

Abstract
Small cell lung cancer (SCLC) is an aggressive disease with one of the highest case-fatality rates among cancer. The recommended therapy for SCLC has not changed significantly over the past 30 years; new therapeutic approaches are a critical need. TP53 is mutated in the majority of SCLC cases and its loss is required in transgenic mouse models of the disease. We synthesized an array of biodegradable poly(beta-amino ester) (PBAE) polymers which self-assemble with DNA and assayed for transfection efficiency in the p53-mutant H446 SCLC cell line using high-throughput methodologies. Two of the top candidates were selected for further characterization and TP53 delivery in vitro and in vivo. Nanoparticle delivery of TP53 resulted in expression of exogenous p53, induction of p21, induction of apoptosis and accumulation of cells in sub-G1 consistent with functional p53 activity. Intratumoral injection of subcutaneous H446 xenografts with polymers carrying TP53 caused marked tumor growth inhibition. This is the first demonstration of TP53 gene therapy in SCLC using non-viral polymeric nanoparticles. This technology may have general applicability as a novel anti-cancer strategy based on restoration of tumor suppressor gene function.
Keywords: p53, PBAE, nanoparticles, gene therapy, SCLC
Introduction
Small cell lung cancer (SCLC) represents 15% of all lung cancer cases and has one of the highest case-fatality rates of all cancers with nearly as many deaths as diagnoses per year. In 2011 over 25,000 deaths were attributable to this disease in the US alone (1). The median survival of patients diagnosed with SCLC is less than 1 year. This statistic has not changed significantly over the past 3 decades despite over 52 randomized phase 3 clinical trials evaluating numerous cytotoxic chemotherapies (2). The combination of etoposide and cisplatin has been the standard first-line therapy for SCLC since the 1980s. In 2003 topotecan became the only drug approved for treatment of patients with relapsed SCLC. New therapeutic approaches are needed to improve long-term survival in this disease.
SCLC exhibits certain recurrent genetic alterations most notably inactivation of the tumor suppressor genes, TP53 and RB. TP53 encodes a transcription factor whose targets regulate cell cycle progression, senescence, DNA repair and apoptosis (3, 4). TP53 mutations are the most common genetic alteration in human cancer, occurring in over 50% of cases (5, 6). Wild-type (WT) p53 activity can also be abrogated by endogenous MDM2 or viral proteins; the human papilloma virus E6 protein, SV-40 large T antigen and adenovirus E1B-55kDa proteins can bind and attenuate p53 activity resulting in cellular transformation (7–9). In transgenic mouse models disruption of TP53 results in increased susceptibility to tumor development, most notably lymphomas and sarcomas. Restoration of p53 in these models results in potent antitumor activity in a cell-type specific manner; TP53 re-expression induces apoptosis in autochthonous lymphomas but senescence in sarcoma and hepatocellular carcinoma models (10, 11).
In SCLC, TP53 alterations are prevalent; among 67 independent SCLC cell lines and 231 primary SCLC tumors TP53 was mutated in 90% and 74% of cases respectively (12). Support for the critical role of TP53 in SCLC pathogenesis also derives from transgenic mouse models, in which Cre-mediated loss of TP53 and RB results in murine SCLC which shares histopathologic features of human SCLC including neural cell adhesion molecule (NCAM; CD56) expression, and elaboration of neuroendocrine (NE) markers such as synaptophysin and chromogranin (13). In this genetic background, AdenoCre placed under the control of the NE cell-specific calcitonin/calcitonin-gene related peptide (CGRP) promoter, but not a Clara-cell specific promoter, resulted in murine SCLC, implicating pulmonary NE cells as the putative cell of origin for SCLC (14). In vitro, Adachi et al, has demonstrated that expression of WT p53 in a p53-null SCLC cell line results in apoptosis (15). As loss of TP53 appears to be critical in SCLC development, restoration of functional p53 may have therapeutic efficacy.
Adenovirus is the one of the most widely studied gene therapy vectors; in non-small cell lung cancer (NSCLC), adenoviral-mediated TP53 (Ad.p53) delivery has been evaluated in several early-phase clinical trials (16, 17). Intratumoral (IT) delivery of Ad.p53 in combination with chemotherapy was found to be safe and histological examination of tumor tissue revealed apoptosis in Ad.p53 treated samples (16). A phase II study, however, failed to show increased response or local benefit of combined Ad.p53 and chemotherapy over chemotherapy alone (17). Adenoviral gene therapy has also been evaluated preclinically in SCLC models. Adenoviral delivery of a siRNA targeting the hepatocyte growth factor receptor, c-Met, in the H446 SCLC cell line resulted in decreased proliferation in vitro and tumor growth inhibition (TGI) in vivo (18). Similarly, adenoviral delivery of fragile histidine triad complex, a putative tumor suppressor gene often mutated in SCLC, induced apoptosis in multiple SCLC cell lines (19).
The use of viral vectors has been limited by safety concerns including insertional mutagenesis and toxicity as well as limited cargo capacity and manufacturing challenges (20, 21). Many patients have pre-existing humoral immunity to adenovirus, or rapidly develop neutralizing antibodies, limiting the potential of adenoviral therapies. Alternative approaches to gene delivery, using non-viral biomaterials such as inorganic nanoparticles, cationic lipids, liposomes, polymers, and peptides, have been limited by low efficicency, resulting in limited efficacy (22–24).
