucose sensors.141 The membranes were applied to the glucose sensors by simply casting the solution over the GOx layer. The authors reported that this membrane allowed for the fabrication of functional glucose sensors in rats for up to 56 days with good sensitivity. However, only one of the nine sensors functioned this long. Indeed, seven of the nine sensors lost functionality in 17 d or less.141
Surface passivation with polyethylene glycol (PEG) has been a widely studied strategy for resisting biofouling.142 With respect to tissue FBR, Quinn and coworkers created co-polymers containing 2-hydroxyethyl methacrylate (HEMA), PEG, and ethylene dimethacrylate and investigated the adsorption to the materials in rat subcutaneous tissue after 3 days.143 Scanning electron microscopy analysis of the implant after explantation revealed that substrates with the hydrogel coating had markedly less fibrous encapsulation relative to controls.143 Subsequent studies of PEG-contatining hydrogels revealed similar beneficial effects at 7 d.144 While Pellethane (i.e., polyurethane) control substrates exhibited significant inflammatory cell adherence and collagen encapsulation, PEG-modified hydrogels were characterized by less cells indicating improved biocompatibility.144
Even without PEG modifications, hydrogels themselves represent an attractive material for fabricating glucose biosensors. Hydrogels have a modulus similar to subcutaneous tissue and absorb water readily allowing easy diffusion of analytes to a sensor.145–146 These properties alone have been shown to reduce fibrous encapsulation in tissue.15,147–148 Related to glucose sensor performance, Yu et al. studied the benefits of hydrogel sensor membranes on implantable glucose sensors.147 Hydrogel-coated biosensors were still functional after 21 days whereas only 13.5% of the controls were still functional.147 Their longevity was attributed to reduced fibrous encapsulation and immune cells adjacent to the hydrogel-coated as revealed by histological analysis of the surrounding tissue.147 Even with these benefits, the accuracy was not examined and it is unclear what impact the hydrogel coating would provide in this respect. Wang and coworkers fabricated similar hydrogel-based glucose sensors and evaluated their biocompatibility in rats.15 Control sensor membranes (epoxy-polyurethane) at 28 days were characterized with a fibrous capsule of ~500 Vm while the capsules around hydrogel-based sensors were roughly 30–60 Vm.15 Ju and associates investigated the use of a collagen hydrogel-formulation as a glucose sensor membrane.148–149 These scaffolds were made using a freeze-drying method in which collagen was dissolved into 3% (v/v) acetic acid, inserted into a polypropylene mold and subsequently freeze-dried. To reduce collaganse degradation in vivo, the hydrogels were crosslinked with nordihydroguaiaretic acid (NDGA) or glutaraldehyde (GA).149 Sensors fabricated with the collagen hydrogels were then implanted into rat tissue for 28 days.148 Histological analysis after the experiment revealed improved biocompatibility of the NDGA-crosslinked compared to the GA-cross-linked scaffolds as evidenced by less inflammatory cell localization and infiltration into the porous membranes. Despite the observed histological benefits, the analytical performance of control sensors (i.e., lacking collagen) was better than that of the collagen-based sensors.148
These results suggest that chemical modifications may improve tissue biocompatibility, but ultimately they have not led to major improvements on sensor performance in vivo. As with previous passivation strategies for blood applications, the body ultimately overwhelms the interface with protein and cell adsorption, resulting in an equally non-native surface.
5.4. Porosity
While chemical surface modification represents one strategy for curtailing the effects of the FBR, more recent work has utilized an implant’s architectural and mechanical properties to control the response.150–151 Although the earliest report of a porous material’s influence on the FBR dates back to 1973,152 several key papers emerged in the mid-1990’s that studied this phenomenon more thoroughly.107,153–155 Brauker et al. implanted polytetrafluoroethylene (PTFE) membranes fabricated with mean pore sizes of either 0.02 or 5.0 μm into the subcutaneous tissue of rats.155 Membranes fabricated with the larger pores were characterized by 80–100x greater vascularization in the tissue surrounding the implant. The membranes with the larger pores also showed increased vascularization at the one year mark. When evaluating other polymeric materials having mean pore sizes from 0.22 to 8.0 μm, the authors observed that this increased vascularization correlated with the ability of inflammatory cells to infiltrate the porous membrane.
A series of studies by Sharkawy and coworkers expanded on Brauker’s work by examining the utility of porous materials for glucose sensor applications.107,153–154 Polyvinyl alcohol (PVA) sponges with mean pore sizes of 60 and 350 μm were implanted into subcutaneous tissue in a rat model for 4 weeks. As hypothesized, the tissue around the porous materials exhibited increased vascularization and decreased collagen capsule thickness.107 To determine the implications of these findings for subcutaneous glucose biosensors, the diffusion coefficient of fluorescein through the surrounding explanted tissue was measured and determined to be approximately two times greater adjacent to porous versus non-porous PVA. Furthermore, the diffusion coefficient of the probe through tissue surrounding porous PVA was statistically equivalent to that of native subcutaneous tissue. To determine the effects of pore sizes on the tissue vasculature adjacent to the implant, PVA sponges with mean pore sizes of 0, 5, 60, or 700 μm were implanted in a similar manner for 3–4 months.153 The observed vascular density adjacent to the porous PVA samples was increased relative to controls, with the 60 μm pores exhibiting the greatest vascular density, again indicating that a pore size on the order of cellular dimensions was optimal for minimizing the FBR.
While Sharkawy’s work revealed the underlying potential of porous materials for improving analyte diffusion to the implant, only more recently have several researchers begun to evaluate the impact of these porous materials on the analytical performance of functional glucose sensors.108–109,121,156 Koschwanez et al. explored the FBR to porous poly-L-lactic acid (PLLA) foam coatings when implanted subcutaneously and percutaneously as part of non-functional and functional glucose biosensors, respectively.121 Amperometric glucose biosensors were inserted into foam sleeves synthesized via a gas foaming/salt leaching method with ammonium bicarbonate as the porogen. When coated onto non-functioning sensors and implanted subcutaneously, the PLLA foams exhibited the expected decrease in collagen capsule thickness and increased capillary density compared to bare non-functional sensors. However, coatings on percutaneous sensors did not lead to similar histological or sensor performance improvements versus control (i.e., bare) sensors. The difference in performance between the two implant modes was attributed by the authors to micromotion. These effects overrode the benefits of porosity due to the continual movement of the percutaneously implanted sensor in the tissue. Thus, incorporating strategies that reduced the effects of micromotion may be critical to fully realize the potential of porous membranes on percutaneous sensors.
Fully subcutaneous glucose biosensors have also been coated with porous membranes in long-term monitoring studies.31,85,156 For example, Updike and coworkers implanted biosensors with an affixed expanded PTFE (ePTFE) membrane having 1–10 μm pores in non-diabetic canines and found they were operable for greater than 160 d with a best-case recalibration interval of 20 d. In contrast, sensors lacking the ePTFE membrane (i.e., having only an underlying layer of unspecified composition) were functional for approximately 94 d with a best-case recalibration interval of 18 d.156 Following subcutaneous implantation into humans, one of five biosensors functioned for ~6 months with ≥90% of all measured glucose concentrations falling within the A and B regions of the resulting Clarke Error Grid (i.e., clinically accurate and acceptable, respectively). However, four of the five sensors failed to produce analytically useful data due to either electronic failure or a lack of integration into the surrounding tissue.85 Further work will need to focus on designing devices that are more robust.
The porous materials described above (i.e., polymer foams and salt-templated polymers) are generally characterized as having broad distributions in pore size. As such, details about the specific pore sizes needed for optimum tissue integration are lacking.. To remedy this, Ratner and coworkers have fabricated porous materials with high precision both in terms of size (±5%) and connectivity.157–158 Specifically, monodisperse poly(methyl methacrylate) (PMMA) beads are used to template materials such as hydrogels (e.g. poly (2-hydroxy)ethyl methacrylate), silicone elastomers, and fibrin.159–160 The template framework is constructed by sieving and shaking the beads, followed by a sintering step that fuses the beads together. Once the polymer is added and the template is removed, well-defined and interconnected pores remain in the space once occupied by the beads. When implanted subcutaneously, materials with 35 μm pores were shown to yield the thinnest and lowest density collagen capsules with a high degree of capillaries in the tissue. Conversely, tissue surrounding materials with either larger or smaller pores were avascular with thicker, denser collagen capsules.157 Based on the results of this study, the pro-angiogenic properties reported previously with either smaller or larger pore-sized materials might be the consequence of their polydispersity. Ratner has hypothesized that the 35 Vm pores force macrophages into a reconstructive phenotype by allowing them to insert into the pores, but not spread or phagocytose the material.158 Consistent with this hypothesis, poly(2-hydroxyethylmethacrylate-co-methacrylic acid) implants with 30–40 μm pores were reported to increase vascularization and reduce capsule thickness when placed into the myocardium of rats over a 4-week period.160 Moreover, porous materials demonstrated a statistically significant increase in total macrophage biomarkers, yet a significant reduction (P < 0.05) in macrophage biomarkers associated with the pro-inflammatory macrophage phenotype. In contrast, macrophages on non-porous implants were more likely to be in the inflammatory phenotype. While these materials have been developed for tissue engineering and drug release applications, future studies should explore the role of specific, well-defined pore size on the performance of both percutaneous and subcutaneous glucose biosensors.
Porous materials have also been prepared via electrospinning.161–162 Formed by elongation and stretching of a polymer-containing liquid droplet under high electrostatic potentials (typically between a high voltage needle and a grounded collector), electrospun fibers have controllable, monodisperse diameters. The resulting fiber mats are porous with high surface areas, thus enabling infiltration of cells.163 The fiber diameter also influences the host response; small fibers (<6 μm in diameter)151,163 reduce the FBR (i.e., capsule formation) more effectively than larger (>6 μm) fibers. The alignment and orientation of the electrospun fibers also modulate the FBR. Cao et al. reported a decrease in collagen capsule thickness for polycaprolactone fibers when their orientation was either aligned (~5 fold reduction) or random (~9 fold reduction) compared to solid films made from the same polymer. Furthermore, the aligned fibers demonstrated clear cellular infiltration both in vivo and in vitro, while randomly-oriented fibers exhibited distinct surface boundaries between the material and the fibrous capsule. The modulus of the fiber material also proved critical, with lower modulus fibers (e.g., polyurethane) showing reduced capsule formation (i.e., size) compared to higher modulus materials (e.g., polyethylene, polyester and poly(L-lactic acid)).151 As described below, a materials’ modulus is an important factor for modulating the FBR to percutaneous biosensors on non-porous materials as well.
