

1. Introduction
Human beings are adept at discerning relevant information from complex systems by processing visual information. Similarly, as scientists labor to understand the fundamental nature of complex biological systems, they have continued to rely on visual information in the form of images to characterize and classify natural phenomena. New technologies designed to produce images of biological specimens have played a key role in the development of our modern understanding of biology. One of the earliest technological examples, the application of light microscopy to the analysis of biological tissue in the 17th century, ultimately led to the discovery of the cell as a key component of biology.1 Fortunately, the ways in which scientists now visualize biological systems have significantly matured. Currently, the methods for imaging biological specimens encompass an extraordinarily large range of technologies, capitalizing on many different measurable physical phenomena to produce images that provide insight into the underlying biology within the specimen. During the previous century, many imaging technologies including microscopy, radiography, ultrasonography, and magnetic resonance imaging have contributed greatly to the visualization of biological processes and to the practice of medicine.2
Each imaging modality has unique advantages and disadvantages that enable them to make contributions to research and clinical practice. One key aspect of imaging that remains a challenge is the effective integration of molecularly specific information as part of the image. Many of the commonly used in vivo imaging technologies produce high quality images, but these cannot be expressed as individual molecular images. Although immunostaining can be used to localize specific molecules within a biological sample, this method depends upon the use of a surrogate marker of the molecule such as an antibody or other specialized reagent and is usually performed on one or at most only a few molecules of interest in a single experiment.
Mass spectrometry (MS) is unique among analytical technologies in its ability to directly measure individual molecular species in complex samples, allowing it to make significant contributions to our understanding of biological molecules. Indeed, the fundamental basis of the dynamic state of living systems was discovered by Rittenberg and Schoenheimer in the 1930's and 1940's through the use of MS and stable isotope tracers.3–5 With the introduction of ionization techniques such as electrospray ionization (ESI)6 and matrix-assisted laser desorption/ionization (MALDI),7 the field of mass spectrometry has grown exponentially in the past 20 years due to the application of MS to biological molecules. These capabilities ushered in a new era of biological research wherein a systems approach can be used to analyze the molecules in living systems in the wake of information provided by the Human Genome Project.8 With the drive to discover new biology has come a concomitant drive for the development of new mass spectrometry instrumentation. The primary benefit of this technology innovation is the ability to measure specific molecular compounds at high structural fidelity with high speed of acquisition, making it possible to perform experiments on biological systems that have not been possible before. Even single experiments have shown near comprehensive coverage of entire proteomes of simple organisms.9–10
Imaging Mass Spectrometry (IMS) is a technology that makes regiospecific molecular measurements directly from biological specimens.11–15 This method of imaging capitalizes on all the advantages of modern mass spectrometers, including high sensitivity, high throughput, and molecular specificity, to produce images that visually represent tissue biology on the basis of specific molecules (e.g. peptides, proteins, lipids, drugs and metabolites). The capabilities of mass spectrometry are unique in the imaging world, providing unique insights into biological systems. The distinguishing principle of imaging mass spectrometry from other mass spectrometric techniques is that the preparation of the sample and the acquisition of the MS data must be performed in a manner that preserves the spatial integrity of the sample within the limits of the spatial resolution of the measurement. Therefore, IMS of a biological sample, such as a tissue section, requires that the mass spectral data be registered to specific spatial locations in order to correlate the molecular information to specific cells or groups of cells commonly visualized by microscopy. Images are reconstructed by plotting the intensities of a given ion on a coordinate system that represents the relative position of the mass spectral acquisition from the biological sample. The resulting images create a visual representation of the sample based on the specific molecular information measured from the sample itself.
IMS has a number of advantages relative to other imaging techniques currently used for biological and clinical studies. First, MS can be used to detect analytes without the need for labeling or otherwise structurally modifying the native compound. This distinction is important for many reasons, but primarily this avoids potential problems if the tagging reagent affects or changes the physical, chemical, or biological function of the molecules of interest or if the reagent has multiple molecular affinities. Second, MS has the capability of monitoring thousands of molecules in a single experiment. From a systems biology perspective, the advantage of the concurrent measurement of whole pathways or components in multiple pathways is crucial to understanding the function of intact cells.
Among the several mass spectrometry ionization techniques that can be used to directly analyze tissues, MALDI has led the way in the development of biological and clinical applications for IMS.16–17 This report describes the essential considerations for performing MALDI IMS experiments on tissue, reviews some of the recent applications to the analysis of clinical specimens, highlights specific contributions of MALDI IMS to our understanding of biology and medicine, and discusses specific advantages and limitations of the technology. This review is not intended to be comprehensive with respect to all aspects of imaging mass spectrometry; rather it focuses on the themes that are essential to the analysis of biological and clinical tissue samples using MALDI IMS. There are excellent reviews that extensively cover both the ionization techniques used in IMS as well as the various mass analyzers that have been adapted for use in IMS and the reader is referred to these for further information.12,18–21
2. Basic Concepts of MALDI Imaging Mass Spectrometry
Although the concept of IMS was introduced over 50 years ago, first using secondary ion mass spectrometry (SIMS)22–23 and later laser ablation mass spectrometry,24–25 widespread biological and clinical applications for IMS were realized after the introduction of MALDI imaging.16 Figure 1 depicts the necessary steps of the MALDI IMS experiment. This general image generation process is common to all IMS platforms, although implementation of different ionization techniques and instrumentation will vary in each specific IMS experiment. MALDI IMS employs a laser to desorb and ionize analytes mixed with the matrix molecule that aids in the desorption/ionization process. This ionization process is influenced by the chemical properties of both the analyte and matrix and on absorption of the energy of the laser; analytes are desorbed and ionized into the gas phase. For imaging, the matrix is applied to the biological specimen that has been placed on the planer surface of a target. The matrix solution acts to extract the analytes from the specimen and thereby aids desorption / ionization. Direct analysis is then performed by a raster of the sample by the laser beam. The MALDI process and proposed ionization mechanisms have been described in detail in several published papers and reviews,26–27 and will not be considered further in this article.
Figure 1. Principle of Imaging Mass Spectrometry.
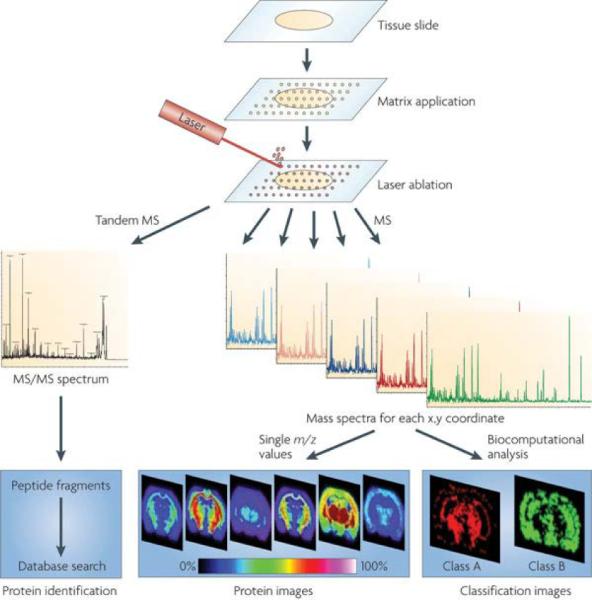
Schematic of a typical workflow for fresh frozen tissue samples. Sample pretreatment steps include cutting and mounting the tissue section on a conductive target. Matrix is applied to the tissue section and mass spectra are generated at each x,y coordinate for protein analysis or tandem MS spectra for protein identification. Further analytical steps include the visualization of the distribution of a single molecule within the tissue (image) or statistical analysis to visualize classification images as well as database searching to identify the protein. Reprinted with permission from Reference 21. Copyright 2010 Macmillan Publishers Limited.
Importantly, MALDI MS has a number of advantages that makes it ideal for imaging biological specimens. First, MALDI is a so called “soft” ionization technique; this allows the analysis of molecules across a wide range of molecular weights, ranging from 100s of Da to beyond 100 kDa. Second, MALDI mass spectra are predominately composed of singly charge molecules, simplifying the analysis of complex mixtures of proteins. Third, since desorption / ionization is accomplished using a laser, the sample can be interrogated in very specific spatially distinct areas by directing the laser to histologically unique regions. Since the laser optics can be optimized by the analyst, for some research grade instruments, the irradiated area can be adjusted from over 100 μm to less than 1 μm.28–29 However, typical commercial instruments permit adjustment of the laser to between 20 and 250 μm for routine analysis. Lastly, the capabilities of modern instrumentation have increased significantly in recent years through the use of a variety of mass analyzers having high mass resolution, high speed, and high sensitivity. Critical to IMS, one of the key advancements has been the application of high frequency, solid-state lasers with repetition rates of 200–5000 Hz. This development has significantly reduced the acquisition times for images of biological specimens to more practical time scales, typically 2–10 minutes for an average size tissue specimen.30
When seeking regiospecific molecular information in tissue, commonly two different approaches can be used. Figure 2 depicts these two distinct data acquisition procedures. The first is to simply image the entire specimen as illustrated in Figure 1, and then to obtain molecular maps at the m/z values of interest. This experiment begins by the application of matrix in a uniform manner either as a continuous coating or a regularly spaced array of spots that defines the spatial resolution. The complete data set is comprised of many hundreds or thousands of m/z values, and the intensities of each ion can be plotted in a false color display. The resulting set of ion images are used to accurately portray the spatial distribution of the molecules that comprise the sample. Figure 3 shows select images extracted from a single MALDI IMS experiment on a mouse brain section, demonstrating the highly dimensional nature of the data collected in MALDI IMS.
Figure 2. Clinically Useful Modalities of MALDI IMS.
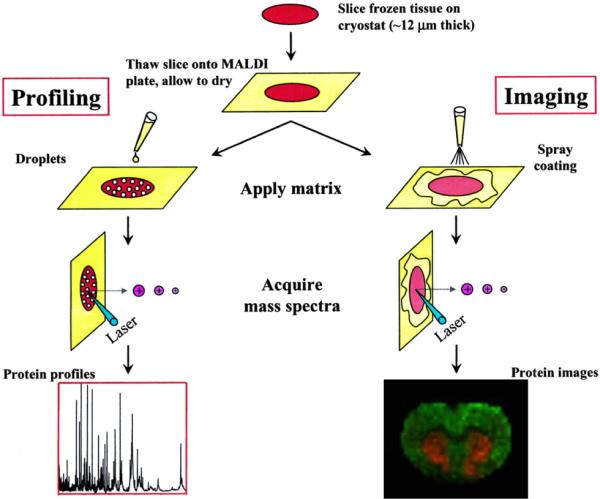
The schematic represents the two most common data acquisition strategies of MALDI MS used for the analysis of clinical specimens. In a profiling mode, data are collected from regions of interest defined by tissue pathology, while in imaging mode, the entire sample surface is raster sampled to reproduce an image of the specimen based ion specific ion intensities. Reprinted with permission from Reference 243. Copyright 2004 American Chemical Society.
Figure 3. Dimensionality of MALDI IMS data sets.
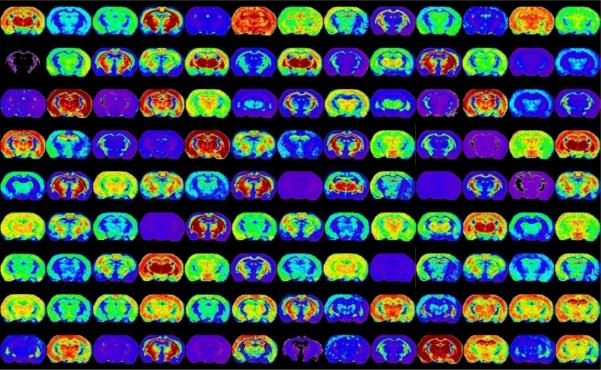
Ion images displayed above originate from a single raster of one section of a mouse brain section. Each image represents a single m/z ion that has a unique distribution in the brain. This figure represents less than 10% of the images collected.
The second approach is to obtain mass spectra only from selected areas of the tissue annotated though microscopy or other imaging modalities. We have termed this approach histology-directed molecular imaging.31 Although this does not produce molecular maps of the whole specimen, it provides molecular data from specific areas, reducing the time of acquisition and data file sizes. Images are then visualized by the superposition of the MS data from many discrete spots on the microscope image (or other image modality). Thus one can target specific cells of interest either within a single specimen or compare those of two different specimens. Matrix is applied only to the areas that are to be compared; therefore, only a minimal number of mass spectra required to compare the selected regions of interest are collected. Figure 4 describes a workflow for histology-directed analysis of a human breast tumor sample. In this example, a hemotoxylin and eosin (H&E) stained serial section was annotated by a pathologist with color coded circles, marking the regions of interest to be analyzed by MALDI IMS (Figure 4A). The coordinates of the annotated spots were transferred to an automated matrix spotter, and matrix was placed precisely in the regions of interest (Figure 4B). Once the matrix has been placed on the tissue, the coordinates of those same locations are loaded into the MALDI mass spectrometer and the data from each position is acquired (Figure 4C). This approach has been successfully applied using both fresh frozen and formalin-fixed tissues.31–40
Figure 4. Workflow for histology-directed profiling.

Two adjacent sections of the sample tissue are required, one for staining and one for MALDI analysis. (a) A pathologist selects cells of interest using the histology image. (As shown here, the cells are colored according to type.) (b) The locations of the selected cells are registered to the matrix spotter that holds the section for MALDI analysis. After spotting with matrix, the coordinates of the matrix spots are registered to the mass spectrometer and spectra are automatically acquired from each location. (c) Spectra demonstrating the cell-specific differences (peaks marked with asterisks) that can be detected. Reproduced with permission from Reference 14. Copyright 2007 Nature Publishing Group.
Since the desired outcomes of the whole specimen and histology-directed imaging experiments differ, the experimental design also differs depending on the biological or clinical question being addressed. If the clinical question to be answered is one of discovery, i.e., a general assessment of the distribution of molecules in the tissue, the image acquisition mode is most commonly used. The histology-directed experiment is often performed in collaboration with a biologist or pathologist who has selected regions that have specific cell types or morphology that are relevant to the question being addressed.
3. Sample Preparation
Sample preparation represents a critical step in the imaging process in order to optimize sensitivity and spatial resolution for the specific analytes of interest. The most common steps for the preparation of samples for IMS are depicted in Figure 5. Careful planning, attention to detail and cleanliness in laboratory practices are required to avoid unwanted loss of sensitivity, loss of spatial resolution, or problems with sample stability.
Figure 5. Key Sample Preparation Steps for MALDI IMS.
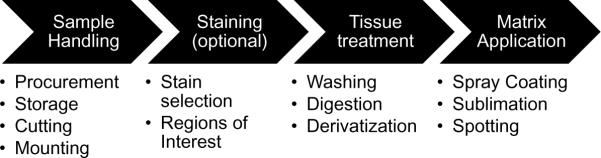
In order to ensure that high quality MALDI IMS data is generated, sample custody is ideally controlled as much as possible through the entire experiment to avoid problems with tissue degradation and contamination. Common considerations for each sample preparation step are listed under each category.
3.1. Sample Handling
The MALDI IMS experiment begins with the collection of the sample. Unlike most analytical technologies where one must account for either the preservation of the molecular or histological integrity of the sample, in MALDI IMS both of these properties of the sample are important considerations. Whenever possible, it is advisable to participate in the planning phase for the sample collection protocol to ensure that the protocol accounts for both of these important factors.
3.1.1. Tissue Procurement and Storage
Fresh frozen tissues
Time is an important factor with respect to sample degradation; therefore, it is necessary to minimize the time between the harvesting of the sample and properly preserving the sample. Failure to preserve the sample in a timely manner exposes the sample to potential changes in cell morphology due to endogenous enzymatic degradation of analytes.41–44 The first processing step is to snap-freeze the sample by exposing the tissue to a cryogen such as liquid nitrogen, liquid pentane, or hexane/dry ice. Generally, the tissue is placed in a loose foil wrapper and immersed in the cryogenic solution slowly to prevent fracturing the tissue. Care must be taken to avoid deformation of the tissue by the container. One useful technique is to fashion a float out of aluminum foil, place the tissue in the float on top of liquid nitrogen, allow the sample to freeze, then wrap and immerse the tissue fully. This technique causes the tissue to freeze more slowly than immediate immersion and can be useful for tissues such as brain that are prone to fracture during the freezing process. This technique also has the added advantage that the morphology is more readily preserved. Depending upon the importance of the sample orientation, it might be necessary to record or mark the specimen orientation before long term storage. The frozen sample can be stored for a minimum of a year (below −80°C) in most cases without any detrimental effects on the quality of the MALDI IMS result;45–46 however, samples vary widely and testing is recommended with each tissue type to ensure that the storage conditions adequately stabilize the sample.
There has emerged a MALDI IMS compatible alternative to flash freezing the freshly harvested sample. Svennsson et al. demonstrated a heat stabilization technology can prevent tissue degradation in a manner that is compatible with LC/MS/MS-based proteomic analyses.47 The device is designed to quickly heat the sample to more than 90° C to deactivate enzyme activity, evacuating the air from the sample in the process. Once preserved, the samples can be stored long term in the freezer. This technology was more recently shown by Goodwin et al. to stabilize specific peptide signals in MALDI IMS experiments.48 Although of interest, further testing and validation will be required before it is widely adopted for use in MALDI IMS studies.
Fixed tissues
Fixation is a method of preserving tissue samples that is used ubiquitously for the preservation of clinical specimens, although many of these protocols are problematic to MALDI IMS studies. The most common method of fixation is by immersion of the fresh tissue in formalin or several kinds of alcohols.49–51 Formalin fixation greatly inhibits the subsequent analysis of proteins by mass spectrometry due to the protein crosslinks that form.52 Once fixed, these tissues are commonly stored in the form of paraffin blocks. Although formalin fixed paraffin embedded (FFPE) samples are not the preferred method of preservation, there does exist a great number of archived tissue specimens that have been preserved in this manner. In order to leverage this vast resource of clinical information, subsequent discussion will describe methods useful for analysis of FFPE samples by MALDI IMS and a number of applications will be discussed.
3.1.2. Sectioning Tissue Specimens
The first step in the preparation of frozen or archived tissues for MALDI IMS is sectioning of the tissue. This process is very straightforward; however, there are a few considerations that are unique to tissue analysis by MALDI IMS. Under normal circumstances, preparation of a tissue for normal histopathology would dictate the sample be embedded in a polymer that aids in the sectioning process. Common examples include optimal cutting temperature (OCT), carboxymethylcellulose (CMC), and agar. Embedding media is used so commonly that frozen sections are often stored by freezing in embedding media. However, many of those polymers are easily ionized by the MALDI process, and due to their abundance in an embedded tissue, are prone to cause contamination and ion suppression.45,53 Figure 6 demonstrates the signal suppression effect of OCT when it is allowed to contaminate the tissue surface. Along with reducing the ion intensity of many species, only approximately 50% of the ions detected in the unadulterated sample are detected when OCT is present.45 Rather than embedding the entire sample, OCT is used to simply affix the tissue block to the cryomicrotome chuck, leaving sufficient distance between the top of the OCT and the plane of the blade passing through the tissue. In this manner, tissue sections can be collected without OCT contamination.