We have developed highly effective biomaterials for non-viral gene delivery to hard-to-transfect cells (23, 25–28). These poly(beta-amino ester) (PBAE) polymers are biodegradable due to ester linkages throughout the polymer backbone which allows for lower toxicity and release of DNA intracellularly. Through their secondary and tertiary amines, these polymers are also able to buffer the endosome, which facilitates endosomal escape (29, 30). Additionally, subtle changes to PBAE structure can improve specificity of transfection and these polymers have been adapted for gene delivery to various cell types including HUVECs, human retinal endothelial cells and human mesenchymal stem cells as well as glioblastoma multiforme, ovarian, prostate and pancreatic cell lines (25, 27, 31–33).
In this study, we sought to develop non-viral nanoparticles which could deliver therapeutic genes with high efficiency to SCLC cells. We synthesized an array of PBAEs using combinatorial chemistry (34) and found several polymers with transfection efficiencies comparable to commercially available agents in SCLC cell lines. These polymers may be generally useful as efficient gene-delivery vectors. As a proof of principle for this approach, we used two PBAE polymers to assess the activity of WT TP53 delivery to the p53-mutant H446 SCLC cell line in vitro and in vivo.
Methods
Plasmids, Chemicals and Reagents
Unless otherwise stated, all reagents were purchased from Sigma Chemical Company (St. Louis, MO USA) or Fisher Scientific (Pittsburgh, PA USA) in the highest available purity. The CMV-GFP (EGFPN1) and CMV-LUC plasmids were purchased from Elim Biopharm (Palo Alto, CA). p53-CMV-GFP plasmid (Addgene, San Diego, CA) contained CMV-promoter regulating the fusion of two gene segments: WT p53 and EGFPN1 backbone.
Monomers Reagents
Monomers used for polymer synthesis were the following: from Acros Organics [1-(3-aminopropyl)pyrrolidine (E8)], Alfa Aesar [1,4-butanediol diacrylate (B4), 1-(3-aminopropyl)-4-methylpiperazine (E7)], Fluka [2-(3-aminopropylamino)ethanol (E6), 3-amino-1-propanol (S3), 4-amino-1-butanol (S4), 5-amino-1-pentanol (S5)], Monomer-Polymer and Dajac Laboratories [1,3-propanediol diacrylate (B3), 1,5-pentanediol diacrylate (B5)], Sigma-Aldrich [1,3-butanediol diacrylate (B3m)], and TCI America [1,3-diaminopentane (E3), 2-methyl-1,5-diaminopentane (E4), (PEO)4-bis-amine (E5)] and as previously described (34).
Preparation of Cy5 labeled dsRED DNA
pDsRed-Max-N1 DNA (Promega, Madison, WI) was labeled with Cy5 fluorophore using the Label IT Cy 5 Tracker kit (Mirus, Madison, WI) as per manufacturer’s protocol and stored at −20°C in light protective conditions.
Cell lines
All cell lines were obtained from American Type Culture Collection (ATCC) and grown in complete RPMI [RPMI 1640 (Quality Biologicals, Gaithersburg, MD) supplemented with 10% fetal bovine serum, L-glutamine, pen/strep, sodium pyruvate, HEPES, and sodium bicarbonate] per ATCC recommendations. The H146, H187 and H446 cell lines were authenticated by STR analysis by the Fragment Analysis Facility at Johns Hopkins University. All cell lines were grown and multiple aliquots were cryopreserved after authentication. All cell lines were used within 6 months of resuscitation.
Synthesis of poly(beta-amino esters) (PBAEs)
The PBAEs were synthesized in a two-step reaction procedure (Figure 1). In the first step, the base polymers were synthesized by mixing diacrylates (B) with amino alcohols (S) at a molar ratio of 1.1:1 or 1.2:1. The reaction was performed in glass scintillating vials with teflon stir bars at 90°C for 24 hours. The base polymer was then dissolved in anhydrous dimethyl sulfoxide (DMSO) to 167 mg/mL. In the second step of the reaction, 480 μL of the base polymer solution is mixed with 320 μL of 0.5 M end-capping amine (E) in 1.5 mL eppendorf tubes and allowed to react in a shaker for 24 hours. Polymers were then divided into smaller volumes and stored at frozen and with desiccant until needed. These polymers were characterized with gel permeation chromatography and proton nuclear magnetic resonance as previously described (34). Select polymers, including polymers used in vivo, were made in larger quantities and further purified. In this case, after the base polymer reaction step, the polymers are dissolved in tetrahydrofuran (THF), instead of DMSO, and then end-capped as above, but with end-capped monomers also diluted in THF. Once the reaction is complete, the polymer is mixed with 4x the volume of ethyl ether. These mixtures were vortexed vigorously and then centrifuged at 4 krpm for 5 minutes, after which the supernatant containing unreacted monomer and organic solvents was removed. This process was then repeated a second time and the purified polymers dried under vacuum for 2 days. The polymers were then dissolved in anhydrous dimethyl sulfoxide and kept frozen with desiccant as described above.
Figure 1. Poly(beta-amino ester) synthesis scheme depicting the conjugate addition of amines to diacrylates in two steps.

The three R groups allow modifications to the polymer backbone (R), side chain (R′) and end-groups (R″). Each monomer comprises a backbone (B), a side chain (S) or an end-group (E).
Nanoparticle Preparation
For each transfection, PBAE:DNA nanoparticles were prepared by separately dissolving PBAE polymer and DNA in 25 mM sodium acetate (pH 5.0). The polymer and DNA solutions were mixed in equal volumes, gently pipetted and incubated for 10 min at RT. A range of w/w ratios were evaluated for the high-throughput screen and are described in the High-throughput screening of PBAE polymer library section found in the Supplemental Data. For all other experiments, PBAE:DNA ratios of 75 w/w and 30 w/w ratios were for in vitro and in vivo studies, respectively. Polymer and DNA stocks were stored at −20°C and thawed on ice before mixing and treating the cells. For Cy5-DsRed experiments, 0.1ug of Cy 5 labeled DsRED was included in the DNA mixtures and PBAE:DNA nanoparticles were prepared as described above.
