

Abstract
Ethanolamine ammonia-lyase (EAL) is a 5’-deoxyadenosylcobalamin (AdoCbl; coenzyme B12) –dependent bacterial enzyme that catalyzes the deamination of the short-chain vicinal amino alcohols, aminoethanol and [S]- and [R]-2-aminopropanol. The coding sequence for EAL is located within the 17-gene eut operon, which codes for the broad spectrum of proteins that comprise the eut metabolosome sub-organelle structure. A high-resolution structure of the ~500 kDa EAL [(EutB-EutC)2]3 oligomer from Escherichia coli has been determined by X-ray crystallography, but high-resolution spectroscopic determinations of reactant intermediate state structures, and detailed kinetic and thermodynamic studies of EAL, have been conducted for the Salmonella typhimurium enzyme. Therefore, a statistically robust homology model for the S. typhimurium EAL is constructed from the E. coli structure. The model structure is used to describe the hierarchy of EutB and EutC subunit interactions that construct the native EAL oligomer, and specifically, to address the long-standing challenge of reconstitution of the functional oligomer from isolated, purified subunits. Model prediction that the (EutB2)3 oligomer assembly will occur from isolated EutB, and that this hexameric structure will template the formation of the complete, native [(EutB-EutC)2]3 oligomer, is verified by biochemical methods. Prediction that cysteine residues on the exposed subunit-subunit contact surfaces of isolated EutB and EutC will interfere with assembly by cystine formation is verified by activating effects of disulfide reducing agents. Ångstrom-scale congruence of the reconstituted and native EAL in the active site region is shown by electron paramagnetic resonance spectroscopy. Overall, the hierarchy of subunit interactions and microscopic features of the contact surfaces, that are revealed by the homology model, guide and provide a rationale for a refined genetic and biochemical approach to reconstitution of the functional [(EutB-EutC)2]3 EAL oligomer. The results establish a platform for further advances toward understanding the molecular mechanism of EAL catalysis, and for insights into therapy-targeted manipulation of the bacterial ethanolamine utilization (eut) metabolosome.
Ethanolamine ammonia-lyase (EAL)1, 2 is a 5’-deoxyadenosylcobalamin (AdoCbl; coenzyme B12; Scheme 1) –dependent bacterial enzyme [EC 4.3.1.7; cobalamin (vitamin B12)-dependent enzyme superfamily3],4-6 that catalyzes the deamination of the short-chain vicinal amino alcohols, aminoethanol and [S]- and [R]-2-aminopropanol.7 The coding sequence for EAL is located on the 17-gene eut operon8 which contains coding regions for the proteins required for bacterial utilization of ethanolamine as a carbon and nitrogen source, in the presence of exogenous AdoCbl.9-11 Studies of the eut metabolosome sub-organellar structure have focused on molecular understanding of the hierarchical assembly,12 and the complex chemistry that is involved in ethanolamine processing.13 Ethanolamine utilization has been identified as a key competitive advantage for colonization of the mammalian gut by bacterial pathogens, and ethanolamine can serve as a trigger for virulence.14, 15 A strong correlation has been found between food-poisoning bacteria and the presence of eut genes.16
Scheme 1.
EAL, the central enzyme in ethanolamine utilization, is a “radical enzyme,” which harnesses the high reactivity of electron-deficient species to perform catalysis.17 Previous detailed studies of EAL structure and mechanism have elucidated the geometry of the reactant centers in the active site,18-21 and determined the kinetics of culled steps in the reaction cycle of EAL,22-26 in the native enzyme from Salmonella typhimurium. X-ray crystallographic structures of the complete EAL from a different bacterium, Escherichia coli, including bound cofactor and substrate have been obtained,27 supplanting an earlier incomplete structure of EutB from Listeria monocytogenes (PDB ID: 2QEZ) and a model for S. typhimurium EutB based on secondary structure-prediction.28 Further advances toward elucidating the molecular mechanism of EAL require a high-resolution model for the S. typhimurium protein, for structure-function correlations, and development of methods for efficient site-directed mutagenesis of EAL. Progress has been thwarted by the large mass (12 subunits, ~500,000 g/mol) and low solubility (<2 mg/ml) of the EAL oligomer,29 and individual subunit instability. Here, we present a robust, homology-modeled structure of EAL from S. typhimurium, which is based on the E. coli structure, and use it to develop and rationalize approaches for expression and reconstitution of functional EAL from purified, individual protein subunits.
EAL is composed of two subunits, coded by the eutB and eutC genes. The larger subunit, EutB (453 residues; 49.4 kDa; S. typhimurium), is a (βα)8, TIM-barrel protein,30 that contains the substrate binding cavity at the C-terminal end of the β-barrel.28 The substrate binding cavity is capped by the AdoCbl binding site. The cofactor site and C-terminal end of the β-barrel are covered by the smaller subunit, EutC (298 residues; 32.0 kDa; S. typhimurium).27 The EutB-EutC heterodimer is the fundamental structural unit of EAL. In the EutB6EutC6, EAL oligomer (488 kDa; S. typhimurium), two EutB-EutC heterodimers are organized as a (EutB-EutC)2 homodimer, and three homodimers are organized into a [(EutB-EutC)2]3 trimer. The native eutb and eutc are under control of the same promoter, and are co-expressed in vivo.
The large molecular mass of the EAL oligomer and associated low solubility29 is linked to the quasi-solid state of the enzyme in the eut metabolosome sub-organelle, in vivo. In vitro, the high aggregation propensity interferes with expression and purification yields of correctly folded, functional EAL (owing to inclusion body formation) during expression of plasmids that include the cloned eutb-eutc coding region, in E. coli vectors. Techniques employed for growth of bacteria in which EAL is overexpressed have used systems designed to lower the rate of gene expression, as follows: (a) Growth and expression under non-inducing conditions,31 (b) use of the BD21 pLysS system,32 and (c) low temperature growth.27 Previous attempts at the alternative approach of reassembling functional EAL from its separately expressed and purified subunits, EutB and EutC, have been unable to recover activity.29 This may, in part, be related to the observed instability of isolated EutC. The Instability Index33 of EutC is 51.4 (a score >40 connotes high instability), compared with 27.0 for EutB. Consistent with these predictions, EutC is subject to rapid trypsinization.34 In addition, the N-terminal region of EutC may drive the formation of inactive EAL aggregates.34
We generate a multi-scale homology model structure of S. typhimurium EAL, which describes the molecular structure of the subunits, including the active site interactions of side chains, substrate and cofactor, and the global subunit interactions in the oligomer, that is based on the X-ray crystallographic structure of E. coli EAL.27 The homology model predicts that a EutB hexamer can form in the absence of EutC, and that cystine disulfide bond formation from cysteine sulfhydryl groups exposed in the isolated subunits will promote protein aggregation. Model predictions in concert with biochemical manipulations lead to development of a low-temperature growth and expression system in NEB T7 Express Iq E. coli, that allows the individual expression of soluble EutB and EutC. Using tandem affinity and size exclusion purification, the EutB subunit can be purified to homogeneity, and EutC can be enriched to >95%. Judicious application of sulfydryl reagents allows the reconstitution of active EAL. Overall, the EAL structure model predictions inform the development of a novel sequential assembly protocol for reconstitution of the functional S. typhimurium EAL oligomer from individual subunits, which is verified by biochemical, spectroscopic, and functional studies.
MATERIALS AND METHODS
Homology Modeling of S. typhimurium EAL
EAL Sequence alignment was performed with the ClustalW2 software35, 36 to map the S. typhimurium EAL sequence onto the E. coli EAL sequence. The complete alignment is given in Figure S1, Supporting Information. Residues 4 to 43 of the N-terminal end of the EutC subunit of the E. coli EAL were truncated for crystallization.27 Therefore, these residues are not present in the X-ray crystallographic structure.27 As a result, the modeled structure also does not contain the first 46 residues of EutC. Five homology models of the [(EutB-EutC)2]3 functional oligomeric structure were generated by using this alignment and the 3ABO E. coli crystal structure with the MODELLER program.27, 37 MODELLER generates homology modeled protein structures by using comparative modeling based on spatial restraints. MODELLER minimizes an effective energy function, which is based not only on alignment to the template structure, but also on statistical experimental restraints from similar structural features, such as main chain bond lengths and dihedral angles from equivalent groups. In addition to these homology-derived restraints, MODELLER also takes into account stereochemical restraints using the CHARMM22 force-field.