5.5. Material modulus
The mechanical properties of a material impact the resulting host response when implanted. External forces exerted on an implanted device can elicit an additional inflammatory response in the surrounding tissue. For percutaneous implants especially, the modulus at the skin-device interface must be considered as mismatches in the elastic modulus between an implant and the skin that surrounds it will result in sheer forces that induce a more severe FBR.164 However, the modulii of the dermis and subcutaneous tissue range from 56–260 kPa and 0.12–23 kPa, respectively,165–168 complicating the design of CGM devices when attempting to match these tissue attributes.32 To investigate how a material’s mechanical properties alter these forces, Subbaroyan and coworkers modeled probes with different elastic modulii (E) implanted into the cerebral cortexes of rats.169 The implanted materials included silicon rubber (E = 200 GPa), polyimide (E = 3 GPa), and a hypothetical material with a modulus of 6 MPa. Through computational modeling studies, the polyimide material demonstrated a 94% reduction in applied strain to the surrounding tissue, while strain with the theoretical 6 MPa material was reduced by two orders of magnitude. Although more empirical evidence is needed, this strategy may prove helpful in reducing chronic inflammation from motion-damaged tissue. In recent work, Irwin and coworkers observed a reduction in macrophage adhesion as the modulus of an interpenetrating polymer network was decreased from 348 to 1.4 kPa.170 While the mechanisms are unclear, biphasic decreases in IL-6 expression (i.e., a pro-inflammatory cytokine) were observed at the lowest and highest modulii. While the modulus associated with these brush-type materials is lower than that of typical bulk polymers, the importance of considering its effect on cellular adhesion is nonetheless important. Alternatively, modifying high-modulus polymers with thin coatings of interpenetrating polymers may alter the modulus that nearby cells experience, thus changing the extent to which they adhere and perhaps the ensuing cytokine expression.171
5.6. Active release materials
While the goal of the prior described studies was primarily to create an inert interface with respect to the host response, physical and chemical alterations to CGM devices at the sensor-tissue interface have not resulted in drastically improved glucose sensor function to date. Taking cues from controlled drug release, researchers have moved to develop active release biomaterials to influence tissue integration.172 Often the drug or small molecules for release is anti-inflammatory and/or angiogenic to address the major hurdles related to the FBR.
5.6.1. Dexamethasone
Dexamethasone (DX) is a glucocorticoid that reduces inflammation. Pharmacologically, glucocorticoids decrease vascular permeability and reduce leukocyte adhesion, recruitment and localization.173 Furthermore, glucocorticoids alter inflammatory cell trafficking, death, and cellular responses, possibly indicating a phenotypic change in the cells.174 The exact mechanism of glucocorticoid action is not completely understood, but 0is believed to involve the inhibition of nuclear factor (NF) κB and activating protein (AP)-1, among other proteins.175–177 Unfortunately, systemic use of DX can cause serious side-effects by depressing the innate immune response, thus increasing the likelihood of infection.178–179
Despite undesirable systemic effects, depression of the immune system may lead to sensor-localized benefits with respect to FBR. Reducing inflammatory cell concentrations at the device could inhibit the associated negative tissue reactions and even induce a more favorable cell phenotype. Local delivery of DX from glucose biosensors was hypothesized to circumvent undesirable systemic side effects of DX. Moussy et al. reported that [3H]DX travelled only an average of 2.70 mm from the point of delivery after 24 h when administered locally.180 Similarly, Ward and coworkers investigated the spatial effects of slow DX delivery in a porcine model using implanted osmotic pumps.181 Histological analysis revealed decreases in macrophage concentrations adjacent to implants 2 mm from the osmotic pump at 4 weeks, while tissue samples ≥17 mm away were not significantly different.181 While macrophage numbers were unaffected far from the pump site, granulocytes were significantly lowered in the tissue 17 mm away from a DX-pump relative to distant tissue (i.e., 3 to 4 cm).181 While these effects are promising, the release rates and amounts of DX must be carefully considered from a safety standpoint. For example, high concentrations of DX over 28 d (i.e., 0.17 mg/kg) led to a systemic reduction in cortisol levels while low concentrations (i.e., <0.10 mg/kg) did not cause such effects.181 In agreement with these results, Dang et al. found that the release of high concentrations of DX could cause systemic immunosuppression.182
The localized effect of low DX concentrations and its potent immunosuppression effects have prompted the development of DX-containing poly(lactic-co-glycolic acid) (PLGA) particles to facilitate controlled DX release from potential biomaterials. These particles are simple to synthesize, enable slow DX release, and biodegrade over 2–3 months.183 Hickey and coworkers described the synthesis of PLGA particles capable of loading and subsequently releasing the glucocorticoid.184 While an initial burst release of ~15% of total DX was observed, the PLGA particles provided approximate zero-order kinetic release of DX over one month, indicating potential for long-term delivery. PLGA particles were also synthesized with 10% (w/w) PEG, resulting in a greater initial release burst (~50%), but then slower DX release until after 4 weeks due to delayed degradation.184 The combination of these two PLGA systems could provide constant DX delivery for more than one month.
The loading of PLGA particles with DX is most commonly accomplished through an oil-in-water emulsion/solvent evaporation technique by combining DX and PLGA in an organic solvent, and mixing in water.184–187 The DX loading efficiency into the PLGA particles can be improved through careful solvent selection. For example, Ju et al. compared the encapsulation efficiency of DX in PLGA particles in oil phases of either 5:1 methylene chloride to methanol or 5:1 methylene chloride to acetone.187 Encapsulation efficiency by the PLGA particles increased from ~8 to ~40% when acetone was used instead of methanol.
The influence of DX release duration on FBR was studied by Bharwaj et al. in a rat model.186 Histological evaluation for short DX delivery (1 week) was accomplished by doping DX-modified PLGA microspheres into PVA hydrogels. The acute inflammatory response was diminished at 3 and 8 d, but inflammation was significantly increased at 30 d compared to the shorter time points (i.e., 3 and 8 d), indicating that DX release simply delayed the immune response.186 This study clearly indicated that DX must be delivered continuously (i.e., for the duration of an implant) in order to manifest the anti-inflammatory benefits.
Implementing the PLGA microsphere substrates, the utility of DX delivery from subcutaneous implants was investigated by Patil and coworkers.188 Dexamethasone-releasing PLGA/PVA hydrogel composites were implanted into the dorsal subcutaneous tissue of a rat with subsequent study of in vivo release kinetics and inflammatory response.188 While the in vivo DX release kinetics were accelerated relative to the release observed in phosphate buffered saline, DX delivery still followed zero-order kinetics. The release of DX reduced the inflammatory response by ~50% at 3 and 7 d compared to control PLGA/PVA hydrogel composites.188 As chronic inflammation set in at ~3 weeks, the DX-releasing composites still exhibited reduced inflammation and fibrosis compared to control hydrogels, demonstrating the ability of local DX release to arrest fibrous encapsulation.188 Later studies by Patil et al. further confirmed reduced fibrotic encapsulation over 4 weeks as a result of DX release from an implanted biomaterial in a rat model.189 Similarly, Sung and coworkers found that local DX release reduced inflammatory cell density adjacent to the hydrogel by ~50% after 8 d in an avian chorioallantoic membrane model.190 Despite the studies that reported favorable (i.e., reduced) FBR for DX-releasing materials, Norton et al. observed no real benefit in reducing the inflammatory response using DX-releasing PLGA microspheres after 2 or 6 weeks in rat subcutaneous tissue.191 The difference in the outcome of these studies may be the result of differences in polymer composition, DX release kinetics, and or total DX payload, clearly indicating the importance of many biomaterial parameters on tissue biocompatibility.
With knowledge that DX release reduces inflammation, Klueh and coworkers examined the influence of locally administered DX on glucose biosensor performance by daily intraperitoneal injections into a mouse model.192 Within 24 h biosensors in control mice suffered large reductions in sensitivity while mice receiving the daily DX injection at the sensor site experienced sustained sensitivity and functionality through 2 weeks, regardless of DX concentration.192 These results highlight the role of DX in improving sensor performance. Unfortunately, systemic administration of DX is not considered a viable approach for treatment. Studying the local DX release from subcutaneous glucose biosensors on sensor performance in canines, Ward and Troupe reported that sensor longevity was enhanced to ~26 d, albeit this result was not statistically significant.193 Furthermore, they did not report on the DX release amounts or kinetics.193 Ju et al. also reported on glucose sensor performance as a function of DX release from collagen scaffolds.187 DX-releasing PLGA particles were embedded into a collagen scaffold and then implanted subcutaneously. Subsequent histology of the surrounding tissue indicated a reduction in inflammatory cell infiltration into the porous scaffolds.187 At 2 weeks, the depth of the dense inflammatory cell band at DX-releasing and control porous scaffolds was 20–100 Vm and 100–150 Vm, respectively.187 At 4 weeks following insertion, DX-releasing substrates had almost no cellular infiltration while control substrates exhibited a cell infiltration depth of 100–150 Vm.187 DX-releasing glucose biosensors retained >50% of their original sensitivity 2 weeks after implantation, the sensitivity of control membranes dropped to 15–42% of their original levels.187 Mou et al. examined the effect of DX delivery (while sampling interstitial fluid) on glucose recovery in a rat microdialysis model.194 Microdialysis probes were perfused for 3 h every other day, which represented the only time in which DX was delivered to tissue. Over a 10-d period, no significant differences were reported in the dialysate glucose concentrations between control and DX-perfused probes, even though a more fragile capsule was observed at DX-perfused probes, preventing histological analysis. The lack of DX activity may have resulted from the minimal and intermittent nature of DX delivery from the probes, further supporting the need to constantly release this anti-inflammatory drug. Overall, these studies suggest that decreased inflammation likely enhances long-term glucose biosensor functionality, though the challenges of achieving long-term DX release while avoiding systemic effects remains.
5.6.2. Vascular endothelial growth factor
Vascular endothelial growth factor, or VEGF, is a cytokine released by macrophages and keratinocytes during wound reconstruction to promote angiogenesis.195–197 The expression of VEGF is induced in response to other growth factors and/or tissue hypoxia, with VEGF ultimately binding to tyrosine kinase receptors to stimulate angiogenesis.195,198 VEGF has also been referred to as vascular permeability factor due to its ability to induce vascular leakage.199 A concern regarding VEGF use is increased local inflammation because cell migration and proliferation also correlate with angiogenesis.200–201 Nevertheless, the potential for greater capillary densities via VEGF release have led researchers to evaluate the utility of this strategy for improving in vivo glucose sensor performance.
Due to a relatively small sphere of influence in subcutaneous tissue, Ward et al. reported that direct delivery of the cytokine to the damaged tissue (via sensor implantation) was necessary to promote improved wound healing.202 An osmotic pump was implanted into rat subcutaneous tissue to enable VEGF delivery at 0.45 Vg/day.202 While tissue 1 and 13 mm from the perfusion site were shown to have enhanced vascularization, no statistical difference between saline and VEGF-osmotic pumps 25 mm away was noted, indicating a highly localized tissue response.202 A follow-up study evaluated the distance-dependence of VEGF-osmotic pumps on implantable glucose sensor performance.203 Biosensors were inserted 1, 13 and 25 mm from the same osmotic pumps in rat subcutaneous tissue.203 The functional lifetime of the sensors near the pump was 63 days whereas control (saline) sensors stopped working accurately at 34 days.203 Both the sensor lag time and the mean absolute relative difference (MARD) for glucose analysis increased with distance away from the VEGF-releasing pumps. However, control (saline) pumps resulted in similar performance.203 Clarke Error Grid Analysis indicated that only sensors 1 mm from the VEGF-eluting pumps showed accuracy improvements versus controls. These results are surprising since the histological analysis revealed increased vascularization in tissue up to 13 mm away from the pumps.202–203 The authors attributed the lack of significant glucose sensor performance improvements to VEGF-induced pro-inflammatory effects.203
Klueh and coworkers also examined the effect of VEGF-induced vascularization on glucose sensor performance in a chick chorioallantoic membrane model.204 To achieve enhanced vascularization, sensors were coated with VEGF-transfected cells prior to implantation. Enhanced vascularization and sensor sensitivity were observed in the tissue adjacent to sensors with the VEGF-transfected cells, although the amount of VEGF release was not confirmed. Furthermore, significant inflammation and the development of FBGCs were reported for VEGF-“producing” biosensors.204 Such inflammatory response would certainly lead to sensor performance issues over time.
