

Abstract
The reactivity of the three distonic isomers of the pyridine radical cation toward tetrahydrofuran is compared in solution and in the gas phase. In solution, the distonic ions were generated by UV photolysis at 300 nm from iodo-precursors in acidic 50:50 tetrahydrofuran/water solutions. In the gas phase, the ions were generated by collisionally activated dissociation (CAD) of protonated iodo-precursors in an FT-ICR mass spectrometer, as described in the literature. The same major reaction, hydrogen atom abstraction, was observed in solution and in the gas phase. Attempts to cleave the iodine atom from the 2-iodopyridinium cation in the gas phase and in solution yielded the 2-pyridyl cation in addition to the desired 2-dehydropyridinium cation. In the gas phase, this ion was ejected prior to the examination of the desired ion’s chemical properties. This was not possible in solution. This study suggests that solvation effects are not significant for radical reactions of charged radicals. On the other hand, the even-electron ion studied, the 2-pyridyl cation, shows substantial solvation effects. For example, in solution, the 2-pyridyl cation forms a stable adduct with tetrahydrofuran, whereas in the gas phase, only addition/elimination reactions were observed.
Introduction
Gas-phase experiments in mass spectrometers are frequently used to explore solution reactions [1]. In solution, the presence of solvent molecules often causes competing reactions with the reagents of interest, which significantly complicates the interpretation of solution results. On the other hand, the capability to isolate ions of interests and the elimination of solvent and other unwanted molecules in mass spectrometers provides a solvent-free environment which is advantageous for studies of reactions of highly reactive species that are difficult to study in solution. Hence, the correlation between reactions occurring in solution and in the gas phase is of great interest.
Since solvation effects are thought to be minor for radical reactions [2], radical reactions provide a good starting point when comparing solution and gas-phase reactions. Aromatic carbon-centered σ-type mono- and biradicals have been studied extensively both in solution [3–5] and in the gas phase [6–8] because of their roles in nonhydrolytic DNA cleavage. In our laboratory, the reactivity of aromatic mono-, bi-, and triradicals (particularly phenyl radicals) has been studied in the gas phase by using Fourier-transform ion cyclotron resonance mass spectrometry (FT-ICR) [9–12]. These studies use the distonic ion approach that is based on phenyl radicals with a chemically inert, positively charged group to allow for their manipulation in the mass spectrometer [13–14]. These gaseous charged radicals have been demonstrated to react by the same pathways as related neutral radicals in solution [13–14].
Polar effects have been determined as a reactivity controlling parameter for polar phenyl radicals in solution [3–4] and in the gas phase [9–10]. Pryor and co-workers [3] have shown that the rate of hydrogen atom abstraction by the phenyl radical in solution is influenced by its substitution. A phenyl radical with a more electron-withdrawing substituent shows greater reactivity because of its ability to better polarize the transition state of its reactions, which lowers the transition state energy. Another study by Takayama and co-workers [4] concluded that increasing the electronegative character of the substituent of a phenyl radical, which increases the polar nature of the radical, increases the rate of hydrogen atom abstraction.
Similar conclusions have been made based on gas-phase studies [9–10]. Literature studies suggest that increasing the polarity of the transition state of reactions of a charged phenyl radical increases its reactivity. The ability of a radical to polarize its transition states can be attributed to a greater electrophilicity of the radical, which is gauged here based on its calculated vertical electron affinity (EA, defined as the energy released when an electron is added to the radical site with no change in geometry). A greater calculated electron affinity usually leads to a more polar transition state and thus, greater reactivity [9]. This finding also rationalizes the substituent effects observed for the phenyl radical in the gas phase. For example, fluorine substitution has been found to increase the electron affinity of a radical, which significantly enhances its reactivity [10].
While the above studies suggest that the reactivity of gaseous phenyl radicals is similar to that reported for phenyl radicals in solution, the comparison is complicated by the fact that charged radicals were studied in the gas phase but neutral radicals in solution. To the best of our knowledge, there is only one study published on the direct comparison of the reactivity of charged phenyl radicals in the gas phase and in solution [15]. Recently, the effect of pH on the efficiency of hydrogen atom abstraction from methanol by isomeric charged phenyl radicals in solution was examined [15]. The efficiency of hydrogen atom abstraction by the charged phenyl radicals in solution was found to decrease as the pH increases, as predicted based on previous gas-phase studies [12, 16]. This inspired the current study. We report here a comparison of the reactivity of the three distonic isomers of the pyridine radical cation (Scheme 1) toward tetrahydrofuran (THF) in solution and in the gas phase.
Scheme 1.