The models generated were first assessed by using the Discrete Optimized Protein Energy (DOPE) profile.38 The DOPE profile allows quick evaluation of a putative structure by distinguishing high energy regions within a structure, which may indicate possible errors. However, it is not an absolute measure and is only useful to rank models generated from the same alignment. The structure with the lowest DOPE score was selected for further analysis. The modeled structure was validated by using the PROCHECK test suite.39 PROCHECK tests the stereochemical quality of protein structures, by assessing main-chain parameters such as bond lengths, bond planarity and Ω, Ф and ψ bond angles, in addition to side-chain parameters such as χ1, χ2, χ3 and χ4 bond angles. The structure was visually examined by using SYBYL 8.0 (Tripos Inc., St. Louis, MO) and PyMol (Delano Scientific LLC, San Fransisco, CA).
The solvent accessibility of residues in the modeled structure was judged using the ASAView web server.40 Briefly, ASAView uses the DSSP program41 to calculate the surface area of each amino acid residue that is accessible to solvent, and scales this solvent accessible surface area by the total area of that residue. Relative solvent accessibilities for EutB and EutC monomers, the EutB-EutC heterodimer, (EutB-EutC)2 homodimer, and [(EutB-EutC)2]3 oligomer were computed, and compared to assess the changes upon hexamer formation.
The electrostatics of the modeled protein surface were calculated by using the Adaptive Poisson-Boltzmann Solver, APBS.42 The PDB2PQR web server,43, 44 a tool that automates the conversion of protein structural coordinates (PDB file) into files containing the calculated charge information (PQR), was used. PDB2PQR builds and assesses the hydrogen bonding network of the protein structure, and estimates protonation states, charge and atomic radii for the biomolecule. APBS uses this input information to solve the Poisson-Boltzmann equation in a parallel manner, allowing the evaluation of the electrostatic potentials for the modeled structure. The results were visualized in PyMol.
Protein Library
A search of the Pfam database45 yielded 693 sequences for EutB and 689 for EutC. A multiple sequence alignment of these sequences was downloaded, and examined with the MATLAB Bioinformatics Toolbox (Mathworks Inc., Natick, MA). In this alignment, EutB residues 11 to 452, and EutC residues 63 to 297 of S. typhimurium were aligned. At each position in the alignment, the most frequent residue was used to build a consensus sequence, which was used to assess the degree of conservation.
Construction of EALH6 plasmid
The 8.5 kbp recombinant wild-type DNA coding for the EutB and EutC subunits of EAL from S. typhimurium in the pBR322 vector was extracted from the E. coli overexpression strain.31 Primers coding an N-terminal (histidine)6 tag complementary to the eutB starting sequence were obtained from Invitrogen (Life Technologies Corp., Carlsbad, CA), as follows: EutBH6 FP – GGG GAA CGA CTT ATG CAT CAC CAT CAC CAT CAC AAA CTA AAG ACC ACA TTG, and EutBH6 BP – CAA TGT GGT CTT TAG TTT GTG ATG GTG ATG GTG ATG CAT AAG TCG TTC CCC.
Mutagenesis was carried out by using Stratagene (Stratagene, La Jolla, CA) QuikChange II XL Site-Directed Mutagenesis kit according to manufacturer’s instructions. Briefly, the wild-type EAL plasmid was used as a template for PCR amplification of the (histidine)6 primers with high fidelity PfuUltra DNA polymerase. The endonuclease, DpnI, was used to digest the methylated template DNA, leaving the new EALH6 DNA intact. The new plasmid was then transformed into XL10 Gold ultracompetent cells (Stratagene, La Jolla, CA), and the sequence was verified by DNA sequencing (performed by Beckman Coulter Genomics, Danvers, MA).
Construction of EutBH6 and EutCH6 plasmids
Preparation of pET28a plasmid
An ampicillin resistant pET28a plasmid with a (histidine)6 tag was used. The plasmid was digested in the multiple cloning site polylinker with the restriction enzymes XhoI and NdeI from New England Biolabs (Ipswich, MA), and the 5 kbp cleaved sticky-ended DNA fragment was purified from a 1% agarose Tris-Acetate-EDTA (TAE) gel by using the QIAquick gel extraction kit (QIAGEN, Venlo, Netherlands).
Preparation of EAL gene
The wild-type EAL pBR322 sequences did not contain any NdeI (CA’T ATG) and XhoI (C’TC GAG) restriction sites. Primers were purchased from Integrated DNA Technologies (Coralville, IA) to insert these restriction sites to the start and end of the EutB and EutC genes separately, as follows: EutBNdeI FP – AAA AAA AAA A CAT ATG AAA CTA AAG ACC ACA TTG TTC GGC AAT G; EutBXhoI BP - AAA AAA AAA A CTC GAG TCA GAA GAA CAA TGA CGG ATC GCC CGC CCG TTT GG; EutCNdeI FP - AAA AAA AAA A CAT ATG GAT CAA AAA CAG ATT GAA GAA ATT GTA CG; EutCXhoI BP - AAA AAA AAA A CTC GAG TTA ACG GGT CAT GTT GAT GCC GGA CGC TTT. PCR was performed with Pfu DNA polymerase, and the eutB (1.5 kbp) and eutC (0.9 kbp) genes were purified from the result by electrophoresis on a 1% agarose TAE gel. DNA was extracted from the gel, and then digested with the restriction enzymes, XhoI and NdeI, to obtain sticky ends complementary to the pET28a plasmid. These sticky ended genes were purified again using agarose gel electrophoresis.
Construction of final plasmids
Sticky-ended pET28a and eutB or eutC were ligated by using T4 DNA ligase. These constructs were transformed into XL10 Gold ultracompetent cells. The plasmid was then transferred into T7 Express Iq competent cells (New England Biolabs, Ipswich, MA), because these cells were found to be optimal for gene expression. The sequences of the EutB and EutC transformants were verified by Beckman Coulter Genomics, and were found to have 100% sequence identity between the restriction sites.
Bacterial Growth and Protein Purification
Growth
Cells were grown from glycerol stocks in 5 mL 2X YT starter cultures with 100 ug/mL ampicillin at 37 °C (30 °C for EutB, and EutC) overnight with shaking. This was used to inoculate 500 mL medium which was allowed to grow until OD600~0.8. Following induction by 0.4 mM IPTG, cells were grown for 5 h, and then harvested by centrifugation at 3,000×g at 4 °C. Cells were washed with 10 mM potassium phosphate (KPi) buffer (pH 7.5), and the pellet was frozen in liquid nitrogen and stored at -80 °C.
Purification
Cells were thawed and resuspended in 5 mL of lysis buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, pH 8.0), which was supplemented with 1 mg/mL lysozyme, 50 uL 100× HALT protease inhibitor cocktail (ThermoFisher Scientific, Waltham, MA), and 2 μL Benzonase Nuclease (Sigma-Aldrich, St. Louis, MO) per gram of wet weight. The cells were then lysed on ice for 1 h, then sonicated at 10 W (10 s on/off cycle) for 3 minutes. The cell debris was separated by centrifugation at 10,000×g at 4 °C for 20 min. The supernatant was filtered with a 0.45 micron filter, then loaded onto a 5 mL HisTrap Fast Flow crude column that was mounted in an AKTA Purifier FPLC (GE Healthcare Life Sciences, Schenectady, NY). The column was washed with lysis buffer, followed by elution buffer (lysis buffer with 250 mM imidazole added) at 1:9 proportion with lysis buffer (34 mM imidazole). EAL was then eluted using 40% elution buffer (106 mM imidazole). At higher elution buffer percentages, the EAL in the eluate was deficient in EutC. For EutBH6 and EutCH6, elution was performed at 100% elution buffer.
The protein-containing fraction (10 mL) was dialyzed in 10 K membrane dialysis tubing against 1 L of 10 mM KPi buffer (pH 7.5) for 6 h, followed by buffer replacement, and dialysis overnight at 4 °C. The protein was then concentrated using aquacide for 1 h, and spin-concentrated using a Pierce 7 mL concentrator with 10 K membrane (ThermoFisher Scientific, Waltham, MA). The resulting protein concentration was checked using the Bradford protein assay,46 by using a 2 mg/mL bovine serum albumin standard (ThermoFisher Scientific, Waltham, MA). For purified EutB and EutC, the concentration was directly assessed using ultraviolet absorption spectroscopy, by using the sequence-predicted extinction coefficients47 at 280 nm of 33,350 M-1cm-1 and 18,450 M-1cm-1, respectively. Absorption spectroscopy measurements were performed on a Shimadzu UV-1601 absorption spectrophotometer.