While the previous studies indicate possible improvements in glucose sensor performance, implementation of VEGF release is not readily accomplished using osmotic pumps or transfected cells. VEGF storage on appropriate materials has thus been pursued to facilitate simpler release mechanisms. For example, PLGA microspheres were loaded with VEGF using an oil-in-water emulsion/solvent evaporation technique,189,205 similar to the DX work described above. These particles are then easily doped into sensor membranes. In this manner, Norton and associates examined the release of VEGF from a hydrogel copolymer of 2-hydroxyethyl methacrylate (HEMA), 1-vinyl-2-pyrrolidinone (VP), and polyethylene glycol acrylate (PEG).206 VEGF release directly from hydrogels loaded with just VEGF was rapid, with almost all release occurring within 1 day.206 The use of PLGA microspheres containing VEGF allowed for much longer release up to ~28 d, demonstrating the ability to extend VEGF release from a sensor surface. Of note, the authors did not attempt to tune the VEGF release (e.g., >1 day or <28 days), suggesting the delivery is less than controllable.206
Others have also reported the benefit of using VEGF-releasing PLGA microspheres to improve vascularization in subcutaneous tissue. Patil et al. implanted VEGF-releasing PLGA/PVA hydrogel composites into rat subcutaneous tissue and noted through histology that capillary density was significantly increased at 3 and 4 weeks after implantation versus controls (PVA hydrogels).189 Ultimately, local inflammation was 2–4 fold greater near the VEGF-releasing materials relative to control substrates over the entire four-week study.189 Similarly, Norton et al. fabricated VEGF-containing PLGA microspheres-doped hydrogels coated onto microdialysis probes, examining tissue histology at 2 and 6 weeks after implanting the probes into rat subcutaneous tissue.191 Vascularity at 2 weeks was reported to be ~300% higher than hydrogel control coatings, though no differences were observed at 6 weeks. Similar to the study by Patil et al.,189 a significant increase in inflammation was observed at 2 weeks relative to controls.191 Sung et al. later characterized the tissue response to VEGF-releasing materials in an avian chorioallantoic membrane model to fully understand neovascularization and inflammation.190 After 8 days of implantation, vascularization was increased by four-fold while inflammation was 50% greater relative to controls. While these studies indicate the potential of VEGF to strongly influence the vasculature adjacent to implanted materials, the corresponding increase in inflammation is particularly troublesome and would likely diminish glucose sensor performance.
5.6.3. Dual DX/VEGF delivery
A dual DX/VEGF release strategy was pursued to limit inflammation and take advantage of the wound healing properties of each. To date, DX has been associated with poor revascularization when applied to wounds. Conversely, VEGF acts to increase local capillary density. The combined use of these factors was thus hypothesized to decrease inflammation and enhance vascularization. Indeed, initial experiments examining the simultaneous delivery of VEGF and DX from PLGA particles showed enhanced tissue integration, decreased inflammation, and increased vascularization in a rat model.189 Following an initial burst of ~20% of the total DX/VEGF storage capacity, the remaining DX and VEGF was released following roughly zero-order kinetics over 4 weeks at rates of 123 ng/day and 21 pg/day, respectively.189 When released separately in vivo over an identical period, the DX reduced the inflammatory response (i.e., cell density) while VEGF enhanced vascularity of the surrounding tissue as expected.189 Histology surrounding PLGA particle-containing hydrogels with combined DX and VEGF release revealed a 50% reduction in inflammation after the first week, similar to that of DX release alone, though inflammation at weeks 2–4 was still greater than DX alone.189 Combined DX and VEGF release also resulted in an enhancement in tissue revascularization compared to DX-releasing and control surfaces, equivalent to the tissue response observed by VEGF release alone.189
While the study by Patil et al. suggests that the improved response/integration may improve in vivo glucose sensor performance, these drugs may still act in an antagonistic manner in vivo. Indeed, glucocorticoids including DX have been shown to decrease the in vivo activity of VEGF.207–209 Norton et al. carefully examined the tissue response to VEGF-, DX-, and VEGF/DX-releasing hydrogels in a rat subcutaneous model,191 observing the expected increase in vascularization due to VEGF but no decrease in local inflammation from DX release. More troubling with respect to the validity of earlier studies, the release of DX and VEGF resulted in no benefit in vascularization or inflammation in the surrounding tissue. In contrast, Sung et al. reported that simultaneous VEGF/DX delivery did not decrease inflammation in an avian chorioallantoic model, but the tissue adjacent to the implant had greater capillary density relative to controls.190 To avoid possible antagonistic behavior between the drugs, DX (1 Vg in 500 VL) was injected prior to implanting the VEGF-releasing substrates. The initial DX treatment and subsequent VEGF release was reported to enhance vascularization and inhibit inflammation.190 The level of decreased inflammation was similar to that achieved by DX injection alone, but the vascularization was lower than VEGF only controls.190 Unfortunately, the above studies contrast significantly with respect to the delivery methods, materials, and animal models, and thus the significance of VEGF/DX dual delivery for tissue reconstruction remains questionable. Furthermore, studies evaluating the benefits of VEGF-, DX-, or VEGF/DX-release on the analytical performance of glucose biosensors have not been evaluated.
5.6.4. Nitric oxide
Since nitric oxide (NO) was identified as the endothelial-derived relaxation factor (EDRF) in 1986, much research has unraveled other key functions of NO in the body.210 Nitric oxide is an endogenously produced free radical synthesized from L-arginine by one of three isoforms of nitric oxide synthase (NOS): neuronal NOS (nNOS), endothelial NOS (eNOS), and inducible NOS (iNOS).211 The nNOS isoform is present in many tissues outside the brain including skeletal muscle and islet cells while eNOS is present almost exclusively in the endothelium.212 These two isoforms create low concentrations (i.e., nM) of NO. Inducible NOS is expressed in multiple cells upon exposure to certain cytokines or lipopolysaccharide (LPS). Macrophages in particular respond to pathogens and foreign objects by synthesizing VM concentrations of NO.212
While the mechanisms have not been completely elucidated, NO influences the FBR in ways that may affect in vivo glucose sensor performance. Multiple reports indicate that NO serves as an angiogenic signaling model, up-regulating VEGF production and increasing blood vessel growth.213–214 VEGF also acts to up-regulate eNOS expression thus inducing vasodilation. The angiogenic behavior exhibited by NO may thus prove helpful for avoiding the avascular encapsulation that plagues glucose sensor performance. Furthermore, NO is also believed to control inflammatory cell recruitment in the early stages of the FBR.215 Localized NO may down-regulate pro-inflammatory cytokine expression (e.g., macrophage chemoattractant protein-1), by nitrosating certain proteins. Carreau et al. reported reduced leukocyte adhesion at elevated NO concentrations, suggesting NO could reduce localization of inflammatory cells at the device-tissue interface.216 Fewer leukocytes may result in reduced inflammation and subsequent FBR. Nitric oxide may therefore be able to accomplish the hypothesized actions of dual DX/VEGF while avoiding the antagonistic behavior between the drugs.
Given NO’s roles in the immune response, favorable tissue reactions to NO-releasing substrates would be expected in vivo.217 However, direct delivery of NO is not straightforward due to NO’s reactivity and gaseous nature. To achieve in vivo release, NO donors have been synthesized as a method to store NO until breakdown of the donor upon some trigger.218–219 The most common NO donors to date include N-diazeniumdiolates and S-nitrosothiols (Figure 3). N-diazeniumdiolates are formed on secondary amines upon exposure to NO gas in basic solution.220 In the presence of a proton source (e.g., water), the N-diazeniumdiolate breaks down to release two molecules of NO and the amine precursor.220 The rate of NO release from this class of donor is highly dependent on the pH, ionic strength, and surrounding chemical environment.220–221 S-nitrosothiols are formed on thiols upon reaction with nitrosating agents (e.g., acidified nitrite), and degrade when exposed to light, copper (I), or heat, releasing one NO molecule per thiol.222 Similar to N-diazeniumdiolates, the NO-release kinetics from S-nitrosothiols are dependent on the structure of the NO donor. The concept of NO release materials for improving the biocompatibility of a material was initially investigated by incorporating low molecular weight (LMW) NO donors into polymer membranes to enable controlled release.223–225 In addition to limited NO release durations and payloads, the LMW NO donors were shown to leach from such coatings with potential cytotoxic effects. Significant effort has focused on the synthesis of specialized macromolecular NO donors to improve NO payloads, durations, and safety.226–237
Figure 3.

N-diazeniumdiolate and S-nitrosothiol nitric oxide (NO) donors with decomposition pathways. Decomposition kinetics are dependent on the chemical structure of the NO donor, pH, temperature, and/or presence of other biological milieu.
Silica-based xerogels and particles have been implemented to investigate the influence of polymeric NO release on tissue biocompatibility. In a seminal study, Hetrick et al. coated 40% (v/v) N-(6-aminohexyl)aminopropyltrimethoxysilane (AHAP3) balance isobutyltrimethoxysilane (BTMOS) xerogels onto medical grade silicone rubber squares and chemically modified them to store NO (via N-diazeniumdiolates).238 The NO payload of these substrates was 1.3 Vmol cm−2 over 3 d. A rat subcutaneous tissue model was used to evaluate the effects of the NO on tissue surrounding the implant after 1, 3, or 6 weeks. In addition to reduced collagen encapsulation (~20–25% vs. controls) observed at 3 and 6 weeks, the NO mitigated inflammation (determined by reduced inflammatory cells near the implant) and appeared to enhance vascular density at 1 and 3 weeks.238 Overall, the results indicated a possible benefit for long-term glucose sensor performance.
Subsequent microdialysis studies were employed in the subcutaneous space of rats to investigate the effects of NO on glucose recovery (i.e., resistance to mass transfer).239 Polyarylethersulfone (PAES) microdialysis probes were perfused with saturated NO solutions for 8 h per day over 14 d to enable consistently high, though intermittent, NO release.239 Using this methodology, zero-order kinetics were achieved during the 8 h perfusion period with a daily NO payload of 4.6 Vmol cm−2. The NO-releasing probes after 7 d were reported to recover larger glucose concentrations compared to controls (Figure 4), indicating lower resistance to mass transfer in the surrounding tissue. Histological analysis of the tissue surrounding the implant after 14 d revealed reduced capsule thickness and inflammatory cell density. The authors concluded that the mitigated FBR by NO release was thus at least partially responsible for the superior glucose recovery.