Figure 6. Effect of OCT on MALDI signal from rat liver.
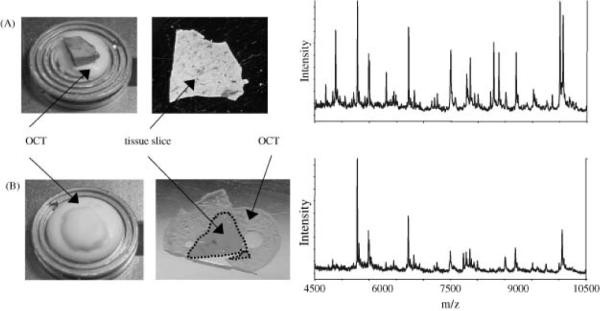
(A) Optimal procedure where OCT is used to adhere the tissue to the sample stage but does not come into contact with the sliced tissue. The resulting spectrum shows many intense signals between m/z 4500 and 10 500. (B) The tissue was embedded in OCT and attached to the sample stage. The resulting tissue slice is surrounded by OCT on the MALDI plate, and the resulting spectrum contains only about half of the signals as that in (A). Reproduced with permission from Reference 45. Copyright 2003 John Wiley & Sons, Ltd.
There are instances where the use of embedding media cannot be avoided due to the fact that the tissue is too fragile to be cut unsupported. Tissue washing with ethanol can be employed to facilitate the removal of residual OCT from the tissue sections and the signal is recovered with minimal effect on the signal from the tissue. As an alternative, a low percentage of CMC can be used if required with minimal effect on the MALDI signal. Gelatin has been shown to be effective as an embedding media for cutting tissue, and the compatibility with MALDI IMS is better than OCT, having fewer background ions originating from the embedding media.54 Another alternative that has been demonstrated useful is ice. This approach is often used in the sectioning whole animals for MALDI IMS.55–56 FFPE samples are already embedded in paraffin and due to the fact that the proteins are highly cross-linked, the paraffin can be removed by washing the section aggressively in organic solvents such as xylene and alcohols.57
Sectioning of the mounted tissues is typically performed between 3 and 20 microns, depending on the tissue. The cutting temperature can vary significantly depending on the type of tissue. Table 1 lists the approximate cutting temperature of many of the common tissues analyzed by MALDI IMS. At these thicknesses, most cells are opened up by the blade to expose the contents of the cell to be extracted and analyzed. Little difference in the spectral features is seen with sections having these thicknesses. However, thicker sections are prone to charge buildup on the surface on irradiation by the laser, affecting the quality of the mass spectrum, and these thicker sections are also prone to crack severely when dried and can easily lift off the MALDI target. Thinner sections can be difficult to handle without tearing the tissue; however, thin sections have been reported to be ideal for proteins58 and matrix precoated targets.59 Optimization of the temperature and the thickness are required to produce high quality sections for imaging.
Table 1.
Recommended Temperatures for Cutting Unfixed Frozen Tissues.60
| Tissue Type | Working Temperature |
|---|---|
| Brain | −12°C |
| Liver | −14°C |
| Lymph Node | −14°C |
| Kidney | −16°C |
| Spleen | −16°C |
| Muscle | −20°C |
| Thyroid | −20°C |
| Skin | −25°C |
| Breast | −25°C |
| Breast with Fat | −30°C or below |
| Adipose Tissue | −30°C or below |
| Fixed Tissue | −12°C to −17°C |
3.1.3 Mounting Tissue for MALDI IMS
Tissue sections can be thaw-mounted onto a solid surface simply by placement of the section onto the surface that is cooled to the same temperature as the tissue section, then slowly warming the surface from the underside. Alternatively, the tissue section can be picked up by a warm slide by inverting the warm slide over the cold sample and slowly lowering the slide until the sample adheres to the surface of the warm slide. In this approach, a thin layer of ice is often left behind on the cold surface below, and this contains some water soluble analytes that may be of interest. Most MALDI time-of-flight mass spectrometers require that the surface of the slide used to mount the tissue be conductive to support the application of the potential required to accelerate the ions in the source of the mass spectrometer. Metal targets, gold coated to enhance photographic imaging of the tissue section, are used because they are conductive. One disadvantage of this approach is that these targets are relatively expensive and therefore cannot be archived or disposed of after use. Another drawback is that metal targets are not transparent, eliminating the possibility of staining and analysis by routine microscopy before or after MALDI IMS. Indium-Tin Oxide (ITO) coated glass slides are ideal for MALDI when microscopic analysis of the section is needed. The ITO coating provides electrical conductance to the top side of the slide sufficient to eliminate sample charging; however, the slide remains transparent making it compatible for microscopy.61
Large sections, such as those of whole body sections, are often fragile and cannot be effectively transferred to a slide by thaw-mounting as described above. In order to maintain the spatial integrity of the sample, the sample is collected onto tape as it is being cut by the cryotome. While the tape is helpful in maintaining the tissue histology, the tape itself is not compatible with MALDI IMS. The acetate tape used to mount the tissue is not conductive, causing the sample surface to charge in MALDI time-of-flight (TOF) mass spectrometers. Successful analysis of these samples have been performed in mass spectrometers whose ion source is decoupled from the mass analyzer such as quadrupole time-of-flight (Q-TOF), ion trap, and fourier transform ion cyclotron resonance (FT-ICR). The tape is fixed to a metal MALDI target using double sided taped. A tape transfer system has been developed that uses a proprietary UV sensitive adhesive to effectively transfer the section from the tape to another substrate.62 This system can be used to transfer the sample to the normal MS target. Recently, the use of conductive carbon tape for the mounting of whole body sections and other fragile samples has been demonstrated.63
3.2. Staining
Staining of the tissue is an optional step in the MALDI IMS process; however, this step is often necessary in order to confidently target regions of the tissue that are of interest. Tissue staining can be incorporated into MALDI IMS workflows in a number of ways. First, staining can be performed prior to application of the matrix onto the exact tissue that is to be analyzed.61 This approach provides the most direct feedback, ensuring that the correct regions of interest are targeted. There are a number of stains that have been tested and found to be compatible with MALDI MS.61 However, direct staining of the tissue before MALDI IMS must be approached with caution because the staining process itself introduces additional variables into the experiment, some of which may create undesirable changes to the molecular composition of the tissue. Most staining protocols require numerous immersion steps in a variety of aqueous and organic solvents. Rigorous testing of the stain compatibility is advised before undertaking a large scale experiment in this manner. Validation of the experimental conditions must be performed to ensure that the method is robust and is not introducing unintended bias. Figure 7 shows the effect of some common histological stains on the quality of MALDI mass spectra taken from tissue. It was found that nuclear-specific stains had the least impact on the mass spectral results. Some alteration of the intensities of the protein spectra were observed; however, most of the ions were conserved across the entire data set.61 Table 2 details the MALDI compatible staining protocols used in that study. It should be noted that this study is specific to larger analytes, and the effect on metabolites, lipids, and drugs would be detrimental using the same conditions.
Figure 7. The effect of common histological stains on MALDI IMS results.

MALDI-MS protein profiles acquired from unstained (Ct: control section rinsed in 70 and 100% ethanol) and stained grade 4 human glioma tissue sections. TP, Terry's Polychrome; TB, Toluidine Blue; NFR, Nuclear Fast Red; CV, Cresyl Violet; MB, Methylene Blue. Reprinted with permission from Reference 61. Copyright 2004 American Chemical Society.
Table 2.
MALDI IMS compatible staining solutions.61
| Stain | Protocol | Stain Time (s)* |
|---|---|---|
| Toluidine Blue (Electron Microscopy Sciences, Catalog No. 22050) | 5% Toluidine Blue in 1% aqueous borax (hydrated sodium borate, Na2B4O7·10 H2O, Fisher Scientific, ACS grade). Dissolve borax in warm distilled water, add Toluidine Blue, stir overnight, filter before use. | 5–10 |
| Nuclear Fast Red (Newcomer Supply, Catalog No. 1255A) | 0.1% Nuclear Fast Red in 5% aqueous aluminum sulfate (AI2(SO4)3). Ready to use. | 45 |
| Methylene Blue (Sigma, Catalog No. MB-1) | 0.15% in 70% ethanol (AAPER alcohol, absolute 200 proof). Dissolve Methylene blue in ethanol, stir overnight, filter before use. | 5–10 |
| Terry's Polychrome | 0.02% aqueous Methylene Blue in 0.02% aqueous potassium carbonate (K2CO3, Merck KGaA, ACS grade). Dissolve potassium carbonate in distilled water, add Methylene Blue, stir overnight, filter before use. | 30 |
| Cresyl Violet (Sigma, Catalog No. C1791) | 0.5% aqueous (deionized water). Stir overnight on low heat, filter solution, add 2 drops of glacial acetic acid to a 100-mL solution, filter before use. | 30 |
Note: Staining solutions were directly deposited on the tissue sections using a Pasteur pipet and allowed to react for the indicated time. Excess stain was removed by successive washing of the plates for 15 s in 70%, followed by 100% ethanol.
When performing MALDI IMS of samples where continuous coatings have been utilized, the tissue morphology can be validated against the ion image after the acquisition of the MALDI data by removal of the matrix followed by H&E staining. Due to the high solubility of most matrices in alcohol-water mixtures, the matrix can be easily removed by immersion in 70% ethanol solution for 1–2 minutes. At this point, the specimen can be stained using the normal H&E protocol.64–65 Although this can be useful, bear in mind that this validation step occurs after the image data has been collected.
Most commonly, the method used for locating the cell-type of interest is the collection and staining of a serial section followed by matching the sections using tissue landmarks that are found in both the stained and unstained tissue.31 This approach does not limit the preferences of the biologist/pathologist for using reagents that are compatible with MALDI IMS analysis. A common stain routinely used in histology is H&E, but unfortunately this stain is poorly compatible with MALDI analysis.61,66 Once the stained section is analyzed for specific cell morphology, the stained section is co-registered with the unstained section to transfer the coordinates for matrix application and analysis.31 It is critically important that the gross shape of the serial sections match as well as possible, avoiding tearing and folding of the tissue section when they are mounted on their respective slides. This will ensure that the tissue images can be accurately co-registered using some common features identifiable on each section.
3.3. Pretreatment of Samples
There are some sample pretreatment steps that, when applied before matrix application, can be useful for enhancing the analytes of interest. Significant gains in sensitivity can be obtained by tailoring a pretreatment protocol to the analytes of interest. The most common pretreatment steps are washing to remove salts and other contaminants, on-tissue digestion, and on-tissue derivatization.
3.3.1. Washing
Biological tissues contain an enormous number and variety of chemical species, ranging in concentration across many orders of magnitude. Some of the most abundant species in the tissue can cause problems for the high sensitivity measurement of some of the less abundant species. For example, some of the chemical species that negatively affect analysis of proteins and peptides are salts and lipids.22,67–68 The problems that may be encountered range from poor crystallization, ion suppression, and gas-phase adduct formation. To overcome these problems, tissue washing procedures have been developed for use prior to the application of matrix in MALDI IMS workflows.
The washing protocol varies depending upon the target class of analytes that are being investigated. In an ideal preparation, the washing procedure would remove all of the unwanted chemical species from the tissue surface without affecting the molecules of interest. When optimizing a washing procedure, it is necessary to test and validate that the procedure developed has minimal effect on both the sensitivity and the localization of the target analytes. The most common washing procedure used for MALDI IMS of proteins is a fixation procedure in increasingly higher percentages of ethanol.45,61,64,69–70 The standard procedure in our laboratory calls for a 30 sec wash in 70% ethanol, a 30 sec wash in 90% ethanol, and a final 30 sec wash in 95% ethanol, followed by drying under ambient conditions. These washing conditions show little delocalization of the proteins and significantly enhance signal-to-noise.45 More aggressive washing procedures are recommended to further remove lipids from tissues having high lipid content. Solvents like chloroform and xylene can be effectively used to delipidate samples in a manner compatible with MALDI IMS resulting in enhanced detection of proteins directly from tissue.46 The tissue surface is simply washed by pipetting 200 microliters of organic solvent over the tissue and allowing it to drain from the surface immediately by placing the tissue on an incline. Recently, an approach that utilizes Carnoy's fluid, a mixture of ethanol, chloroform and acetic acid, has been developed that yields excellent results for imaging proteins.65,71 Aqueous washing has been shown to promote the detection of integral membrane proteins. Grey et al. demonstrated that extensive washing of the tissue with water removes many of the soluble proteins, allowing for the imaging of two variant forms of Aquaporin-0 in ocular and retina tissue.72
Although conventional wisdom suggests that washing would be detrimental to low molecular weight analytes, recent work suggests that there is a benefit of washing for the detection of both peptides37,73–75 and lipids.76–78 For example, it has been demonstrated that washing in graded ethanol solutions is useful for the removal of high abundance lipids while preserving the spatial organization of neuropeptides related to Parkinson's disease.73–75 Washing in aqueous buffered solutions has been shown by multiple groups to enhance the detection of lipids in MALDI IMS.76–78In situ tryptic digestions are enhanced by washing in a solution containing 90% ethanol, 9% glacial acetic acid, and 1% water.37,73 Table 3 summarizes some of the commonly used tissue washing procedures. Although it is known that washing improves MALDI IMS results of many analytes, there is currently no extensive resource of tissue washing methods that can be referenced. As the community continues to develop MALDI IMS protocols for more analytes, one would expect that greater numbers of analytes will be detectable with improved sensitivities.
Table 3.
Tissue Washing Procedures for MALDI IMS.
| Analyte | Protocol |
|---|---|
| Proteins | 70% ethanol, 90% ethanol ,95% ethanol, 3 wash steps, 30 sec each step. |
| Proteins65,71 | 70% ethanol, 100% ethanol, Carnoy's fluid (2 min.), 100% ethanol, H2O, 100% ethanol. 6 wash steps, 30 sec each step unless noted. Carnoy's fluid is composed of 6:3:1 (v:v:v) ethanol/chloroform/glacial acetic acid. |
| Proteins/in situ trypsin digest37,73 | 70% ethanol, 95% ethanol (2×), final wash in solution of 90% ethanol, 9% glacial acetic acid, 1% H2O, 4 wash steps, 30 sec each step. |
| Peptides73–75 | 70% ethanol, 95% ethanol (2×), 3 wash steps, 10 sec each step. |
| Lipids76 | 50 mM ammonium formate (pH 6.4, 4°C) for 15 sec. |
3.3.2. On-Tissue Digestion
In some cases, the analysis of proteins is best done by enzymatically digesting proteins on the tissue surface, followed by the analysis of the peptide products. This method is particularly advantageous for the analysis of FFPE tissues because the digestion liberates peptide fragments from the chemically cross-linked tissue. The approach used most often involves the application of trypsin to the tissue surface using an automated spotting or spraying instrument. The tissue is prepared for trypsin application by first performing an antigen retrieval step.38,79 The basic steps of an on-tissue digestion protocol is that trypsin solution is applied to the surface; the wetted surface is maintained in a humid environment at ambient or elevated temperature (37–50°C) to maintain the solution on the surface and allow sufficient time for the digestion reaction to occur.36–38,57,79–82 Figure 8 shows an image of tryptic fragment peptides from a FFPE tissue specimen prepared by spray coating of trypsin onto the tissue surface. In this example, a 0.66 ng/μL solution of modified porcine trypsin in 10 mM NH4HCO3 and 5% (v/v) acetonitrile was deposited in 25 spray coating cycles using an ImagePrep station (Bruker Daltonics). Tissue was incubated for 4 min and dried for 60 s between cycles.79 In another example, an automated spotter was used to deposit a solution containing trypsin onto the rat brain tissue in an array with a 250 μm center-to-center spacing between individual spots. The trypsin was spotted over a series of 30 iterations by depositing 5 drops (100 pl per drop) per iteration to achieve a total spot volume of 15 nL (5 drops of the trypsin solution were deposited at each position in 8 min time intervals). Each digest spot was permitted to dry completely after each spotting iteration. The trypsin spotting proceeded at room temperature (21°C) over a period of 4 h, allowing time for digestion to take place.37 For a complete review of the current procedures used in our laboratory for the preparation of FFPE for MALDI IMS by in situ tryptic digestion tissues, consult the review by Casadonte et al.36 Once in situ digestion has been performed, MALDI matrix is applied to the tissue to facilitate the analysis of the peptide fragments produced.
Figure 8. MALDI IMS of FFPE tissue by in situ enzymatic digestion.

Ion intensity maps generated for peptides observed on citric acid antigen retrieval (CAAR)-treated FFPE tissue. An archived FFPE section of human ovarian cancer (a–f, top row) was mounted onto a slide and treated with CAAR. A fresh-frozen section of ovarian cancer tissue (a–f, bottom row) from the same patient was mounted onto a separate slide. H&E stains of the FFPE section and fresh-frozen section (a) are included as well as ion maps (b–f). An (g) enlarged H/E stain and (h) ion intensity map for m/z 1267 are also included. Scale bars = 2 mm. Reprinted with permission from Reference 79. Copyright 2010 American Chemical Society.
3.3.3. Derivatization
MALDI IMS of drugs and metabolites within tissue sections can be challenging due to low molecular weight background signals, poor ionization efficiency and ion suppression of some drugs, and potential isobaric interferences of endogenous molecules. Chemical derivatization is routinely used to improve the detection of drugs by a variety of analytical techniques, including gas chromatography (GC), high-performance liquid chromatography (HPLC), and increasingly, MALDI MS. Recent studies have applied different derivatization strategies to facilitate the detection and localization of several low molecular weight compounds through the modification of analytes in the tissue.83–85 For example, studies of the distribution of 3-methoxysalicylamine (3-MoSA), a scavenger of levuglandins, in intact tissue by MALDI IMS are very limited by matrix ion interference and very low sensitivity.84 A method for the derivatization of 3-MoSA directly on tissue with the reagent 1,1'-thiocarbonyldiimidazole (TCDI) was developed to improve detection of this compound (Scheme 1). 3-MoSA reacts with TCDI under basic conditions to form a product with a protonated molecular ion at m/z 196, which fragments to an ion at m/z 137 upon collisionally activated dissociation. The resulting 3-MoSA-TCDI derivative has drastically improved sensitivity compared to underivatized 3-MoSA. Fragment ions from the MS/MS analysis of 3-MoSA-TCDI were easily distinguished from background ions and thus produced distinct images for the localization of 3-MoSA on tissues from dosed animals. On-tissue derivatization was carried out by manually spray-coating TCDI in ammonium bicarbonate buffer onto tissue sections, followed by the application of matrix (α-cyano-4-hydroxycinnamic acid, CHCA) onto the tissue sections by sublimation. Figure 9 shows images for serial kidney sections from a mouse dosed at 300 mg/kg and sacrificed 30 min post-dose. These results show a clear image for the 3-MoSA-TCDI derivative with localization towards the center of the kidney where the renal medulla is found. Figure 9 images correlate well with LC-ESIMS/MS quantitation from the same tissue homogenates.84
Scheme 1.