The structure of pyridine radical cation and its three distonic isomers
Experimental
2-Iodopyridine (98%), 3-iodopyridine (98%), 4-iodopyridine (97%), d5-pyridine (99.5%), THF (99%), and d8-THF (99%) were purchased from Sigma-Aldrich and used as received. In solution, the distonic ions were generated by photolysis of the iodopyridine precursors in a Rayonet chamber reactor (model RPR-200, Southern New England Ultraviolet Company) equipped with 16 UV lamps. The lamps are broadband. The iodopyridine precursors (10 mM) were dissolved in a mixture of THF and HPLC grade water (1:1, by volume) and the pH was adjusted by adding hydrochloric acid. The solution was then subjected to UV irradiation (λ = 300 nm) for 30 min. The reaction chamber was kept at the room temperature by boiling off liquid nitrogen into the chamber. Dark control experiments were performed.
Product analysis was carried out by using a Surveyor Plus High Performance Liquid Chromatograph (Thermo Scientific) coupled with a Thermo Scientific Linear Quadrupole Ion Trap (LQIT) mass spectrometer, specifically, a Thermo LTQ, equipped with an electrospray ionization (ESI) source. The reaction mixtures were diluted 100 times with HPLC grade water and spiked with d5-pyridine, which was used as an internal standard. Twenty five µL of the solution was injected into the HPLC column and eluted for 20 min. The column used was Aquasil C18 (dimensions 100×2.1 mm, particle size 5 µm, Thermo Scientific). The mobile phase was 0.1% HCOOH in CH3OH/0.1% HCOOH in H2O, eluted at a flow rate of 200 µL/min. The detailed elution conditions are shown in Table 1. The ESI conditions were as follows: spray voltage 5 kV, sheath gas flow 30 (arbitrary units), auxiliary gas 5 (arbitrary units), capillary temperature 275°C.
Table 1.
Gradient elution timetable for HPLC/MS analysisa
| Time (min) | A (%) | B (%) |
|---|---|---|
| 0.0 | 90 | 10 |
| 14.0 | 10 | 90 |
| 15.8 | 10 | 90 |
| 16.0 | 90 | 10 |
| 20.0 | 90 | 10 |
A: 0.1% (v/v) HCOOH in H2O, B: 0.1% (v/v) HCOOH in CH3OH
The reaction products were identified by comparing the mass spectra measured for mixtures with and without photolysis (dark control experiments). The structures of the products were confirmed by studying reactions with d8-THF, as well as performing collisionally activated dissociation (CAD) on the ionized products. Typical CAD conditions were as follows: isolation widow 2 Da, normalized collision energy 25–30 (arbitrary units), and activation time 30 ms. During CAD, the precursor ions are subjected to resonance excitation for 30 ms, during which time they undergo collisions with helium buffer gas in the linear quadrupole ion trap. Upon collisions, some of the ions’ kinetic energy is converted into internal energy, which causes the ions to undergo unimolecular dissociation to form fragment ions. The fragmentation patterns allow for structural determination of the precursor ions.
In the gas phase, the distonic ions were studied in a Finnigan Model 2001 FTMS dualcell 3 Tesla Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer by using literature methods [12, 17].
All density functional theory (DFT) calculations were performed by using the Gaussian 03 [18] electronic structure program suite.
Results and Discussion
Reactivity of 2-, 3-, and 4-dehydropyridinium cations toward THF in solution
UV absorption spectra of solutions containing 2-, 3-, and 4-iodopyridine in 50:50 THF/water at pH 1 are shown in Figure 1. For all of the isomeric iodopyridines, similar UV absorption intensity at 300 nm was observed. While this is not the wavelength where maximum UV absorption occurs, a previous gas-phase study has shown that 355 nm UV light has enough energy to homolytically cleave the C-I bond in a benzene derivative to generate a radical site. Furthermore, previous study has reported successful generation of 2-, 3-, and 4-dehydropyridinium cations via photolysis of 2-, 3-, and 4- iodopyridine in 50:50 CH3OH/water solution at 300 nm [15]. Therefore, based on these observations, photolysis of 2-, 3-, and 4- iodopyridine in 50:50 THF/water solution at 300 nm should yield the corresponding dehydropyridines.
Figure 1.