Figure 4.

Glucose recovery at nitric oxide (NO)-releasing and control polyarylethersulfone (PAES) microdialysis probes implanted into rat subcutaneous tissue for up to 14 d. Nitric oxide release was accomplished daily by perfusing saturated NO solutions for 8 h at a flow rate of 2 VL/min resulting in 4.6 Vmol of NO release daily. Significant differences (*) in glucose recovery were observed after 7 d. Reprinted with permission from ref 229. Copyright 2011 by American Chemical Society.
Only one report to date has appeared evaluating the effect of NO release on in vivo glucose sensor performance. Gifford et al. doped (Z)-1-[N-methyl-N-[6-(Nbutylammoniohexyl)amino]]-diazen-1-ium-1,2-diolate (DBHD/NO) into a polyurethane/polydimethylsiloxane glucose sensor membrane.103 The NO release was fully expired after only 18 h. Nevertheless, the analytical performance of percutaneous NO-releasing sensors in rats was improved by 2.4 and 2.1% (Clarke Error Grid Analysis) for the NO-releasing sensors relative to controls on days 1 and 3, respectively. Histological analysis of the tissue adjacent to the sensors revealed decreased inflammation at 24 but not 48 h.103 The anti-inflammation correlated with the NO-release duration indicating that the observed benefits may only persist as long as the interface is releasing NO, in contrast to observations by Hetrick et al. where long-term reduction in inflammation were observed after cessation of NO release.103,238 This work suggests that NO release may represent an important strategy for improving in vivo glucose sensor performance.
While these studies highlight the effect of NO release on the FBR and its potential to improve glucose sensor performance, research to date is still in early stages with respect to NO storage and release, and combining the chemistry of NO release with enzymatic glucose sensing. Work by Nichols et al. examined the influence of NO-release duration and payload ranging from 6 h to 14 d and 2.7 to 9.3 Vmol cm−2, respectively, on the FBR using 316L stainless steel wires as model sensors (with respect to size) coated with different NO release formulations.240 Collagen encapsulation was reduced at 3 and 6 weeks, and the greatest effect was observed for the largest payloads and longest release. In contrast to the capsule thickness, inflammation was only affected by substrates still actively releasing NO at time of explants. These results indicate that the short-term benefits reported by Gifford et al. may be further enhanced with a greater and more sustained NO release profile.103,240 Based on studies to date, the anti-inflammatory influence by localized (i.e., surface) NO release persist only during the release event. Although further study is required, such results stress the need to develop long-term NO-releasing materials.
6. Conclusions
Continuous glucose monitoring devices would dramatically alter the treatment of diabetes and the quality of life of those afflicted by this disease. Unfortunately, the development of analytically accurate in vivo sensors that function for extended (i.e., at least weeks) periods remains a shortcoming and insurmountable challenge due to the FBR. Recent insights regarding specific in vivo impediments to sensor function have helped form strategies to reduce inflammation and improve tissue integration of sensor materials, particularly the outer sensor membrane that serves to protect the enzyme, tune response, and ultimately is responsible for the magnitude of the FBR. New strategies for directing rather than avoiding the FBR by the active release of drugs and/or the body’s own signaling mediators of wound healing have created a unique path to alter the natural host response. Eventually such research should result in improved in vivo sensor performance. Old and new researchers in this area must first appreciate traditional biosensor designs, the inherent and often complex FBR concomitant with sensor insertion into native tissue, and the requirement that any alterations to the outer sensor membrane must be carried out with special attention to sensor design requirements with respect to both analytical merits and in vivo performance/utility.
Acknowledgments
The authors acknowledge research support from the National Institutes of Health (NIH Grant EB000708).
Biographies

Scott P. Nichols received his BS in Chemistry from the University of Missouri-Columbia in 2007 and his PhD in Chemistry from the University of North Carolina at Chapel Hill in 2012. He is currently a Postdoctoral Research Associate at Duke University under Bruce Klitzman and a consultant for PROFUSA. His current research interests include evaluating the biocompatibility and analytical performance of implantable glucose sensors.

Ahyeon Koh is a PhD candidate in Chemistry at the University of North Carolina at Chapel Hill. She received her BS and MS in Chemistry from Sogang University in 2004 and 2007, respectively, where she performed research with Prof. Woonsup Shin on chemically amplified electrochemical detection of ferrocenemethanol by ferrocyanide on 4-nitrophenyl grafted electrodes. Her graduate research has focused on developing nitric oxide-releasing polyurethane glucose sensor membranes to mitigate the foreign body response in vivo.

Wesley Storm is a PhD candidate in Chemistry at the University of North Carolina at Chapel Hill. He earned his BS in Chemistry at James Madison University where he carried out research with Daniel M. Downey, analyzing trace metal concentrations in fish otoliths as a method to assess pollutants in freshwater streams. His graduate research has focused on the development of antimicrobial surfaces for biomedical and environmental applications.

Jae Ho Shin is an Assistant Professor in the Department of Chemistry at Kwangwoon University in Seoul, South Korea, where he received his BS and MS in Chemistry. He earned his PhD in Chemistry from the University of North Carolina at Chapel Hill in 2006 under the direction of Dr. Mark Schoenfisch. He was a Carolina Center of Cancer Nanotechnology Excellence Postdoctoral Fellow at UNC-Chapel Hill for one year. His research interests include sol-gel-derived chemical and biological sensors, electrochemical immunosensing platforms, and nitric oxide-releasing silica scaffolds.

Mark Schoenfisch is a Professor of Chemistry in the Department of Chemistry at the University of North Carolina at Chapel Hill. Dr. Schoenfisch received undergraduate degrees in Chemistry (BA) and Germanic Languages and Literature (BA) at the University of Kansas prior to attending the University of Arizona for graduate studies in Chemistry (PhD). Before starting at UNC-Chapel Hill, he spent two years as a National Institutes of Health Postdoctoral Fellow at the University of Michigan. His research interests include analytical sensors, biomaterials, and the development of nitric oxide release scaffolds as new therapeutics.
Contributor Information
Scott P. Nichols, Email: snichols@unc.edu.
Ahyeon Koh, Email: ahyeon@email.unc.edu.
Wesley L. Storm, Email: wstorm@email.unc.edu.
Jae Ho Shin, Email: jhshin@kw.ac.kr.
References
- 1.WHO. World Health Statistics 2012. World Health Organization; 2012. [Google Scholar]
- 2.CDC. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention; 2012. [Google Scholar]
- 3.Newman JD, Turner APF. Biosens Bioelectron. 2005;20:2435. doi: 10.1016/j.bios.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 4.Wang J. Chem Rev. 2008;108:814. doi: 10.1021/cr068123a. [DOI] [PubMed] [Google Scholar]
- 5.Heller A. Annu Rev Biomed Eng. 1999;1:153. doi: 10.1146/annurev.bioeng.1.1.153. [DOI] [PubMed] [Google Scholar]
- 6.Wilson GS, Hu YB. Chem Rev. 2000;100:2693. doi: 10.1021/cr990003y. [DOI] [PubMed] [Google Scholar]
- 7.Williams DF. Biomaterials. 2008;29:2941. doi: 10.1016/j.biomaterials.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Clark LC, Lyons C. Ann NY Acad Sci. 1962;102:29. doi: 10.1111/j.1749-6632.1962.tb13623.x. [DOI] [PubMed] [Google Scholar]
- 9.Updike SJ, Hicks GP. Nature. 1967;214:986. doi: 10.1038/214986a0. [DOI] [PubMed] [Google Scholar]
- 10.Guilbault GG, Lubrano GJ. Anal Chim Acta. 1973;64:439. doi: 10.1016/S0003-2670(01)82476-4. [DOI] [PubMed] [Google Scholar]
- 11.Shichiri M, Yamasaki Y, Kawamori R, Hakui N, Abe H. Lancet. 1982;320:1129. doi: 10.1016/s0140-6736(82)92788-x. [DOI] [PubMed] [Google Scholar]
- 12.Wang J. Electroanalysis. 2001;13:983. [Google Scholar]
- 13.Park S, Boo H, Chung TD. Anal Chim Acta. 2006;556:46. doi: 10.1016/j.aca.2005.05.080. [DOI] [PubMed] [Google Scholar]
- 14.Park S, Park S, Jeong R-A, Boo H, Park J, Kim HC, Chung TD. Biosens Bioelectron. 2012;31:284. doi: 10.1016/j.bios.2011.10.033. [DOI] [PubMed] [Google Scholar]
- 15.Wang C, Yu B, Knudsen B, Harmon J, Moussy F, Moussy Y. Biomacromolecules. 2008;9:561. doi: 10.1021/bm701102y. [DOI] [PubMed] [Google Scholar]
- 16.Shoji E, Freund MS. J Am Chem Soc. 2001;123:3383. doi: 10.1021/ja005906j. [DOI] [PubMed] [Google Scholar]