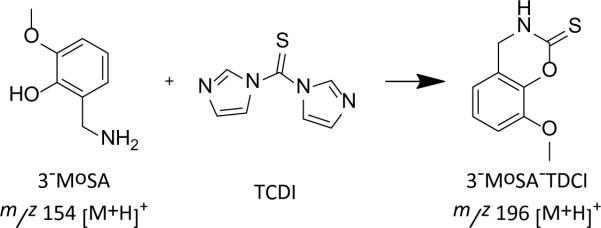
On-tissue chemical derivatization of 3-methoxysalicylamine for MALDI-imaging mass spectrometry. Reaction of 3-MoSA with TCDI.
Figure 9. MALDI-IMS performed using on tissue derivatization.
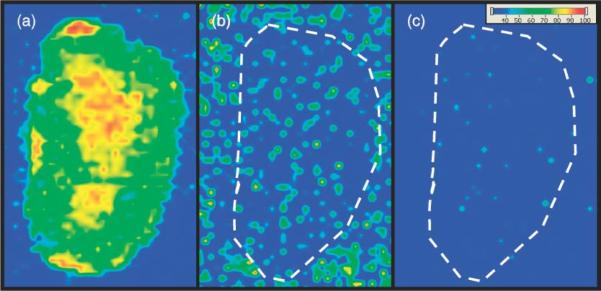
Serial kidney sections from mouse dosed with 3-MoSA at 300 mg/kg and sacrificed at 30 min post-dose. (a) TCDI-treated tissue, plotting m/z 196 → 137 for 3-MoSA-TCDI. (b) Tissue not treated with TCDI, plotting 154 → 137 (underivatized 3-MoSA). (c) Tissue not treated with TCDI, plotting 196 → 137. Images are plotted as m/z 137 TIC. Reproduced with permission from Reference 84. Copyright 2011 John Wiley & Sons, Ltd.
N-terminal derivatization methods have been shown useful for the enhancement of peptide and protein de novo sequencing from tissue samples.85 This method utilizes N-succinimidyloxycarbonylmethyl)-tris-(2,4,6-trimethoxyphenyl) phosphonium bromide (TMPP) to modify the proteins contained within a tissue section. TMPP has the advantage that the derivatives can form on tissue under ambient conditions. Another recent example demonstrates that derivatization reagents can be incorporated into pre-coated MALDI targets that also contain MALDI matrix. Reagents such as these remove much of the difficulty of sample preparation, making it possible for the technology to be used by non-specialists.83 While the examples above clearly indicate that derivatization is highly beneficial to the MALDI IMS of certain analyte classes, this approach is relatively new to the field and one must remember that reaction yields can vary and side-reactions can occur. Nevertheless, derivatization will undoubtedly play an important role in the growing adoption of MALDI IMS for the analysis of biologically important compounds.
3.4. Application of Matrix for MALDI IMS
3.4.1 Matrix Selection and Solvent Conditions
The matrix absorbs the laser energy and transfers the analyte to the gas phase, promoting ionization in the process.26,86 The selection of the matrix plays an important role in the successful analysis of specific analyte classes. Table 4 lists some of the more useful matrices for MALDI IMS of tissue specimens. Also listed are the analyte classes for which they are commonly used. This table represents a starting point in the selection of the best matrix for your application. The three most commonly used matrices for MALDI IMS are 2,5-Dihydroxybenzoic acid (DHB),87 α-cyano-4-hydroxycinnamic acid (CHCA),88 and 3,5-Dimethoxy-4-hydroxycinnamic acid (SA, sinapinic acid).89 However, there are many other matrices that have been found to be useful for the various analytes that are encountered in tissue specimens. These matrices have been developed for a variety of analyte classes and each have strengths and weaknesses both for the preparation of tissue samples and for the analyte classes for which they have been optimized.
Table 4.
Common organic MALDI matrices used of the direct analysis of biological tissues.
| Matrix | Common Analytes |
|---|---|
| 2,5-Dihydroxybenzoic acid (DHB)45,87,90 | peptides, proteins, lipids, drugs |
| α-cyano-4-hydroxycinnamic acid (CHCA)45,88,90 | peptides, proteins, lipids, drugs |
| 3,5-Dimethoxy-4-hydroxycinnamic acid (SA, sinapinic acid)45,89–90 | proteins |
| 3-hydroxypicolinic acid (3-HPA)90,99–100 | peptides, oligonucleotides |
| 2,4-dinitrophenylhydrazine (2,4-DNPH)57,101 | peptides |
| 4,6-trihydroxyacetophenone (THAP)90,102 | lipids, oligonucleotides, drugs |
| 2,6-dihydroxyacetophenone (DHA)90,103 | lipids |
| 1,5-Diaminonaphthalene (DAN)104 | lipids |
Recent work has been demonstrated the utility of ionic liquid matrices for MALDI IMS.90–93 These matrices have some advantage in that there are no crystals to limit the spatial resolution. Inorganic matrices such as metal particles have a long history of being useful for MALDI MS, first demonstrated using cobalt metal particles; for this work Koichi Tanaka was awarded the Nobel Prize in chemistry in 2001.94 There are now a number of approaches to the use of inorganic matrices and surfaces that can promote desorption ionization. These include matrices such as gold nanoparticles,95–96 colloidal graphite,97 and clathrate nanostructures.98 Although nontraditional matrices have demonstrated limited utility, their use in routine IMS has not been widely adopted due to limitations in sensitivity and the number of analytes for which they can be used.
The solvent system used in the application of matrix plays a very important role in the sample preparation. Analytes are often prepared for MALDI MS analysis by pre-mixing the analyte solution with the matrix and drying the mixture to a crystalline state before irradiation with the laser;105–106 however, this method is not readily applicable to the analysis of tissue directly. In MALDI IMS, the tissue section containing the analytes is mounted to the MALDI target before the matrix is applied to the tissue section. For those matrices requiring crystallization, the solubility of the analytes and the solvent system must be compatible to promote mixing and recrystallization of the two. This is usually ensured by adjusting the type and amount of organic solvent used as well as the pH of the solution. The choice of solvent for the application of matrix can cause significant differences in the properties of crystallization on the tissue.67,106 Crystal formation is influenced by tissue content including lipid content, salt content, and by the choice of additives such as acetic acid, trifluoroacetic acid, as well as others. The matrix preparation must produce a uniform coating of matrix crystals that have sufficient crystal density to permit sampling of the surface without having regions that are absent of signal. The size of the individual crystal should be small relative to the raster step size to avoid creating images that contain artifacts generated by intermittent sampling of crystals. When applying uniform matrix coatings (i.e. spray coating or sublimation), minimal solvent is applied to the tissue surface to avoid lateral diffusion of analytes across the surface. Under these conditions, it may be necessary to recrystallize the matrix by placement of the coated sample in a humidified chamber for a few minutes. This step can induce many times more signal intensity and analytes in MALDI IMS. In one example, protein images are significantly enhanced by the recrystallization of sublimated sinapinic acid using 5% acetic acid vapor for 2 minutes.71
A useful starting point for proteins and peptides is a matrix concentration of 10–20 mg/mL in a 50–70% organic solvent (e.g. methanol, ethanol, acetonitrile) having also 0.1% trifluoroactic acid (TFA).45 Although these conditions will also work very well for some small molecule drugs, metabolites, and lipids, the chemical diversity of these analyte classes is very large relative to the 20 amino acids that commonly comprise proteins. Optimum results may only be attained for these low molecular weight analytes by making adjustments to the matrix and solvent conditions.
Some analytes, such as hydrophobic proteins or transmembrane proteins, cannot be solubilized using the typical conditions for MALDI sample preparation. For these molecules, it may not be feasible to solubilize these in significant concentrations without the use of detergents. For this purpose, there have been developed a number of MALDI compatible detergents that can be applied. These include nonionic detergents such as n-octyl glucoside107 and triton X-100,108 low percentages of ionic detergents such as sodium dodecyl sulfate (SDS),108 and cleavable detergents like 3-[3-(1,1-bisalkoxyethyl)pyridine-1-yl]propane-1-sulfonate (PPS).109
3.4.2. Matrix Application for Imaging
When the biological task necessitates imaging of the whole tissue specimen, matrix can be applied uniformly to the entire surface to be imaged in one of two ways. First, and most commonly, matrix is applied to the sample in order to produce a continuous coating of matrix and has the greatest flexibility for the acquisition of the image data. The disadvantage of the continuous coating is that lateral migration of the analytes may occur. The second approach is to coat the tissue with discrete matrix droplets using automated matrix spotting instruments. The spotting approach has the advantage that the lateral migration of the analytes is confined within each droplet spot on the tissue.
Manual Spray
Manual spraying of matrix has the advantage that it is the least expensive and easiest to implement in the laboratory although the most subjective. Early work utilized an artist airbrush to coat samples with matrix. These sprayers can make very fine droplets in an even spray; however, these sprayers typically have metal surfaces that can corrode, changing the performance over time. The tool most commonly used for the manual application of matrix is a glass reagent sprayer often used for the deposition of reagents on a TLC plate.45 Matrix is deposited into the reagent reservoir and the tissue is lightly sprayed with matrix, wetting the tissue minimally with each spray. The matrix is permitted to dry on the surface, and the process is then repeated some 10–20 times until the amount of matrix is sufficient for the analysis of the analytes. For the trained analyst, this technique can be used to generate very high resolution images of the tissue. The disadvantage of the manual technique is that it is subject to the experience of the person who is spraying the matrix making it difficult to standardize across multiple samples and laboratories. It can be easy to over wet the tissue using this technique, and the conditions such as drying between passes can change based on atmospheric conditions such as humidity, leading to delocalization and degradation of image quality.45 Also, the spray characteristics of glass reagent sprayers can vary greatly.
Robotic Sprayers
Matrix can be applied in a more controlled manner using an automated device to spray the matrix solution onto the sample in a repeatable, programmable way. There currently exist several commercial solutions that are dedicated to the application of matrix onto tissue. Two used in the authors' laboratory include the ImagePrep (Bruker Daltonics)110–111 and TM Sprayer (HTX Technologies).112 Although these technologies differ in mechanism, the basic principle is the same; the matrix is sprayed as a very fine aerosol or mist which is allowed to deposit of the surface of the sample. The matrix layer is dried and multiple coating steps may be used to deposit enough matrix to successfully carry out the analysis. The advantage of these systems over the manual technique is primarily that the conditions of the coating can be adjusted in a systematic way to optimize the coating procedure. Once the methodology is optimized for a tissue type, the program can be recalled and applied to another sample, even if the analyses are performed on different days. Automation provides the necessary rigor to be able to reproducibly carry out IMS studies on large sample sets. The matrix crystals that are formed using spray devices are typically 5 to 25 μm, but this is highly subject to the solvent system chosen. If the crystal size needs to be reduced, the matrix can be deposited using a solvent system having a high percentage of organic solvent. These coatings can result in a matrix crystal size of less than 5 μm; however, the crystals will need to be further conditioned for IMS by recrystallization to increase the amount of analyte incorporated.66
Sublimation
Sublimation is an approach used to make uniform matrix coatings first described by Hankin et al.113 for the analysis of lipids and has since been expanded to include many other analyte classes.71,76,104,114 Many of the matrix compounds used for MALDI IMS are amenable to sublimation at temperatures (c.a. 125°C) and pressures (10−4 torr) that are easily achieved in the laboratory with commonly available equipment. The matrix is placed inside a sublimation chamber while the sample plate with the tissue to be imaged is placed inverted over the top of the matrix crystal bed and attached to a cold finger. Once the chamber is evacuated, heat is applied to the matrix, causing it to sublime and condense on the surface of the cold sample. The technique is controlled by limiting the heating time, stopping when the desired thickness has been reached. Sublimation has the desired effect of purifying the matrix of any non-volatile impurities during the coating process.113 Additionally, the crystals produced using sublimation and rehydration are among the smallest that can be achieved, making it ideal for high spatial resolution imaging. Figure 10 demonstrates the quality of resolution of lipid imaging at 10 μm spatial resolution using 1,5-Diaminonaphthalene (DAN), a matrix that is useful for the analysis of lipids in both positive and negative ion mode.104 The technique has been demonstrated numerous times for lipids76,104,113 as well as proteins71 and small molecules.114 Methods that utilize sublimation for protein analysis require additional recrystallization of the matrix for best results.71
Figure 10. Sublimation enables lipid imaging at high spatial resolution.

IMS of lipids in the negative ionization mode from a transversal mouse cerebellum region coated with DAN by sublimation and acquired with a lateral resolution of 10 μm. In the H&E staining, a, b, and c represent white matter and the molecular and granular layers, respectively. Reprinted with permission from Reference 104. Copyright 2012 American Chemical Society.
Automated Spotting
Automated spotters are used to prepare samples for imaging by coating the sample with a regularly spaced array of droplets. In this case, the spacing of the droplets, rather than the raster spacing of the mass spectrometer define the image resolution. Reports in the literature describe a number of printing devices that have been adapted for MALDI matrix deposition onto tissue. In one example, a common desktop inkjet printer was modified for matrix deposition.115 Generally, there is a wide array of liquid handling instruments that could be used to accomplish this task; however, out of concerns for cross-contamination of spots and for reasons of speed, the authors prefer non-contact printers.
Commercial automated matrix spotters combine optical scanners with a software system that can be used to mark the regions for matrix placement. Two different commercial systems used by the authors for the deposition of matrix are the Portrait® 630 Spotter (Labcyte, Inc.)69 and the ChIP-1000 (Shimadzu Scientific Instruments). These systems have similar specification, ejecting approximately 100 pL droplets using solvents that are approximately 50% organic solvent. These droplets dry on the tissue surface to yield spots that are less than 150 μm. Multiple droplet passes, as those required to typically analyze most tissues, yield spots that are approximately 180–200 μm that can be placed at a spacing of approximately 200–250 μm center-to-center.
The Portrait 630 Spotter uses focused acoustic energy to transfer droplets from a matrix reservoir to the sample surface.69 Figure 11 shows the schematic of the acoustic spotter prototype device upon which the Portrait 630 Spotter was based. The acoustic transducer is focused on the liquid surface, ejecting droplets from the surface that deposit on the sample target that is mounted in an inverted position over the matrix reservoir. An x-y translation stage moves the sample relative to the droplet ejection site to print matrix in different locations on the sample. The advantage of the acoustic tranducer is that there are no moving parts or nozzles that the matrix contacts, eliminating clogging of the system. The Portrait 630 is robust and can operate many hours continuously. Figure 12 demonstrates the use of the Portrait 630 to print an array of matrix spots on the lens of a human eye at 200 μm spatial resolution along with the associated MALDI IMS results for the variant forms of the protein Aquaporin-0 (AQP0).72 An optical image of the prepared sample that was spotted using the Portrait 630 Spotter is shown in Figure 12a.
Figure 11. Schematic of the acoustic reagent multispotter with translational stage.
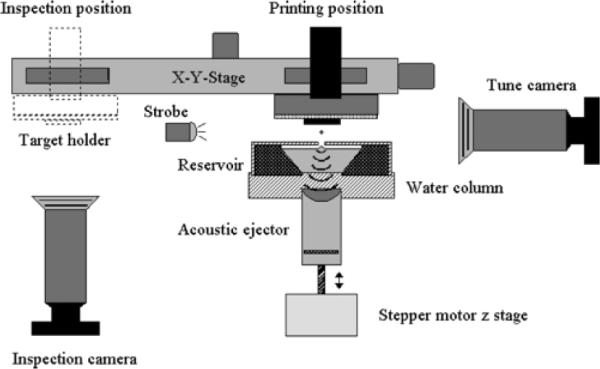
Reprinted with permission from Reference 69. Copyright 2006 American Chemical Society.
Figure 12. MALDI imaging of AQP0 in a human lens prepared using an acoustic matrix spotter (Portrait 630 Spotter).
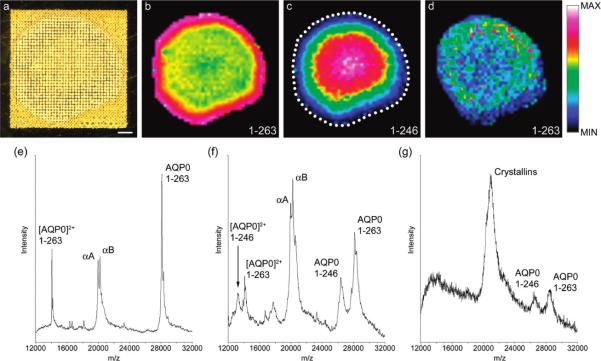
(a) Optical scan of an equatorial section from an 11 year-old human lens prepared for integral membrane protein imaging with high-density matrix microdroplet array applied at 200 μm spot spacing. (b) Signal for full-length AQP0 (1–263) (plotted m/z 28 129) is most intense around the edge of the lens, and persists in the lens core. (c) The major truncation product in the human lens AQP0 (1–246) (plotted m/z 26 378) increases in intensity in the core of the lens. (d) Signal for full-length AQP0 (1–263) in unwashed tissue sections, indicating the importance of the washing step. (e) Extracted spectrum from the lens periphery showing singly- and doubly charged AQP0 signals and signals for the abundant soluble proteins αA- and αB-Crystallin. (f) Extracted spectrum from the lens core, showing singly- and doubly charged full-length AQP0 and AQP0 (1–246). Signals for Crystallin proteins are also abundant. (g) Extracted spectrum from the lens cortex of unwashed tissue, showing noisy, poorly resolved mass spectral signals for AQP0 1–263 and 1–246, and abundant Crystallin signal. Scale bar = 1 mm. The dotted line indicates the edge of the tissue. Reprinted with permission from Reference 72. Copyright 2009 American Chemical Society.
The ChIP-1000 chemical printer has been adapted for use with MALDI matrix.37–38,73,116–118 It utilizes a piezoelectric dispenser to dispense matrix droplets that are 100 pL in volume. The print head can be filled by the analyst making it customizable to many solutions. The disadvantage of this system is that the nozzle can easily clog when concentrated solutions of matrix are used, leading to frequent and thorough cleaning of the system.
3.4.3. Matrix Application for Histology-Directed Profiling
The histology directed profiling modality of MALDI IMS, as described earlier in Figure 4, requires the placement of discrete spots of matrix onto regions of interest identified by a biologist or pathologist. Using one of the staining approaches previously mentioned, regions of interest are annotated and the coordinates of those annotations are transferred to the unstained section to facilitate the placement of matrix onto the tissue.31 There are two primary methods of placing discrete matrix spots onto clinical samples, manual spotting and spotting using automated spotting instruments. The selection of the method of matrix deposition is dependent on the spatial resolution and the accuracy of reagent placement required to target the cells of interest.
The most cost efficient method for deposition of matrix is the manual placement into discrete locations on the tissue using a pipette or capillary. This is a variation of the dried-droplet method applied to tissue analysis.7 The size of the spots tend to be quite large (>500 μm) and the crystal density can be sparse. For the purpose of the experiment, one should consider that anything covered by the droplet is homogeneously distributed within the resulting dried-droplet although this may not be necessarily true. Many early examples demonstrate the usefulness of this approach to find interesting molecular changes in tissues.119–121 However, as regions of interest become increasingly small, this method becomes more difficult for even the most skilled person to place the matrix in a precise location. Additionally, this method is labor intensive, especially for multiple sections. Due to the inhomogeneity of the spots, manual acquisition is often the best approach to the analysis of manually placed matrix spots for applications involving low spatial resolution analysis. In cases where the droplet placement is important and the size of the feature is small (< 300 μm), then automated spotting instruments are used for the placement of matrix onto the tissue. Matrix can be placed with higher spatial accuracy than can be achieved by manual spot placement. Placement accuracies less than 20 μm can be achieved with multiple spotting passes using the ChIP-1000 chemical printer and the Portrait 630 Spotter. Furthermore, the spot homogeneity is much higher using automated spotting instruments, increasing the reproducibility of automated acquisition of MALDI MS data.