Gel filtration
Gel filtration was performed on an analytical Superdex 200 10/300 GL column from (GE Healthcare Life Sciences, Schenectady, NY) by using the AKTA Purifier FPLC system. Protein Enzyme in a volume of 100 μL was injected into the column, which was pre-equilibriated with 10 mM KPi buffer (pH 7.5), and run at a flow rate of 0.5 mL/min. Absorbance was monitored at 280 nm.
SDS and Native PAGE
Polyacrylamide Gel Electrophoresis (PAGE) was performed by using 10% Mini Protean TGX gels from BioRad Laboratories (Hercules, CA) according to known protocols.48, 49 Coomassie Brilliant Blue dye was used for staining.
Enzyme Activity Assay
Enzyme activity was determined by using the coupled alcohol dehydrogenase assay, essentially as described.50 The assay mixture contained 10 mM KPi buffer (pH 7.5), 2:1 5’-deoxyadenosylcobalamin/active sites, substrate, 120 μM NADH and 4.3 U of alcohol dehydrogenase at 25 °C. For EAL reconstitution, 2:1 5’-deoxyadenosylcobalamin/active sites was added to EutB in a 10 mM KPi buffer (pH 7.5) with 5 mM reductant, while EutC was incubated separately. Incubations prior to assay were performed for 1h on ice. The two subunits were mixed and enzyme activity was assayed, as described above.
Electron Paramagnetic Resonance Spectroscopy
Samples for EPR spectroscopy were prepared under dim red-safe lighting, and protected from light during the experiment to prevent photo-degradation of 5’-deoxyadenosylcobalamin. All preparations were performed on ice. A proportion of 2:1 5’-deoxyadenosylcobalamin/active sites was added to 50 μM enzyme (active site concentration; corresponding EAL concentration, 8.3 μM) in 10 mM KPi buffer (pH 7.5) with 5 mM TCEP. [S]-2-aminopropanol (30 mM) was added to this mixture to start the reaction. The sample was mixed, transferred to a 2 mm OD quartz epr tube and flash frozen in liquid nitrogen. Less than 15 seconds elapsed between mixing and freezing. EPR spectra were obtained by using a Bruker E500 ElexSys EPR spectrometer that was equipped with a Bruker ER4123 SHQE cavity. Temperature was controlled by using a Bruker ER4131VT liquid nitrogen/gas flow cryostat system. All spectra were collected at a microwave power of 2 mW and microwave frequency of 9.45 GHz, at 120 K.
RESULTS AND DISCUSSION
Homology modeling of Salmonella typhimurium EAL
EAL from E. coli and S. typhimurium display 98% sequence identity in the EutB subunit and 91% sequence identity in the EutC subunit (Figure S1, Supporting Information).27, 31 In EutB, there are 9 non-conserved positions (Figure S2, Supporting Information). Two of these positions, EutB322 [I (E. coli)→L (S. typhimurium)] and EutB159 [A (E. coli)→C (S. typhimurium)] are on the interior β-strands of the (βα)8 barrel structure. These residues lie near to the N-terminal end of the barrel, and are not predicted to make substrate contacts. Two other non-conserved positions, EutB203 [S (E. coli)→T (S. typhimurium)] and EutB209 [I (E. coli)→V (S. typhimurium)] are on the α-helices on the exterior of the barrel, and another position, EutB185 [V (E. coli)→A (S. typhimurium)] is in the loop region, that connects barrel α-helix 1 to β-strand 2 at the N-terminal end of the barrel. EutB87 [Q (E. coli)→R (S. typhimurium)] is involved in the binding interface between EutB subunits. There are a larger number, 26, of non-conserved residues in EutC. Of these, only EutC78 [V (E. coli)→A (S. typhimurium)] is involved in the EutC-EutB binding interface. The high sequence identity allowed S. typhimurium EAL to be modeled directly from the template structure of the E. coli enzyme,27 following sequence alignment.35, 36 The selected template was the X-ray crystallographic structure of EAL, Protein Data Bank code, 3ABO. The 3ABO structure includes the complete oligomer structure [(EutB-EutC)2]3, and the EutB-EutC dimer units contain the bound substrate, ethanolamine, and the cobalamin cofactor analog, cyanocobalamin. Five homology models were generated using MODELLER.51 The models were analyzed by using the Discrete Optimized Protein Energy (DOPE), which is an effective energy function that allows comparison between modeled structures.38 The model with the lowest DOPE score was selected. The selected model was also validated by using the PROCHECK model stereochemistry test suite (Figure S3, Supporting Information).39 The model showed 99.9% of residues in allowed regions of the ϕ, ψ-, or Ramachandran, plot, of which 93.5% were in favored regions. The 2 residues that are in disallowed regions (EutC(L197), EutC(S156)) are present in loop segments outside of the α- and β-secondary structure, and therefore do not have a significant impact on the structure of the model. The model structure was also assessed by using the G-factor, a log-odds score based on observed distributions52 of stereochemical parameters. Values of the G-factor were all significantly larger than the limits of -0.5 (probability of being incorrect) (Table S1, Supporting Information).
The homology model of the S. typhimurium EAL oligomer structure is presented in Figure 1. An overlay of the homology model and E. coli secondary structures is shown in Figure S4, Supporting Information. The positions of non-homologous residues are highlighted in Figure S5, and the root mean square deviation of backbone Cα atoms is presented in Figure S6 of Supporting Information.
Figure 1.
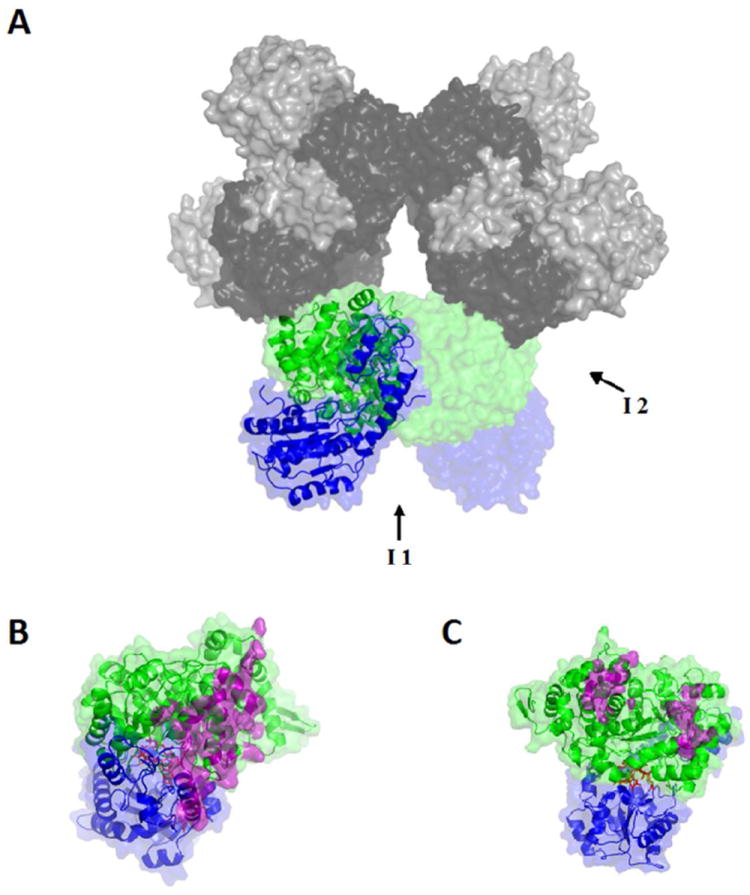
Surface and secondary structure representations of the homology modeled structure of S. typhimurium EAL. (A) The [(EutB-EutC)2]3 EAL oligomer. One (EutB-EutC)2 heterodimer is shown as green (EutB subunits) and blue (EutC subunits), and other two heterodimers in grey tone (EutB, dark grey; EutC, light grey). (B and C) Two EutB-EutC heterodimers, that compose one (EutB-EutC)2 homodimer. EutB is colored green and EutC is blue. (B) The intra-heterodimer interface (denoted I1, in panel A), with residues involved in the contact surface represented in purple. (C) The inter-heterodimer interface (denoted I2, in panel A), with residues involved in the contact surface represented in purple.