Nanoparticle characterization
Plasmid DNA and PBAEs were separately diluted in 25 mM sodium acetate buffer to 0.06 mg/mL and either 1.8, 3.6, or 4.5 mg/mL, respectively (corresponding to PBAE:DNA weight/weight, wt/wt, ratios of 30, 60, or 90). Equal volumes of the DNA and PBAE solutions were mixed and incubated for 10 minutes at room temperature. The resulting solution of nanoparticles was then used either for sizing or DNA complexation studies. In the case of sizing, the samples were further diluted in PBS and loaded into a Nanosight NS500 nanoparticle tracking analysis (NTA) instrument. The solutions were diluted anywhere from 50x to 200x, so that the nanoparticle concentrations were appropriate for NTA analysis (35). NTA videos were captured for 60 seconds and analyzed using the NTA software, version 2.1. To further examine the ability of the polymers to bind to the DNA, the nanoparticle solutions were run on a gel using electrophoresis and compared to naked DNA. A 1% agarose gel with 0.1 μL/mL ethidium bromide was made. The naked DNA solution was mixed with a 30% glycerol loading buffer with bromophenol blue dye, while the nanoparticle solutions were mixed with the loading buffer without the dye to prevent interference between the dye and PBAE-DNA interaction. The gel was run for 30 min at 100V and imaged using the UVP BioDoc-It Imaging Center.
Transfection protocol
Adherent cells
H446 cells were seeded at low density and allowed to adhere overnight. PBAE:DNA nanoparticles were added to cells followed by gentle shaking. After 4 hours, the media was replaced with fresh media and the cells were incubated at 37°C for 44 h.
Suspension cells (H146, H187)
Cells were seeded in complete RPMI overnight then incubated with PBAE/DNA nanoparticles for 4 h with gentle shaking. After 4 h, cells were centrifuged at low speed for 2 min, fresh media was added and the cells further incubated at 37°C for next 44 hours.
Fluorescence microscopy
GFP positive cells were analyzed using a Motic AE31 inverted microscope using a FITC 480nm filter. Cy5 labeled cells were visualized and analyzed using a Zeiss Axio Observer microscope fitted with a Cy5 specific filter (emission wavelength of 690nm).
Flow cytometry
GFP fluorescence was used to indicate successful transfection, propidium iodide (PI) was used for dead cell discrimination, and Cy5 fluorescence to indicate successful nanoparticle association using a FACSCalibur flow cytometer (BD Biosciences, Rockville, MD). Briefly, adherent cells were trypsinized, centrifuged and resuspended in 500 μL of fresh media and analyzed by flow cytometry. A total of 10,000 cells were acquired per analysis. All analyses were run with triplicate samples. Data analysis was performed using BD CellQuest Pro software (BD Biosciences).
Western blot
Whole cell extracts were prepared using RIPA buffer containing protease and phosphatase inhibitors and clarified by low-speed centrifugation. The protein extracts were quantitated by BCA assay, resolved by SDS-PAGE on 4–12% Bis-Tris gels and transferred to PVDF membranes. Membranes were blotted with a 1:500 dilution of primary antibodies to p53 (Santa Cruz Biotechnologies), p21 (Calbiochem), anti-GFP (Abcam) and actin (Santa Cruz) and detected using HRP-conjugated secondary antibodies and visualized by ECL per protocol (Pierce).
Cell cycle analysis
Cells were prepared at 48 and 72 hrs post-treatment for cell cycle analysis following a standard PI staining protocol and analyzed by flow cytometry using a FACSCalibur flow cytometer and CellQuest Pro software. Each assay was run in triplicate and all data is presented as the mean +/− SEM.
Annexin V staining
To determine early apoptotic events, cells were assessed for Annexin V positivity. Cells were plated and transfected as described above. At the indicated time after transfection cells were detached from the tissue culture plates using Accutase (Sigma), stained with Annexin V Cy5 (BD Biosciences) per manufacturer’s directions and analyzed on a FACSCalibur flow cytometer. Cisplatin (10uM) treated cells were used as a positive control. Each assay was run in triplicate and all data is represented as the mean +/− SEM.
Fluorescence-activated cell sorting (FACS)
Cells were sorted using a using a FACS Aria II (BD Biosciences) into GFP-positive and GFP-negative populations 18 hrs post-transfection. Untreated cells passed through the sorting machine were used as an additional control. Post-sort analysis was completed within 2 hrs of collection, cells were replated in complete RPMI and assessed at 48 and 72hrs post-transfection.
In vivo experiments
Three-to-four week old athymic female nude mice were injected subcutaneously with 1.5x106 H446 cells suspended in PBS and mixed 1:1 in matrigel (BD Biosciences). Once tumor volumes reached 200 mm3, the mice were randomized [N=3–4] into three groups: 1) Untreated, 2) 457:CMV-LUC, 3) 457:CMV-p53-LUC. The PBAE:DNA nanoparticles were prepared at 30 w/w; 50 μg of DNA was used per IT injection and the mice were treated twice a week for 3 weeks. Tumor measurements, using a digital caliper, and weights were collected twice a week. Tumor volume was calculated as follows: tumor volume (mm3) = [length (mm) x width (mm)2]/2. Toxicity was monitored by weight loss and animal activity following ACUC protocols. Tumor growth curves are presented as mean +/− SEM. The area under curve (AUC) was calculated using GraphPad Prism estimating total AUC from day 1 to day 18 for respective treatment groups. The percentage inhibition in tumor growth was compared among all the treatment groups over the total time course of the study. Statistical significance was then determined by two-way ANOVA followed by Bonferroni post-hoc test. (* p ≤ 0.001 CMV-p53-LUC vs. CMV-LUC; # p ≤ 0.01 CMV-p53-LUC vs. untreated). All animal experiments were performed following JHU Animal Care and Use Committee regulations.