4. Data Acquisition and Analysis in MALDI IMS
Owing to the numerous academic groups and companies that have focused on instrument and software development in the previous decade, there now exists many MALDI mass spectrometers that have IMS capabilities. In general, there are three components to the IMS platform: 1) the instrument hardware that comprises the MALDI source and mass analyzer, 2) the software platform designed for the acquisition of IMS data, and 3) the visualization and analysis software. A number of instrument manufacturers provide IMS solutions that integrate these components into a single platform. Given all the options that are now available, it is necessary to carefully consider exactly what capabilities are important for each specific IMS application. These requirements can be quite different for proteins when compared to small molecules. This section discusses some of the most important factors to consider during selection of the IMS platform for IMS for a particular experiment.
4.1. Impact of Mass Spectrometer Performance on MALDI IMS
The metrics that are used to compare and contrast mass spectrometer performance include mass resolving power, mass accuracy, sensitivity, dynamic range, and tandem MS capabilities. Table 5 summarizes some of these attributes for the most common mass analyzers used for MALDI IMS. Just as in other mass spectrometry experiments, these parameters have significant implications for the IMS experiment and ultimately impact image quality and the biological conclusions drawn from the images. A number of considerations are most important for IMS that are often not as critical for routine mass spectrometry, such as spatial resolution, throughput, and data storage. These parameters affect instrument selection and planning of IMS experiments. Previous publications have extensively reviewed each of the unique mass analyzers used for MALDI IMS,11–13,18–20 and this section will only briefly mention the most important metrics for instrument selection for MALDI IMS studies.
Table 5.
Characteristics of MALDI Mass Spectrometers used for IMS.
| Mass Analyzer | Mass Resolving Power (m/Δm) | m/z Range | Throughput (pixels/sec) | Tandem MS Capabilities | Application |
|---|---|---|---|---|---|
| Linear TOF | 103 | 0 – 106 | 2 – 5 | No | Peptides/ Proteins |
| Reflectron TOF | 103 – 104 | 0 – 105 | 2 – 5 | MS2 (post-source decay) | Drug/Metabolites/Peptides/Lipids |
| TOF/TOF | 103 – 104 | 0 – 104 | 2 – 5 | MS2 | |
| Q-TOF | 103 – 104 | 100 – 6k | < 1 | MS2 | |
| Linear Ion Trap | 102 – 103 | 50 – 3k | < 1 | MSn | |
| Triple Quadrupole | 102 – 103 | 50 – 5k | > 100 | MS2 | |
| FT-Orbitrap | 104 – 105 | 50 – 10k | < 1 | MSn | |
| FT-ICR | 104 – 106 | 100 – 10k | 1 – 3 | MSn |
4.1.1. Spatial Resolution
Spatial resolution significantly impacts the nature of the molecular information that can be derived from an IMS experiment. Figure 13 demonstrates the effect that a change in spatial resolution has on the quality of the resulting image. Ultimately, the biological or clinical problem and the nature of the sample dictate the minimum spatial resolution of the experiment. However, since the increase in spatial resolution negatively impacts other parameters such as sample throughput, sensitivity, and the amount of data that must be managed, it is not beneficial to acquire data at spatial resolutions that exceed that which is necessary. These tradeoffs are important considerations and should be thoughtfully balanced when choosing experimental parameters for MALDI IMS.
Figure 13. Comparison of serial mouse brain images acquired at different spatial resolutions.
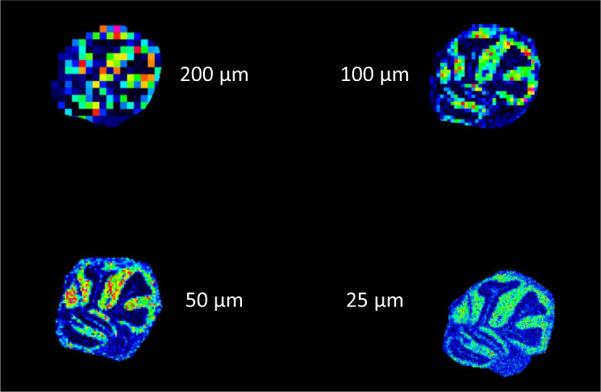
Ion images (m/z 6765) of mouse cerebellum imaged by MALDI IMS at spatial resolutions of 200, 100, 50, and 25 μm.
Two important parameters must be set to specify the spatial resolution of the imaging experiment; the laser beam diameter and the pitch between the ablation spots (pixels). At its highest spatial resolution, MALDI IMS has been demonstrated at subcellular resolutions.28–29 These examples utilize advanced optics to focus the laser beam to diameters less than 1 μm.29,122 Commercial instruments provide solutions that are configured with a range of laser focus, ranging from approximately 200 μm down to 20 μm. This range of laser beam settings is not available on all instruments, and one must also consider that the actual performance can differ markedly from the specification depending upon the experimental conditions. The SmartBeam® laser provided by Bruker as part of the TOF, TOF/TOF and FT-ICR systems has the option to adjust the laser spot size to 5 different preconfigured focuses, with a minimum focus of 20 μm.123 Some other instruments have limited or no adjustments that can be routinely made on the beam size. Imaging at spatial resolutions less than the size of the beam diameter have been reported using an oversampling method, acquiring data at a spatial resolution less than the measured beam diameter.30 When utilizing this type of acquisition, significant validation of the acquisition parameters must be performed to avoid degradation of the signal quality of each pixel due to undersampling or excessively oversampling each pixel.
4.1.2. Sensitivity and Dynamic Range
Analysis of clinical specimens at high spatial resolutions often comes at the expense of sensitivity, obviously the result of the smaller area being ablated and the limited number of ions produced. The method of sample preparation can also dramatically alter the sensitivity of the assay as well. These practical considerations play a very important role in the selection of the mass spectrometer to be used. The sensitivity for a specific analyte measured out of the context of a complex biological tissue can be less sensitive that the standard compound because of ion suppression effects and interfering ions. It is therefore important to evaluate instruments using representative samples and at representative spatial resolution before selecting the platform used for MALDI IMS experiment when a choice is available.
Dynamic range, the range of analyte concentration that can be detected, is a property of the mass spectrometer that can be very limiting for sample analysis. Intact tissues are molecularly highly complex samples that contain analytes whose concentrations and masses vary over many orders of magnitude. For example, physiological salts and blood proteins are commonly found in fresh tissues and can contain molecular constituents that span more than 12 orders of magnitude in concentration.124 Limitations in sensitivity and dynamic range of IMS remain a challenge for many analytes; therefore, proper selection of the instrument for each experiment and optimization of the sample preparation are necessary to maximize success. Many of the methods that were reviewed in the section on Sample Preparation (Section 3.0) were developed specifically to maximize sensitivity and dynamic range. For the higher molecular weight range analytes (>5 kDa), MALDI TOF remains the instrument that offers the best all-around sensitivity for these experiments.
FT-ICR MS instruments have some unique capabilities to increase sensitivity and dynamic range. Ion accumulation techniques like In-Cell ion Accumulation (ICA)125 and Continuous Accumulation of Selected Ions (CASI)126–127 that enrich specific ion populations have been shown to have impressive benefits for MALDI IMS. Using a quadrupole and hexapole to select and accumulate ions prior to detection by the ICR cell, essentially all of the ions generated from and area on the surface at a given location can be ablated and analyzed. Since the ion population is enriched for the molecule of interest, the ICR mass analyzer performance is maximized by reducing potential space-charging effects that may occur in the analysis of complex samples. This ensures that the maximum dynamic range can achieved for the m/z window selected.126 Due to the limitations of the ion optics of these instruments, these technologies are practically limited to m/z under 6,000.
4.1.3. Tandem MS Capabilities
Targeted analyses for specific analytes by tandem MS, as is typically performed by LC/MS/MS, have become routine for the analysis of small molecules in MALDI IMS. This then is a targeted approach having increased sensitivity and specificity. Currently, MALDI IMS has been implemented on all types of the commercially available tandem mass spectrometers, including time-of-flight (TOF/TOF), quadrupole time-of-flight (Q-TOF), linear ion trap (LIT), and fourier transform ion-cyclotron resonance (FT-ICR) and orbital ion traps. These instruments permit the isolation of specific ions based on m/z, followed by fragmentation and detection of the fragment ions. The characteristic fragments ions from the parent ion add selectively while effectively increasing the signal-to-noise and dynamic range for the detection of that selected ion. Multiple analytes can be measured using this method; however, the detection bandwidth is limited by the scan rates of the instrument. The reader should consult the specification for each specific instrument for more information on these limitations.
Detection is sequential in tandem MS operation because each parent ion is isolated individually. However, modern instruments can monitor many molecules (>5 parent ions) in a single experiment, allowing some degree of multiplexing termed multiple reaction monitoring (MRM). Tandem mass spectrometry in IMS is essential for many applications in the m/z range below 2000 due to the diversity of chemical species in each nominal m/z region. It is not uncommon to have many overlapping analyte signals, requiring the added specificity of tandem MS.
4.1.4. Mass Accuracy and Resolving Power
Mass accuracy and resolving power are related topics in that increases in mass accuracy often come as a result of increase mass resolving power; however, there are distinct benefits that these attributes bring MALDI IMS. Using low resolution instrumentation, signals from nominally isobaric species are averaged and can be difficult to deconvolute. However, these molecules can have unique structures, biological functions, or may originate from background peaks such as the matrix itself. Therefore, the spatial distribution of each of these compounds can be different but can be distinguished depending on the mass resolution of the instrument. High resolution MALDI IMS is normally accomplished using one of three different mass spectrometers, Q-TOF (m/Δm ~ 30,000), FT-orbitrap (m/Δm – 30,000–100,000), and FT-ICR (m/Δm >> 100,000). Figure 14 demonstrates the benefits of high mass resolution in the analysis of a drug metabolite, 2-hydroxymethyl olanzapine.128 In this example, there are 3 nominally isobaric species detected in the m/z window shown. Using a low resolution instrument, these signals cannot be resolved and the distinct distribution of each metabolite is then averaged among the species present. However, the mass resolving power of the FT-ICR can distinguish these ions based on accurate mass measurements, providing additional specificity to the IMS experiment through accurate determination of the elemental composition of the analyte and any fragment ions that originate from tandem MS. FT-ICR and orbitrap instruments are capable of measuring ions with mass accuracies to 0.1 and 3 ppm, respectively, directly from tissue sections.129–133 Accurate mass measurement, coupled with tandem MS, enable small endogenous compounds to be unambiguously identified directly from the tissue. Figure 15 demonstrates one such analysis in which an orbitrap mass spectrometer was used to image and identify a phosphatidylcholine (PC), phosphatidylserine (PS), and cerebroside all having the same nominal mass.134
Figure 14. The Benefit of High Mass Resolution in MALDI IMS.
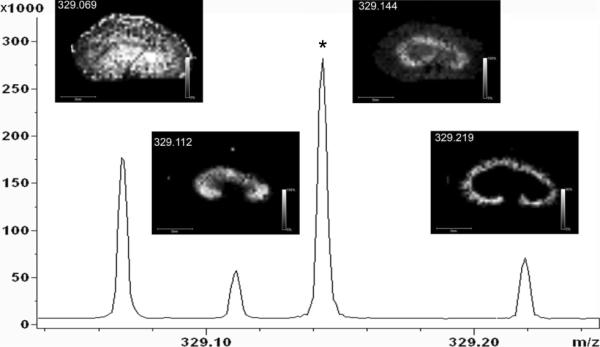
FTICR images of the drug metabolite, 2-hydroxymethyl olanzapine ([M+H]+ = 329.1431) shows distinct localization when compared to three nominally isobaric ions. The asterisk indicates the peak corresponding to the drug metabolite. Reprinted with permission from Reference 128. Copyright 2008 American Chemical Society.
Figure 15. Identification of Lipids Species using Accurate Mass in MALDI IMS.

(A) Mass spectrum of m/z region 848–849 acquired on an Orbitrap analyzer. Mass spectrometric images B–D correspond to major ions detected in A. Fragmentation of nominal m/z 848 in the linear ion trap produces the MS/MS spectrum seen in E. Images observed in F and G corresponds to the two major isobars found at 848. Matching images produced by the full scan and MS/MS spectra and exact mass calculations identify the ions in the full scan mass spectrum as: 848.5603 [PC(18:0,20:4)+K]+; 848.6417 [Cer(d18:1,24:1)+K]+; 848.6617 [Cer(d18:1,24:1h)+Na]+; 848.6975 [Cer(d18:1,25:0)+Na]+. NL: neutral loss; PC: phosphatidylcholine; PS: phosphatidylserine; Cer: cerebroside. Reprinted with permission from Reference 134. Copyright 2009 American Chemical Society.
4.1.5. Throughput
Early in its development, speed of acquisition has been one of the challenges of MALDI IMS. With the instrumentation available ten years ago, it was difficult to imagine widespread application of the technique for research or clinical purposes. However over the past few years, commercially available MALDI instruments have been equipped with lasers operating at frequencies of 1–2 kHz; 100 μm spatial resolutions images for average sized samples (~ 1 cm diameter) can be performed in 1–2 hours. For histology-directed experiments where only select regions of interest are profiled, this analysis time can be mere minutes. Even so, throughput remains a challenge for applications that demand high spatial resolution or necessitate the analysis of many replicate specimens for statistically meaningful results. Fortunately, technology developments continue to increase the speed of acquisition. Simultof Systems (Sudbury, MA) has recently commercialized a series of MALDI instruments that acquire images at frequencies up to 5 kHz. These instruments reduce the acquisition time up to 50-fold relative to earlier generation instrumentation having 100 Hz lasers. Since the mass spectral acquisition rates are so fast, the sample translation stage is operated in a continuous manner rather than stopping and starting. The result is that the lipid analysis of a mouse brain at 100 μm spatial resolution can be accomplished in less than 10 minutes (Figure 16).30 Sample throughput has obvious implications for the analysis of clinical samples. To date, MALDI IMS has been most used in the context of clinical research. In order for MALDI IMS analyses to be meaningful for diagnostic purposes, the result must be generated within a time that clinical decisions can be impacted by the result of the diagnostic assay. As sample acquisition rates improve and the rate limiting step becomes sectioning and preparing the sample, MALDI IMS then becomes an attractive technology for clinical diagnostic applications.
Figure 16. High-Speed MALDI IMS.
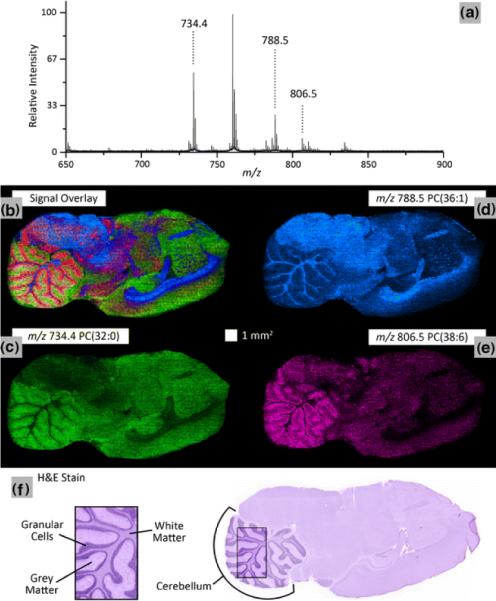
A 100 μm spatial resolution lipid ion image of a sagittal rat brain tissue section using continuous laser raster sampling acquired in 10 min. (a) Representative spectrum is shown. The ion image overlay (b) of signal from m/z 734.4 PC(32:0) (c), m/z788.5 PC(36:1) (d), and m/z 806.5 PC(38:6) (e) highlights the differentiation of the spatial distributions for the selected ions. These results correlate to the H and E stained serial tissue section highlighting structural difference between grey matter, white matter and granular cells in the cerebellum (f). Important instrumental parameters: 3 kHz laser repetition rate, 5 mm/s sample stage velocity, and 60 laser shots/spectrum hardware average. Reprinted with permission from Reference 30. Copyright 2011 Springer.
4.2. Data Analysis
Due to the variations in specimen size, image resolution, and spectral resolution, individual MALDI IMS data sets can be many 10s to 100s of gigabytes in total. The high dimensionality of the data can present a challenge to the individuals and the software programs attempting to interpret and manage the data. In recent years there have been a number of software programs introduced to process MALDI IMS data. Due to the time and expense, it is difficult to comprehensively test each of the software packages to compare the benefits and disadvantages of each, so the subsequent section lists and briefly describes the software packages as a reference to the reader to find tools for visualization and analysis.
4.2.1. Data acquisition software
Data acquisition software is generally separate from the standard control software found on most commercial instruments. MALDI IMS acquisition software is designed to register the target location to an optical image of the specimen that is to be imaged. Using tools similar to those found in many image markup software programs, the region of interest is defined by the user. This consists of drawing a shape around the area of interest, defining the spatial resolution within the area of interest, then an automated acquisition is initiated to collect mass spectra at each position in the raster pattern. Since these software packages are highly integrated with the control software of the instrument, instrument vendors almost universally provide MALDI imaging software with the instrument. Some examples include, flexImaging™ (Bruker), TissueView™ (ABSciex), and ImageQuest™ (Thermo). Additionally, integrated software solutions are provided by Waters, Simultof Systems, and Shimazu that facilitate MALDI IMS data acquisition using their instruments.
For instruments whose commercial release predates the widespread adoption of MALDI IMS (prior to 2002), there exist some third party software packages that can be used to acquire images on these instruments. The web resource, www.maldi-msi.org, manages a repository of software packages that can be downloaded without charge. Among these tools are image acquisition tools for previous generation instruments from Applied Biosystem (Voyager and 4000 series).
4.2.2. Image Analysis
MALDI imaging data can be generated from a variety of samples, sizes, and instrument platforms. Each vendor's platform has customized tools for the acquisition of MALDI IMS data. In addition, there are several free and third party software packages available.
Data Formats
The format of the data is important with respect to its portability and the way in which appropriate metadata is stored and recalled. To date, there are not rigorous standards for data formats, and there have been numerous formats proposed in the literature. Most mass spectrometers have a proprietary data format that is controlled by the manufacturers. This trend has carried forward into the native IMS data generated by these instruments as well. However, options exist for readily exporting and viewing data using third party software. Two of the most common open source formats for IMS data are Analyze 7.5 and imzML.
Analyze 7.5 is a file format borrowed and modified from MRI data. This data structure was developed for MALDI IMS to facilitate the use of the BioMap software (described below). Due to the introduction of this format early and the free viewing software to visualize the data, this format has been widely adopted and many of the vendor software packages support the export of raw data to Analyze 7.5 format. One disadvantage is the limited amount of metadata associated with this file format.