- 17.Shoji E, Freund MS. J Am Chem Soc. 2002;124:12486. doi: 10.1021/ja0267371. [DOI] [PubMed] [Google Scholar]
- 18.Wang J. Chem Rev. 2008;108:814. doi: 10.1021/cr068123a. [DOI] [PubMed] [Google Scholar]
- 19.Vaddiraju S, Burgess DJ, Tomazos I, Jain FC, Papadimitrakopoulos F. J Diabetes Sci Technol. 2010;4:1540. doi: 10.1177/193229681000400632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Degani Y, Heller A. J Phys Chem. 1987;91:1285. [Google Scholar]
- 21.Pishko MV, Katakis I, Lindquist SE, Ye L, Gregg BA, Heller A. Angew Chem-Int Edit. 1990;29:82. [Google Scholar]
- 22.Wilson GS, Hu Y. Chem Rev. 2000;100:2693. doi: 10.1021/cr990003y. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen JK, Freckmann G, Kapitza C, Ocvirk G, Koelker KH, Kamecke U, Gillen R, Amann-Zalan I, Jendrike N, Christiansen JS, Koschinsky T, Heinemann L. Diabetic Med. 2009;26:714. doi: 10.1111/j.1464-5491.2009.02750.x. [DOI] [PubMed] [Google Scholar]
- 24.Oliver NS, Toumazou C, Cass AEG, Johnston DG. Diabetic Med. 2009;26:197. doi: 10.1111/j.1464-5491.2008.02642.x. [DOI] [PubMed] [Google Scholar]
- 25.Mou X, Lennartz MR, Loegering DJ, Stenken JA. Biomaterials. 2010;31:4530. doi: 10.1016/j.biomaterials.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Potts RO, Tamada JA, Tierney MJ. Diabetes Metab Res. 2002;18:S49. doi: 10.1002/dmrr.210. [DOI] [PubMed] [Google Scholar]
- 27.Rao G, Glikfeld P, Guy RH. Pharm Res. 1993;10:1751. doi: 10.1023/a:1018926215306. [DOI] [PubMed] [Google Scholar]
- 28.Tamada JA, Bohannon NJV, Potts RO. Nat Med. 1995;1:1198. doi: 10.1038/nm1195-1198. [DOI] [PubMed] [Google Scholar]
- 29.Ferrante do Amaral CE, Wolf B. Med Eng Phys. 2008;30:541. doi: 10.1016/j.medengphy.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Vashist SK. Anal Chim Acta. 2012;750:16. doi: 10.1016/j.aca.2012.03.043. [DOI] [PubMed] [Google Scholar]
- 31.Gough DA, Kumosa LS, Routh TL, Lin JT, Lucisano JY. Sci Transl Med. 2010;2:42ra53. doi: 10.1126/scitranslmed.3001148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helton KL, Ratner BD, Wisniewski NA. J Diabetes Sci Technol. 2011;5:632. doi: 10.1177/193229681100500317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shichiri M, Asakawa N, Yamasaki Y, Kawamori R, Abe H. Diabetes Care. 1986;9:298. doi: 10.2337/diacare.9.3.298. [DOI] [PubMed] [Google Scholar]
- 34.Bindra DS, Zhang Y, Wilson GS, Sternberg R, Thevenot DR, Moatti D, Reach G. Anal Chem. 1991;63:1692. doi: 10.1021/ac00017a008. [DOI] [PubMed] [Google Scholar]
- 35.Moschou EA, Sharma BV, Deo SK, Daunert S. J Fluoresc. 2004;14:535. doi: 10.1023/b:jofl.0000039341.64999.83. [DOI] [PubMed] [Google Scholar]
- 36.Pickup JC, Hussain F, Evans ND, Rolinski OJ, Birch DJS. Biosens Bioelectron. 2005;20:2555. doi: 10.1016/j.bios.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 37.Badugu R, Lakowicz JR, Geddes CD. Curr Opin Biotech. 2005;16:100. doi: 10.1016/j.copbio.2004.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shibata H, Heo YJ, Okitsu T, Matsunaga Y, Kawanishi T, Takeuchi S. Proc Natl Acad Sci USA. 2010;107:17894. doi: 10.1073/pnas.1006911107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heo YJ, Shibata H, Okitsu T, Kawanishi T, Takeuchi S. Proc Natl Acad Sci USA. 2011;108:13399. doi: 10.1073/pnas.1104954108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pickup J, McCartney L, Rolinski O, Birch D. BMJ. 1999;319:1289. doi: 10.1136/bmj.319.7220.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tura A, Maran A, Pacini G. Diabetes Res Clin Pract. 2007;77:16. doi: 10.1016/j.diabres.2006.10.027. [DOI] [PubMed] [Google Scholar]
- 42.Arnold MA, Olesberg JT, Small GW. In: In Vivo Glucose Sensing. Cunningham DD, Stenken JA, editors. John Wiley & Sons, Inc; 2010. [Google Scholar]
- 43.Barman I, Kong CR, Singh GP, Dasari RR, Feld MS. Anal Chem. 2010;82:6104. doi: 10.1021/ac100810e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lyandres O, Yuen JM, Shah NC, VanDuyne RP, Walsh JT, Glucksberg MR. Diabetes Technol Ther. 2008;10:257. doi: 10.1089/dia.2007.0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shafer-Peltier KE, Haynes CL, Glucksberg MR, Van Duyne RP. J Am Chem Soc. 2003;125:588. doi: 10.1021/ja028255v. [DOI] [PubMed] [Google Scholar]
- 46.Yonzon CR, Haynes CL, Zhang XY, Walsh JT, Van Duyne RP. Anal Chem. 2004;76:78. doi: 10.1021/ac035134k. [DOI] [PubMed] [Google Scholar]
- 47.Alexeeva NV, Arnold MA. J Diabetes Sci Technol. 2010;4:1041. doi: 10.1177/193229681000400503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pfützner A, Caduff A, Larbig M, Schrepfer T, Forst T. Diabetes Technol Ther. 2004;6:435. doi: 10.1089/1520915041705839. [DOI] [PubMed] [Google Scholar]
- 49.Caduff A, Dewarrat F, Talary M, Stalder G, Heinemann L, Feldman Y. Biosens Bioelectron. 2006;22:598. doi: 10.1016/j.bios.2006.01.031. [DOI] [PubMed] [Google Scholar]
- 50.Vashist SK. Anal Chim Acta. 2012;750:16. doi: 10.1016/j.aca.2012.03.043. [DOI] [PubMed] [Google Scholar]
- 51.BT012: Industry Experts. Hyperabad, India: Mar, 2012. Biosensors - A Global Market Overview. [Google Scholar]
- 52.GDME0130MAR. GlobalData; New York: Jan, 2012. Continuous Glucose Monitoring (CGM) Systems - Global Pipeline Analysis, Competitive Landscape and Market Forecasts to 2017. [Google Scholar]
- 53.Henning T. In: In Vivo Glucose Sensing. Cunningham DD, Stenken JA, editors. John Wiley & Sons, Inc; Hoboken, NJ: 2010. [Google Scholar]
- 54.Klonoff D, Bernhardt P, Ginsberg BH, Joseph J, Mastrototaro J, Parker DR, Vesper H, Vigersky R. Performance Metrics for Continuous Interstitial Glucose Monitoring, Approved Guideline; Wayne, PA. 2008. [Google Scholar]
- 55.McEnroe RJ, Burritt MF, Powers DM, Rheinheimer DW, Wallace BH. Interference Testing in Clinical Chemistry, Approved Guideline; Wayne, PA. 2005. [Google Scholar]
- 56.Heller A, Feldman B. Chem Rev. 2008;108:2482. doi: 10.1021/cr068069y. [DOI] [PubMed] [Google Scholar]
- 57.Wang J. Talanta. 2008;75:636. doi: 10.1016/j.talanta.2007.10.023. [DOI] [PubMed] [Google Scholar]
- 58.Wilson GS, Gifford R. Biosens Bioelectron. 2005;20:2388. doi: 10.1016/j.bios.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 59.Barlett PN, Cooper JM. Journal of Electroanalytical Chemistry. 1993;362:1. [Google Scholar]
- 60.Chen X, Matsumoto N, Hu Y, Wilson GS. Anal Chem. 2002;74:368. doi: 10.1021/ac015628m. [DOI] [PubMed] [Google Scholar]
- 61.Myrer JW, Measom GJ, Durrant E, Fellingham GW. J Athl Training. 1997;32:238. [PMC free article] [PubMed] [Google Scholar]
- 62.Wilson GS, Johnson MA. Chem Rev. 2008;108:2462. doi: 10.1021/cr068082i. [DOI] [PubMed] [Google Scholar]
- 63.Wilson GS, Zhang Y. In: In Vivo Glucose Sensing. Cunningham DD, Stenken JA, editors. John Wiley & Sons, Inc; Hoboken, NJ: 2009. [Google Scholar]
- 64.Cambiaso A, Delfino L, Grattarola M, Verreschi G, Ashworth D, Maines A, Vadgama P. Sens Actuat B-Chem. 1996;33:203. [Google Scholar]
- 65.Mano N, Mao F, Heller A. J Electroanal Chem. 2005;574:347. [Google Scholar]
- 66.Galeska I, Chattopadhyay D, Moussy F, Papadimitrakopoulos F. Biomacromolecules. 2000;1:202. doi: 10.1021/bm0002813. [DOI] [PubMed] [Google Scholar]
- 67.Ishihara K, Tanaka S, Furukawa N, Nakabayashi N, Kurita K. J Biomed Mater Res. 1996;32:391. doi: 10.1002/(SICI)1097-4636(199611)32:3<391::AID-JBM12>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 68.Tipnis R, Vaddiraju S, Jain F, Burgess DJ, Papadimitrakopoulos F. J Diabetes Sci Technol. 2007;1:193. doi: 10.1177/193229680700100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moussy F, Harrison DJ, O'Brien DW, Rajotte RV. Anal Chem. 1993;65:2072. doi: 10.1021/ac00063a023. [DOI] [PubMed] [Google Scholar]
- 70.Koschwanez HE, Reichert WM. Biomaterials. 2007;28:3687. doi: 10.1016/j.biomaterials.2007.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tse PHS, Gough DA. Biotechnol Bioeng. 1987;29:705. doi: 10.1002/bit.260290607. [DOI] [PubMed] [Google Scholar]
- 72.Miron J, Gonzalez MP, Vazquez JA, Pastrana L, Murado MA. Enzyme Microb Tech. 2004;34:513. [Google Scholar]
- 73.Zhao Q, Topham N, Anderson JM, Hiltner A, Lodoen G, Payet CR. J Biomed Mater Res. 1991;25:177. doi: 10.1002/jbm.820250205. [DOI] [PubMed] [Google Scholar]
- 74.Wisniewski N, Reichert M. Colloid Surface B. 2000;18:197. doi: 10.1016/s0927-7765(99)00148-4. [DOI] [PubMed] [Google Scholar]
- 75.Kovatchev B, Anderson S, Heinemann L, Clarke W. Diabetes Care. 2008;31:1160. doi: 10.2337/dc07-2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clarke WL, Kovatchev BP. J Diabetes Sci Technol. 2007;1:669. doi: 10.1177/193229680700100510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kovatchev BP, Gonder-Frederick LA, Cox DJ, Clarke WL. Diabetes Care. 2004;27:1922. doi: 10.2337/diacare.27.8.1922. [DOI] [PubMed] [Google Scholar]