UV absorbance spectra of solutions containing 2-. 3-, and 4-iodopyridine in 50:50 THF/water at pH 1. UV absorbance at 300 nm is indicated for each spectrum.
The same major reaction, hydrogen atom abstraction, was observed for the three isomeric radicals (2–4) in solution and in the gas phase. Hence, the primary reaction products are a THF radical and protonated pyridine. However, some of the primary products underwent a further reaction in solution, namely, addition of the THF radical to the protonated pyridine, followed by the loss of a hydrogen atom (Table 2, Scheme 2). This was not unexpected as addition of a THF radical to protonated pyridine followed by the loss of a hydrogen atom in 50:50 THF/water solution has been reported in the literature [19].
Table 2.
Reactions of radicals 2–4 with THF and their products’ branching ratios in the gas phasea and in 50:50 THF/water solution
| Gas phase | 50:50 THF/water solution | |||
|---|---|---|---|---|
 |
H abstraction (m/z 80) | 100% | H abstraction (m/z 80) | 98% |
| H abstraction, addition of THF radical to protonated pyridine at the ortho-position, loss of a H atom (m/z 150) |
2% | |||
| Efficiency = 76% | ||||
 |
H abstraction (m/z 80) | 100% | H abstraction (m/z 80) | 99% |
| H abstraction, addition of THF radical to protonated pyridine at the ortho- and para-positions, loss of a H atom (m/z 150) |
1% | |||
| Efficiency = 38% | Yield = 28% | |||
 |
H abstraction (m/z 80) | 81% | H abstraction (m/z 80) | 98% |
| CH2 abstraction (m/z 93) | 8% | |||
| C2H3 abstraction (m/z 106) | 6% | H abstraction, addition of THF radical to protonated pyridine at the ortho- and para-positions, loss of a H atom (m/z 150) |
2% | |
| CHO abstraction (m/z 108) | 3% | |||
| C2H3O abstraction (m/z 122) | 2% | |||
| Efficiency = 28% | Yield = 62% | |||
Reference 12
Scheme 2.

Proposed mechanisms for reaction of radical 4 with THF in 50:50 THF/water solution. Radical 3 is proposed to follow analogous reaction mechanisms.
The observation of the addition reaction only in solution is rationalized by the ability of the solvent cage to hold the product complex together while it cools by collisions with solvent molecules before it has time to dissociate to the primary products, and hence keeps the THF radical in the proximity of the protonated pyridine molecule for long enough for the addition reaction to take place. In the gas phase, this reaction was not observed. This can be rationalized by the high exothermicity of the hydrogen atom abstraction reaction (−26.0 kcal/mol for radical 3) [11], which leads to fast dissociation of the gas-phase product complex. The structures of the products of the addition of the THF radical to protonated pyridine followed by loss of a hydrogen atom will be discussed in more detail below.
Enhanced reactivity has been reported for gaseous neutral and charged radicals with greater electron affinities, as expected based on polar effects [9, 12, 15, 20]. In agreement with this expectation, the activation enthalpies for hydrogen atom abstraction by radicals 2 – 4 from the α- carbon of THF in the gas phase have been calculated to be –17.4 kcal/mol, –10.1 kcal/mol, and –8.3 kcal/mol, respectively [12] (these values are negative due to the initial formation of a high-energy collision complex in the gas phase). Indeed, the reactivity ordering of the radicals in the gas phase is 2 > 3 > 4 (Table 2). However, the yields of the solution reactions indicate a reactivity ordering 4 > 3which is opposite to the gas-phase result. For radical 2the yield of the solution reaction cannot be determined due to unwanted formation of 2-pyridyl cation upon photolysis of 2-iodopyridinium cation, in addition to the desired radical 2. In solution, these ions cannot be studied independently. This finding has been reported in an earlier study [21] and will be discussed in more detail below.
The difference in the observed reactivity between radicals 3 and 4 in solution and gas phase can be rationalized based on the different protonated states of the radicals in solution at the given pH. The pKa of the protonated 2-, 3- and 4-iodopyridines have been reported to be 1.1, 2.3 and 2.9, respectively [22]. Hence, at pH 1, the ratio of protonated molecules to unprotonated molecules for the 2-iodopyridine is 1.3, 3-iodopyridine 20, and 4-iodopyridine 63. Photolysis of these molecules leads to a much larger fraction of protonated radicals for the 4-isomer than the 3- isomer. Further, the proton affinity (PA) values of 3- and 4-dehydropyridines have been calculated to be 218.2 kcal/mol and 219.5 kcal/mol, respectively [23]. The pKa values of the protonated radicals are likely to have the same ordering as the PA of their conjugate bases (for example, for 2-bromopyridine, PA = 216.3 kcal/mol, and for protonated 2-bromopyridine, pKa = 0.9, whereas for 3-bromopyridine, the corresponding values are 217.5 kcal/mol and 2.84, respectively; the same situation is also true for 2- and 3-chloropyridines [24]). Hence, even if there is time for the radicals to equilibrate their protonation state after formation and during the reaction, the 4-isomer is likely to have a greater proportion of protonated forms than the others. Therefore, at a given time, more protonated 4-dehydropyridine molecules (radical 4) exist in the solution than for 3-dehydropyridine. Since the reactivity of the radicals is significantly increased upon protonation, a larger proportion of protonated radicals in the case of 4 should result in a greater reactivity.
Structural delineation of products formed upon addition of the THF radical to protonated pyridine
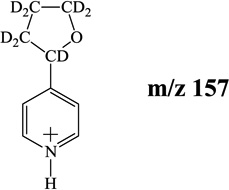
By considering the resonance structures of the pyridinium cation, the addition of THF radical should occur predominantly at the ortho- and para-positions of the pyridine ring, as reported in a previous study [19]. This was confirmed for 3 and 4 by the observation of two isomeric products (two HPLC peaks) for molecules with MW of 149 Da that correspond to the deprotonated product ions of addition of THF radical to protonated pyridine followed by the loss of a hydrogen atom (Scheme 2). The identity of the isomeric products was probed by examining the reaction of d8-THF with the radicals. For radical 3the product ions of addition of deuterated THF radical to protonated pyridine at the ortho- and para-positions must be followed by the loss of a hydrogen atom and hence yield ions of the same m/z (m/z 158), which was confirmed by observation of two HPLC peaks for molecules with MW of 157 Da. For radical 4the two addition reactions yield product ions of different m/z. Addition of deuterated THF radical at the ortho-position must be followed by the loss of a hydrogen atom to yield ions of m/z 158, while addition at the para-position must be followed by loss of a deuteron atom to yield ions of m/z 157 (Scheme 3). The observation of ions of different m/z values for radical 4 allows for the identification of the isomeric addition products based on their retention times in the HPLC.
Scheme 3.