The imzML format was developed to conform to the same standards proposed by the Human Proteome Organization-Proteomics Standards Initiative for storing mass spectrometry data.135 The goals for the development of this file format were to completely describe the IMS experiment, minimize the file size, ensure fast and flexible handling of the data, and remain consistent with the existing mzML format.136 This format consists of two separate files. One binary file (*.ibd) stores the spectral data and one XML file (*.imzML) is used to store all associated metadata. Both file types are required to fully describe the data. A controlled vocabulary that is used for complete description of the information is contained in the XML file. For a complete description of the details of the imzML file format, the reader is referred to a recent review136 or the website (www.imzML.org). The imzML format has been adopted by a growing number of instrument vendors, and continues to be used by developers of open source software for conversion of IMS data and for visualization.
Biomap
Biomap is an open source platform for the visualization and processing of IMS data. The concept for this software package began with work in the authors' laboratory and was integrated into an imaging software package for the visualization of other imaging data sets including magnetic resonance (MR), positron emission tomography (PET), computed tomography (CT) and optical imaging.137 The platform for this method can be extended to include the visualization of multiple data sources provided that an import filter is created. The software was written in IDL and visualization is based on mutiplaner reconstruction, supporting the extraction of slices from 3D data sets and displaying them in 2-dimensions. The software permits the selection of regions of interest within the image from which the mass spectral data can be plotted. From the spectrum view, navigation tools permit integration of signals that can be extracted to create new ion images from the data set. The software also supports registration of images, overlays of ion images with optical images, and contains tools for the processing of MALDI IMS data on the mass spectral level, including tools for baseline correction and spectral averaging. Biomap provides database functionality to facilitate organization and storage of image files. This software continues to be important and remains popular due to the fact that it is a free download (www.maldi-msi.org), and facilitates sharing of data with collaborators that do not otherwise have resources to invest in third party MALDI IMS software.
Datacube Explorer
This software was designed and created by the FOM Institute AMOLF expressly for visualization of IMS data. This software package offers many similar functions to Biomap software, navigation of the spectrum and image, selection of regions of interest, and spectral processing tools. Datacube Explorer also has a tool for the automated extraction of images from the data set. This software is available for free download (www.maldi-msi.org).
MALDI Imaging Team Imaging Computing System (MITICS)
The MITICS software package consists of both a control software that works with Applied Biosystems brand mass spectrometers and a visualization software that is compatible with other platforms, provided the data is converted to imzML.138 MITICS stores the XML data in a database to facilitate browsing and display of the data. Since the databanks can be distributed across a network for computers, several images can be reconstructed simultaneously, making the software faster to use. MITICS can be obtained upon request by contacting the authors of the software (www.maldi-imaging.com).138
FlexImaging
Among the commercially available software solutions for MALDI IMS, flexImaging is an integrated package for MALDI image acquisition and image viewing provided as part of the suite of tools for Bruker mass spectrometers. The acquisition software works by first uploading an optical image of the sample to be imaged followed by registration with the stage of the mass spectrometer using fiducial registration points and selection of regions of interest for imaging. The software controls data acquisition and subsequent display as visual images. This allows for clear registration of the ion images with the tissue histology. The flexImaging software also incorporates a variety of spectral processing tools that allow for the correction of baseline noise and normalization of spectral data. FlexImaging is available for purchase and is often bundled with the purchase of the manufacturer's instruments. A limitation of the software is that only data acquired using Bruker instruments can be read and displayed.
Quantinetix
This is a vendor neutral software package that is designed for applications that utilize MALDI IMS for the quantitiation of drugs in tissue samples. It is the first commercial software developed for use in this area and is designed to use standards doped onto tissue,139 isotopically labeled standard compounds,140–142 or an in-house developed technique that uses ion suppression calculations to quantitate the amount of analyte in the tissue.143 The software is available for purchase from ImaBiotech SAS (http://www.imabiotech.com/Quantinetix-TM-Maldi-Imaging.html).
4.2.3. Biomarker Discovery Software
Histology-directed analysis requires a different set of tools for data analysis than those required for imaging whole sections, although there are areas of complementarity that are highlighted below. The data analysis process can generally be divided into two separate processes, data preprocessing and analysis.
Spectral Preprocessing
The concept of data preprocessing is to remove sources of variation or noise other than the biologically relevant information in the MALDI spectra.144–147 These processes include baseline correction, normalization, alignment and calibration, smoothing, and peak detection. Baseline correction is a process by which the chemical noise that is prominent in the low mass regions of the MALDI mass spectrum is computationally removed by algorithms that estimate and subtract the noise from the spectrum. This process flattens the baseline so that calculation of the area under the peak more accurately represents the true biological contribution to the signal measured. When performing baseline correction, the parameters for fitting the baseline are optimized for the instrument spectral resolution to avoid over fitting the data and subtracting biologically relevant information. Numerous methods have been developed for baseline subtraction of MALDI data.148–149
Normalization transforms the peak heights of a set of spectra taken from a similar biological context and places them on a common intensity scale. The normalization process normally works best when there are common features found in the mass spectra. In IMS, metrics such as total ion current are often used as scaling factors to accomplish normalization. Attention must be given to ensure that the parameters for data acquisition are matched as closely as possible across the data set and that there are no spectra in which detector saturation or any significant ion suppression due to highly abundant proteins are present. These phenomena can cause the data to be severely skewed by the normalization process. Many algorithms have been developed for spectral normalization of MALDI data.141,143,146,150–152
In a large data set that has been acquired over the course of many months or years, slight variations in mass calibration can cause difficulties for determining which peaks are common across the data set. A shift in m/z can create difficulties when extracting peak areas and can create the false positive or negative result. Commonly a recalibration is performed to align the spectra to a common m/z scale.146
Peak detection and integration is one of the more important steps of the process; however, this process has proven to be also the most difficult to perform in an automated manner. One key issue is the presence of unresolved peaks, making it difficult for peak picking algorithms to be blindly applied to the data set without taking time to visualize the result and manually correct any inappropriately labeled peaks. Recent papers have evaluated peak-picking algorithms for use with MALDI mass spectra.150,152–153 Many of the manufacturer provided solutions have peak picking algorithms embedded that are proprietary. It is recommended that the peak picking algorithms selected and their corresponding integration functions be carefully examined and understood before applying these tools to a MALDI data set for biomarker discovery.
Matlab (Mathworks, Inc., Natick, MA) provides a suite of MALDI spectral preprocessing tools as part of the Matlab Bioinformatics Toolbox.154 This toolbox consists of a set of functions that can be used for all of the preprocessing steps mentioned above. The documentation provided is complete, detailing the many options available to adjust parameters to meet the needs of data processing. In addition, there are free packages available in the statistical package R that are varied in their functionality and the types of algorithms used (see www.r-project.org).
Spectral Classification
Data mining techniques for MALDI IMS data are generally categorized as unsupervised or supervised methods of analysis. The output of the preprocessing steps previously described is a table for m/z values and the corresponding intensity or peak areas extracted from each spectrum in the data set. These data can be subject to a number of data mining algorithms designed to select signals that best discriminate the biological task being investigated. These methods are not unique to MALDI data, and have been applied to numerous multivariate data sets. Several recent reviews detail these approaches for MALDI data.150,155
The most common unsupervised data analysis approach is the use of principle component analysis (PCA). PCA groups together peaks whose intensities are correlated in certain areas of the tissue. The output of the analysis are a set of principle components that are rank ordered according to the amount of variance that they describe in the data. Analysis by PCA provides a measure of similarity that can be used to estimate the areas of the tissue that are most alike on a molecular basis. These principle components can be mapped back to the tissue surface to visualize the molecular similarity of different regions and are useful for finding tissue substructures described by the molecular information.156 There are other unsupervised classification approaches that can also be applied to MALDI IMS data. A recent paper compared four of these approaches and demonstrates the application of different classification algorithms on the same data set to provide unique insight into the tissue substructure.157
Supervised data analysis is effective where a set of spectra collected from externally validated origin or disease state is used to build a model for the classification of unknown spectra that have been collected from similar tissues. This method requires a rigorously curated sample set for the training phase of the experiment in order to produce a model that is predictive of the disease state when tested on blinded samples. The algorithms used for these types of supervised classifications include genetic algorithms, support vector machine, random forest, and supervised neural network.155 These approaches have been applied in a number of studies to classify samples according to their molecular profiles.158–166
Software
There are commercially available software packages that are available that have incorporated tools for analysis of MALDI data to determine the ions that best discriminate disease states. One of the more effective solutions is ClinProTools (Bruker Daltonics). This software incorporates all of the preprocessing functions with algorithms described for classification (both supervised and unsupervised). This software also works in tandem with flexImaging software, the IMS acquisition and visualization tool, to correlate the spectral classifications back to the ion image, described in the literature as Class Imaging.156,167–168 Other commercial software solutions include Progenesis (Nonlinear Dynamics, Durham, NC) and Expressionist (Genedata, Basal, Switzerland). Both of these solutions include extensively developed algorithms for the preprocessing of MALDI mass spectra. Progenesis has basic statistical tools to compare spectra on a peak by peak basis to find fold changes that are relevant to the groups being compared. Expressionist has advanced tools for both supervised and unsupervised classification.
4.2.4. Three-Dimensional Image Visualization
3D MALDI imaging involves registration and combination of multiple tissue section in a single visual representation. Although conceptually simple, important issues apply during sample preparation to generate registration points that can be used to plot the 2-dimensional planes in correct registration. Details of these steps have been described for a variety of sample types.73,169–177 The different ion planes can be displayed in three dimensions in order to create a volume that can be correlated with other types of 3D imaging data such as MRI. Figure 17 shows the rendering of a 3-dimensional MALDI IMS data set measured in the substantia nigra of a mouse brain, highlighting the distribution of two ions.169 This example demonstrates that the volume can be viewed from virtually any angle to gain a better appreciation of the distribution of the molecules in the sample.
Figure 17. Three-Dimensional Rendering of MALDI IMS Data.
Four different views of the 3D construction of the substantia nigra and interperduncular nucleus. PEP-19 is shown in green, and a protein with m/z 7416 is shown in red. The brightest colors correspond to highest intensity. Reprinted with permission from Reference 169. Copyright 2012 American Chemical Society.
The software used for rendering 3D volumes includes a variety of commonly used software packages such as Image J (http://rsbweb.nih.gov/ij/, NIH), Matlab (Mathworks, Natick, MA), and Amira (Visage Imaging, Inc.). Recently, unsupervised classification techniques similar in principle to those mentioned in the previous section have been applied to the analysis of 3D MALDI data sets.178
5. Selected Biological and Clinical Applications
The application of MALDI IMS to problems of biological and clinical significance has increased exponentially since the introduction of the technology over a decade ago. The following sections highlight four areas where MALDI IMS is impacting our understanding of human disease and contributing to the development of better diagnostic and therapeutic approaches to these diseases. These examples intentionally exclude methodology papers and are intended to highlight studies in which MALDI IMS data provided further understanding of biological processes.
5.1. Molecular Physiology of the Eye
The eye has been the subject of numerous studies that have utilized MALDI IMS. These methods have focused on a range of analytes that include the characterization of proteins,72,179–185 lipids,186–193 metabolites,194–195 and drugs.196–197 Examination of the proteome of the eye using MALDI IMS began with work by Schey and co-workers. The seminal publication examined the distribution of crystalline proteins in the lens,179 a highly abundant set of proteins comprising more than 90% of the proteins present in the lens. There is a limited amount of protein turnover in the eye; consequently, the crystalline proteins are subject to a variety of posttranslational modifications over their lifetime. With age, these modified proteins can build up in the eye and can be correlated with clinical conditions, including the formation of cataracts. MALDI IMS analysis revealed different distributions for the two subunits of α-crystallin and their modified forms. Subsequent work further demonstrated that MALDI IMS can be employed to characterize modified α-crystallin forms. Aside from several truncated forms, the study confirms the phosphorylation of α-crystallin occurs preferentially in the middle cortex of the lens.180 MALDI IMS has been used to characterize modified forms of α-crystallin in both cataractous rat models184 and human samples.182–183 Methodology for the analysis of membrane proteins was developed to profile retinal and lens tissue using a histology-directed approach.181 Following extensive washing in water, then matrix was spotted manually with a mixture of formic acid and hexafluoroisopropanol. The membrane proteins aquaporin-0 (AQP0) and MP20 were detected in the lens, while opsin was measured in the retinal tissue. AQP0 and opsin imaging was performed in a related study (Figure 12).72
MALDI IMS methodologies for small molecules in the eye have been developed for a variety of lipid classes and metabolites. The measurement of phospholipids184,188–189 and cholesterol and sphingolipdids185,187 has been described in detail by multiple groups. Using silver nanoparticles as a matrix, fatty acid imaging has also been accomplished.189 This revealed distribution zones of fatty acids in different layers of the retina demonstrating the potential of the technique to reveal the physiology of a tissue sample by molecular measurements. MALDI IMS has also been used to study the influence that lipids in the membranes have on proteins, as for example, in a study of phosphatidyl choline distribution in the role of membrane composition on the conformation of rhodopsin.186 Recently, metabolite analysis has been shown useful in furthering the understanding of age-related macular degeneration (AMD).194–195 Lipofuscin, a fluorescent orange accumulation of degraded chemical species, is correlated with the advancement of AMD. Previous studies identified the cytotoxic metabolite bis-retinoid A2E as a primary component of the retinal pigment epithelium. Using MALDI IMS, A2E was confirmed as a major component of lipofuscin, in addition to other bis-retinoid compounds. Figure 18 shows the correlation of A2E signals from MALDI IMS studies with the lipofuscin localization as measured by fluorescence microscopy.194 These findings were confirmed and further validated using LC/MS/MS methods.195 These results and the methods for metabolite imaging of the eye open the field to new discoveries by making it possible to map molecular components of the disease.
Figure 18. Lipofuscin fluorescence and MALDI images of A2E in mouse RPE.

RPE tissue images from 6-month-old Sv129 and Abca4−/− mice. Left panels: fluorescence intensity images; right panels: MALDI images of A2E in the same tissue. Fluorescence images were acquired as micrographs (λexc = 488 nm, λem = 565–725 nm, 10× objective, NA = 0.3) of individual fields and joined by overlapping areas. Images are shown at the same intensity scaling. The MALDI images were acquired after the tissue was spotted with MALDI matrix at 150 m resolution. The pixel intensity is proportional to A2E quantity, with the scale normalized to total ion current. All images are oriented as follows: dorsal (top); ventral (bottom); nasal (left, Sv129; right, Abca4−/−); temporal (right, Sv129; left, Abca4−/−). Scale bar, 1 mm.
5.2. Cancer Research
Cancer research occupies a prominent position in the development MALDI IMS technologies. Indeed, one of the first examples of the application of MALDI IMS to biological research was to study human glioblastoma,17,119,198 demonstrating the power of the technology to both visualize the biological processes that are related to cancer, but also the capability for classifying samples and predicting patient prognosis. This work catalyzed numerous other applications of MALDI IMS to the field of cancer.
One recent application to cancer research is the determination of human epidermal growth factor receptor 2 (HER2) status in cancer tissue.199–200 HER2 expression characterizes a population of patients that have a high likelihood to respond to treatment with trastuzumab (Herceptin). Since 2006, the American Society of Clinical Oncology (ASCO) and the College of American Pathologists (CAP) recommends that HER2 status be determined for all invasive breast cancer.201 The two tests for HER2 status, immunohistochemistry (IHC) and fluorescence in situ hybridization (FISH), are used effectively, but these methods suffer from subjective interpretation, high cost, and are time-consuming to perform. Using MALDI IMS, researchers have been able to characterize breast and gastric tumor HER2 status using mass spectral data acquired directly from the tissue section. Walch and colleagues constructed a model for the classification of breast tumors based on a set of 48 fresh frozen human breast tumors with predefined HER2 status confirmed with both IHC and FISH assays.199 This work resulted in a model that used 7 m/z values taken from the MALDI mass spectra to classify the tumor specimens based on HER2 status with a sensitivity of 83% and specificity of 92%. One of the proteins, m/z 8404, was identified as cysteine-rich intestinal protein 1 (CRIP1). Further analysis of the distribution of this protein in tumor tissue demonstrates the utility of this marker in HER2-positive breast cancer tissue. Figure 19 shows the MALDI ion images of CRIP1 which is clearly more highly expressed in the HER2 positive tumor tissue but is not found in HER2-negative tumors or the surrounding stroma.
Figure 19. Distribution of cysteine-rich intestinal protein 1 in HER2-positive breast cancer.

m/z 8404 (shown in red) is up-regulated in HER2-positive breast cancer tissues: optical microscopic images of the MALDI-IMS measured and subsequently H&E stained original tissue sections of a HER2-positive (A) and a HER2-negative (B) case. Immunohistochemistry for HER2 of the respective case on a consecutive serial section is shown on the very right side. Note the strong immunoreactivity (score 3+) of the positive case (A), while the reaction in the negative case indicates a score 0 (B). The visualization of m/z 8404 by MALDI-IMS (shown in red) shows up-regulation in HER2-positive tissues (A) and clearly shows that this spectral feature is specific for cancer cells in this HER2-positive case but absent in cancer cells of the HER2-negative case (B). The spectral feature of m/z 6225 (shown in yellow) is an example specific for cancer cells but not distinguishing between HER2-positive and HER2-negative tissues and is therefore present in both cases (A and B); m/z 4969 (shown in blue) is a m/z species specific for tumor stroma and thus also present in both cases (A and B). Scale bars ) 1 mm. Reprinted with permission from Reference 199. Copyright 2010 American Chemical Society.
In a more recent study, HER2 status was also predicted in gastric tumors as well as breast cancer using the same algorithm.200 The general MALDI IMS classifier for breast and non-breast cancer HER2 status has a sensitivity of 78% and a specificity of 88%. The specific molecular species that are predictive of HER2 status are conserved across multiple tissue types and are useful for the prediction of HER2 status in both breast and gastric cancers. More recent results in the area of gastric cancer have employed MALDI IMS to distinguish early-stage and late-stage cancer patients into different prognostic groups.202 This seven protein signature identified the overexpression of proteins HNP-1 and S100-A6 as an indicator of poor prognosis. MALDI IMS was also used to identify the proteins COX7 A2, TAGLN2, and S100-A10 as prognostic markers in Barrett's adenocarcinoma. Interestingly, the over expression of S100-A6 and S100-A10 have previously observed in late-stage glioma patients198 and even more recently in metastatic papillary thyroid carcinoma (PTC)203 using MALDI IMS. These studies show that high expression levels of S100-A6 was correlated with poor prognosis for glioma patients and also metastatic potential in PTC. The role of S100-A6 in cancer is not fully known; however, it is clear from these studies that the presence of this protein in cancer is correlated with more aggressive disease states.