Results
We generated a polymer array of 30 structurally distinct PBAEs and over 120 nanoparticle formulations (Fig 1). This array contained base polymers composed of four backbone monomers that differed by single carbons between the acrylate groups (B3, B3m, B4, and B5 corresponded to 3 carbons, 3 carbons + 1 methyl group, 4 carbons, and 5 carbons respectively) and three side chain monomers (S3, S4, and S5 that differed by having 3, 4, and 5 carbons respectively between amine and alcohol groups). These base polymers were terminated with one of six amine-containing small molecule end-groups. Structures were validated by GPC and NMR (34). We performed a high-throughput luciferase-based screen of the PBAE array to identify leading polymers optimized for efficient transfection of SCLC cells. Gene delivery of exogenous luciferase plasmid resulted in luminescence that was quantified and normalized to untreated cells (Supplementary Fig. 1). The leading candidate polymers, such as 456 (Fig. 1), were able to transfect H446 cells several orders of magnitude better than other nanoparticle formulations, and resulted in transfection efficiencies as high or higher than both positive controls, FuGENE HD and Lipofectamine 2000. The size of the leading PBAE/DNA nanoparticles was examined using nanoparticle tracking analysis (35) and quantified by nanoparticle number-average size (Fig. 2A). DNA encapsulation and binding by the leading polymers was determined by gel electrophoresis, demonstrating that at the optimized PBAE:DNA weight:weight ratios used in our study, the DNA is completely complexed by the polymers (Fig 2B). In general, the nanoparticle diameters range from 100–200 nm, with the range for the leading polymers, a more narrow 100–150 nm. It has been previously reported that within this range, the size of the particle does not correlate strongly with transfection efficacy (29).
Figure 2. Nanoparticle characterization.
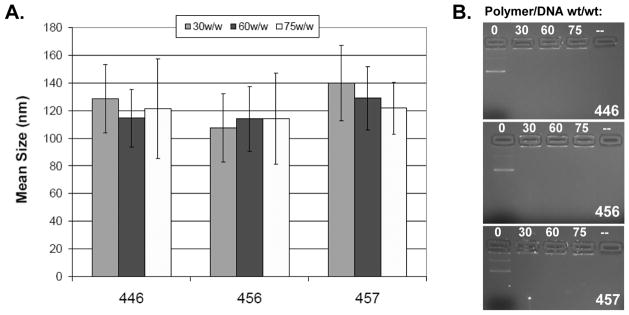
The ability of the PBAEs to form nanoparticles was examined using NTA to determine the nanoparticle size (A) and by gel electrophoresis (B). Gel electrophoresis of the nanoparticles shows that at the proper PBAE:DNA weight:weight ratios, the DNA is completely complexed by the polymers. In general, the nanoparticle diameters range from 100 nm to 200 nm, with the range for the select polymers shown from 100 – 150 nm.
The extensive luciferase-based screen identified PBAEs that could induce high levels of expression, but to identify those PBAEs which could transfect the highest percentage of cells, we used a GFP-based analysis. We selected 32 of the top candidates from the initial screen and quantitated the transfection efficiency by fluorescence microscopy and flow cytometry (Fig. 3A–B). Two of the top polymers, 456 and 457, exhibited transfection efficiencies of approximately 40%, comparable to the commercially available transfection agent, Fugene-HD (Fig. 3C, grey bars). The majority of the polymers also induced higher level GFP expression, quantitated by geometric mean fluorescence, compared with Fugene HD (Fig. 3C, black bars). We adapted our transfection protocol for suspension cells and found that 456 polymers could transfect H146 and H187 SCLC cell lines with effieciencies of 33 ±3% and 22±2%, respectively (Fig. 3D–F). As suspension cells are notoriously difficult to transfect, we found these results encouraging. We further assessed the ability of 456 polymers to transfect non-transformed cell lines. The transfection efficiency of our nanopartcles in the WI-38 and IMR-90 human lung fibroblast lines was 13% and 11%, respectively; therefore, the 456 polymers appeared to be fairly selective for SCLC over non-transformed cells (Supplementary Fig. 2). To determine whether the transfection efficiency was limited by poor nanoparticle association and uptake by SCLC cells, we transfected H446 cells with PBAEs complexed with a Cy5-labeled plasmid. We observed that >95% of cells were Cy5-positive (Supplementary Fig. 3) suggesting that events downstream of nanoparticle uptake such as intracellular trafficking, endosomal release or nuclear uptake and processing may be additional determinants of PBAE transfection.
Figure 3. A GFP-based secondary screen identifies several PBAE polymers that can deliver genes to adherent and suspension SCLC cell lines at efficiencies comparable to commercially available reagents.

(A–B) H446 cells were transfected with PBAEs complexed with CMV-GFP DNA then analyzed by microscopy and flow cytometry. (C) The transfection efficiency and geometric mean fluorescence of 15 PBAE polymers are shown; the PBAE polymers are indicated across the x-axis. Percent transfection is presented as the mean +/− SEM of triplicate runs and the geometric mean fluorescence is the geometric mean FL1 signal +/− SEM of triplicate runs. Fugene HD was used as a control. Statistical significance of geometric mean fluorescence was determined by one-way ANOVA comparing each group with Fugene-HD (*: p ≤ 0.01). (D–F) Two SCLC suspension cell lines, H146 and H187 cells were transfected with 456:CMV-GFP and analyzed by fluorescence microscopy and flow cytometry.