Proposed mechanism for reaction of radical 4 with d8-THF in 50:50 d8-THF/water solution. Addition of the deuterated THF radical to protonated pyridine at the ortho- and para-position yields ions of different m/z values.
In contrast to radicals 3 and 4addition of THF radical to the pyridine ring of radical 2 after hydrogen atom abstraction, followed by the loss of a hydrogen atom, only yields a single product, based on the observation of only a single HPLC peak for molecules with MW of 149 Da. Based on the retention time of the molecule, this product arises from the addition of the THF radical to the pyridine ring at the ortho-position, followed by loss of a hydrogen atom. Further, when the reaction was performed with d8-THF, the product formed upon addition of the deuterated THF radical to the pyridine ring of radical 2 after deuterium atom abstraction, followed by the loss of a deuteron atom, yields a molecule with MW of 156 Da, which indicates that addition must have occurred at the ortho-position (Scheme 4). Addition of the THF radical to the pyridine ring at the para-position was not observed, likely due to the stabilizing hydrogen bonding interaction that lowers the TS energy for addition at the ortho-position in radical 2 [12]. This finding is supported by the calculation of the transition state structures for hydrogen atom abstraction from THF by radicals 2 - 4 (Scheme 5). The calculated transition state structure for radical 2 reveals a hydrogen bonding interaction between the protonated nitrogen atom and the oxygen atom of the THF molecule. This interaction keeps the THF molecule in the proximity of the ortho-carbon of the pyridine ring, thus facilitating addition of the THF radical to the orthorather than the para-position.
Scheme 4.

Proposed mechanism for reaction of radical 2 with d8-THF in 50:50 d8-THF/water solution. Addition of the deuterated THF radical to protonated pyridine occurs exclusively at the ortho-position.
Scheme 5.

Calculated (MPW1K/6-31+G(d,p) transition state structures for hydrogen atom abstraction from THF by radicals 2 – 4 [12].
The structures of the isomeric products formed upon addition of the THF radical to the pyridine ring of radicals 2–4 after hydrogen atom abstraction, followed by the loss of a hydrogen atom, were confirmed by CAD on the protonated products after chromatographic separation. An ion of m/z 150 that had been identified as the product of the addition of the THF radical to the pyridine ring at the ortho-position fragments predominantly by loss of H2O to yield an ion of m/z 132. The resulting ion readily fragments by loss of H2 and CH3 to give ions of m/z 130 and m/z 117, respectively (Figure 2). This was confirmed by isolating the fragment ion of m/z 132 and subjecting it to CAD, which yielded ions of m/z 130 and m/z 117 (Table 3).
Figure 2.

The CAD mass spectrum of one of the isomeric product ions of m/z 150 formed upon addition of the THF radical to the pyridine ring followed by the loss of a hydrogen atom. The likely structure for the ion of m/z 150 is indicated.
Table 3.
The m/z values and branching ratios of the fragment ions formed upon CAD of the protonated products of addition of the THF radical (or deuterated THF radical) to the pyridine ring at the ortho-position, followed by loss of a hydrogen atom
| Production | MS2 fragment ions and their branching ratios |
MS3 fragment ions and their branching ratios |
||
|---|---|---|---|---|
 |
m/z 132 (−H2O) | 80% | m/z 130 (−H2) | 90% |
| m/z 117 (−CH3) | 10% | |||
| m/z 120 (−CH2O) | 17% | |||
| m/z 106 (−C2H4O) | 3% | |||
 |
m/z 138 (−DHO) | 78% | m/z 134 (−D2) | 87% |
| m/z 120 (−CD3) | 13% | |||
| m/z 125 (−CD2O) | 19% | |||
| m/z 109 (−C2D4O) | 3% | |||
 |
m/z 139 (−DHO) | 80% | m/z 135 (−D2) | 93% |
| m/z 121 (−CD3) | 7% | |||
| m/z 126 (−CD2O) | 17% | |||
| m/z 110 (−C2D4O) | 3% | |||
 |
m/z 139 (−DHO) | 79% | m/z 135 (−D2) | 86% |
| m/z 121 (−CD3) | 14% | |||
| m/z 126 (−CD2O) | 19% | |||
| m/z 110 (−C2D4O) | 2% | |||
The proposed mechanisms for H2O and H2 losses upon CAD from ions of m/z 150 are shown in Scheme 6. The reaction is initiated by proton transfer from the nitrogen atom of the pyridine ring to the oxygen atom of the THF molecule, followed by a heterolytic bond cleavage between carbon 1 and oxygen of the THF ring. Proton transfer from carbon 2 to the hydroxyl-group of the ring-opened structure of the THF molecule takes place. Nucleophilic attack by the nitrogen atom of the pyridine ring at carbon 4 of the ring-opened structure of the THF molecule causes cleavage of the carbon-oxygen bond and loss of H2O molecule to yield an ion of m/z 132. Upon the second CAD, this bicyclic ion losses a H2 molecule to yield an aromatic bicyclic ion of m/z of 130.
Scheme 6.