Subunit interaction hierarchy from the homology model of Salmonella typhimurium EAL
Figure 1 illustrates the different hierarchies of organization of the EutB and EutC subunits in the oligomer: (a) EutB and EutC monomers form the EutB-EutC heterodimer. (b) Two heterodimers form a (EutB-EutC)2 homodimer, and (c) three (EutB-EutC)2 homodimers form the biologically active oligomer, which is described as a trimer of dimers, [(EutB-EutC)2]3. Thus, there are two different types of contact faces between EutB-EutC units. The (EutB-EutC)2 intra-homodimer contact face has an area of 1293 Å2, and is shown in Figure 1B. The edge-to-edge contact from inter-homodimer formation of the trimer, shown in Figure 1C, has a smaller surface area of 804 Å2. The primary contacts for both the intra- and inter-homodimer interfaces are on the EutB subunits. The model shows that the EutC subunit is not involved in the edge-to-edge, intra-homodimer interaction. Therefore, the model predicts that the hexameric (EutB2)3 trimer will form in solution, in the absence of EutC.
Figure 2 shows the common contact face between EutB and EutC, which has a surface area of 2080 Å2. The EutC contact face has a higher incidence of positively charged residues (23% positive, 10% negative), compared with the corresponding face on EutB, which carries the opposite charge (6% positive, 20% negative), where the percentages are relative to all contact residues. Figures S7 and S8 (Supporting Information) present the classes of contact surface residues and the location in EutB and EutC, respectively. In addition, two salt bridges are identified in this binding interface: EutB-D398 – EutC-R80 and EutB-D411 – EutC-R89. These results suggest that static charge-charge interactions contribute significantly to EutB-EutC heterodimer assembly, and therefore, that low ionic strength conditions will favor inter-subunit interactions.
Figure 2.
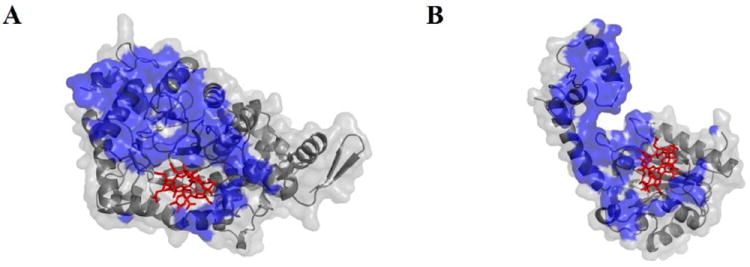
Contact surfaces of the EutB and EutC subunits in the EutB-EutC heterodimer, in the modeled structure of S. typhimurium EAL. (A) EutB subunit. (B) EutC subunit. The contact face is shown as a blue surface. The 5’-deoxyadenosylcobalamin cofactor is represented as red sticks.
The EAL from S. typhimurium contains 16 cysteine residues. Eight of these cysteine residues (EutB: C223, C361, C363, C364, C388; EutC: C158, C243, C261) are highly conserved, based on a survey of 693 EutB sequences and 689 EutC sequences from different bacterial species and strains.45 Figure 3 illustrates the positions of the cysteine residues in the EutB and EutC structures. The conserved residues include an unusual CDCC motif, in the active site region in EutB (residues C361, C363, C364), and C261 of EutC, which is in the cofactor binding site. Both of these active site regions become solvent exposed in the isolated subunits, and therefore, during the separate expression of EutB and EutC. Native EAL appears to be devoid of cystine disulfide bonds, with the possible exception of cysteines in the active site, and C37 and C46 of EutC, which were not modeled, because they are present on a cleaved leader sequence in E. coli. However, the EAL homology model indicates that, when the subunits are grown separately, opportunistic disulfide linkages may form, which indicates a requirement for a disulfide-reducing environment, during the reconstitution of isolated EutB and EutC subunits.
Figure 3.
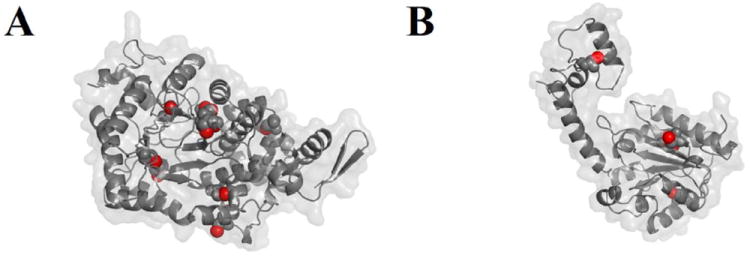
Positions of cysteine side chain sulfhydryl groups (red spheres) in the EutB and EutC subunits in the modeled structure of S. typhimurium EAL. (A) EutB subunit. (B) EutC subunit. The subunit orientations are the same as shown in Figure 2.
Expression and purification of wild-type EAL
The (histidine)6–tagged, wild-type EAL construct, EALH6, allows expression of EAL at 37 °C without significant inclusion body formation. An SDS-PAGE gel of overexpressed EAL purified from this construct is shown in Figure 4. The EutB and EutC protein subunits have calculated masses of 48 kDa, and 30 kDa, which are comparable to the expected masses of 50 kDa and 32 kDa, respectively. The differences in masses between EutB and EutC from the expressed EAL oligomer and the individually purified subunits are explained by the presence of the pET28 (histidine)6–tag sequence in the plasmids used for the purification of the individual protein subunits.
Figure 4.
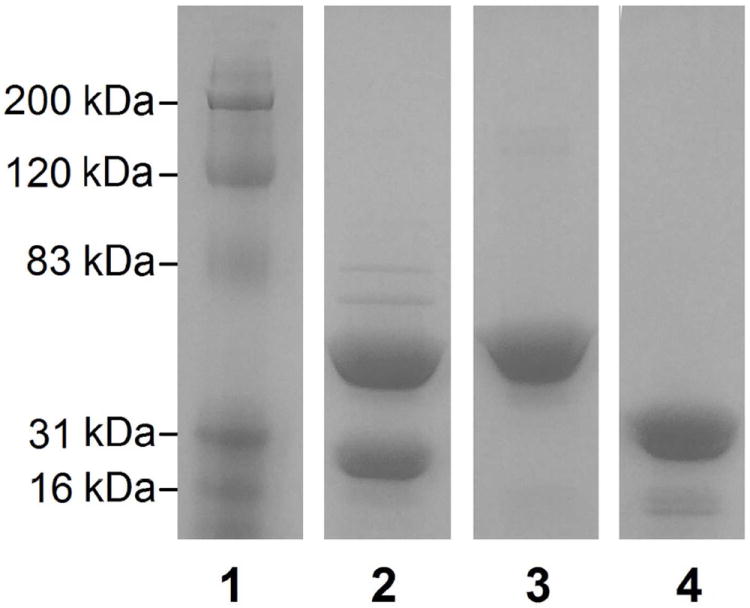
Sodium dodecyl sulfate polyacrylamide gel electrophoresis of purified proteins. Lanes contain the following: (1) Protein molecular mass ladder. (2) EAL purified from the EALH6 construct. (3) EutB. (4) EutC. Lane 4 was adjusted to correct for dye front curvature.
EAL purified from the EALH6 construct had a higher proportion of the EutB protein, relative to EutC. This phenomenon has been previously noted in other EAL overexpression systems, and attributed to more efficient expression of genes that are closer to the promoter region, relative to genes further downstream.34 In the EALH6 pBR322-based construct, eutB precedes eutC, and is therefore closer to the lac promoter. Alternative explanations are that incompletely constructed EAL has a higher affinity for the nickel column than the complete oligomer, or that EutC is released from column-bound EutB during the washing procedures. In order to accomplish the purification of the homogeneous [(EutB-EutC)2]3 complex, elution from the nickel affinity column was performed at a relatively low imidazole concentration (100 mM). At higher imidazole concentrations (250 mM), the eluted EAL was deficient in EutC (Figure S9, Supporting Information). Densitometric analysis53 of SDS PAGE gels showed that the subunit molar ratio of the purified EAL was 1.00:1.01, following elution at 100 mM imidazole, compared to a molar ratio of 1.00:0.32, for elution with 250 mM imidazole (Table S2, Supporting information). In addition, native PAGE and gel filtration experiments show no evidence of excess EutB or EutC in purified EALH6 (Figure 5 and Figure S10, Supporting Information). In summary, the results indicate the following: (a) EutC-deficient EutB-EutC oligomer-like complexes bind with higher affinity to the nickel affinity column. (b) The purified [(EutB-EutC)2]3 complex can be obtained by elution with 100 mM imidazole.