- 78.Cengiz E, Tamborlane WV. Diabetes Technol Ther. 2009;11:S11. doi: 10.1089/dia.2009.0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Keenan DB, Mastrototaro JJ, Voskanyan G, Steil GM. J Diabetes Sci Technol. 2009;3:1207. doi: 10.1177/193229680900300528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choleau C, Klein JC, Reach G, Aussedat B, Demaria-Pesce V, Wilson GS, Gifford R, Ward WK. Biosens Bioelectron. 2002;17:647. doi: 10.1016/s0956-5663(01)00304-9. [DOI] [PubMed] [Google Scholar]
- 81.Rebrin K, Sheppard NF, Steil GM. J Diabetes Sci Technol. 2010;4:1087. doi: 10.1177/193229681000400507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choleau C, Klein JC, Reach G, Aussedat B, Demaria-Pesce V, Wilson GS, Gifford R, Ward WK. Biosens Bioelectron. 2002;17:641. doi: 10.1016/s0956-5663(01)00306-2. [DOI] [PubMed] [Google Scholar]
- 83.Groenendaal W, Basum Gv, Schmidt KA, Hilbers PAJ, Riel NAWv. J Diabetes Sci Technol. 2010;4:1032. doi: 10.1177/193229681000400502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gibney MA, Arce CH, Byron KJ, Hirsch LJ. Curr Med Res Opin. 2010;26:1519. doi: 10.1185/03007995.2010.481203. [DOI] [PubMed] [Google Scholar]
- 85.Gilligan BC, Shults M, Rhodes RK, Jacobs PG, Brauker JH, Pintar TJ, Updike SJ. Diabetes Technol Ther. 2004;6:378. doi: 10.1089/152091504774198089. [DOI] [PubMed] [Google Scholar]
- 86.Castle JR, Ward WK. J Diabetes Sci Technol. 2010;4:221. doi: 10.1177/193229681000400127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Helton KL, Ratner BD, Wisniewski NA. J Diabetes Sci Technol. 2011;5:647. doi: 10.1177/193229681100500318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Petrofsky JS. J Diabetes Sci Technol. 2011;5:657. doi: 10.1177/193229681100500319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ratner BD. Polym Int. 2007;56:1183. [Google Scholar]
- 90.Anderson JM. Ann Rev Mater Res. 2001;31:81. [Google Scholar]
- 91.Anderson JM, Rodriguez A, Chang DT. Semin Immunol. 2008;20:86. doi: 10.1016/j.smim.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wilson CJ, Clegg RE, Leavesley DI, Pearcy MJ. Tissue Eng. 2005;11:1. doi: 10.1089/ten.2005.11.1. [DOI] [PubMed] [Google Scholar]
- 93.Luttikhuizen DT, Harmsen MC, Van Luyn MJA. Tissue Eng. 2006;12:1955. doi: 10.1089/ten.2006.12.1955. [DOI] [PubMed] [Google Scholar]
- 94.Zhao QH, McNally AK, Rubin KR, Renier M, Wu Y, Rosecaprara V, Anderson JM, Hiltner A, Urbanski P, Stokes K. J Biomed Mater Res. 1993;27:379. doi: 10.1002/jbm.820270311. [DOI] [PubMed] [Google Scholar]
- 95.Mosser DM, Edwards JP. Nat Rev Immunol. 2008;8:958. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jones JA, Chang DT, Meyerson H, Colton E, Kwon IK, Matsuda T, Anderson JM. J Biomed Mater Res Part A. 2007;83A:585. doi: 10.1002/jbm.a.31221. [DOI] [PubMed] [Google Scholar]
- 97.Anderson JM, Defife K, McNally A, Collier T, Jenney C. J Mater Sci-Mater Med. 1999;10:579. doi: 10.1023/a:1008976531592. [DOI] [PubMed] [Google Scholar]
- 98.Kao WYJ, Zhao QH, Hiltner A, Anderson JM. J Biomed Mater Res. 1994;28:73. doi: 10.1002/jbm.820280110. [DOI] [PubMed] [Google Scholar]
- 99.Sieminski AL, Gooch KJ. Biomaterials. 2000;21:2233. doi: 10.1016/s0142-9612(00)00149-6. [DOI] [PubMed] [Google Scholar]
- 100.Thomé-Duret V, Gangnerau MN, Zhang Y, Wilson GS, Reach G. Diabetes Metab. 1996;22:174. [PubMed] [Google Scholar]
- 101.Gerritsen M, Jansen JA, Kros A, Vriezema DM, Sommerdijk N, Nolte RJM, Lutterman JA, Van Hovell S, Van der Gaag A. J Biomed Mater Res. 2001;54:69. doi: 10.1002/1097-4636(200101)54:1<69::aid-jbm8>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 102.Gifford R, Kehoe JJ, Barnes SL, Kornilayev BA, Alterman MA, Wilson GS. Biomaterials. 2006;27:2587. doi: 10.1016/j.biomaterials.2005.11.033. [DOI] [PubMed] [Google Scholar]
- 103.Gifford R, Batchelor MM, Lee Y, Gokulrangan G, Meyerhoff ME, Wilson GS. J Biomed Mater Res Part A. 2005;75A:755. doi: 10.1002/jbm.a.30359. [DOI] [PubMed] [Google Scholar]
- 104.Wisniewski N, Klitzman B, Miller B, Reichert WM. J Biomed Mater Res. 2001;57:513. doi: 10.1002/1097-4636(20011215)57:4<513::aid-jbm1197>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 105.Lowry JP, O'Neill RD, Boutelle MG, Fillenz M. J Neurochem. 1998;70:391. doi: 10.1046/j.1471-4159.1998.70010391.x. [DOI] [PubMed] [Google Scholar]
- 106.Klueh U, Kaur M, Qiao Y, Kreutzer DL. Biomaterials. 2010;31:4540. doi: 10.1016/j.biomaterials.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sharkawy AA, Klitzman B, Truskey GA, Reichert WM. J Biomed Mater Res. 1997;37:401. doi: 10.1002/(sici)1097-4636(19971205)37:3<401::aid-jbm11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 108.Dungel P, Long N, Yu B, Moussy Y, Moussy F. J Biomed Mater Res Part A. 2008;85A:699. doi: 10.1002/jbm.a.31593. [DOI] [PubMed] [Google Scholar]
- 109.Koschwanez HE, Reichert WM, Klitzman B. J Biomed Mater Res Part A. 2010;93A:1348. doi: 10.1002/jbm.a.32630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jablecki M, Gough DA. Anal Chem. 2000;72:1853. doi: 10.1021/ac991018z. [DOI] [PubMed] [Google Scholar]
- 111.Novak MT, Yuan F, Reichert WM. Anal Bioanal Chem. 2010;398:1695. doi: 10.1007/s00216-010-4097-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhang YN, Bindra DS, Barrau MB, Wilson GS. Biosens Bioelectron. 1991;6:653. doi: 10.1016/0956-5663(91)87018-7. [DOI] [PubMed] [Google Scholar]
- 113.Marchant R, Hiltner A, Hamlin C, Rabinovitch A, Slobodkin R, Anderson JM. J Biomed Mater Res. 1983;17:301. doi: 10.1002/jbm.820170209. [DOI] [PubMed] [Google Scholar]
- 114.Schutte RJ, Xie L, Klitzman B, Reichert WM. Biomaterials. 2009;30:160. doi: 10.1016/j.biomaterials.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ward WK, Slobodzian EP, Tiekotter KL, Wood MD. Biomaterials. 2002;23:4185. doi: 10.1016/s0142-9612(02)00160-6. [DOI] [PubMed] [Google Scholar]
- 116.Makale MT, Chen PC, Gough DA. Am J Physiol-Heart C. 2005;289:H57. doi: 10.1152/ajpheart.01001.2004. [DOI] [PubMed] [Google Scholar]
- 117.Ertefai S, Gough DA. J Biomed Eng. 1989;11:362. doi: 10.1016/0141-5425(89)90097-6. [DOI] [PubMed] [Google Scholar]
- 118.Koschwanez HE, Klitzman B, Reichert WM. J Diabetes Sci Technol. 2008;2:977. doi: 10.1177/193229680800200608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Klueh U, Kreutzer DL. Diabetes Technol Ther. 2005;7:727. doi: 10.1089/dia.2005.7.727. [DOI] [PubMed] [Google Scholar]
- 120.Mang A, Pill J, Gretz N, Kränzlin B, Buck H, Schoemaker M, Petrich W. Diabetes Technol Ther. 2005;7:163. doi: 10.1089/dia.2005.7.163. [DOI] [PubMed] [Google Scholar]
- 121.Koschwanez HE, Yap FY, Klitzman B, Reichert WM. J Biomed Mater Res Part A. 2008;87A:792. doi: 10.1002/jbm.a.31824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Moussy F, Harrison DJ, Rajotte RV. Int J Artif Organs. 1994;17:88. [PubMed] [Google Scholar]
- 123.Wagner JG, Schmidtke DW, Quinn CP, Fleming TF, Bernacky B, Heller A. Proc Natl Acad Sci U S A. 1998;95:6379. doi: 10.1073/pnas.95.11.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Valdes TI, Klueh U, Kreutzer D, Moussy F. J Biomed Mater Res Part A. 2003;67A:215. doi: 10.1002/jbm.a.10055. [DOI] [PubMed] [Google Scholar]
- 125.Valdes TI, Kreutzer D, Moussy F. J Biomed Mater Res. 2002;62:273. doi: 10.1002/jbm.10152. [DOI] [PubMed] [Google Scholar]
- 126.Wisniewski N, Rajamand N, Adamsson U, Lins PE, Reichert WM, Klitzman B, Ungerstedt U. Am J Physiol-Endocrinol Metab. 2002;282:E1316. doi: 10.1152/ajpendo.00259.2001. [DOI] [PubMed] [Google Scholar]
- 127.Blakytny R, Jude E. Diabetic Med. 2006;23:594. doi: 10.1111/j.1464-5491.2006.01773.x. [DOI] [PubMed] [Google Scholar]
- 128.Gerritsen H, Lutterman JA, Jansen JA. J Biomed Mater Res. 2000;52:135. doi: 10.1002/1097-4636(200010)52:1<135::aid-jbm17>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 129.Le NN, Rose MB, Levinson H, Klitzman B. J Diabetes Sci Technol. 2011;5:605. doi: 10.1177/193229681100500315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Li DJ, Ohsaki K, Ii K, Cui PC, Ye Q, Baba K, Wang QC, Tenshin S, Takano-Yamamoto T. J Biomed Mater Res. 1999;45:322. doi: 10.1002/(sici)1097-4636(19990615)45:4<322::aid-jbm6>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 131.Helton KL, Ratner BD, Wisniewski NA. J Diabetes Sci Technol. 2011;5:647. doi: 10.1177/193229681100500318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Picha GJ, Drake RF. J Biomed Mater Res. 1996;30:305. doi: 10.1002/(SICI)1097-4636(199603)30:3<305::AID-JBM5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 133.Klueh U, Liu Z, Feldman B, Henning TP, Cho B, Ouyang T, Kreutzer D. J Diabetes Sci Technol. 2011;5:583. doi: 10.1177/193229681100500313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Turner RFB, Harrison DJ, Rojotte RV. Biomaterials. 1991;12:361. doi: 10.1016/0142-9612(91)90003-s. [DOI] [PubMed] [Google Scholar]
- 135.Mercado RC, Moussy F. Biosens Bioelectron. 1998;13:133. doi: 10.1016/s0956-5663(97)00112-7. [DOI] [PubMed] [Google Scholar]
- 136.Zhang Y, Wilson GS. Anal Chim Acta. 1993;281:513. [Google Scholar]
- 137.Ward WK, Jansen LB, Anderson E, Reach G, Klein J-C, Wilson GS. Biosens Bioelectron. 2002;17:181. doi: 10.1016/s0956-5663(01)00268-8. [DOI] [PubMed] [Google Scholar]