We next evaluated the ability of the 456 polymer to deliver functional p53. Transfection of H446 cells with 456:CMV-p53-GFP, but not 456:CMV-GFP, induced morphologic changes and punctate GFP localization (Fig. 4A). At 48 hrs post-transfection, 22% of cells transfected with 456:CMV-p53-GFP were strongly GFP-positive, compared to 41% of those transfected with 456:CMV-GFP (Fig. 4B–C). p53-GFP expression was seen as early as 2 hrs and peaked at 18–24 hrs post-transfection (Fig. 4D). Consistent with functional p53 activity, we observed p21 induction at 18–24 hrs, a significant increase of Annexin V positive cells at 48 hrs and cellular accumulation in sub-G1 at 72 hrs post-transfection with 456:CMV-p53-GFP (Fig. 4D–F). To ascertain the effect of WT p53 restoration in more homogeneous populations, we sorted H446 cells transfected with either 456:CMV-GFP or 456:CMV-p53-GFP into GFP-positive and GFP-negative populations (Fig. 5A). Post-sort (20 hrs post-transfection) flow cytometry confirmed that the sorted cells were relatively homogeneous (>80%) and GFP expression was maintained over 72 hrs (Fig. 5B). At 48 and 72 hrs, we observed robust p53-GFP expression and p21 induction in GFP-positive cells transfected with 456:CMV-p53-GFP (population 4), but in none of the other sorted populations (Fig. 5C). Cell cycle analysis also revealed > 40% of the population 4 cells had accumulated in sub-G1 at 48 and 72 hrs, consistent with functional p53 activity (Fig. 5D–E).
Figure 4. Delivery of p53-GFP by 456 polymers induces morphologic changes, p21 expression, apoptosis and sub-G1 accumulation in bulk transfected cells.

(A–B) H446 cells were transfected with 456 polymers complexed with either CMV-GFP plasmid or a CMV-p53-GFP and visualized by microscopy (A) and flow cytometry (B). (C) Percent transfection is presented as the mean +/− SEM of triplicate runs. (D) Cells were harvested at serial time points after transfection; lysates were examined for p53-GFP, p53 and p21 expression using anti-p53, anti-p21, anti-GFP (to visualize p53-GFP) antibodies. Actin was used as a loading control. (E) Annexin V staining was performed at 48 hours after transfection, cisplatin treated cells were used as a positive control for the assay. (F) Cell cycle analysis at 72 hours post-transfection.
Figure 5. Cells sorted for p53-GFP expression exhibit marked morphologic changes, p21 induction and accumulation in sub-G1 consistent with functional p53 activity.

(A) H446 cells were transfected with 456 polymers complexed with either CMV-GFP or CMV-p53-GFP and sorted for GFP positive and negative populations (populations 2–5) and plated in fresh media. Untreated, sorted cells were used as a control for changes due to the sorting procedure (population 1). (B) GFP expression was quantitated by flow cytometry immediately post-sort (20H), 48 and 72 hours post-transfection. (C) Western analysis confirmed p53-GFP expression and p21 induction primarily in the p53-GFP positive (4) population. (D–E) Morphologic changes were observed by brightfield microscopy and cell cycle analysis demonstrated accumulation of cells in sub-G1. Statistical significance was determined by one-way ANOVA comparing all groups at 48 and 72 hours (***: p ≤ 0.0001 population 4 vs. 2 or 1)
Successful nanoparticle delivery of WT p53 would be expected to result in cell cycle arrest, induction of apoptosis and inhibition of tumor progression in vivo. To determine whether PBAE-mediated p53 delivery had antitumor activity in vivo, we administered nanoparticles carrying CMV-p53-LUC intratumorally (IT) into subcutaneous H446 xenografts. For this proof of principal analysis, we selected another leading PBAE polymer from our in vitro studies, 457, as this polymer appeared to better target subcutaneous tumors in pilot in vivo experiments (data not shown). Nude mice bearing H446 xenografts received twice weekly IT injections of 457:CMV-p53-LUC, 457:CMV-LUC, or neither nanoparticle formulation, and the tumors were serially measured (Fig. 6). We observed > 50% tumor growth inhibition with IT injection of 457:CMV-p53-LUC relative to either 457:CMV-LUC or no treatment control (p ≤ 0.01 for comparison to either control). Thus, when effectively delivered to tumors, PBAE-mediated nanoparticle delivery and exogenous expression of WT p53 can inhibit tumor growth of human SCLC xenografts.
Figure 6. Intratumoral delivery of 457:CMV-p53-LUC causes tumor growth inhibition.
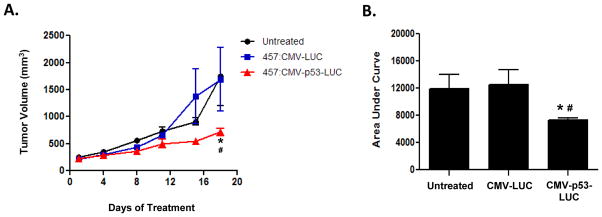
Nude mice bearing H446 xenografts received twice weekly IT injections of 457:CMV-LUC nanoparticles (n=3), 457:CMV-p53-LUC nanoparticles (n=4), or neither agent (n=3). Tumors were measured twice weekly. Group means and SEM were calculated and are shown. (A) IT injection of 457:CMV-p53-LUC resulted in statistically significant tumor growth inhibition compared with controls. (B) Area under the tumor growth curve by day 18. Statistical significance was determined by two-way ANOVA at day 18 (**: p ≤ 0.001 CMV-p53-LUC vs. CMV-LUC; #: p ≤ 0.01 CMV-p53-LUC vs. untreated).