Figure 5.
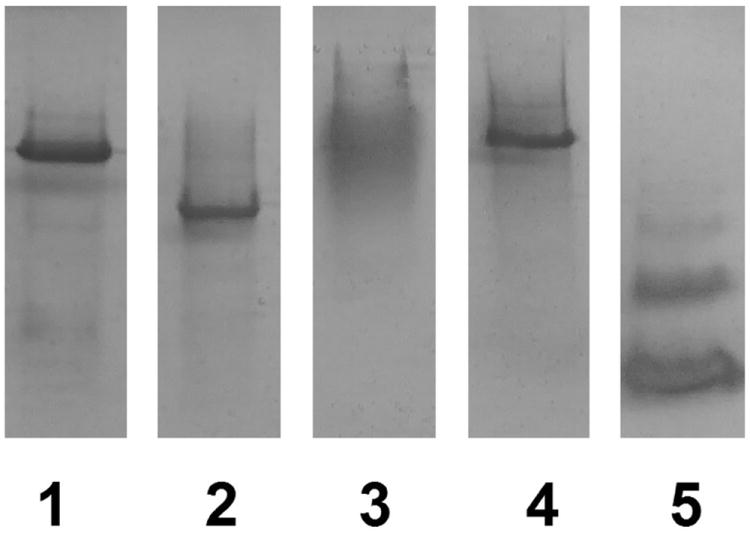
Native polyacrylamide gel electrophoresis of purified proteins. Lanes contain the following: (1) EAL purified from the EALH6 construct. (2) EutB. (3) EutC. (4) EAL reconstituted from the purified EutB and EutC subunits. (5) Bovine serum albumin standard. Lane 5 was adjusted to correct for dye front curvature.
Expression and purification of individual EutB and EutC proteins
The formation of insoluble aggregates during the separate, individual expression of EutB and EutC proteins from the respective constructs, was prevented by expression at low temperature (30 °C) in NEB T7 Express Iq E. coli cells. The tight control of EAL expression by the upregulated lac repressor in this strain54 prevents the formation of inclusion bodies, and leads to formation of soluble EutB and EutC. Using a single step of nickel affinity column chromatography, over 90% pure protein could be obtained as judged by densitometric analysis of SDS-PAGE gels. The EutB and EutC proteins were purified further, by using size exclusion chromatography. EutB was purified to homogeneity, and EutC was purified to >95%. An overloaded SDS-PAGE gel of the purified protein preparations is shown in Figure 4. The mass of the EutB protein was calculated to be 50 kDa, which agrees well with the sequence-predicted mass of 52 kDa. The EutC protein had a calculated mass of 33 kDa, which is comparable to the sequence-predicted value of 34 kDa. Following purification, the EutC protein showed visible aggregation over the course of 2 h, when at high concentrations (> 5 mg/mL). Therefore, care was taken to ensure that EutC was not kept at high concentration for periods > 15 min. The EutB protein was not observed to form higher molecular mass aggregates, up to a protein concentration of 40 mg/mL, over the course of 2 h. The results show that the individual EutB and EutC proteins can be expressed and purified, separately, by the developed protocol.
Characterization of oligomeric state of individually expressed and purified EutB and EutC
Figure 5 shows non-denaturing PAGE results for protein purified from expression of the EALH6, EutB and EutC constructs. The EALH6 protein oligomer (494 kDa; lane 1) migrates as a single dominant band, with a relative mobility (Rf) of 0.187. The purified EutB protein (Figure 5, lane 2) displays a single dominant band with a larger Rf value (0.247). The presence of a single band for purified EutB is consistent with the presence of EutB as the 310 kDa (EutB2)3, or EutB6 hexameric, oligomer. This interpretation is supported by gel filtration experiments (Figure S10, Supporting Information), which show that purified EutB elutes closer to the authentic EALH6 oligomer (+1.0 ml), than to purified EutC (+5.5 mL). Although differences in protein size, structure and charge influence Rf values in non-denaturing PAGE, so that comparisons of different proteins are approximate, at best, we note that the bovine serum albumin control in Figure 5 (lane 5), shows characteristic monomer (66 kDa), dimer (132 kDa) and trimer (198 kDa; Rf = 0.265) bands,55 which display relatively large Rf values.
Figure 5 also shows that the purified EutC subunit migrates as a diffuse band in the non-denaturing gel. Under denaturing and reducing conditions, EutC migrates as a single band in SDS-PAGE, as shown in Figure 4, and under reducing conditions, it elutes in a single fraction in Gel Filtration (Figure S10, Supporting Information). Therefore, the diffuse band on the non-denaturing gel is caused by non-specific aggregation of isolated EutC. The first 27 residues of EutC have been associated with the low solubility and aggregation of wild-type EAL in E. coli.34 It was found that these residues are not essential for activity, and can be removed.34 The first 19 residues of E. coli EutC are homologous to those in S. typhimurium, and have been identified as a signaling sequence that targets EAL to the eut metabolosome microcompartment, where EAL is found in vivo.56 The residues in the N-terminal region of EutC may induce the formation of aggregates of varying sizes and conformations.
Assembly of EAL from individually expressed and purified EutB and EutC
Figure 5 shows the non-denaturing PAGE results for reconstitution of 1:1 mol/mol purified EutB and EutC proteins. The reconstituted EAL (lane 4) has Rf =0.178, compared with Rf=0.187 for wild type EAL (lane 1). The closely similar Rf values indicate that the [(EutB-EutC)2]3 EAL oligomer is reconstituted from its component subunits. The reconstituted EAL shows some evidence of diffuse staining at Rf values less than the principal band. This may represent improperly constructed EAL, owing to aggregation, because EutB is present in stoichiometric amounts with EutC. In summary, the results demonstrate that the developed purification and reconstitution protocols lead to the native [(EutB-EutC)2]3, oligomeric structure of EAL, from isolated EutB and EutC subunits.
Steady-state enzyme activity of EAL reconstituted from isolated EutB and EutC
The homology model structures in Figure 3 show that cysteine residues are exposed on the contact surfaces of both the EutB and EutC subunits. Thus, EutB and EutC subunits, that are expressed and purified individually, will have solvent exposed cysteine residues in the active site region, and, in the absence of the reducing intra-cellular environment,57 these cysteines may undergo oxidative reactions to form intra- and/or inter-subunit disulfide bond formation. In-line with this prediction, cystine disulfide bond reducing agents were found to be crucial for the functional reconstitution of EAL from isolated EutB and EutC subunits, as shown by the kcat values for different reconstitution conditions in Figure 6. A kcat value of 0.51 ± 0.09 s-1 is obtained, when no cystine disulfide reducing agent is added to the post-affinity column eluate, prior to loading onto the gel filtration column, and when no cystine disulfide reducing agent is present in the kinetic assay buffer. Figure 6 shows that addition of the disulfide reducing agent, tris-(2-carboxyethyl) phosphine (TCEP), to the sample, prior to loading on the gel filtration column, leads to 25-fold increase in the kcat value to 12.6 ±0.7 s-1. An additional increment to 15.6 ±0.3 s-1 is achieved when TCEP is added to the separate, isolated EutB and EutC, and incubated for 2-5 min, prior to mixing the subunits for the steady-state kinetic assay. Figure 6 shows that pre-incubation with TCEP prior to the steady-state kinetic assay mixture is more effective in raising kcat than the disulfide reducing agents, β-mercaptoethanol (β-ME) and D,L-dithiothreitol (DTT) at equivalent concentrations. β-ME is a monothiol reductant, and is therefore less effective than the dithiol, DTT.58, 59 From the selectivity of TCEP for reducing cysteine residues in the protein,60 we can conclude that the primary mechanism of inactivation of EAL is through oxidation of cysteines, rather than other pathways, such as proteolytic degradation.
Figure 6.