- 138.Mathur AB, Collier TO, Kao WJ, Wiggins M, Schubert MA, Hiltner A, Anderson JM. J Biomed Mater Res. 1997;36:246. doi: 10.1002/(sici)1097-4636(199708)36:2<246::aid-jbm14>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 139.Anderson JM, Hiltner A, Wiggins MJ, Schubert MA, Collier TO, Kao WJ, Mathur AB. Polym Int. 1998;46:163. [Google Scholar]
- 140.Jones JA, Dadsetan M, Collier TO, Ebert M, Stokes KS, Ward RS, Hiltner PA, Anderson JM. Journal of Biomaterials Science, Polymer Edition. 2004;15:567. doi: 10.1163/156856204323046843. [DOI] [PubMed] [Google Scholar]
- 141.Yu B, Long N, Moussy Y, Moussy F. Biosens Bioelectron. 2006;21:2275. doi: 10.1016/j.bios.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 142.Hetrick EM, Schoenfisch MH. Chem Soc Rev. 2006;35:780. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]
- 143.Quinn CP, Pathak CP, Heller A, Hubbell JA. Biomaterials. 1995;16:389. doi: 10.1016/0142-9612(95)98856-9. [DOI] [PubMed] [Google Scholar]
- 144.Quinn CAP, Connor RE, Heller A. Biomaterials. 1997;18:1665. doi: 10.1016/s0142-9612(97)00125-7. [DOI] [PubMed] [Google Scholar]
- 145.Jhon MS, Andrade JD. J Biomed Mater Res. 1973;7:509. doi: 10.1002/jbm.820070604. [DOI] [PubMed] [Google Scholar]
- 146.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 147.Yu BZ, Wang CY, Ju YM, West L, Harmon J, Moussy Y, Moussy F. Biosens Bioelectron. 2008;23:1278. doi: 10.1016/j.bios.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 148.Ju YM, Yu B, West L, Moussy Y, Moussy F. J Biomed Mater Res Part A. 2010;92A:650. doi: 10.1002/jbm.a.32400. [DOI] [PubMed] [Google Scholar]
- 149.Ju YM, Yu B, Koob TJ, Moussy Y, Moussy F. J Biomed Mater Res Part A. 2008;87A:136. doi: 10.1002/jbm.a.31756. [DOI] [PubMed] [Google Scholar]
- 150.van Tienen TG, Heijkants RGJC, Buma P, de Groot JH, Pennings AJ, Veth RPH. Biomaterials. 2002;23:1731. doi: 10.1016/s0142-9612(01)00280-0. [DOI] [PubMed] [Google Scholar]
- 151.Sanders JE, Cassisi DV, Neumann T, Golledge SL, Zachariah SG, Ratner BD, Bale SD. J Biomed Mater Res Part A. 2003;65A:462. doi: 10.1002/jbm.a.10525. [DOI] [PubMed] [Google Scholar]
- 152.Karp RD, Johnson KH, Buoen LC, Ghobrial HK, Brand I, Brand KG. J Natl Cancer Inst. 1973;51:1275. doi: 10.1093/jnci/51.4.1275. [DOI] [PubMed] [Google Scholar]
- 153.Sharkawy AA, Klitzman B, Truskey GA, Reichert WM. J Biomed Mater Res. 1998;40:586. doi: 10.1002/(sici)1097-4636(19980615)40:4<586::aid-jbm10>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 154.Sharkawy AA, Klitzman B, Truskey GA, Reichert WM. J Biomed Mater Res. 1998;40:598. doi: 10.1002/(sici)1097-4636(19980615)40:4<598::aid-jbm11>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 155.Brauker JH, Carr-Brendel VE, Martinson LA, Crudele J, Johnston WD, Johnson RC. J Biomed Mater Res. 1995;29:1517. doi: 10.1002/jbm.820291208. [DOI] [PubMed] [Google Scholar]
- 156.Updike SJ, Shults MC, Gilligan BJ, Rhodes RK. Diabetes Care. 2000;23:208. doi: 10.2337/diacare.23.2.208. [DOI] [PubMed] [Google Scholar]
- 157.Marshall AJ, Irvin CA, Barker T, Sage EH, Hauch KD, Ratner BD. ACS Polym Prepr. 2004;45:100. [Google Scholar]
- 158.Ratner BD. ACS Polym Prepr. 2009;50:286. [Google Scholar]
- 159.Linnes MP, Ratner BD, Giachelli CM. Biomaterials. 2007;28:5298. doi: 10.1016/j.biomaterials.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Madden LR, Mortisen DJ, Sussman EM, Dupras SK, Fugate JA, Cuy JL, Hauch KD, Laflamme MA, Murry CE, Ratner BD. Proc Natl Acad Sci USA. 2010;107:15211. doi: 10.1073/pnas.1006442107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ramakrishna S, Fujihara K, Teo WE, Yong T, Ma ZW, Ramaseshan R. Mater Today. 2006;9:40. [Google Scholar]
- 162.Greiner A, Wendorff JH. Angew Chem, Int Ed. 2007;46:5670. doi: 10.1002/anie.200604646. [DOI] [PubMed] [Google Scholar]
- 163.Cao H, McHugh K, Chew SY, Anderson JM. Journal of Biomedical Materials Research Part A. 2010;93A:1151. doi: 10.1002/jbm.a.32609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Holt B, Tripathi A, Morgan J. J Biomech. 2008;41:2689. doi: 10.1016/j.jbiomech.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hendriks FM, Brokken D, Van Eemeren JTWM, Oomens CWJ, Baaijens FPT, Horsten JBAM. Skin Research and Technology. 2003;9:274. doi: 10.1034/j.1600-0846.2003.00019.x. [DOI] [PubMed] [Google Scholar]
- 166.Van Houten EEW, Doyley MM, Kennedy FE, Weaver JB, Paulsen KD. J Magn Reson Im. 2003;17:72. doi: 10.1002/jmri.10232. [DOI] [PubMed] [Google Scholar]
- 167.Diridollou S, Black D, Lagarde JM, Gall Y, Berson M, Vabre V, Patat F, Vaillant L. Int J Cosmetic Sci. 2000;22:421. [PubMed] [Google Scholar]
- 168.Barel A, Courage WPC. In: Handbook of non-invasive methods and the skin. Serup J, Jemec G, editors. CRC Press; Boca Raton (FL): 1995. [Google Scholar]
- 169.Subbaroyan J, Martin DC, Kipke DR. Journal of Neural Engineering. 2005;2:103. doi: 10.1088/1741-2560/2/4/006. [DOI] [PubMed] [Google Scholar]
- 170.Irwin EF, Saha K, Rosenbluth M, Gamble LJ, Castner DG, Healy KE. J Biomat Sci-Polym E. 2008;19:1363. doi: 10.1163/156856208786052407. [DOI] [PubMed] [Google Scholar]
- 171.Engler AJ, Griffin MA, Sen S, Bonnetnann CG, Sweeney HL, Discher DE. J Cell Biol. 2004;166:877. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Koh A, Nichols SP, Schoenfisch MH. J Diabetes Sci Technol. 2011;5:1052. doi: 10.1177/193229681100500505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Perretti M, Ahluwalia A. Microcirculation. 2000;7:147. [PubMed] [Google Scholar]
- 174.Coutinho AE, Chapman KE. Mol Cell Endocrinol. 2011;335:2. doi: 10.1016/j.mce.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Clark AR. Mol Cell Endocrinol. 2007;275:79. doi: 10.1016/j.mce.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 176.De Bosscher K, Haegeman G. Mol Endocrinol. 2009;23:281. doi: 10.1210/me.2008-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rhen T, Cidlowski JA. N Engl J Med. 2005;353:1711. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 178.Ziesche E, Scheiermann P, Bachmann M, Sadik CD, Hofstetter C, Zwissler B, Pfeilschifter J, Muhl H. Clin Exp Immunol. 2009;157:370. doi: 10.1111/j.1365-2249.2009.03969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Jamieson AM, Yu S, Annicelli CH, Medzhitov R. Cell Host Microbe. 2010;7:103. doi: 10.1016/j.chom.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Moussy Y, Hersh L, Dungel P. Biotechnol Prog. 2006;22:819. doi: 10.1021/bp060031j. [DOI] [PubMed] [Google Scholar]
- 181.Ward WK, Hansen JC, Massoud RG, Engle JM, Takeno MM, Hauch KD. J Biomed Mater Res Part A. 2010;94A:280. doi: 10.1002/jbm.a.32651. [DOI] [PubMed] [Google Scholar]
- 182.Dang TT, Bratlie KM, Bogatyrev SR, Chen XY, Langer R, Anderson DG. Biomaterials. 2011;32:4464. doi: 10.1016/j.biomaterials.2011.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Burgess D, Hickey A. In: Encyclopedia of pharmaceutical technology. Swarbrick J, Boylon J, editors. Marcel Dekker; New York, Basel: 1994. [Google Scholar]
- 184.Hickey T, Kreutzer D, Burgess DJ, Moussy F. Biomaterials. 2002;23:1649. doi: 10.1016/s0142-9612(01)00291-5. [DOI] [PubMed] [Google Scholar]
- 185.Eroglu H, Kas HS, Oner L, Turkoglu OF, Akalan N, Sargon MF, Ozer N. J Microencapsul. 2001;18:603. doi: 10.1080/02652040010019587. [DOI] [PubMed] [Google Scholar]
- 186.Bhardwaj U, Sura R, Papadimitrakopoulos F, Burgess DJ. J Diabetes Sci Technol. 2007;1:8. doi: 10.1177/193229680700100103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ju YM, Yu BZ, West L, Moussy Y, Moussy F. J Biomed Mater Res Part A. 2010;93A:200. doi: 10.1002/jbm.a.32512. [DOI] [PubMed] [Google Scholar]
- 188.Patil SD, Papadimitrakopoulos F, Burgess DJ. Diabetes Technol Ther. 2004;6:887. doi: 10.1089/dia.2004.6.887. [DOI] [PubMed] [Google Scholar]
- 189.Patil SD, Papadmitrakopoulos F, Burgess DJ. J Control Release. 2007;117:68. doi: 10.1016/j.jconrel.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 190.Sung JY, Barone PW, Kong HJ, Strano MS. Biomaterials. 2009;30:622. doi: 10.1016/j.biomaterials.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 191.Norton LW, Koschwanez HE, Wisniewski NA, Klitzman B, Reichert WM. J Biomed Mater Res Part A. 2007;81A:858. doi: 10.1002/jbm.a.31088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Klueh U, Kaur M, Montrose DC, Kreutzer DL. J Diabetes Sci Technol. 2007;1:496. doi: 10.1177/193229680700100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ward WK, Troupe JE. Asaio J. 1999;45:555. doi: 10.1097/00002480-199911000-00009. [DOI] [PubMed] [Google Scholar]
- 194.Mou X, Lennartz MR, Loegering DJ, Stenken JA. J Diabetes Sci Technol. 2011;5:619. doi: 10.1177/193229681100500316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Werner S, Grose R. Physiol Rev. 2003;83:835. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 196.Brown LF, Yeo KT, Berse B, Yeo TK, Senger DR, Dvorak HF, Vandewater L. J Exp Med. 1992;176:1375. doi: 10.1084/jem.176.5.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Frank S, Hubner G, Breier G, Longaker MT, Greenhalgh DG, Werner S. J Biol Chem. 1995;270:12607. doi: 10.1074/jbc.270.21.12607. [DOI] [PubMed] [Google Scholar]