Discussion
Here we report on a non-viral, biodegradable PBAE nanoparticle that self-assembles with DNA and can deliver WT TP53 to SCLC cells. Using a luciferase-based high throughput screening approach, we were able to identify PBAE polymers that could transfect SCLC lines at efficiencies comparable to commercially available transfection reagents. We showed that the 456 polymer could deliver functional p53 to SCLC cell lines resulting in p21 expression, induction of apoptosis and accumulation in sub-G1. Finally, we demonstrated that IT delivery of 457:CMV-p53-LUC significantly suppressed tumor growth while injection of the polymer with CMV-LUC had no effect. Thus, when effectively delivered, exogenously expressed WT p53 suppresses tumor growth of H446 xenografts.
Loss of p53 tumor-suppressor activity is a critical event in cancer development across multiple tumor types. In transgenic SCLC models, TP53 loss is required for tumor development (13, 36). Re-expression of WT p53 has potent efficacy in established p53-null lymphomas, sarcomas and hepatocellular carcinomas (10, 11, 37). Nanoparticle delivery of TP53 using (D,L-lactide-co-glycolide) (PLGA) polymers caused growth inhibition in p53-null PC-3 prostate cancer xenografts (38).
In addition to p53 deletion or inactivation, some p53 mutations result in dominant negative (DN) or gain-of-function activities. The ability of WT p53 to overcome a gain-of-function p53 mutation is unclear. In spontaneous p53-mutant lymphoma and sarcoma models, WT p53 restoration resulted in tumor growth arrest but not tumor regression (39). In these experiments, p53 was restored to endogenous levels, and the authors suggested that the ability of p53 to overcome a DN p53 mutant may be dose-dependent. As an alternative approach, small molecules that can restore mutant p53 to its WT conformation have been explored. One such molecule, PRIMA-1 (p53-dependent reactivation of massive apoptosis), is able to restore the transcriptional activity of mutant p53 and has efficacy in several preclinical cancers models including SCLC (40). A primary limitation is the requirement for mutant p53; PRIMA-1 would not be effective in tumors harboring a null mutant or that are driven by overexpression of p53 binding proteins such as MDM2. The H446 SCLC cell line harbors a G154V alteration in the DNA binding domain of TP53 and expresses high basal levels of the mutant protein. We demonstrated that CMV-driven expression of p53 was effective in this p53-mutant cell line; our cell sorting experiments show that H446 cells that expressed high-level exogenous WT p53 had robust p21 induction and accumulated in sub-G1.
This study demonstrates that PBAE-mediated gene transfection is feasible both in vitro and, importantly, in tumor-bearing animals. Further, this work shows that this approach can be used to restore activity of a silenced or absent tumor suppressor, resulting in specific inhibition of tumor growth. This same approach could be used to restore multiple silenced genes or to deliver other therapeutic agents. These polymers are amenable to a variety of modifications that may improve their efficiency in targeted delivery of an anti-cancer payload.
A better understanding of the limitations on DNA delivery is required to improve the utility of this non-viral gene transfer system. While nearly 100% of the cells take up the particles, about 40% of the cells successfully expressed the target protein in our initial experiments. This indicates that events downstream may be limiting transfection efficiency PBAEs are able to successfully escape endosomes (41), thus cytosolic transport and nuclear import are possible barriers and are the subject of active investigation(23). Delivery of therapeutic agents that are not reliant on 100% transfection, that can induce a bystander effect, may be well-suited for this system. For example, nanoparticle delivery of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces apoptosis and tumor regression in the A549 NSCLC cell line (42). TRAIL-induced death, however, is reliant on host factors, including the presence of TRAIL receptors, DR4 or DR5, and caspase 8. Agents such as mitomycin C, doxorubicin, etoposide and epigenetic modulators are able to sensitize cells to TRAIL by upregulating of DR4/5 or caspase 8 (43–45). Another approach, gene-directed enzyme prodrug therapy (GDEPT), is based on cell-specific delivery of an enzyme which can covert a systemically administered prodrug into a toxin. While the direct effects are cell-specific, this system could be adapted to induce a bystander effect by selecting a drug whose metabolite can traverse gap junctions.
Our in vivo data demonstrate that if effectively delivered to tumors, PBAE nanoparticles can successfully transfect SCLC xenografts and inhibit tumor growth. Our work is further supported by Huang et al. demonstrating that PBAE nanoparticles can deliver diptheria toxin and induce tumor regression when delivered locally in an ovarian cancer xenograft model (46). Systemic delivery and in vivo stability are major challenges in the clinical development of these nanoparticles. PBAE particles are readily amenable to approaches to add electrostatic coatings to improve these properties (42, 47, 48). In SCLC, cell-surface expression of CD56 is nearly universal and can be targeted effectively by antibodies. A mantaysinoid-conjugated anti-CD56 antibody is being evaluated clinically in SCLC and other tumors (49, 50). Coatings which incorporate NCAM-binding moieties may enhance SCLC specificity and improve in vivo stability of PBAE particles.
Therapeutic gene delivery has held great promise for decades but has been limited by the lack of stable, safe delivery vehicles. Here we report the first demonstration to our knowledge of effective non-viral polymeric delivery of therapeutic genes in SCLC in vivo. The ability to deliver high level gene expression utilizing a non-toxic biodegradable nanoparticle has multiple advantages over viral-based delivery systems, including flexibility in cargo capacity, ability to deliver mixed nucleic acids targets, lack of insertional mutagenesis and minimal immunogenicity. PBAEs fulfill all of these criteria and, with optimization of DNA delivery and systemic stability, may ultimately provide a powerful platform for therapeutic restoration of gene expression.
Supplementary Material
Acknowledgments
The authors would like to thank Gary L. Gallia, MD, PhD, Timothy Burns, MD, PhD and John Poirier, PhD for scientific discussions and manuscript review.