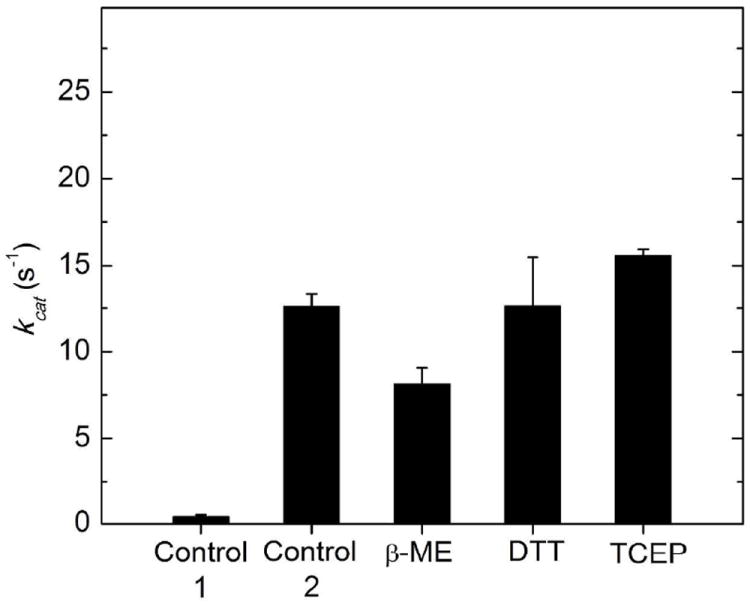
Dependence of the activity of reconstituted EAL on the presence of different disulfide reducing agents. Activity reprents the maximum turnover number (kcat) attained at excess ethanolamine substrate (concentration≫KM), under the assay conditions described in Methods. The EutB and EutC proteins were present at stoichiometric concentrations. The bars correspond to the following conditions: Control 1: Reductant was omitted during the purification procedure and during activity measurement. All other samples were purified in the presence of 5 mM TCEP reductant, which was removed as a consequence of the gel filtration step of the purification procedure. Control 2: No reductant added to the kinetic assay mixture. β-ME: β-mercaptoethanol (5 mM) added to kinetic assay mixture. DTT: Dithiothreitol (5 mM) added to assay mixture. TCEP: Tris-2-carboxyethyl phosphine (5 mM) added to assay mixture. Values represent the averages and standard deviation (error bars) of three measurements.
Although the enhancement of kcat by DTT is comparable to TCEP, to within the standard deviation of the activity measurements, the stablity of DTT is reduced by nickel contamination from the affinity purification step.59 In addition, TCEP is compatible with sulfhydryl-specific spin labeling reagents at the TCEP concentration used,61 which is a consideration for downstream EPR spectroscopy-associated experiments. Therefore, TCEP was selected as the cystine disulfide reductant for use in the purification and manipulation of EutB and EutC.
Table 1 shows the steady-state enzyme kinetic parameters for the EutB/EutC- reconstituted EAL and the histidine6-tagged EALH6 oligomers. The kcat value of reconstituted EAL is (70 ±16)% of the kcat for EALH6, and the KM value was reduced by 1.4-fold. The difference in KM between EALH6 and reconstituted EAL may be caused by by a subtle effect on the protein structure, that arises from the different His6 tags. In EALH6, there is one His6 tag, which is located on the EutB subunit. The individual EutB and EutC subunits each have a His6 tag, which is adjacent to a linker region. Thus, the reconstituted EAL includes two His6 tags. The reduction in kcat is consistent with the level of functional reconstitution from isolated subunits, as described in more detail, below. The catalytic efficiency, or “specificity constant,”62 which is given by kcat/KM, does not differ significantly for the two EAL oligomers. Isolated, purified EutB and EutC showed no detectable activity (< 0.03 s-1). In summary, the steady-state enzyme kinetics results demonstrate that the developed purification and reconstitution protocol leads to the formation of a functional EAL, staring from the isolated EutB and EutC protein subunits. This result is consistent with the formation of the [(EutB-EutC)2]3 EAL oligomer structure, which is evidenced by the gel electrophoresis results in Figure 5.
Table 1.
Steady-state enzyme kinetic parameters for the purified EAL oligomer and EAL oligomer reconstituted from EutB and EutC subunits. Results represent the average and standard deviation for at least three measurements.
| kcat (s-1) | KM (μM) | kcat/KM (M-1 s-1) | |
|---|---|---|---|
| EAL | 22.7 ± 0.3 | 31.6 ± 4.5 | 7.2 (± 1.1) × 105 |
| Reconstituted EAL | 16.0 ± 2.4 | 22.0 ± 4.2 | 7.3 (± 2.5) × 105 |
EPR spectroscopy of EAL reconstituted from isolated EutB and EutC
The cob(II)alamin-substrate radical pair intermediate accumulates to a level of >90%, relative to active sites, in EAL during steady-state turnover on the substrate, (S)-2-aminopropanol.63 The substrate radical accumulates, because the radical rearrangement is the rate-limiting step in the catalytic cycle.63 The cob(II)alamin-substrate radical pair intermediate can be cryotrapped, and the characteristic radical pair line shape can be detected by using low temperature (120 K), continuous-wave (CW) EPR spectroscopy.18, 63, 64 The line shape arises from the isotropic J-coupling and anisotropic dipolar coupling of the two unpaired electron spins, for a defined, rigid orientation of the substrate radical and cob(II)alamin over an 11 Å electron-electron separation,18, 65 and is therefore exquisitely sensitive to the structure of the protein in the active site region.18, 64 Figure 7 shows the characteristic native cob(II)alamin-substrate radical pair line shape, with features at g⊥ =2.35 for cob(II)alamin and around g=2.00 for the substrate radical, as reported previously.18 Figure 7 shows that the characteristic cob(II)alamin-substrate radical pair line shape is also present in the reconstituted EAL sample, and that the individual EutB and EutC proteins did not support the formation of paramagnetic species. The spectra of wild type EAL and reconstituted EAL overlap, upon rescaling the wild type spectrum by a factor of 0.73. The results suggest the following: (a) The functional reconstitution of EAL from isolated EutB and EutC subunits is achieved at a level of 73%, relative to the EALH6 enzyme. This value is consistent with the reduction in kcat of 70% (Table 1). The origin of the 70% level of functional reconstitution may be the partial aggregation of EutC, as evidenced on the non-denaturing PAGE gel (Figure 5, lane 4). (b) The appearance of the radical pair indicates that the free energy surface for the reaction, and specifically, rate limitation by the radical rearrangement step, is the same in the reconstituted and control EAL oligomers. (c) The structure of EAL in the active site region of the reconstituted enzyme is comparable to the structure in native EAL.
Figure 7.
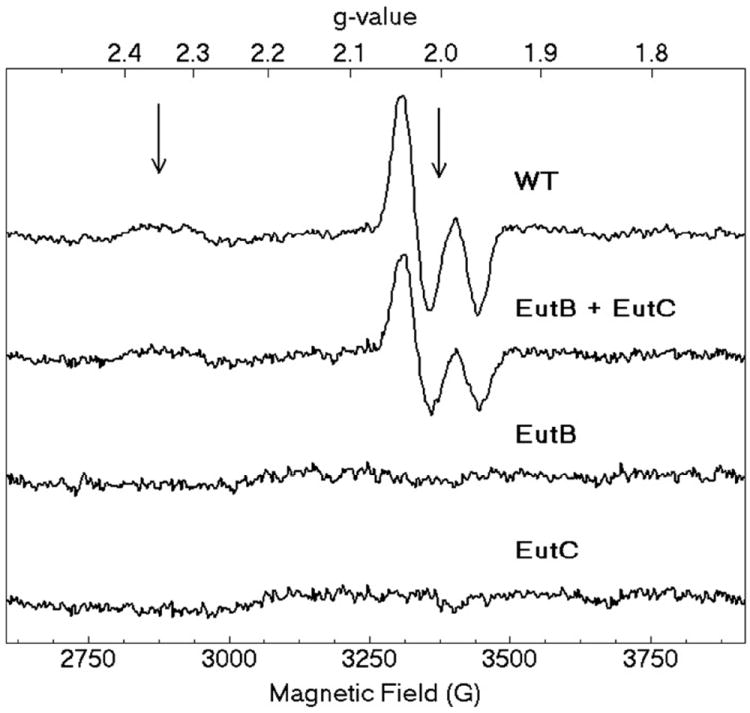
X-band CW-EPR spectra of the cryotrapped cob(II)alamin-substrate radical pair in EAL in different protein samples. From top, spectra shown are as follows: WT: EALH6 protein. EutB+EutC: EAL enzyme reconstituted from purified EutB and EutC subunits. EutB: Purified EutB subunit, alone. EutC: Purified EutC subunit, alone. Arrows show g = 2.35 (g⊥ for cob(II)alamin) and g=2.0023 (free electron g-value). Experimental conditions: microwave frequency, 9.458 GHz; temperature, 120 K; microwave power, 2.04 mW; magnetic field modulation, 10.0 G; modulation frequency, 100 kHz; scan rate, 71 G/s; time constant, 10.24 ms.