MALDI IMS has been applied to a number of other cancer types in a similar manner to that just described for breast and gastric cancers. For example, MALDI IMS was applied to the classification of papillary non-invasive bladder cancer. In 2004, the classical method of grading papillary non-invasive bladder cancer into three diagnostic groups (G1, G2, and G3) was replaced in favor of a system consisting of two groups (low grade and high grade) due to a high degree of interobserver variability with the old method. This policy change led to difficulty in reassigning previously categorized tumors into two rather than 3 groups, particularly for the G2 cases. In this study, MALDI IMS was proposed as means to resolve discordance in these types of reassignments. In G2 tumors graded as high grade or low grade by two independent pathologists, MALDI IMS classification achieved 87.5% and 78.3% accordance, respectively.204 Another recent example of the application of MALDI IMS to the diagnosis of cancer is the distinction of primary tumors from metastatic tumors in terms of the origin of the disease. For a large number of patients, the primary tumor cannot be identified. It was demonstrated that MALDI IMS can distinguish metastatic liver cancer from a set of six primary adenocarcinomas from different organs (esophagus, breast, colon, liver, stomach, and thyroid). Further analysis of the MALDI IMS data also demonstrated that metastatic colon cancer tumors in the liver could be classified successfully using a classifier constructed from primary colon cancer tumors.205
Molecular analysis of tumor margins of clear cell renal carcinoma (ccRCC) has been performed using MALDI IMS.206 Due to a statistically high incidence of recurrence, it has been suggested that the conventional methods of determining the margin of excised tumors based on histological examination may not adequately describe the boundary between normal and diseased tissue. The results of these experiments indicate that there are protein signatures in the tumor microenvironment surrounding the tumor, in cells outside the histological margin that are molecularly abnormal although normal to classical histology. For example, proteins involved in the mitochondrial electron transport system were under-expressed in the ccRCC tumor. These studies found that this pattern of under-expression was present in the histologically normal tissue surrounding the tumor and relatively far removed from the histologically determined margin. This molecular influence of the tumor outside the histological margin may be the result of secretions of exosomes and proteins or micro-foci of tumor cells that have either infiltrated the adjacent normal tissue. It is suggested that there is a population of cells in the tumor microenvironment that are undergoing malignant transformation at some level, but have not yet affected the overall histology of the cell. Further investigations are needed to better understand this invasive process.
Methodologies for the analysis of FFPE tissues have also been useful in the study of cancer samples. This approach combines in situ tryptic digestion of proteins with MALDI analysis to analyze the peptide fragments liberated form the tissue.37,57,207 One of the first applications of this technology reported was in the analysis of tissue microarrays (TMA) of biopsies from adenocarcinoma and squamous cell carcinoma.38 Using automated spotting instruments, an array of trypsin was deposited onto a tissue microarray that contained 112 needle core biopsies from lung tumor patients. The spatial resolution, established by the spacing for the trypsin/matrix spot placement, was 250 μm that resulted in approximately 25 separate analysis from each core. The samples were imaged using MALDI IMS, and a subset of the resulting data were used to build a model for classification of the biopsies using a support vector machine algorithm. The model was validated with the remaining sample set. Comparison of the results from MALDI IMS with results from pathology, showed 100% correlation for patient classifications for both adenocarcinoma and squamous cell carcinoma. Figure 20 shows partial results from the analysis of TMA cores using MALDI IMS; the class imaging results are compared with the histology results. Further work was performed to identify as many as 50 proteins from these tissues using a MALDI TOF/TOF. Although the total number of patient samples was small in this example, this study demonstrates the feasibility of the approach for the discovery of new diagnostic molecular signatures of cancer. More recent work combines MALDI ion mobility measurements with PCA to discriminate pancreatic tumor biopsies from normal tissue.208 The increased specificity of ion mobility separation allowed for better discrimination of peptide signals from interference peaks, ultimately leading to more confident identifications of the proteins from the tissue and less background in the resulting ion images. PCA analysis demonstrated that the data can distinguish cancer from normal tissue based upon the molecular profile.
Figure 20. MALDI IMS of FFPE Tissue Microarrays.
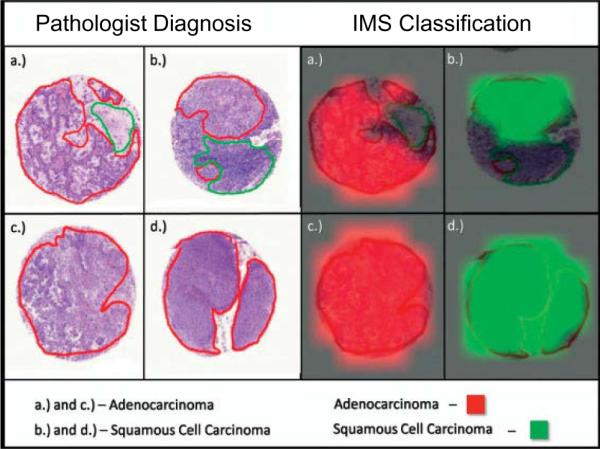
Visual representation of the statistical classification of four biopsies compared to the marking and diagnosis based on histology. Cancerous regions are outlined in red in the histological sections, whereas normal tissue is outlined in green. Reprinted with permission from Reference 38. Copyright 2008 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Recently, techniques for the analysis of FFPE samples were applied to the study of skin cancer. Spitzoid neoplasms encompass a spectrum of disease states that range from benign melanocytic lesions called Spitz Nevi (SN) to Spitzoid Malignant Melanoma (SMM). These two diagnoses represent two diseases that have quite different prognoses; and therefore, accurate diagnosis is critical to guide the appropriate therapy decision. The standard of clinical diagnosis of SN and the differentiation from SMM is examination of the histopathology and applying established criteria. In spite of the vastly different prognosis of these diseases, a large group of melanocytic lesions have conflicting histopathologies, making it difficult to distinguish between SN and SMM. Therefore, there exists a remarkable discordance concerning the correct classification among expert dermatopathologist. Due to the potential to misclassify a potentially deadly disease as benign, pathologists are incentivized to very conservative diagnoses to avoid legal risk, classifying ambiguous lesions as malignant melanoma. Histology-directed analysis was also employed to characterize the molecular changes observed when comparing SN and SMM in FFPE tissue samples.33 A total set of 114 unequivocal archived FFPE samples, including 56 SN and 58 SMM were analyzed in this study. The samples were randomly assigned to either a training (26 SN/25 SMM) or testing cohort (30 SN/33 SMM). Serial sections of each sample were prepared, one of which was H&E stained. Annotation of the stained section was performed by a board-certified dematopatholgist, then digestion and application for matrix was performed on the unstained section (Figure 21a). MALDI analysis was performed at each region of interest, followed by statistical analysis of the data and construction of a classification model using the training set data which was then validated using the validation cohort. Using this analytical approach, a classification model that consisted of 5 differentially expressed peptides was validated (Figure 21b). Using this signature measured from the tumor, SN was classified correctly in the validation cohort with 97% sensitivity and 90% specificity. This study demonstrates that MALDI IMS is a viable technology for the objective classification of SN with equivocal histopathology and may lead to better understanding of the disease at the molecular level. Histology-directed analysis has been recently applied to the study of other cancers, including Wilms tumors,32,209 lung,210 kidney,211 and breast cancers.212
Figure 21. Analysis of Spitzoid Lesions.

A) Spitzoid melanoma (Top) and Spitz nevi (Bottom) were annotated for areas of tumor (blue) and dermis (yellow) and targeted for on-tissue tryptic digestion and mass spectrometry. B) Average spectra from tumor regions from Spitzoid melanoma (red) and Spitz nevi (green). Two of the 5 peptides that are part of the classifier are marked with an asterisk. Adapted from Reference 33.
5.3. Drug Distribution
The application of MALDI IMS to the imaging of pharmaceutical compounds has been of great interest since the introduction of the technology. During drug development, multiple imaging technologies are used to fully characterize candidate drugs with some of these requiring the synthesis of drug analogues that are labeled to facilitate detection. Although this approach can be effective, it often comes at great cost and requires a significant amount of time. Furthermore, imaging techniques that require labeling of the compound often lack the molecular specificity to differentiate both the parent drug and any metabolized forms of the drug in the same experiment. MALDI IMS is an attractive alternative technology because it measures the unlabeled compound and can be used to simultaneously monitor multiple molecular species, including both endogenous and exogenous molecules in a single experiment. The standard approaches for the analysis of drug distribution, such as quantitative whole-body autoradiography (QWBA), are not practical for the measurement of multiple analytes in a single experiment.
Although some early work with the MALDI profiling approach demonstrate the analysis of drugs from tissue sections,213 the first application of whole section imaging of drugs administered systemically to an animal was reported in 2003.214 This study established that drugs present in biological tissue could be measured from intact tissue sections, and furthermore, it established that MALDI IMS measurements could be quantitatively correlated with dose. Later it was shown that drug distribution measurements made using MALDI IMS result are highly correlated to those generated by QWBA.215 The introduction of whole-body animal imaging by MALDI IMS was an important step forward for the application of the technology to problems in drug development.56,216 These studies established the efficiency of imaging whole body sections of rodents for the analysis of both proteins and drugs.56 These data were used to reconstruct a molecular image of the entire rat. In the same study, imaging of the antipsychotic drug olanzapine (OLZ) and major metabolites (N-desmethylolanzapine and 2-hydroxymethyl olanzapine) was performed. After a single dose of OLZ, the distribution of the OLZ and metabolized forms could be determined at time points ranging from 2 to 6 hours post dose. Figure 22 shows the MALDI images of OLZ and metabolites 6 hours post-dose alongside the optical image of the whole-body section annotated for reference. At this time point, OLZ has already started to clear from the target organs, brain and spinal cord, and is largely metabolized and concentrated in the liver and bladder. Likewise, the metabolites were measured simultaneously and were shown to localize in the lungs, liver, and bladder. Overall, the metabolized form of the drug comprised 28% of the total signal that could be attributed to the OLZ. Previous studies regarding the metabolism of OLZ were found to support the MALDI imaging results. In a subsequent study, OLZ imaging was performed using MALDI FTICR MS.128 The high resolution capability of FTICR allows OLZ to be measured distinctly from other interfering ions (Figure 14) and provides structural confirmation through exact mass measurement. Accurate mass measurements have proven to be valuable in the analysis of low molecular weight compounds such as drugs, and these methods continue to be developed on a variety of instrumentation.217–218
Figure 22. MALDI IMS of drug and metabolite distribution at 6 h post-dose in a whole rat sagittal tissue section by a single IMS analysis.
Optical image of a 6 h post OLZ dosed rat tissue section across four gold MALDI target plates (A). Organs outlined in red. MS/MS ion image of OLZ (m/z 256) (B). MS/MS ion image of N-desmethyl metabolite (m/z 256) (C). MS/MS ion image of 2-hydroxymethyl metabolite (m/z 272) (D). Bar, 1 cm. Reprinted with permission from Reference 56. Copyright 2006 American Chemical Society.
MALDI IMS has since been used in a variety of studies that are designed to investigate the distribution of drugs in tissue specimens. For example, it has been applied to the imaging of MRI contrast agents in both fresh as well fixed tissues.219 The drug, B22956/1, was administered in vivo and the liver was removed and subjected to imaging by MALDI. The use of MRI contrast agents allowed the MALDI IMS results to be validated by the MRI result. Furthermore, the results were further validated using inductively coupled plasma atomic emission spectroscopy (ICP-AES) to quantitatively measure this agent, a Gd-containing compound. The results demonstrated that the three analytical techniques generated data that were in agreement although from different molecular perspectives, and the sensitivity of MALDI imaging was at the same level as that obtained using the other techniques.
Direct analysis of pharmaceutical tablet formulations was studied using MALDI IMS to assess the homogeneity of active drug compounds in the tablet and to improve the manufacturing process.220 MALDI IMS has also been applied to the study of implanted medical devices to study the controlled release of drugs by these products.221–222 In one example, drug-eluting coronary stents (CYPHER® and NEVO®) were characterized.222 Restenosis, the narrowing of the artery after the placement of a stent, is the a major problem that can arise following a stenting procedure. Drugs that inhibit restenosis are incorporated into the structure of the stent, embedded in a controlled release polymer coating on the stent surface. The purpose of this study was to better understand the release of the drug, sirolimus, from the stent and the distribution of the drug into the tissue adjacent to the stent. The stents were implanted into porcine subjects, and explanted at time points of 1, 8, and 30 days. It was found that approximately 50% of the drug remained in the device at 14 days and the amount of drug in the stent was below the limit of detection at 30 days. These data align with previous studies for the controlled release of sirolimus using conventional methods for the study of drug release and metabolism.
MALDI IMS has been found useful for forensic applications that monitor the use of illicit drugs in human hair. In one study, the incorporation of methamphetamine (MA) was monitored in a growing human hair.223 A single human hair taken from a chronic MA user was sectioned lengthwise, coated with CHCA, and analyzed using MALDI FTICR MS. The analysis produced a profile of the pattern of use of the drug. This study clearly demonstrated that MALDI IMS has the potential to offer important forensic insight into the investigation of patterns of drug use.
Solvent-free matrix application has been applied for the analysis of small molecules by MALDI IMS.224–226 The analysis of clozapine and 4-bromophenyl-1,4- diazabicyclo(3.2.2)nonane-4-carboxylic acid (BDNC) has been reported in rat tissues. Unlike most analytes, the detection of these two compounds is compromised when using a more conventional application of matrix that is applied in a solvent system to assist in the extraction and recrystallization of the matrix. In this case, the matrix, finely ground CHCA, was applied to the sample using a sieve with the excess matrix removed by a stream of oxygen-free nitrogen.224 It was found that BDNC was detected in higher concentration in the white matter of the brain, and the identity of compound was verified by direct tandem mass spectrometry from the tissue section.226 In order to further validate the results obtained by MALDI IMS, regions of the tissue were dissected using laser microdissection and the amount of drug in those regions were measured using a quantitative LC/MS method. It was determined that the drug amount was 2-fold higher in the white matter of the brain relative to neighboring grey matter and correlates well with the findings from the MALDI IMS results. Solvent-free application methods have been recently applied to the analysis of positron emission tomography ligands, raclopride and SCH 23390 in rat brain tissue sections.225 Quantitative data was generated by applying the standard compound to an adjacent vehicle control section during the same experiment and comparing the dose tissue to a standard curve derived from the doped tissue. The sensitivity of this assay is not sufficient to measure these PET ligands at concentrations low enough to avoid oversaturation of the D2/D3 receptors that they target. However, IMS can be useful in the discovery and development of novel ligands even if not effective for receptor binding.
MALDI IMS has been applied to the study of drug distribution in lung tissue.130,133,139,227 The distribution of a tiotropium, a bronchiodialator used in the management of chronic obstructive pulmonary disease that is delivered via inhaled delivery, was imaged in lung tissue to better understand the distribution in lung tissue of rats.139 The transport of tiotropium out of the central conducting airways was observed in a relatively uniform manner. Using LC/MS/MS methods, the concentration of tiotropium in lung tissue 15 minutes after inhalation was determined to be 8.9 ± 3.0 mmol/g, corresponding to 8% of the lung dose. These results correlate well to the quantitative results determined by MALDI IMS and are explained by the rapid adsorption of tiotropium in lung tissue. Similar methodologies were applied to study the distribution of another inhaled brochiodialator, ipratropium, in human biopsy tissue.130 This study demonstrates that the technique is applicable to drug distribution in human tissues at physiologically relevant concentrations. Ipratropium bromide was administered to patients 10 minutes before bronchoscopy, during which bronchial biopsies were collected. The lung tissue was analyzed by MALDI IMS, and it was found that the higher level of drug signal was associated with areas rich in smooth muscle and in areas where evidence of inflammation was present. Ipratropium concentrations were determined to be between 3 and 30 nM in lung tissue. It is not known at this time whether the localization of the drug is related to the binding of the drug to target receptors, or if there is a secondary mechanism of transport that also contributes to the localization of the drug in these tissues. However, these findings were consistent with the known expression of the muscarinic receptors (M2 and M3) in the smooth muscle tissue. Reliable assays for the assessment of drug levels in human tissue biopsies could contribute to clinical indices that would be of great value in titrating drug therapies for individual patients.
5.4. Neuroscience
The study of the brain using MALDI IMS can be traced back to some of the first experiments performed using the technology. The brain has been an often used organ in the development of methodologies for MALDI IMS because the inherent bilateral symmetry in normal brains of model animals provides an internal measure of quality control in the ion image and for its extensive substructure. Many of the seminal papers that describe the development of the technology17 and establish its utility for biological and clinical applications,119,198 do so using brain as the model organ. One early example used a histology-directed approach to identify phenotypic markers of Parkinson's disease in an experimental animal model treated unilaterally with 6-hydroxydopamine.120 Protein expression changes in the striatum were compared between the dopamine depleted and normal hemispheres in the same section. The expression profiles reveal several statistically significant protein changes in the dopamine depleted striatum as well an increase in protein acetylation. The changes were decreased upon treatment with l-Dopa, further indicating that the observed protein changes are disease related. Another example is the examination of protein distribution in the brain to study patterns of myelination.228 This work was performed to develop a better understanding the changes in myelin composition and the changes that these proteins undergo in demyelination mechanisms of diseases like multiple sclerosis. The detection of myelin by MALDI IMS was reported in 2005 as part of the work demonstrating 3-dimensional MALDI IMS.175 The myelin basic protein isoforms, MBP 8 and MBP 5, were imaged and the distributions were validated using immunohistochemical stains.
Neuropeptide discovery and the mapping of their spatial distribution by MALDI IMS has been the subject of several investigations.54,74–75,125,229–235 Much of this work was enabled by the methodologies for neuropeptide analysis developed by Sweedler and coworkers.236–237 An early study looked at the neuropeptides distribution in the crustacean Cancer borealis.229 A number of peptide isoforms belonging to 10 different neuropeptide families were investigated. It was found that certain peptide isoforms had distinctly different distributions that the other members of the neuropeptide family. A following study extending this work in the same animal model to include the profiling and imaging of lipids and neuropeptides in 3-dimensions.54 More recently, MALDI IMS was used to identify a novel Tachikinin-related peptide (TRP) in the crustacean, Callinectes sapidus.233 This novel peptide was shown in this study to regulate feeding in this crustacean. TRP was also found to co-localize with another previously known neuropeptide with the same function. Functional electrophysiology studies that tested the peptide's ability to stimulate the gastric mill (food chewing) rhythm and the pyloric (food filtering) rhythm corroborated these finding. Another recent study has examined neuropeptide distributions in FFPE tissues of crustaceans.230 MALDI IMS was used to confirm the presence of several peptide families (tachykinin, allatostatin, periviscerokinin, pyrokinin, FLRFamide, and neuropeptide F) in the central complex of an insect brain.232 The expression and distribution of each was found to be uniquely distributed in the central complex, and furthermore each showed differing expression at different stages of embryonic development. This study was an important step in the molecular characterization of the developing central complex.