Grant Support
This research was supported by Barbara’s Fund at the Community Foundation for the National Capital Region (C.L. Hann), the Cigarette Restitution Fund (C.L. Hann), and NIH (R21CA152473) (J.J. Green).
Abbreviations
- CMV, FACS, GFP, HTS, IT, LUC, NCAM, NSCLC, PBAE, SCLC, TGI
Footnotes
Relevant conflicts of interest: None
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Oze I, Hotta K, Kiura K, Ochi N, Takigawa N, Fujiwara Y, et al. Twenty-seven years of phase III trials for patients with extensive disease small-cell lung cancer: disappointing results. PloS one. 2009;4:e7835. doi: 10.1371/journal.pone.0007835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 4.Junttila MR, Evan GI. p53--a Jack of all trades but master of none. Nat Rev Cancer. 2009;9:821–9. doi: 10.1038/nrc2728. [DOI] [PubMed] [Google Scholar]
- 5.Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic acids research. 1994;22:3551–5. [PMC free article] [PubMed] [Google Scholar]
- 6.Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Human mutation. 2002;19:607–14. doi: 10.1002/humu.10081. [DOI] [PubMed] [Google Scholar]
- 7.Li M, Brooks CL, Wu-Baer F, Chen D, Baer R, Gu W. Mono- versus polyubiquitination: differential control of p53 fate by Mdm2. Science. 2003;302:1972–5. doi: 10.1126/science.1091362. [DOI] [PubMed] [Google Scholar]
- 8.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–36. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 9.Yew PR, Liu X, Berk AJ. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev. 1994;8:190–202. doi: 10.1101/gad.8.2.190. [DOI] [PubMed] [Google Scholar]
- 10.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 11.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.COSMIC. Wellcome Trust Sanger Institute Cancer Genome Project web site. [Google Scholar]
- 13.Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9. doi: 10.1016/s1535-6108(03)00220-4. [DOI] [PubMed] [Google Scholar]
- 14.Sutherland KD, Proost N, Brouns I, Adriaensen D, Song JY, Berns A. Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell. 2011;19:754–64. doi: 10.1016/j.ccr.2011.04.019. [DOI] [PubMed] [Google Scholar]
- 15.Adachi J, Ookawa K, Shiseki M, Okazaki T, Tsuchida S, Morishita K, et al. Induction of apoptosis but not G1 arrest by expression of the wild-type p53 gene in small cell lung carcinoma. Cell Growth Differ. 1996;7:879–86. [PubMed] [Google Scholar]
- 16.Nemunaitis J, Swisher SG, Timmons T, Connors D, Mack M, Doerksen L, et al. Adenovirus-mediated p53 gene transfer in sequence with cisplatin to tumors of patients with non-small-cell lung cancer. J Clin Oncol. 2000;18:609–22. doi: 10.1200/JCO.2000.18.3.609. [DOI] [PubMed] [Google Scholar]
- 17.Schuler M, Herrmann R, De Greve JL, Stewart AK, Gatzemeier U, Stewart DJ, et al. Adenovirus-mediated wild-type p53 gene transfer in patients receiving chemotherapy for advanced non-small-cell lung cancer: results of a multicenter phase II study. J Clin Oncol. 2001;19:1750–8. doi: 10.1200/JCO.2001.19.6.1750. [DOI] [PubMed] [Google Scholar]
- 18.Wang ZX, Lu BB, Yang JS, Wang KM, De W. Adenovirus-mediated siRNA targeting c-Met inhibits proliferation and invasion of small-cell lung cancer (SCLC) cells. The Journal of surgical research. 2011;171:127–35. doi: 10.1016/j.jss.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 19.Zandi R, Xu K, Poulsen HS, Roth JA, Ji L. The effect of adenovirus-mediated gene expression of FHIT in small cell lung cancer cells. Cancer Invest. 2011;29:683–91. doi: 10.3109/07357907.2011.626475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Check E. Gene therapy put on hold as third child develops cancer. Nature. 2005;433:561. doi: 10.1038/433561a. [DOI] [PubMed] [Google Scholar]
- 21.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001;7:33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 22.Putnam D. Polymers for gene delivery across length scales. Nature materials. 2006;5:439–51. doi: 10.1038/nmat1645. [DOI] [PubMed] [Google Scholar]
- 23.Sunshine JC, Bishop CJ, Green JJ. Advances in polymeric and inorganic vectors for nonviral nucleic acid delivery. Therapeutic delivery. 2011;2:493–521. doi: 10.4155/tde.11.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Partridge KA, Oreffo ROC. Gene delivery in bone tissue engineering: Progress and prospects using viral and nonviral strategies. Tissue engineering. 2004;10:295–307. doi: 10.1089/107632704322791934. [DOI] [PubMed] [Google Scholar]
- 25.Shmueli RB, Sunshine JC, Xu Z, Duh EJ, Green JJ. Gene delivery nanoparticles specific for human microvasculature and macrovasculature. Nanomedicine. 2012 doi: 10.1016/j.nano.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhise NS, Gray RS, Sunshine JC, Htet S, Ewald AJ, Green JJ. The relationship between terminal functionalization and molecular weight of a gene delivery polymer and transfection efficacy in mammary epithelial 2-D cultures and 3-D organotypic cultures. Biomaterials. 2010;31:8088–96. doi: 10.1016/j.biomaterials.2010.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tzeng SY, Hung BP, Grayson WL, Green JJ. Cystamine-terminated poly(beta-amino ester)s for siRNA delivery to human mesenchymal stem cells and enhancement of osteogenic differentiation. Biomaterials. 2012;33:8142–51. doi: 10.1016/j.biomaterials.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Green JJ. 2011 Rita Schaffer lecture: nanoparticles for intracellular nucleic acid delivery. Annals of biomedical engineering. 2012;40:1408–18. doi: 10.1007/s10439-012-0550-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Green JJ, Langer R, Anderson DG. A combinatorial polymer library approach yields insight into nonviral gene delivery. Accounts of chemical research. 2008;41:749–59. doi: 10.1021/ar7002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonawane ND, Szoka FC, Jr, Verkman AS. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J Biol Chem. 2003;278:44826–31. doi: 10.1074/jbc.M308643200. [DOI] [PubMed] [Google Scholar]