- 198.Ferrara N. Endocr Rev. 2004;25:581. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 199.Nagy JA, Benjamin L, Zeng HY, Dvorak AM, Dvorak HF. Angiogenesis. 2008;11:109. doi: 10.1007/s10456-008-9099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Zhao Q, Egashira K, Inoue S, Usui M, Kitamoto S, Ni W, Ishibashi M, Hiasa K-i, Ichiki T, Shibuya M, Takeshita A. Circulation. 2002;105:1110. doi: 10.1161/hc0902.104718. [DOI] [PubMed] [Google Scholar]
- 201.Reinders MEJ, Sho M, Izawa A, Wang P, Mukhopadhyay D, Koss KE, Geehan CS, Luster AD, Sayegh MH, Briscoe DM. J Clin Invest. 2003;112:1655. doi: 10.1172/JCI17712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Ward WK, Quinn MJ, Wood MD, Tiekotter KL, Pidikiti S, Gallagher JA. Biosens Bioelectron. 2003;19:155. doi: 10.1016/s0956-5663(03)00180-5. [DOI] [PubMed] [Google Scholar]
- 203.Ward WK, Wood MD, Casey HM, Quinn MJ, Federiuk IF. Diabetes Technol Ther. 2004;19:155. doi: 10.1089/152091504773731320. [DOI] [PubMed] [Google Scholar]
- 204.Klueh U, Dorsky DI, Kreutzer DL. Biomaterials. 2005;26:1155. doi: 10.1016/j.biomaterials.2004.04.017. [DOI] [PubMed] [Google Scholar]
- 205.Kim T-K, Burgess DJ. JPharm Pharmacol. 2002;54:897. doi: 10.1211/002235702760089009. [DOI] [PubMed] [Google Scholar]
- 206.Norton LW, Tegnell E, Toporek SS, Reichert WM. Biomaterials. 2005;26:3285. doi: 10.1016/j.biomaterials.2004.07.069. [DOI] [PubMed] [Google Scholar]
- 207.Edelman JL, Lutz D, Castro MR. Exp Eye Res. 2005;80:249. doi: 10.1016/j.exer.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 208.Machein MR, Kullmer J, Ronicke V, Machein U, Krieg M, Damert A, Breier G, Risau W, Plate KH. Neuropathol Appl Neurobiol. 1999;25:104. doi: 10.1046/j.1365-2990.1999.00166.x. [DOI] [PubMed] [Google Scholar]
- 209.Wu WS, Wang FS, Yang KD, Huang CC, Kuo YR. J Invest Dermatol. 2006;126:1264. doi: 10.1038/sj.jid.5700274. [DOI] [PubMed] [Google Scholar]
- 210.Ignarro LJ. Angew Chem-Int Edit. 1999;38:1882. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<1882::AID-ANIE1882>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 211.Walford G, Loscalzo J. J Thromb Haemost. 2003;1:2112. doi: 10.1046/j.1538-7836.2003.00345.x. [DOI] [PubMed] [Google Scholar]
- 212.Griffith OW, Stuehr DJ. Annu Rev Physiol. 1995;57:707. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 213.Cooke JP. Atheroscler Suppl. 2003;4:53. doi: 10.1016/s1567-5688(03)00034-5. [DOI] [PubMed] [Google Scholar]
- 214.Dulak J, Jozkowicz A. Antioxid Redox Signal. 2003;5:123. doi: 10.1089/152308603321223612. [DOI] [PubMed] [Google Scholar]
- 215.Schwentker A, Vodovotz Y, Weller R, Billiar TR. Nitric Oxide-Biol Chem. 2002;7:1. doi: 10.1016/s1089-8603(02)00002-2. [DOI] [PubMed] [Google Scholar]
- 216.Carreau A, Kieda C, Grillon C. Exp Cell Res. 2011;317:29. doi: 10.1016/j.yexcr.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 217.Carpenter AW, Schoenfisch MH. Chem Soc Rev. 2012;41:3742. doi: 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Wang PG, Xian M, Tang XP, Wu XJ, Wen Z, Cai TW, Janczuk AJ. Chem Rev. 2002;102:1091. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 219.Jen MC, Serrano MC, van Lith R, Ameer GA. Adv Funct Mater. 2012;22:239. doi: 10.1002/adfm.201101707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hrabie JA, Keefer LK. Chem Rev. 2002;102:1135. doi: 10.1021/cr000028t. [DOI] [PubMed] [Google Scholar]
- 221.Davies KM, Wink DA, Saavedra JE, Keefer LK. J Am Chem Soc. 2001;123:5473. doi: 10.1021/ja002899q. [DOI] [PubMed] [Google Scholar]
- 222.Williams DLH. Acc Chem Res. 1999;32:869. [Google Scholar]
- 223.Amadeu TP, Seabra AB, de Oliveira MG, Costa AMA. J Eur Acad Dermatol Venereol. 2007;21:629. doi: 10.1111/j.1468-3083.2006.02032.x. [DOI] [PubMed] [Google Scholar]
- 224.Amadeu TP, Seabra AB, de Oliveira MG, Monte-Alto-Costa A. J Surg Res. 2008;149:84. doi: 10.1016/j.jss.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 225.Coneski PN, Nash JA, Schoenfisch MH. ACS Appl Mater Interfaces. 2011;3:426. doi: 10.1021/am101010e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Shin JH, Metzger SK, Schoenfisch MH. J Am Chem Soc. 2007;129:4612. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 227.Shin JH, Schoenfisch MH. Chem Mat. 2008;20:239. doi: 10.1021/cm702526q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Riccio DA, Nugent JL, Schoenfisch MH. Chem Mat. 2011;23:1727. doi: 10.1021/cm102510q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Riccio DA, Dobmeier KP, Hetrick EM, Privett BJ, Paul HS, Schoenfisch MH. Biomaterials. 2009;30:4494. doi: 10.1016/j.biomaterials.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hetrick EM, Schoenfisch MH. Biomaterials. 2007;28:1948. doi: 10.1016/j.biomaterials.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 231.Nablo BJ, Rothrock AR, Schoenfisch MH. Biomaterials. 2005;26:917. doi: 10.1016/j.biomaterials.2004.03.031. [DOI] [PubMed] [Google Scholar]
- 232.Marxer SM, Rothrock AR, Nablo BJ, Robbins ME, Schoenfisch MH. Chem Mat. 2003;15:4193. [Google Scholar]
- 233.Nablo BJ, Schoenfisch MH. J Biomed Mater Res Part A. 2003;67A:1276. doi: 10.1002/jbm.a.20030. [DOI] [PubMed] [Google Scholar]
- 234.Robbins ME, Schoenfisch MH. J Am Chem Soc. 2003;125:6068. doi: 10.1021/ja034019o. [DOI] [PubMed] [Google Scholar]
- 235.Nablo BJ, Chen TY, Schoenfisch MH. J Am Chem Soc. 2001;123:9712. doi: 10.1021/ja0165077. [DOI] [PubMed] [Google Scholar]
- 236.Riccio DA, Coneski PN, Nichols SP, Broadnax AD, Schoenfisch MH. ACS Appl Mater Interfaces. 2012;4:796. doi: 10.1021/am201443r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Riccio DA, Schoenfisch MH. Chem Soc Rev. 2012;41:3731. doi: 10.1039/c2cs15272j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Hetrick EM, Prichard HL, Klitzman B, Schoenfisch MH. Biomaterials. 2007;28:4571. doi: 10.1016/j.biomaterials.2007.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Nichols SP, Le NN, Klitzman B, Schoenfisch MH. Anal Chem. 2011;83:1180. doi: 10.1021/ac103070t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Nichols SP, Koh A, Brown NL, Rose MB, Sun B, Slomberg DL, Riccio DA, Klitzman B, Schoenfisch MH. Biomaterials. 2012;33:6305. doi: 10.1016/j.biomaterials.2012.05.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Sachedina N, Pickup JC. Diabetic Med. 2003;20:1012. doi: 10.1046/j.1464-5491.2003.01037.x. [DOI] [PubMed] [Google Scholar]
- 242.Weinstein RL, Schwartz SL, Brazg RL, Bugler JR, Peyser TA, McGarraugh GV. Diabetes Care. 2007;30:1125. doi: 10.2337/dc06-1602. [DOI] [PubMed] [Google Scholar]
- 243.Linke B, Kerner W, Kiwit M, Pishko M, Heller A. Biosens Bioelectron. 1994;9:151. doi: 10.1016/0956-5663(94)80107-x. [DOI] [PubMed] [Google Scholar]
- 244.Group DRiCNDS. Diabetes Care. 2004;27:722. [Google Scholar]
- 245.Tierney MJ, Tamada JA, Potts RO, Jovanovic L, Garg S. Biosens Bioelectron. 2001;16:621. doi: 10.1016/s0956-5663(01)00189-0. [DOI] [PubMed] [Google Scholar]
- 246.Rossetti P, Porcellati F, Fanelli CG, Bolli GB. Diabetes Technol Ther. 2006;8:326. doi: 10.1089/dia.2006.8.326. [DOI] [PubMed] [Google Scholar]
- 247.Barone PW, Parker RS, Strano MS. Anal Chem. 2005;77:7556. doi: 10.1021/ac0511997. [DOI] [PubMed] [Google Scholar]
- 248.Barone PW, Strano MS. Angew Chem-Int Edit. 2006;118:8318. [Google Scholar]
- 249.Liang F, Pan T, Sevick-Muraca EM. Optical Diagnostics and Sensing. San Jose, CA: 2005. p. 7. [Google Scholar]
- 250.Larin KV, Eledrisi MS, Motamedi M, Esenaliev RO. Diabetes Care. 2002;25:2263. doi: 10.2337/diacare.25.12.2263. [DOI] [PubMed] [Google Scholar]
- 251.Weiss R, Yegorchikov Y, Shusterman A, Raz I. Diabetes Technol Ther. 2007;9:68. doi: 10.1089/dia.2006.0059. [DOI] [PubMed] [Google Scholar]
- 252.Malchoff CD, Shoukri K, Landau JI, Buchert JM. Diabetes Care. 2002;25:2268. doi: 10.2337/diacare.25.12.2268. [DOI] [PubMed] [Google Scholar]
- 253.Rawer R, Stork W, Müller-Glaser K-D. Biomed Tech. 2002;47:186. doi: 10.1515/bmte.2002.47.s1a.186. [DOI] [PubMed] [Google Scholar]
- 254.Cameron BD, Anumula H. Diabetes Technol Ther. 2006;8:156. doi: 10.1089/dia.2006.8.156. [DOI] [PubMed] [Google Scholar]
- 255.Enejder AMK, Scecina TG, Oh J, Hunter M, Shih W-C, Sasic S, Horowitz GL, Feld MS. J Biomed Opt. 2005;10:031114. doi: 10.1117/1.1920212. [DOI] [PubMed] [Google Scholar]
- 256.Alexeev VL, Das S, Finegold DN, Asher SA. Clin Chem. 2004;50:2353. doi: 10.1373/clinchem.2004.039701. [DOI] [PubMed] [Google Scholar]
- 257.Gourzi M, Rouane A, Guelaz R, Alavi M, McHugh M, Nadi M, Roth P. J Med Eng Technol. 2005;29:22. doi: 10.1080/03091900410001720247. [DOI] [PubMed] [Google Scholar]
- 258.Wentholt I, Hoekstra J, Zwart A, DeVries J. Diabetologia. 2005;48:1055. doi: 10.1007/s00125-005-1754-y. [DOI] [PubMed] [Google Scholar]