MALDI IMS has been used to study the opioid peptide changes in the brain that are associated with l-DOPA-induced dyskinesia (LID).74–75,231 This debilitating disorder is common in patients that are receiving treatment for parkinson's disease. Previous studies have established by the measurement of mRNA that the levels of prodynorphin and and preproenkephalin are increased in the striatum of animal models with LID. MALDI IMS was used to characterize the products of the opiod precursors and the distribution of these peptides in the striatum. It was found that a number of dynorphins and enkephalins were detectable by MALDI IMS. In particular it was found that dynorphin B, α-neoendorphin, substance P, and PEnk (220–229) had elevated abundance in the dorsolateral striatum of high-dyskinetic animals when compared to low-dyskinetic and control animals. The expression levels of these peptides are correlated with LID severity. A novel des-tyrosine dynorphin peptide was also characterized in the same study. Removal of the N-terminal tyrosine is known to reduce binding to the opioid receptor and is an important processing step for the modulation of biological activity that may have important applications for the remediation of maintenance of LID. In a related study, elevated levels of dynorphin B, α-neoendorphin, and the major metabolite of these peptides Leu-Enk-Arg localized in the lateral sustatia nigra and were correlated with higher levels of LID.75 Figure 23 shows a statistically significant expression increase in the level of dynorphin B in the lateral substantia nigra in the high-dyskinetic animals. The authors were able to demonstrate by MALDI IMS that the presence of dynorphin B and Leu-Enk-Arg were anti-correlated at specific sites, suggesting that the dynorphin B has been released in these areas and is metabolized by enzymatic processes. Clearly, MALDI IMS will continue to play an important role in understanding the molecular mechanisms underlying the motor impairment and improvement observed in LID.
Figure 23. Elevated dynorphin B in the lateral substatia nigra of high dyskinetic mice.
(A) Average MS traces show a higher dynorphin B peak in high dyskinetic (HD) group (average MS ± SEM, n = 5 and 5 in low dyskinetic (LD) and HD group, respectively). (B) MALDI IMS of DynB ion images of animals from control (n = 4), LD (n = 5) and HD (n = 5) group. The HD group display clearly elevated DynB in both high medial and lateral substatia nigra (SN) (arrows). (C) The increase in DynB peak intensity was most pronounced in the lateral tier of SN. Values are expressed as mean % peak area of intact SN ± SEM (p<0.01 HD vs. LD, HD vs. control). (D) Correlation of Dyn B peak intensities and the cumulative dyskinesia score. Scale bar = 2 mm. Reprinted with permission from Reference 75. Copyright 2011 Ljungdahl et al.
MALDI IMS has been used to study changes in lipid expression that occur as a result of traumatic brain injury.238–242 Lipids are important structural components of the brain, and are present in great abundance. A significant disruption to the normal brain chemistry, such as that caused by brain injury is reflected by changes in the lipid profiles in the damaged tissue. In particular, lipids are subject to oxidative modification.238 Ischemic brain injury in rats studied by MALDI IMS revealed that PC (16:0/18:1) was reduced, and that complementary to this reduction, lyso-PC (16:0) increased. Figure 24 shows the result of the analysis of the ischemic brain by MALDI IMS, and the corresponding changes in lipid signals as a function of the position on the tissue. It is known that the hydrolysis of PC (16:0/18:1) at the sn-2 position is catalyzed by phospholipase A2 (PLA2), generating a lyso-PC and a free fatty acid. However, Western blot analysis did not show significant induction of PLA2 in the ischemic region. Further studies are necessary to confirm if lyso-PC (16:0) detected is produced by the hydrolysis of PC (16:0/18:1) and if PLA2 plays a role in the apparent hydrolysis. In a separate study, MALDI IMS has been used to study changes in the ganglioside distribution of ischemic brains in mouse.241 Glangliosides, a glycosphingolipid abundant in the cell membranes of the central nervous system, are known to be involved in the development diseases in the brain. The results showed that, following injury, that unique distribution patterns of GM1, GD1, and GT1b were observed that correlated with the area of tissue damage. Interestingly, the results also showed that GM2 and GM3 were transiently induced in the tissue bordering the infarcted region. More recent MALDI IMS studies examined the changing distribution of ceramides240 and N-acylphosphatidylethanolamines242 in ischemic brain tissue. Taken together, there is significant evidence that the remodeling of neuronal lipids is important in the brains response to traumatic injury, and further work will seek to connect these findings to the mechanism of disease.
Figure 24. Lipid expression changes in ischemic rat brain.
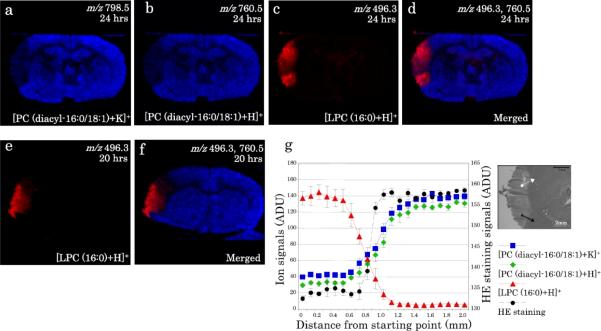
Distributions of PC (16:0/18:1) and LPC (16:0) in the ischemic brain. (a–d) Distributions of [PC (16:0/18:1)+K]+ (a), [PC (16:0/18:1)+H]+ (b) and [LPC (16:0)+H]+ (c) in the ischemic brain at 24 h after injury as well as a merged image (d) are presented. In (d), ion signals of [PC (16:0/18:1)+H]+ and [LPC (16:0)+H]+ are indicated by blue and red, respectively. (e, f) Distributions of [LPC (16:0)+H]+ (e) in the ischemic brain at 20 h after injury as well as a merged image (f) are presented. In (f), ion signals of [PC (16:0/18:1)+H]+ and [LPC (16:0)+H]+ are indicated by blue and red, respectively. (g) The intensities of each ion were plotted across the border from the ischemic area to the normal area, as indicated. The inset indicates the measured area on the adjacent section stained by H&E. To indicate the border of the injured area, the H&E staining signals at the corresponding position were also quantified and plotted on the graph. Data were corrected from five adjacent zones and the normalized intensities are presented as means ± 1 SD. Reprinted with permission from Reference 239. Copyright 2010 IBRO. Published by Elsevier, Ltd.
6. Perspective and Concluding Remarks
MALDI IMS is a unique and powerful method for the analysis of biological and clinical samples. Leveraging the speed, sensitivity, and molecular specificity of modern mass spectrometers to create molecular images, IMS is an analytical imaging tool that provides unique and insightful views of biologically and clinically important processes. MALDI IMS data sets are enormously complex, being comprised of thousands of unique ion images. These images can be registered to histology images, correlating molecular information and adding a new dimension of understanding to the histopathology of the tissue. Unlike most clinically useful imaging methods, mass spectrometry can measure endogenous analytes without the requirement of target specific molecular labeling reagents. This capability provides the means for highly multiplexed analyses that can be used for both targeted/diagnostic assays and as a platform for the discovery of new biology. MALDI IMS is capable of analyzing numerous analyte classes, including proteins and peptides as well as both endogenous and exogenous metabolites such as lipids and drugs. The technology for MALDI IMS has developed significantly in the previous 15 years, with numerous commercial mass spectrometers now equipped for MALDI IMS and having advancements through both software and hardware. The speed of acquisition of MALDI IMS has increased by orders of magnitude in the past decade, making it possible to acquire images of typical samples in just a few minutes. Despite the enormous capabilities of MALDI IMS technology, challenges remain in the areas of higher spatial resolution, higher molecular weight compounds, improved quantitation, higher sensitivity, and more global molecular coverage. Furthermore, additional requirements such as reproducibility, platform stability, and ease of use are necessary bring it to routine use for clinical diagnostics.
Sensitivity remains a never-ending challenge as we learn of the intracacies of the molecular events within cells. The applications described in the literature to date demonstrate the use of the technology for the analysis of only a relatively small fraction of the molecules in the sample. Limitations of dynamic range for global analyses can be overcome by moving to more targeted analyses; however, there are limits to the number of biologically significant analytes that can be measured. Some of the crucial chemical changes in human disease occur in either a small population of cells, or in relatively low copy numbers per cell, placing them below current detection limits. This situation is exacerbated for cases when higher spatial resolution is achieved through smaller pixels. New methods for the preparation of samples that are optimized for specific analytes, more sensitive mass spectrometers, and better instrumental techniques to perform targeted analysis or enrichment in the gas phase are needed to further improve the sensitivity of IMS.
The most usable mass range for MALDI IMS, with current instrumentation, is below 30 kDa and to date only a few examples of MALDI IMS of proteins above 100 kDa have been reported. Advances in detector technology that reduce or eliminate mass bias and novel methods of sample preparation that focus on enhanced ionization efficiency of large analytes are needed to substantially improve this issue. In addition, the resolving power of current instrument for proteins limits the information that can be obtained. For example, the difference of a single methylation or phosphorylation can have enormous biological impact, but for intact high molecular weight proteins, such a measurement would be difficult. Although modified protein forms can be imaged by in situ digestion and high-resolution analysis using FT-ICR, significantly improved mass spectral resolution for these species is needed to image intact posttranslationally modified proteins.
The clinical use of MALDI IMS and its implication on health remains an enormous potential especially for anatomic pathology. In order for this potential to be realized, further development and validation of this technology must be performed. First, the preparation of samples has in the past occupied a great deal of time and effort. Sample preparation technologies, including spotting robotics, spray coaters, and matrix pre-coated slides, have developed along with protocols for use. However, little work has been done to standardize these technologies for use across platforms and laboratories. Before diagnostic assays can be performed, rigorously validated methodologies must be developed for a sample set of statistically significant size to characterize the performance of the test. Second, methods for the identification of analytes must be improved. Validation of the analytical methodology across platforms will require that the specificity of the assay for the target analyte is confirmed. Identification of the analyte in situ is an important and final confirmation that the assay has adequate specificity. Furthermore, it may be necessary in cases where the assay cannot be performed at the appropriate scales by mass spectrometry that the assay is transferred to a more traditional, immuno-based platform. Development of antibodies will require that the analyte is accurately identified. Strategies that permit rapid and accurate identification and structural confirmation of the analytes discovered to be important using MALDI IMS need to be further developed. Third, there must be improvements in quantitation. While in many cases the qualitative assessment of the molecular information will suffice to distinguish disease states based on the presence and absence of specific ions, there also are applications where more subtle changes in abundance are important or cases where absolute quantitation is required in order to make a clinical decision. Improved methods for sample preparation, including the addition of standards to the tissue, better instrument dynamic range, and algorithms for the analysis of the data must be developed to improve quantitation by MALDI IMS. Finally, vendors must create and support a complete, fully integrated, system for MALDI IMS. While there are some vendors working toward that end, there is not yet a complete technology `solution' that includes a fully integrated sample preparation, analysis, and informatics platform from a single vendor.
Despite the challenges and current limitations, MALDI ISM has seen an exponential growth in both technology development and applications over the past 5–6 years and promises to continue to do so. MALDI IMS now joins a powerful suite of imaging modalities such as microscopy, MRI, PET, radiography, ultrasonography, as well as many others. IMS brings to this suite its exquisite molecular specificity and broad analyte coverage for the analysis of cells and tissue. Extensive proofs of concept applications have demonstrated the capabilities of MALDI IMS for biological discovery and to provide clinical value. We believe that as the technology advances, MALDI IMS will greatly accelerate in its use in biological research and will further take on its greatest role in the chemical and pathology worlds as we move increasingly close to the realization of personalized medicine.
Acknowledgements
This work was supported by grants from the National Center for Research Resources (5 P41 RR031461) and the National Institute of General Medical Sciences (8 P41 GM103391) from the National Institutes of Health. The authors acknowledge M. Reid Groseclose, Jamie Allen, and Erin H. Seeley for contributing data for Figures 3, 13, and 21, respectively. The authors further acknowledge Angus C. Grey, Rosalie K. Crouch, Yiannis Koutalos, Kevin L. Schey, and Zsolt Ablonczy for contributing data for Figure 18.
7. Abbreviations
- AMD
Age-Related Macular Degeneration
- AQP0
Aquaporin-0
- ASCO
American Society of Clinical Oncology
- A2E
bis-Retinoid A2E
- BDNC
4-bromophenyl-1,4- diazabicyclo(3.2.2)nonane-4-carboxylic acid
- CAP
College of American Pathologists
- CASI
Continuous Accumulation of Selected Ions
- ccRCC
Clear Cell Renal Carcinoma
- CHCA
α-cyano-4-hydroxycinnamic acid
- CMC
Carboxymethylcellulose
- CRIP1
Cysteine-Rich Intestinal Protein 1
- CT
Computed Tomography
- DAN
1,5-Diaminonaphthalene
- DHA
2,6-dihydroxyacetophenone
- DHB
2,5-Dihydroxybenzoic acid
- ESI
Electrospray Ionization
- FFPE
Formalin Fixed Paraffin Embedded
- FISH
Fluorescence in situ Hybridization
- FT
Fourier Transform
- GC
Gas Chromatography
- H&E
Hemotoxylin and Eosin
- HER2
Human Epidermal Growth Factor Receptor 2
- HPLC
High Performance Liquid Chromatography
- ICA
In-Cell Ion Accumulation
- ICR
Ion Cyclotron Resonance
- ICP-AES
Inductively Couple Plasma Atomic Emission Spectroscopy
- IHC
Immunohistochemistry
- IMS
Imaging Mass Spectrometry
- ITO
Indium-Tin Oxide
- LC
Liquid Chromatography
- LID
L-Dopa-Induced Dyskinesia
- LIT
Linear Ion Trap
- MA
Methamphetamine
- MALDI
Matrix Assisted Laser Desorption/Ionization
- MR
Magnetic Resonance
- MRM
Multiple Reaction Monitoring
- MS
Mass Spectrometry
- OCT
Optimal Cutting Temperature
- OLZ
Olanzapine
- PCA
Principle Component Analysis
- PC
Phosphatidylcholine
- PET
Positron Emission Tomography
- PLA2
Phospholipase A2
- PPS
3-[3-(1,1-bisalkoxyethyl)pyridine-1-yl]propane-1-sulfonate
- PS
Phosphatidylserine
- PTC
Papillary Thyroid Carcinoma
- Q-TOF
Quadrupole Time-of-Flight
- QWBA
Quantitative Whole-Body Radiography
- SA
3,5-Dimethoxy-4-hydroxycinnamic acid
- SDS
Sodium Dodecyl Sulfate
- SIMS
Secondary Ion Mass Spectrometry
- SMM
Spitzoid Malignant Melanoma
- SN
Spitz Nevi
- TCDI
1,1'-Thiocarbonyldiimidazole
- TFA
Trifluoroacetic Acid
- THAP
4,6-trihydroxyacetophenone
- TMA
Tissue Microarray
- TMPP
N-Succinimidyloxycarbonyl-methyl)-tris-(2,4,6-trimethoxyphenyl) phosphonium bromide
- TOF
Time-of-Flight
- TRP
Tachikinin-Related Peptide
- 2,4-DNPH
2,4-dinitrophenylhydrazine
- 3-HPA
3-hydroxypicolinic acid
- 3-MoSA
3-Methoxysalicylamine
Biographies

Jeremy L. Norris earned his B.S in Chemistry from the University of Tennessee in 1998 and his Ph.D. from Vanderbilt University in 2003 and has been working professionally in mass spectrometry, separation science, and organic chemistry since that time. Until 2011, Dr. Norris was Vice President of Research and Development at Protein Discovery, Inc. During his 6 year tenure at Protein Discovery, Dr. Norris developed and commercialized more than 12 products in the area of mass spectrometry and protein research including two instrument platforms for preparative protein separations. Currently, these instruments are installed in more than 150 laboratories worldwide and provide key capabilities in the area of top-down proteomics. Dr. Norris returned to Vanderbilt University in 2011 as Managing Director of the National Resource for Imaging Mass Spectrometry where his current research interests include the development of novel instrumentation, methodologies and applications of MALDI imaging mass spectrometry.

Richard M. Caprioli is Professor of Biochemistry, Chemistry, Medicine and Pharmacology at Vanderbilt University. Dr. Caprioli received his B.S. in 1965 from Columbia University in New York, NY, and his Ph.D. in 1969 in Biochemistry, also at Columbia University. Dr. Caprioli has been President of ASMS; has been Editor-in-Chief of the Journal of Mass Spectrometry since 1990; and is currently serving a 3-year term on the Board of Directors of HUPO. Dr. Caprioli received the Thomson Medal Award from the International Mass Spectrometry Society in 2003, the Field and Franklin Award from the American Chemical Society in April, 2006, and the HUPO Distinguished Achievement Award in Proteomic Sciences for 2010. Professor Caprioli's general research interests lie in discovery of temporal and spatial processes in biological systems using mass spectrometry. Recent work involves the development of Imaging Mass Spectrometry, a technology whereby molecular images of peptides, proteins, drugs, and other compounds are localized in tissue sections with molecular weight specificity. He has published over 300 peer-reviewed articles.