- 31.Sunshine J, Green JJ, Mahon K, Yang F, Eltoukhy A, Nguyen DN, et al. Small molecule end group of linear polymer determine cell-type gene delivery efficacy. Advanced Materials. 2009;21(48):4947–51. doi: 10.1002/adma.200901718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tzeng SY, Guerrero-Cazares H, Martinez EE, Sunshine JC, Quinones-Hinojosa A, Green JJ. Non-viral gene delivery nanoparticles based on Poly(beta-amino esters) for treatment of glioblastoma. Biomaterials. 2011;32:5402–10. doi: 10.1016/j.biomaterials.2011.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Showalter SL, Huang YH, Witkiewicz A, Costantino CL, Yeo CJ, Green JJ, et al. Nanoparticulate delivery of diphtheria toxin DNA effectively kills Mesothelin expressing pancreatic cancer cells. Cancer Biol Ther. 2008;7:1584–90. doi: 10.4161/cbt.7.10.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sunshine JC, Akanda MI, Li D, Kozielski KL, Green JJ. Effects of Base Polymer Hydrophobicity and End-Group Modification on Polymeric Gene Delivery. Biomacromolecules. 2011 doi: 10.1021/bm200807s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhise NS, Shmueli RB, Gonzalez J, Green JJ. A novel assay for quantifying the number of plasmids encapsulated by polymer nanoparticles. Small. 2012;8:367–73. doi: 10.1002/smll.201101718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaffer BE, Park KS, Yiu G, Conklin JF, Lin C, Burkhart DL, et al. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010;70:3877–83. doi: 10.1158/0008-5472.CAN-09-4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127:1323–34. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 38.Sharma B, Ma W, Adjei IM, Panyam J, Dimitrijevic S, Labhasetwar V. Nanoparticle-mediated p53 gene therapy for tumor inhibition. Drug delivery and translational research. 2011;1:43–52. doi: 10.1007/s13346-010-0008-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Suh YA, Fuller MY, Jackson JG, Xiong S, Terzian T, et al. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J Clin Invest. 2011;121:893–904. doi: 10.1172/JCI44504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011;17:2830–41. doi: 10.1158/1078-0432.CCR-10-3168. [DOI] [PubMed] [Google Scholar]
- 41.Akinc A, Langer R. Measuring the pH environment of DNA delivered using nonviral vectors: implications for lysosomal trafficking. Biotechnology and bioengineering. 2002;78:503–8. doi: 10.1002/bit.20215. [DOI] [PubMed] [Google Scholar]
- 42.Zhou J, Liu J, Cheng CJ, Patel TR, Weller CE, Piepmeier JM, et al. Biodegradable poly(amine-co-ester) terpolymers for targeted gene delivery. Nature materials. 2012;11:82–90. doi: 10.1038/nmat3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng H, Hong B, Zhou L, Allen JE, Tai G, Humphreys R, et al. Mitomycin C potentiates TRAIL-induced apoptosis through p53-independent upregulation of death receptors: Evidence for the role of c-Jun N-terminal kinase activation. Cell Cycle. 2012;11 doi: 10.4161/cc.21670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaculova A, Kaminskyy V, Jalalvand E, Surova O, Zhivotovsky B. Doxorubicin and etoposide sensitize small cell lung carcinoma cells expressing caspase-8 to TRAIL. Mol Cancer. 2011;9:87. doi: 10.1186/1476-4598-9-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaminskyy VO, Surova OV, Vaculova A, Zhivotovsky B. Combined inhibition of DNA methyltransferase and histone deacetylase restores caspase-8 expression and sensitizes SCLC cells to TRAIL. Carcinogenesis. 2011;32:1450–8. doi: 10.1093/carcin/bgr135. [DOI] [PubMed] [Google Scholar]
- 46.Huang YH, Zugates GT, Peng W, Holtz D, Dunton C, Green JJ, et al. Nanoparticle-delivered suicide gene therapy effectively reduces ovarian tumor burden in mice. Cancer Res. 2009;69:6184–91. doi: 10.1158/0008-5472.CAN-09-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harris TJ, Green JJ, Fung PW, Langer R, Anderson DG, Bhatia SN. Tissue-specific gene delivery via nanoparticle coating. Biomaterials. 2010;31:998–1006. doi: 10.1016/j.biomaterials.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shmueli RB, Anderson DG, Green JJ. Electrostatic surface modifications to improve gene delivery. Expert Opin Drug Deliv. 2010;7:535–50. doi: 10.1517/17425241003603653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Aletsee-Ufrecht MC, Langley K, Rotsch M, Havemann K, Gratzl M. NCAM: a surface marker for human small cell lung cancer cells. FEBS letters. 1990;267:295–300. doi: 10.1016/0014-5793(90)80948-i. [DOI] [PubMed] [Google Scholar]
- 50.Fossella F, Woll P, Lorigan P, Tolcher A, O’Brien M, O’Keefe J. Clinical Experience of Imgn901 (Bb-10901) in patients with small cell lung carcinoma (SCLC). 13th World Conference on Lung Cancer; 2009; San Francisco. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.