Proposed mechanism for H2O and H2 loss upon CAD of the protonated product ion of m/z 150 formed upon addition of the THF radical to the ortho-position in the pyridine ring followed by loss of a hydrogen atom, followed by CAD of the fragment ion of m/z 132.
Analogous fragmentation behavior yielding product ions with similar branching ratios was observed for the corresponding deuterated1 product ions (ions of m/z 157 for radical 2 and ions of m/z 158 for radicals 3 and 4) upon CAD, as shown in Table 3. The deuterated product ions fragment predominantly by a major loss of DHO, followed by loss of D2 or CD3. These findings provide evidence of hydrogen bonding interaction between the nitrogen atom of the pyridine ring and the oxygen atom of the THF molecule. The above fragmentation behavior, as well as the presence of hydrogen bonding interaction between the nitrogen atom of the pyridine ring and the oxygen atom of the THF molecule, suggest that this ion (m/z 157 for radical 2 and m/z 158 for radicals 3 and 4) is formed upon addition of THF radical to the pyridine ring at the ortho- rather than para-position.
In contrast to the above product, the product ion formed upon addition of the THF radical to the pyridine ring at the para-position fragments predominantly via homolytic cleavage of the carbon-carbon bond between the α-carbon of THF and the γ-carbon of the protonated pyridine, followed by hydrogen atom abstraction to give protonated pyridine (m/z 80) and 2,3- dihydrofuran (Figure 3; Scheme 7). These products then undergo proton transfer to yield tetrahydrofuran-2-ylium cation (m/z 71). This fragmentation behavior is in best agreement with a structure wherein the THF radical has added to the para-position of the pyridine ring. Analogous fragmentation behavior with similar product branching ratios was observed for the corresponding deuterated product ions (ion of m/z 158 for radical 3 and ion of m/z 157 for radical 4) upon CAD, as shown in Table 4.
Figure 3.

The CAD mass spectrum of the product ion formed upon addition of THF radical to the pyridine ring at the para-position followed by the loss of a hydrogen atom.
Scheme 7.

Proposed mechanism for CAD of the product ion formed upon addition of THF radical to the pyridine ring at the para-position followed by the loss of a hydrogen atom.
Table 4.
The m/z values and branching ratios of the fragment ions formed upon CAD of protonated products of addition of the THF radical (or deuterated THF radical) to the pyridine ring at the para-position, followed by loss of a hydrogen atom
| Product ion | MS2 fragment ions and their branching ratios |
||
|---|---|---|---|
 |
m/z 71 (−C5H5N) | 43% | |
| m/z 80 (−C4H6O) | 54% | ||
| m/z 122 (−C2H4) | 3% | ||
 |
m/z 77 (−C5H4DN) | 46% | |
| m/z 81 (−C4D6O) | 29% | ||
| m/z 82 (−C4HD5O) | 23% | ||
| m/z 125 (−C2D4) | 2% | ||
 |
m/z 77 (−C5H3D2N) | 47% | |
| m/z 82 (−C4D6O) | 30% | ||
| m/z 83 (−C4HD5O) | 21% | ||
| m/z 126 (−C2D4) | 2% | ||
Unusual reactivity of the 4-dehydropyridinium cation toward THF
In addition to hydrogen atom abstraction, radical 4 undergoes slow electrophilic addition/elimination reactions with THF in the gas phase, i.e., CH2C2H3CHO, and C2H3O abstraction [12]. This unusual reactivity has been proposed to arise from an ionized carbene-type resonance structure that facilitates nucleophilic addition to the most electrophilic carbon atom in the para-position of 4 (for a mechanism proposed for CH2 abstraction, see Scheme 8) [12]. These nucleophilic addition/elimination reactions were not observed for radical 4 in solution. Calculations indicate that hydrogen bonding interaction between the protonated nitrogen atom and a water molecule (solvent) causes the carbon at the para-position to be less electrophilic than in the gas phase (Scheme 9). This may explain the absence of nucleophilic addition to the paraposition in 4 in solution.
Scheme 8.