CONCLUSIONS
A homology model for the S. typhimurium EAL is constructed from the X-ray crystallographic structure of EAL from E. coli,27 and demonstrated to be robust by statistical criteria. The model structure is used to describe the hierarchy of EutB and EutC subunit interactions that construct the native EAL oligomer, and specifically, to address the long-standing29, 31 challenge of reconstitution of the functional oligomer from isolated, purified subunits. The model predicts that (EutB2)3 oligomer assembly will occur from isolated EutB, and that this hexameric structure will template the formation of the complete, native [(EutB-EutC)2]3 oligomer. SDS- and non-denaturing PAGE and gel filtration verify this prediction. The homology model also shows multiple cysteine residues in the subunit-subunit contact surfaces of EutB and EutC, which predicts that formation of non-native, cystine disulfide bond interferes with reconstitution. This is verified by the activation of steady-state enzyme activity, by addition of disulfide reducing agents during purification of the isolated EutB and EutC, and during reconstitution of the EAL oligomer. The significant static charge interactions at the interface between EutB (net negative) and EutC (net positive) in the heterodimer interface dictate low ionic strength conditions during reconstitution. The Ångstrom-scale congruence of the reconstituted and native EAL in the active site region, which is revealed by EPR spectroscopy, demonstrates correct assembly of the catalytic machinery. Overall, the hierarchy of subunit interactions and microscopic features of the contact surfaces, that are revealed by the homology model, guide the establishment of a refined genetic and biochemical approach to reconstitution of the functional [(EutB-EutC)2]3 EAL oligomer, and provide a rationale for the structure. This is a prerequisite for further advances toward understanding of the molecular mechanism of EAL catalysis, by using site-directed mutagenesis, and for insight into approaches to therapeutic manipulation of eut-associated metabolosome and disease pathways.
Supplementary Material
Acknowledgments
We are grateful to Dr. Kim Gernert, Director of the Biomolecular Computing Resource at Emory University School of Medicine (BimCore), for protein modeling advice and assistance.
Funding
This work was supported by NIH/NIDDK grant DK54514 (K. W.).
ABBREVIATIONS
- AdoCbl
5’-deoxyadenosylcobalamin
- β-ME
β-mercaptoethanol
- CW
continuous-wave
- DTT
dithiothreitol
- EAL
ethanolamine ammonia-lyase
- eut
ethanolamine utilization
- EPR
electron paramagnetic resonance
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- KPi
potassium phosphate
- PDB
protein data bank
- TAE
tris-Acetate-EDTA
- TCEP
tris-(2-carboxyethyl) phosphine
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Suporting Information
Figures S1-S10 and Tables S1 and S2. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.Bandarian V, Reed GH. Ethanolamine ammonia-lyase. In: Banerjee R, editor. Chemistry and Biochemistry of B12. Wiley; New York: 1999. [Google Scholar]
- 2.Frey PA. Cobalamin coenzymes in enzymology. In: Mander L, Lui H-W, editors. Comprehensive Natural Products II Chemistry and Biology. Elsevier; Oxford UK: 2010. pp. 501–546. [Google Scholar]
- 3.Hubbard BK, Gulick AM, Babbitt PC, Rayment I, Gerlt JA. Evolution of enzymatic activities in the enolase superfamily: Mechanism, structure, and metabolic context of glucarate dehydratase from Escherichia coli. Faseb J. 1999;13:A1446–A1446. [Google Scholar]
- 4.Banerjee R. Chemistry and Biochemistry of B12. Wiley; New York: 1999. [Google Scholar]
- 5.Brown KL. Chemistry and Enzymology of Vitamin B12. Chem Rev. 2005;105:2075–2149. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 6.Toraya T. Radical Catalysis in Coenzyme B12-Dependent Isomerization (Eliminating) Reactions. Chem Rev. 2003;103:2095–2127. doi: 10.1021/cr020428b. [DOI] [PubMed] [Google Scholar]
- 7.Bradbeer C. Clostridial Fermentations of Choline and Ethanolamine. I. Preparation and Properties of Cell-Free Extracts. J Biol Chem. 1965;240:4669–4674. [PubMed] [Google Scholar]
- 8.Kofoid E, Rappleye C, Stojiljkovic I, Roth J. The 17-gene ethanolamine (eut) operon of Salmonella typhimurium encodes five homologues of carboxysome shell proteins. J Bacteriol. 1999;181:5317–5329. doi: 10.1128/jb.181.17.5317-5329.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brinsmade SR, Paldon T, Escalante-Semerena JC. Minimal functions and physiological conditions required for growth of Salmonella enterica on ethanolamine in the absence of the metabolosome. J Bacteriol. 2005;187:8039–8046. doi: 10.1128/JB.187.23.8039-8046.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roof DM, Roth JR. Ethanolamine Utilization in Salmonella-Typhimurium. J Bacteriol. 1988;170:3855–3863. doi: 10.1128/jb.170.9.3855-3863.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roof DM, Roth JR. Functions Required for Vitamin-B12-Dependent Ethanolamine Utilization in Salmonella-Typhimurium. J Bacteriol. 1989;171:3316–3323. doi: 10.1128/jb.171.6.3316-3323.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penrod JT, Roth JR. Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol. 2006;188:2865–2874. doi: 10.1128/JB.188.8.2865-2874.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garsin DA. Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat Rev Microbiol. 2010;8:290–295. doi: 10.1038/nrmicro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kendall MM, Gruber CC, Parker CT, Sperandio V. Ethanolamine controls expression of genes encoding components involved in interkingdom signaling and virulence in enterohemorrhagic Escherichia coli O157:H7. mBio. 2012;3 doi: 10.1128/mBio.00050-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Srikumar S, Fuchs TM. Ethanolamine utilization contributes to proliferation of Salmonella enterica serovar Typhimurium in food and in nematodes. Appl Environ Microbiol. 2011;77:281–290. doi: 10.1128/AEM.01403-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Kristensen DM, Coleman MK, Mushegian A. Detection of biochemical pathways by probabilistic matching of phyletic vectors. PloS one. 2009;4:e5326. doi: 10.1371/journal.pone.0005326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stubbe J, van der Donk W. Protein radicals in enzyme catalysis. Chem Rev. 1998;98:705–762. doi: 10.1021/cr9400875. [DOI] [PubMed] [Google Scholar]
- 18.Canfield JM, Warncke K. Geometry of reactant centers in the Co-II-substrate radical pair state of coenzyme B-12-dependent ethanolamine deaminase determined by using orientation-selection-ESEEM spectroscopy. J Phys Chem B. 2002;106:8831–8841. doi: 10.1021/jp046167m. [DOI] [PubMed] [Google Scholar]
- 19.Canfield JM, Warncke K. Active site reactant center geometry in the Co(II)-product radical pair state of coenzyme B12-dependent ethanolamine deaminase determined by using orientation-selection electron spin-echo envelope modulation spectroscopy. J Phys Chem B. 2005;109:3053–3064. doi: 10.1021/jp046167m. [DOI] [PubMed] [Google Scholar]
- 20.LoBrutto R, Bandarian V, Magnusson OT, Chen X, Schramm VL, Reed GH. 5’-Deoxyadenosine contacts the substrate radical intermediate in the active site of ethanolamine ammonia-lyase: 2H and 13C electron nuclear double resonance studies. Biochemistry. 2001;40:9–14. doi: 10.1021/bi001865s. [DOI] [PubMed] [Google Scholar]
- 21.Warncke K, Utada AS. Interaction of the substrate radical and the 5’-deoxyadenosine-5’-methyl group in vitamin B-12 coenzyme-dependent ethanolamine deaminase. J Am Chem Soc. 2001;123:8564–8572. doi: 10.1021/ja003658l. [DOI] [PubMed] [Google Scholar]