Proposed mechanism for CH2 abstraction from THF by radical 4 in the gas phase [12].
Scheme 9.

Calculated Mulliken atomic charges of radical 4 with and without hydrogen bonding with a water molecule [27].
In contrast to radical 4the influence of the ionized carbene-type resonance structure was not obvious for radical 2 in the gas-phase or solution experiments. This is most likely due to a stabilizing hydrogen bonding interaction in the transition state of radical 2 with THF (Scheme 5), which helps to catalyze hydrogen atom abstraction [12].
Reactivity of the 2-pyridyl cation toward THF in solution
As mentioned above, photolysis of 2-iodopyridine in solution has been reported to yield the 2-pyridyl cation in addition to 2-dehydropyridine [21]. At high pH, formation of 2-pyridyl cation was suggested to occur via electron transfer within the initially formed 2-dehydropyridine and iodine radicals after photolysis of 2-iodopyridine (Scheme 10) [21]. At low pH, formation of 2-pyridyl cation most likely occurs via loss of HI from protonated 2-iodopyridine (possibly as shown in Scheme 10), as observed in the gas phase upon sustained off-resonance irradiation collision-activated dissociation (SORI-CAD) of 2-iodopyridinium cation [26]. The same cation can also be generated upon fragmentation of 2-substituted pyridine radical cations [27].
Scheme 10.

Proposed mechanisms for the formation of 2-pyridyl cation upon photolysis of 2-iodopyridine in solution at high [21] and low pH.
Formation of pyridyl cations was not observed for 3- and 4-dehydropyridines in solution or in the gas phase. Elimination of HI upon SORI-CAD from gaseous 3- and 4-iodopyridinium ions would require the iodine atom to abstract a hydrogen atom bound to carbon, or the product complex to have a long enough lifetime for the cleaved iodine atom to travel near the NH group and abstract a hydrogen atom from this site. Clearly, neither process is feasible. In solution, the absence of formation of pyridyl cations from 3 and 4 is most likely due to the high ionization energies of the 3- and 4-dehydropyridines, which hinders electron transfer to the iodine atom (EA = 3.06 eV) [28]. This hypothesis is supported by the calculated ionization energies (IE) of 2, 3-, and 4-dehydropyridines (7.63 eV, 8.07 eV, and 8.33 eV, respectively) [23].
When photolysis of 2-iodopyridine was performed in 50:50 THF/H2O solution at pH 1, the 2-pyridyl cation was found to react rapidly with H2O to form 2-hydroxypyridinium cation and only a minor reaction product with THF was observed. Therefore, to eliminate unwanted reaction of the 2-pyridyl cation with H2O molecules, the photolysis was performed in 100% THF solution at pH 7.
In 100% THF solution, reaction of the 2-pyridyl cation with THF yields product ions with m/z of 150. When the reaction was performed with d8-THF, the m/z of this product ion shifted to 158. The structure of this ion was identified by performing CAD. Upon CAD, the ion with m/z of 150 fragments predominantly by loss of C4H6(m/z 96) as well as by loss of C3H6 (m/z 108), loss of C5H5ON (m/z 55), and loss of C4H6O (m/z 80), as shown in Figure 4. Analogous fragmentation behavior with similar product branching ratios was observed for CAD of the deuterated product ion with m/z 158 (Table 5). The observed fragmentation behavior for both of the undeuterated and the deuterated product ions formed upon reaction of the 2-pyridyl cation with THF (or d8-THF) suggests that this ion corresponds to the adduct of 2-pyridyl cation with THF (or d8-THF).
Figure 4.