- 22.Bender G, Poyner RR, Reed GH. Identification of the Substrate Radical Intermediate Derived from Ethanolamine during Catalysis by Ethanolamine Ammonia-Lyase. Biochemistry. 2008;47:11360–11366. doi: 10.1021/bi801316v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poyner RR, Anderson MA, Bandarian V, Cleland WW, Reed GH. Probing nitrogen-sensitive steps in the free-radical-mediated deamination of amino alcohols by ethanolamine ammonia-lyase. J Am Chem Soc. 2006;128:7120–7121. doi: 10.1021/ja060710q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang M, Warncke K. Kinetic and thermodynamic characterization of Co-II-substrate radical pair formation in coenzyme B-12-dependent ethanolamine ammonia-lyase in a cryosolvent system by using time-resolved, full-spectrum continuous-wave electron paramagnetic resonance spectroscopy. J Am Chem Soc. 2008;130:4846–4858. doi: 10.1021/ja710069y. [DOI] [PubMed] [Google Scholar]
- 25.Zhu C, Warncke K. Reaction of the Co(II)-Substrate Radical Pair Catalytic Intermediate in Coenzyme B(12)-Dependent Ethanolamine Ammonia-Lyase in Frozen Aqueous Solution from 190 to 217 K. Biophys J. 2008;95:5890–5900. doi: 10.1529/biophysj.108.138081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu C, Warncke K. Kinetic Isolation and Characterization of the Radical Rearrangement Step in Coenzyme B-12-Dependent Ethanolamine Ammonia-lyase. J Am Chem Soc. 2010;132:9610–9615. doi: 10.1021/ja907769g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shibata N, Tamagaki H, Hieda N, Akita K, Komori H, Shomura Y, Terawaki S, Mori K, Yasuoka N, Higuchi Y, Toraya T. Crystal Structures of Ethanolamine Ammonia-lyase Complexed with Coenzyme B-12 Analogs and Substrates. J Biol Chem. 2010;285:26484–26493. doi: 10.1074/jbc.M110.125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun L, Warncke K. Comparative model of EutB from coenzyme B12-dependent ethanolamine ammonia-lyase reveals a beta8alpha8, TIM-barrel fold and radical catalytic site structural features. Proteins. 2006;64:308–319. doi: 10.1002/prot.20997. [DOI] [PubMed] [Google Scholar]
- 29.Faust LP, Babior BM. Overexpression, Purification, and Some Properties of the Adocbl-Dependent Ethanolamine Ammonia-Lyase from Salmonella-Typhimurium. Arch Biochem Biophys. 1992;294:50–54. doi: 10.1016/0003-9861(92)90135-j. [DOI] [PubMed] [Google Scholar]
- 30.Gerlt JA, Raushel FM. Evolution of function in (βα)8-barrel enzymes. Curr Op Chem Biol. 2003;7:252–264. doi: 10.1016/s1367-5931(03)00019-x. [DOI] [PubMed] [Google Scholar]
- 31.Faust LRP, Connor JA, Roof DM, Hoch JA, Babior BM. Cloning, Sequencing, and Expression of the Genes Encoding the Adenosylcobalamin-Dependent Ethanolamine Ammonia-Lyase of Salmonella-Typhimurium. J Biol Chem. 1990;265:12462–12466. [PubMed] [Google Scholar]
- 32.Bandarian V, Reed GH. Hydrazine cation radical in the active site of ethanolamine ammonia-lyase: Mechanism-based inactivation by hydroxyethylhydrazine. Biochemistry. 1999;38:12394–12402. doi: 10.1021/bi990620g. [DOI] [PubMed] [Google Scholar]
- 33.Guruprasad K, Reddy BVB, Pandit MW. Correlation between Stability of a Protein and Its Dipeptide Composition - a Novel-Approach for Predicting Invivo Stability of a Protein from Its Primary Sequence. Protein Eng. 1990;4:155–161. doi: 10.1093/protein/4.2.155. [DOI] [PubMed] [Google Scholar]
- 34.Akita K, Hieda N, Baba N, Kawaguchi S, Sakamoto H, Nakanishi Y, Yamanishi M, Mori K, Toraya T. Purification and some properties of wild-type and N-terminal-truncated ethanolamine ammonia-lyase of Escherichia coli. J Biochem. 2010;147:83–93. doi: 10.1093/jb/mvp145. [DOI] [PubMed] [Google Scholar]
- 35.Goujon M, McWilliam H, Li WZ, Valentin F, Squizzato S, Paern J, Lopez R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010;38:W695–W699. doi: 10.1093/nar/gkq313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 37.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007;Chapter 2(Unit 2):9. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 38.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. Procheck - a Program to Check the Stereochemical Quality of Protein Structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 40.Ahmad S, Gromiha M, Fawareh H, Sarai A. ASAView: database and tool for solvent accessibility representation in proteins. BMC bioinformatics. 2004;5:51. doi: 10.1186/1471-2105-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 42.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. P Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–W525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bradford MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 47.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein Identification and Analysis Tools on the ExPASy Server. In: Walker JM, editor. The Proteomics Protocols Handbook. Humana Press; 2005. pp. 571–607. [Google Scholar]
- 48.Laemmli UK. Cleavage of Structural Proteins during Assembly of Head of Bacteriophage-T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 49.Ornstein L. Disc Electrophoresis. I. Background and Theory. Ann N Y Acad Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 50.Kaplan BH, Stadtman ER. Ethanolamine Deaminase a Cobamide Coenzyme-Dependent Enzyme. I. Purification Assay and Properties of Enzyme. J Biol Chem. 1968;243:1787. [PubMed] [Google Scholar]
- 51.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 52.Engh RA, Huber R. Accurate Bond and Angle Parameters for X-Ray Protein-Structure Refinement. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 53.Mitov MI, Greaser ML, Campbell KS. GelBandFitter--a computer program for analysis of closely spaced electrophoretic and immunoblotted bands. Electrophoresis. 2009;30:848–851. doi: 10.1002/elps.200800583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Samuelson JC. Recent developments in difficult protein expression: a guide to E. coli strains, promoters, and relevant host mutations. Method Mol Biol. 2011;705:195–209. doi: 10.1007/978-1-61737-967-3_11. [DOI] [PubMed] [Google Scholar]
- 55.Arakawa T, Kita Y. Protection of bovine serum albumin from aggregation by tween 80. J Pharm Sci. 2000;89:646–651. doi: 10.1002/(SICI)1520-6017(200005)89:5<646::AID-JPS10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 56.Choudhary S, Quin MB, Sanders MA, Johnson ET, Schmidt-Dannert C. Engineered protein nano-compartments for targeted enzyme localization. PloS one. 2012;7:e33342. doi: 10.1371/journal.pone.0033342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Derman AI, Prinz WA, Belin D, Beckwith J. Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science. 1993;262:1744–1747. doi: 10.1126/science.8259521. [DOI] [PubMed] [Google Scholar]
- 58.Cleland WW. Dithiothreitol New Protective Reagent for Sh Groups. Biochemistry. 1964;3:480–482. doi: 10.1021/bi00892a002. [DOI] [PubMed] [Google Scholar]
- 59.Jocelyn PC. Chemical-Reduction of Disulfides. Method Enzymol. 1987;143:246–256. doi: 10.1016/0076-6879(87)43048-6. [DOI] [PubMed] [Google Scholar]
- 60.Burns JA, Butler JC, Moran J, Whitesides GM. Selective Reduction of Disulfides by Tris(2-Carboxyethyl)Phosphine. J Org Chem. 1991;56:2648–2650. [Google Scholar]
- 61.Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. A comparison between the sulfhydryl reductants tris(2-carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal Biochem. 1999;273:73–80. doi: 10.1006/abio.1999.4203. [DOI] [PubMed] [Google Scholar]
- 62.Fersht A. Structure and mechanism in protein science. W H Freeman and Company; New York: 1999. [Google Scholar]
- 63.Babior BM, Moss TH, Orme-Johnson WH, Beinert H. Mechanism of Action of Ethanolamine Ammonia-Lyase, a B12-Dependent Enzyme - Participation of Paramagnetic Species in Catalytic Deamination of 2-Aminopropanol. J Biol Chem. 1974;249:4537–4544. [PubMed] [Google Scholar]
- 64.Gerfen GJ. EPR spectroscopy of B12-dependent enzymes. In: Banerjee R, editor. Chemistry and Biochemistry of B12. John Wiley and Sons; New York: 1999. pp. 165–195. [Google Scholar]
- 65.Pilbrow J. EPR of B12-Dependent Enzyme Reactions and Related Systems. In: Dolphin D, editor. B12. Wiley; New York: 1982. pp. 431–462. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.