The CAD mass spectrum of the product ion with m/z of 150 formed upon reaction of the 2-pyridyl cation with a THF molecule in solution. The likely structure for the ion of m/z 150 is indicated.
Table 5.
Products and their branching ratios for the reaction of the 2-pyridyl cation in 100% THF solution, in 100% d8-THF solution, and in the gas phase [23]. The m/z values and the branching ratios of the fragment ions formed upon CAD of the adduct of the 2-pyridyl cation with THF (or d8-THF) formed in solution are also listed.
| Reaction products and their branching ratios |
MS2 fragments ions and their branching ratios |
|||
|---|---|---|---|---|
|
100% THF solution |
Adduct (m/z 150) | 100% | Adduct – C4H6 (m/z 96) | 60% |
| Adduct – C3H6 (m/z 108) | 30% | |||
| Adduct – C5H5ON (m/z 55) | 8% | |||
| Adduct – C4H6O (m/z 80) | 2% | |||
|
100% d8- THF solution |
Adduct (m/z 158) | 100% | Adduct – C4D6 (m/z 98) | 50% |
| Adduct – C3D6 (m/z 110) | 36% | |||
| Adduct – C5H4DON (m/z 62) | 10% | |||
| Adduct – C4D6O (m/z 82) | 4% | |||
| Gas phase | Adduct – C4H6 (m/z 96) | 62% | ||
| Adduct – C3H6 (m/z 108) | 34% | |||
| Adduct – C2H4O (m/z 106) | 3% | |||
| Adduct – C4H6O (m/z 80) | 1% | |||
| Efficiency = 78% | ||||
While formation of a stable adduct between the 2-pyridyl cation and THF molecule was observed in solution, in the gas phase, only addition/elimination reactions were observed. Interestingly, the major addition/elimination reaction products that were observed upon reaction of the 2-pyridyl cation with THF in the gas phase have similar branching ratios as the fragment ions that were observed upon CAD of the stable adduct of the 2-pyridyl cation and THF molecule formed in solution (Table 5). Hence, the difference in the reactivity of 2-pyridyl cation toward THF in the gas phase and in solution can be rationalized by the presence of nearby solvent molecules in solution which can accept energy from the adduct and keep the product complex together for a longer time than in the gas phase, and eventually cool the adduct. On the other hand, the exothermicity of addition of a THF molecule to the 2-pyridyl cation leads to fast dissociation of the adduct in the gas phase.
Conclusions
The same major reaction, hydrogen atom abstraction, was observed in solution and in the gas phase for the three distonic isomers of the pyridine radical cation. Minor products formed by addition of the initially formed THF radical to pyridine (at the ortho- and para-positions for the 3- and 4-isomers but only ortho-position for the 2-isomer) followed by hydrogen atom loss were also observed in solution but not in the gas phase, likely due to the cooling and trapping ability of the solvent cage. These results suggest that solvation effects may not be significant for reactions of radicals that are charged. On the other hand, the even-electron 2-pyridyl cation shows much greater solvation effects, indicated by a significant difference in its reactivity in solution and in the gas phase. In solution, 2-pyridyl cation forms a stable adduct with a THF molecule, whereas in the gas phase, addition/elimination reactions were observed.
Acknowledgments
The project described was supported by Grant Number R01GM052418 from the National Institute of General Medical Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medical Sciences or the National Institutes of Health. F.W. also thanks the Frederick N. Andrews endowment for a Graduate Fellowship.
References
- 1.Gronert S. Mass Spectrometric Studies of Organic Ion/Molecule Reactions. Chem. Rev. 2001;101:329–360. doi: 10.1021/cr9900836. [DOI] [PubMed] [Google Scholar]
- 2.Kanabus-Kaminska JM, Gilbert BC, Griller BC. Solvent Effects on the Thermochemistry of Free-Radical Reactions. J. Am. Chem. Soc. 1989;111:3311–3314. [Google Scholar]
- 3.Pryor WA, Echols JT, Jr, Smith K. Rates of the Reactions of Substituted Phenyl Radicals with Hydrogen Donors. J. Am. Chem. Soc. 1966;88:1189–1199. [Google Scholar]
- 4.Takayama K, Masanori K, Migita T. Reactivities of Aryl Radicals in Hydrogen Abstraction and Addition. Chem. Lett. 1973:193–195. [Google Scholar]
- 5.Migita T, Takayama K, Abe Y, Kosugi M. Relative Reactivities of Substituted Phenyl Radicals in Elementary Reactions. J. Chem. Soc. Perkin Trans. 1979;2:1137–1142. [Google Scholar]
- 6.Fahr A, Stein SE. Gas-phase Reactions of Phenyl Radicals with Aromatic Molecules. J. Phys. Chem. 1988;92:4951–4955. [Google Scholar]
- 7.Chen RH, Kafafi A, Stein SE. Reactivity of Polycyclic Aromatic Aryl Radicals. J. Am. Chem. Soc. 1989;111:1418–1423. [Google Scholar]
- 8.Kopinke F-D, Zimmermann G, Anders K. Relative Reactivities of Carbon-Hydrogen Bonds in Hydrogen Atom Abstraction by Phenyl Radicals. J. Org. Chem. 1989;54:3571–3576. [Google Scholar]
- 9.Tichy SE, Thoen KK, Price JM, Ferra JJ, Petucci CJ, Kenttämaa HI. Polarity of the Transition State Controls the Reactivity of Related Charged Phenyl Radicals Toward Atom and Group Donors. J. Org. Chem. 2001;66:2726–2733. doi: 10.1021/jo001634r. [DOI] [PubMed] [Google Scholar]
- 10.Li R, Smith R, Kenttämaa HI. Fluorine Substitution Enhances the Reactivity of Substituted Phenyl Radicals toward Organic Hydrogen Atom Donors. J. Am. Chem. Soc. 1996;118:5056–5061. [Google Scholar]
- 11.Petucci C, Nyman M, Guler L, Kenttämaa HI. Hydrogen Atom Abstraction Reactions of Charged Polyaromatic σ-Radicals Related to the Active Intermediates of the Enediyne Antitumor Drugs. J. Am. Chem. Soc. 2002;124:4108–4115. doi: 10.1021/ja012243c. [DOI] [PubMed] [Google Scholar]
- 12.Adeuya A, Price JM, Jankiewicz BJ, Nash JJ, Kenttämaa HI. Gas-phase Reactivity of Protonated 2-, 3-, and 4-Dehydropyridine Radicals Toward Organic Reagents. J. Phys. Chem. A. 2009;113:13663–13674. doi: 10.1021/jp901380y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith RL, Kenttämaa HI. A General Method for the Synthesis of Charged Phenyl Radicals in the Gas Phase. J. Am. Chem. Soc. 1995;117:1393–1396. [Google Scholar]
- 14.Thoen K, Smith RL, Nousiainen JJ, Nelson ED, Kenttämaa HI. Charged Phenyl Radicals. J. Am. Chem. Soc. 1996;118:8669–8676. [Google Scholar]
- 15.Widjaja F, Jin Z, Nash JJ, Kenttämaa HI. Direct Comparison of Solution and Gas- Phase Reactions of the Three Distonic Isomers of the Pyridine Radical Cation with Methanol. J. Am. Chem. Soc. 2012;134:2085–2093. doi: 10.1021/ja207899j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heidbrink JL, Ramirez-Arizmendi LE, Thoen KK, Guler L, Kenttämaa HI. Polar Effects Control Hydrogen-Abstraction Reactions of Charged, Substituted Phenyl Radicals. J. Phys. Chem. A. 2001;105:7875–7884. [Google Scholar]
- 17.Thoen KK, Perez J, Ferra JJ, Kenttämaa HI. Synthesis of Charged Phenyl Radicals and Biradicals by Laser Photolysis in a Fourier-Transform Ion Cyclotron Resonance Mass Spectrometer. J. Am Soc. Mass Spectrom. 1998;9:1135–1140. doi: 10.1016/S1044-0305(98)00097-X. [DOI] [PubMed] [Google Scholar]
- 18.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision D.01. Wallingford CT: Gaussian, Inc; 2004. [Google Scholar]
- 19.Minisci F, Vismara E, Fontana F, Morini G, Serravalle M. Polar Effects in Free- Radical Reactions. Solvent and Isotope Effects and Effects of Base Catalysis on the Regioand Chemoselectivity of the Substitution of Protonated Heteroaromatic Bases by Nucleophilic Carbon-centered Radicals. J. Org. Chem. 1987;52:730–736. [Google Scholar]
- 20.Donahue NM, Clarke JS, Anderson JG. Predicting Radical-Molecule Barrier Heights: The Role of the Ionic Surface. J. Phys. Chem. A. 1998;102:3923–3933. [Google Scholar]
- 21.Ohkura K, Seki K, Terashima M, Kanaoka Y. The Pyridyl Cation as a Reactive Intermediate in the Photoreaction of Iodopyridines with Benzenes. Tett. Lett. 1989;30:3433–3436. [Google Scholar]
- 22.Holubek J, Volke J. Polarography of Hetereocyclic Aromatic Compounds. 13. Polarography Fission of Carbon-Halogen Bonds in Monohalogenopyridines. Collect. Czech. Chem. C. 1962;27:680–692. [Google Scholar]
- 23.Calculated at the G3MP2B3 level of theory
- 24.Hunter EP, Lias SG. Evaluated Gas Phase Basicities and Proton Affinities of Molecules: An Update. J. Phys. Chem. Ref. Data. 1998;27:413–656. [Google Scholar]
- 25.Calculated with the CHELPG procedure and fitting to the molecular dipole moment at the UBPW91/cc-pVDZ//UBPW91/cc-pVDZ level of theory
- 26.(a) Jankiewicz BJ. Gas-Phase Studies on the Reactivity of Charged, Aromatic σ,σ,σ- Triradicals by Using Distonic Ion Approach and Fourier Transform Ion Cyclotron Resonance (FT-ICR) Mass Spectrometry. Ph. D. Thesis. Purdue University; 2008. [Google Scholar]; (b) Kirkpatrick L. Gas-Phase Studies on the Reactivity and Formation of para-Benzynes and Carbon-Nitrogen (CN) ortho-Benzynes in a Fourier Transform Ion Cyclotron Resonance (FT-ICR) Mass Spectrometer. Ph. D. Thesis. Purdue University; 2010. [Google Scholar]
- 27.Corilo YE, Eberlin MN. Recognizing α-, β- or γ-Substitution in Pyridines by Mass Spectrometry. J. Mass Spectrom. 2008;43:1636–1640. doi: 10.1002/jms.1442. [DOI] [PubMed] [Google Scholar]
- 28.Hanstorp D, Gustafsson M. Determination of the Electron Affinity of Iodine. J. Phys. B: At. Mol. Opt. Phys. 1992;25:1773–1783. [Google Scholar]