9. References
- (1).Goodwin RJA, Iverson SL, Andren PE. Rapid Commun. Mass Spectrom. 2012;26:494. doi: 10.1002/rcm.6125. [DOI] [PubMed] [Google Scholar]
- (2).Kreye F, Hamm G, Karrout Y, Legouffe R, Bonnel D, Siepmann F, Siepmann J. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.04.017. [DOI] [PubMed] [Google Scholar]
- (3).Schoenheimer R, Rittenberg D. Science. 1935;82:156. doi: 10.1126/science.82.2120.156. [DOI] [PubMed] [Google Scholar]
- (4).Schoenheimer R. The Dynamic State of Body Constituents. Harvard University Press; Cambridge, MA: 1942. [Google Scholar]
- (5).Schoenheimer R, Rittenberg D. Physiol. Rev. 1940;20:218. [Google Scholar]
- (6).Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- (7).Karas M, Hillenkamp F. Anal. Chem. 1988;60:2299. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- (8).Ideker T, Galitski T, Hood L. Annu. Rev. Genom. Hum. G. 2001;2:343. doi: 10.1146/annurev.genom.2.1.343. [DOI] [PubMed] [Google Scholar]
- (9).de Godoy LMF, Olsen JV, Cox J, Nielsen ML, Hubner NC, Frohlich F, Walther TC, Mann M. Nature. 2008;455:1251. doi: 10.1038/nature07341. [DOI] [PubMed] [Google Scholar]
- (10).Picotti P, Bodenmiller B, Mueller LN, Domon B, Aebersold R. Cell. 2009;138:795. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).McDonnell LA, Heeren RMA. Mass Spectrom. Rev. 2007;26:606. doi: 10.1002/mas.20124. [DOI] [PubMed] [Google Scholar]
- (12).Chughtai K, Heeren RMA. Chem. Rev. 2010;110:3237. doi: 10.1021/cr100012c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Amstalden v. H. E. R., Smith DF, Heeren RMA. J. Chromatogr., A. 2010;1217:3946. doi: 10.1016/j.chroma.2010.01.033. [DOI] [PubMed] [Google Scholar]
- (14).Cornett DS, Reyzer ML, Chaurand P, Caprioli RM. Nat. Methods. 2007;4:828. doi: 10.1038/nmeth1094. [DOI] [PubMed] [Google Scholar]
- (15).Pacholski ML, Winograd N. Chem. Rev. 1999;99:2977. doi: 10.1021/cr980137w. [DOI] [PubMed] [Google Scholar]
- (16).Caprioli RM, Farmer TB, Gile J. Anal. Chem. 1997;69:4751. doi: 10.1021/ac970888i. [DOI] [PubMed] [Google Scholar]
- (17).Stoeckli M, Chaurand P, Hallahan DE, Caprioli RM. Nat. Med. 2001;7:493. doi: 10.1038/86573. [DOI] [PubMed] [Google Scholar]
- (18).Chaurand P. J. Proteomics. 2012;75:4883. doi: 10.1016/j.jprot.2012.04.005. [DOI] [PubMed] [Google Scholar]
- (19).Jungmann JH, Heeren RM. J. Proteomics. 2012;75:5077. doi: 10.1016/j.jprot.2012.03.022. [DOI] [PubMed] [Google Scholar]
- (20).Watrous JD, Alexandrov T, Dorrestein PC. J. Mass Spectrom. 2011;46:209. doi: 10.1002/jms.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Schwamborn K, Caprioli RM. Nat. Rev. Cancer. 2010;10:639. doi: 10.1038/nrc2917. [DOI] [PubMed] [Google Scholar]
- (22).Castaing R, Slodzian G. J. Microscop. 1962;1:395. [Google Scholar]
- (23).Liebl H. J. Appl. Phys. 1967;38:5277. [Google Scholar]
- (24).Vaeck LV, Struyf H, van Roy W, F. A. Mass Spectrom. Rev. 1994;13:189. [Google Scholar]
- (25).Vaeck LV, Struyf H, van Roy W, Adams F. Mass Spectrom. Rev. 1994;13:209. [Google Scholar]
- (26).Karas M, Kruger R. Chem. Rev. 2003;103:427. doi: 10.1021/cr010376a. [DOI] [PubMed] [Google Scholar]
- (27).Jaskolla TW, Karas M. J. Am. Soc. Mass Spectrom. 2011;22:976. doi: 10.1007/s13361-011-0093-0. [DOI] [PubMed] [Google Scholar]
- (28).Spengler B, Hubert M. J. Am. Soc. Mass Spectrom. 2002;13:735. doi: 10.1016/S1044-0305(02)00376-8. [DOI] [PubMed] [Google Scholar]
- (29).Zavalin A, Yang J, Caprioli RM. 60th ASMS Conference on Mass Spectrometry and Allied Topics; Vancouver, Canada. 2012. [Google Scholar]
- (30).Spraggins JM, Caprioli RM. J. Am. Soc. Mass Spectrom. 2011;22:1022. doi: 10.1007/s13361-011-0121-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Cornett DS, Mobley JA, Dias EC, Andersson M, Arteaga CL, Sanders ME, Caprioli RM. Mol. Cell. Proteomics. 2006;5:1975. doi: 10.1074/mcp.M600119-MCP200. [DOI] [PubMed] [Google Scholar]
- (32).Axt J, Murphy AJ, Seeley EH, Martin CA, Taylor C, Pierce J, Caprioli RM, Whiteside M, Lovvorn HN., 3rd. J. Surg. Res. 2011;170:112. doi: 10.1016/j.jss.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lazova R, Seeley EH, Keenan M, Gueorguieva R, Caprioli RM. Am. J. Dermatopathol. 2012;34:82. doi: 10.1097/DAD.0b013e31823df1e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Oswald-Richter KA, Beachboard DC, Seeley EH, Abraham S, Shepherd BE, Jenkins CA, Culver DA, Caprioli RM, Drake WP. J. Clin. Immunol. 2012;32:1129. doi: 10.1007/s10875-012-9700-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Burnum KE, Tranguch S, Mi D, Daikoku T, Dey SK, Caprioli RM. Endocrinol. 2008;149:3274. doi: 10.1210/en.2008-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Casadonte R, Caprioli RM. Nat. Protoc. 2011;6:1695. doi: 10.1038/nprot.2011.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Groseclose MR, Andersson M, Hardesty WM, Caprioli RM. J. Mass Spectrom. 2007;42:254. doi: 10.1002/jms.1177. [DOI] [PubMed] [Google Scholar]
- (38).Groseclose MR, Massion PP, Chaurand P, Caprioli RM. Proteomics. 2008;8:3715. doi: 10.1002/pmic.200800495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hardesty WM, Kelley MC, Mi D-M, Low RL, Caprioli RM. J. Proteomics. 2011;74:1002. doi: 10.1016/j.jprot.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Taverna D, Nanney LB, Pollins AC, Sindona G, Caprioli R. Exp. Dermatol. 2011;20:642. doi: 10.1111/j.1600-0625.2011.01289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Franzen B, Yang Y, Sunnemark D, Wickman M, Ottervald J, Oppermann M, Sandberg K. Proteomics. 2003;3:1920. doi: 10.1002/pmic.200300535. [DOI] [PubMed] [Google Scholar]
- (42).Ferrer I, Santpere G, Arzberger T, Bell J, Blanco R, Boluda S, Budka H, Carmona M, Giaccone G, Krebs B, Limido L, Parchi P, Puig B, Strammiello R, Strobel T, Kretzschmar H. J. Neuropathol. Exp. Neurol. 2007;66:35. doi: 10.1097/nen.0b013e31802c3e7d. [DOI] [PubMed] [Google Scholar]
- (43).Crecelius A, Gotz A, Arzberger T, Frohlich T, Arnold GJ, Ferrer I, Kretzschmar HA. Proteomics. 2008;8:1276. doi: 10.1002/pmic.200700728. [DOI] [PubMed] [Google Scholar]
- (44).Skold K, Svensson M, Norrman M, Sjogren B, Svenningsson P, Andren PE. Proteomics. 2007;7:4445. doi: 10.1002/pmic.200700142. [DOI] [PubMed] [Google Scholar]
- (45).Schwartz SA, Reyzer ML, Caprioli RM. J. Mass Spectrom. 2003;38:699. doi: 10.1002/jms.505. [DOI] [PubMed] [Google Scholar]
- (46).Lemaire R, Wisztorski M, Desmons A, Tabet JC, Day R, Salzet M, Fournier I. Anal. Chem. 2006;78:7145. doi: 10.1021/ac060565z. [DOI] [PubMed] [Google Scholar]
- (47).Svensson M, Boren M, Skold K, Falth M, Sjogren B, Andersson M, Svenningsson P, Andren PE. J. Proteome Res. 2009;8:974. doi: 10.1021/pr8006446. [DOI] [PubMed] [Google Scholar]
- (48).Goodwin RJ, Lang AM, Allingham H, Boren M, Pitt AR. Proteomics. 2010;10:1751. doi: 10.1002/pmic.200900641. [DOI] [PubMed] [Google Scholar]
- (49).Gillespie JW, Best CJ, Bichsel VE, Cole KA, Greenhut SF, Hewitt SM, Ahram M, Gathright YB, Merino MJ, Straus berg RL, Epstein JI, Hamilton SR, Gannot G, Baibakova GV, Calvert VS, Flaig MJ, Chuaqui RF, Herring JC, Pfeifer J, Petricoin EF, Linehan WM, Duray PH, Bova GS, Emmert-Buck MR. Am. J. Pathol. 2002;160:449. doi: 10.1016/S0002-9440(10)64864-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ahram M, Flaig MJ, Gillespie JW, Duray PH, Linehan WM, Ornstein DK, Niu S, Zhao Y, Petricoin EF, 3rd, Emmert-Buck MR. Proteomics. 2003;3:413. doi: 10.1002/pmic.200390056. [DOI] [PubMed] [Google Scholar]
- (51).Chaurand P, Latham JC, Lane KB, Mobley JA, Polosukhin VV, Wirth PS, Nanney LB, Caprioli RM. J. Proteome Res. 2008;7:3543. doi: 10.1021/pr800286z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Metz B, Kersten GF, Hoogerhout P, Brugghe HF, Timmermans HA, de Jong A, Meiring H, ten Hove J, Hennink WE, Crommelin DJ, Jiskoot W. J. Biol. Chem. 2004;279:6235. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- (53).Crecelius AC, Cornett DS, Caprioli RM, Williams B, Dawant BM, Bodenheimer B. J. Am. Soc. Mass Spectrom. 2005;16:1093. doi: 10.1016/j.jasms.2005.02.026. [DOI] [PubMed] [Google Scholar]
- (54).Chen R, Hui L, Sturm RM, Li L. J. Am. Soc. Mass Spectrom. 2009;20:1068. doi: 10.1016/j.jasms.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Reyzer ML, Chaurand P, Angel PM, Caprioli RM. Meth. Mol. Biol. 2010;656:285. doi: 10.1007/978-1-60761-746-4_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Khatib-Shahidi S, Andersson M, Herman JL, Gillespie TA, Caprioli RM. Anal. Chem. 2006;78:6448. doi: 10.1021/ac060788p. [DOI] [PubMed] [Google Scholar]
- (57).Lemaire R, Desmons A, Tabet JC, Day R, Salzet M, Fournier I. J. Proteome Res. 2007;6:1295. doi: 10.1021/pr060549i. [DOI] [PubMed] [Google Scholar]
- (58).Sugiura Y, Shimma S, Setou M. J. Mass Spectrom. Soc. Jpn. 2006;54:45. [Google Scholar]
- (59).Grove KJ, Frappier SL, Caprioli RM. J. Am. Soc. Mass Spectrom. 2011;22:192. doi: 10.1007/s13361-010-0013-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Methods and Techniques for Frozen Tissue Sections. IHC WORLD, LLC.; 2003–2011. http://www.ihcworld.com/_protocols/histology/frozen_section.htm. [Google Scholar]
- (61).Chaurand P, Schwartz SA, Billheimer D, Xu BJ, Crecelius A, Caprioli RM. Anal. Chem. 2004;76:1145. doi: 10.1021/ac0351264. [DOI] [PubMed] [Google Scholar]
- (62).Nissanov J, Bertrand L, Tretiak O. Microsc. Res. Tech. 2001;53:239. doi: 10.1002/jemt.1089. [DOI] [PubMed] [Google Scholar]
- (63).Goodwin RJ, Nilsson A, Borg D, Langridge-Smith PR, Harrison DJ, Mackay CL, Iverson SL, Andren PE. J. Proteomics. 2012;75:4912. doi: 10.1016/j.jprot.2012.07.006. [DOI] [PubMed] [Google Scholar]
- (64).Chaurand P, Norris JL, Cornett DS, Mobley JA, Caprioli RM. J. Proteome Res. 2006;5:2889. doi: 10.1021/pr060346u. [DOI] [PubMed] [Google Scholar]
- (65).Deutskens F, Yang J, Caprioli RM. J. Mass Spectrom. 2011;46:568. doi: 10.1002/jms.1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Caldwell RL, Caprioli RM. Mol. Cell. Proteomics. 2005;4:394. doi: 10.1074/mcp.R500006-MCP200. [DOI] [PubMed] [Google Scholar]
- (67).Bornsen KO. Meth. Mol. Biol. 2000;146:387. doi: 10.1385/1-59259-045-4:387. [DOI] [PubMed] [Google Scholar]
- (68).Goodwin RJ, Pennington SR, Pitt AR. Proteomics. 2008;8:3785. doi: 10.1002/pmic.200800320. [DOI] [PubMed] [Google Scholar]
- (69).Aerni H-R, Cornett DS, Caprioli RM. Anal. Chem. 2006;78:827. doi: 10.1021/ac051534r. [DOI] [PubMed] [Google Scholar]
- (70).Seeley EH, Oppenheimer SR, Mi D, Chaurand P, Caprioli RM. J. Am. Soc. Mass Spectrom. 2008;19:1069. doi: 10.1016/j.jasms.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Yang J, Caprioli RM. Anal. Chem. 2011;83:5728. doi: 10.1021/ac200998a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Grey AC, Chaurand P, Caprioli RM, Schey KL. J. Proteome Res. 2009;8:3278. doi: 10.1021/pr800956y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Andersson M, Groseclose MR, Deutch AY, Caprioli RM. Nat. Methods. 2008;5:101. doi: 10.1038/nmeth1145. [DOI] [PubMed] [Google Scholar]
- (74).Hanrieder J, Ljungdahl A, Faelth M, Mammo SE, Bergquist J, Andersson M. Mol. Cell. Proteomics. 2011;10:M111.009308. doi: 10.1074/mcp.M111.009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Ljungdahl A, Hanrieder J, Faelth M, Bergquist J, Andersson M. PLoS One. 2011;6:e25653. doi: 10.1371/journal.pone.0025653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Angel PM, Spraggins JM, Baldwin HS, Caprioli R. Anal. Chem. 2012;84:1557. doi: 10.1021/ac202383m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Wang HY, Liu CB, Wu HW. J. Lipid Res. 2011;52:840. doi: 10.1194/jlr.D013060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Wang HY, Wu HW, Tsai PJ, Liu CB. Anal. Bioanal. Chem. 2012;404:113. doi: 10.1007/s00216-012-6077-5. [DOI] [PubMed] [Google Scholar]
- (79).Gustafsson JOR, Oehler MK, McColl SR, Hoffmann P. J. Proteome Res. 2010;9:4315. doi: 10.1021/pr9011766. [DOI] [PubMed] [Google Scholar]
- (80).Aoki Y, Toyama A, Shimada T, Sugita T, Aoki C, Umino Y, Suzuki A, Aoki D, Daigo Y, Nakamura Y, Sato T-A. Proc. Jpn. Acad., Ser. B. 2007;83:205. doi: 10.2183/pjab/83.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Stauber J, MacAleese L, Franck J, Claude E, Snel M, Kaletas BK, Wiel IMVD, Wisztorski M, Fournier I, Heeren RMA. J. Am. Soc. Mass Spectrom. 2010;21:338. doi: 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
- (82).Wisztorski M, Franck J, Salzet M, Fournier I. Meth. Mol. Biol. 2010;656:303. doi: 10.1007/978-1-60761-746-4_18. [DOI] [PubMed] [Google Scholar]
- (83).Manier ML, Reyzer ML, Goh A, Dartois V, Via LE, Barry CE, 3rd, Caprioli RM. J. Am. Soc. Mass Spectrom. 2011;22:1409. doi: 10.1007/s13361-011-0150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Chacon A, Zagol-Ikapitte I, Amarnath V, Reyzer ML, Oates JA, Caprioli RM, Boutaud O. J. Mass Spectrom. 2011;46:840. doi: 10.1002/jms.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Franck J, El Ayed M, Wisztorski M, Salzet M, Fournier I. Anal. Chem. 2009;81:8305. doi: 10.1021/ac901043n. [DOI] [PubMed] [Google Scholar]
- (86).Jaskolla TW, Karas M. J. Am. Soc. Mass Spectrom. 2011;22:976. doi: 10.1007/s13361-011-0093-0. [DOI] [PubMed] [Google Scholar]
- (87).Strupat K, Karas M, Hillenkamp F. Int. J. Mass Spectrom. 1991;111:89. [Google Scholar]
- (88).Beavis RC, Chaudhary T, Chait BT. Org. Mass Spectrom. 1992;27:156. [Google Scholar]
- (89).Beavis RC, Chait BT. Rapid Commun. Mass Spectrom. 1989;3:436. doi: 10.1002/rcm.1290031208. [DOI] [PubMed] [Google Scholar]
- (90).Tholey A, Heinzle E. Anal. Bioanal. Chem. 2006;386:24. doi: 10.1007/s00216-006-0600-5. [DOI] [PubMed] [Google Scholar]
- (91).Chan K, Lanthier P, Liu X, Sandhu JK, Stanimirovic D, Li J. Anal. Chim. Acta. 2009;639:57. doi: 10.1016/j.aca.2009.02.051. [DOI] [PubMed] [Google Scholar]
- (92).Chan K, Lanthier P, Liu X, Sandhu JK, Stanimirovic D, Li J. Anal. Chim. Acta. 2009;639:57. doi: 10.1016/j.aca.2009.02.051. [DOI] [PubMed] [Google Scholar]
- (93).Meriaux C, Franck J, Wisztorski M, Salzet M, Fournier I. J. Proteomics. 2010;73:1204. doi: 10.1016/j.jprot.2010.02.010. [DOI] [PubMed] [Google Scholar]
- (94).Tanaka K, Waki H, Ido Y, Akita S, Yoshida Y, Yoshida T. Rapid Commun. Mass Spectrom. 1988;2:151. [Google Scholar]
- (95).McLean JA, Stumpo KA, Russell DH. J. Am. Chem. Soc. 2005;127:5304. doi: 10.1021/ja043907w. [DOI] [PubMed] [Google Scholar]
- (96).Su CL, Tseng WL. Anal. Chem. 2007;79:1626. doi: 10.1021/ac061747w. [DOI] [PubMed] [Google Scholar]
- (97).Cha S, Zhang H, Ilarslan HI, Wurtele ES, Brachova L, Nikolau BJ, Yeung ES. Plant J. 2008;55:348. doi: 10.1111/j.1365-313X.2008.03507.x. [DOI] [PubMed] [Google Scholar]
- (98).Northen TR, Yanes O, Northen MT, Marrinucci D, Uritboonthai W, Apon J, Golledge SL, Nordstrom A, Siuzdak G. Nature. 2007;449:1033. doi: 10.1038/nature06195. [DOI] [PubMed] [Google Scholar]
- (99).Tang K, Allman SL, Chen CH. Rapid Commun. Mass Spectrom. 1993;7:943. doi: 10.1002/rcm.1290071016. [DOI] [PubMed] [Google Scholar]
- (100).Pan C, Xu S, Zhou H, Fu Y, Ye M, Zou H. Anal. Bioanal. Chem. 2007;387:193. doi: 10.1007/s00216-006-0905-4. [DOI] [PubMed] [Google Scholar]
- (101).Fenaille F, Tabet JC, Guy PA. Anal. Chem. 2004;76:867. doi: 10.1021/ac0303822. [DOI] [PubMed] [Google Scholar]
- (102).Papac DI, Wong A, Jones AJ. Anal. Chem. 1996;68:3215. doi: 10.1021/ac960324z. [DOI] [PubMed] [Google Scholar]
- (103).Gorman JJ, Ferguson BL, Nguyen TB. Rapid Commun. Mass Spectrom. 1996;10:529. doi: 10.1002/(SICI)1097-0231(19960331)10:5<529::AID-RCM522>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- (104).Thomas A, Charbonneau JL, Fournaise E, Chaurand P. Anal. Chem. 2012;84:2048. doi: 10.1021/ac2033547. [DOI] [PubMed] [Google Scholar]
- (105).Beavis RC, Chait BT. Meth. Enzymol. 1996;270:519. doi: 10.1016/s0076-6879(96)70024-1. [DOI] [PubMed] [Google Scholar]
- (106).Cohen SL, Chait BT. Anal. Chem. 1996;68:31. doi: 10.1021/ac9507956. [DOI] [PubMed] [Google Scholar]
- (107).Djidja MC, Francese S, Loadman PM, Sutton CW, Scriven P, Claude E, Snel MF, Franck J, Salzet M, Clench MR. Proteomics. 2009;9:2750. doi: 10.1002/pmic.200800624. [DOI] [PubMed] [Google Scholar]
- (108).Mainini V, Angel P, Magni F, Caprioli RM. Rapid Commun. Mass Spectrom. 2011;25:199. doi: 10.1002/rcm.4850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Norris JL, Porter NA, Caprioli RM. Anal. Chem. 2003;75:6642. doi: 10.1021/ac034802z. [DOI] [PubMed] [Google Scholar]
- (110).Schuerenberg M, Luebbert C, Deininger S, Ketterlinus R, Suckau D. Nat. Methods. 2007;4:iii. [Google Scholar]
- (111).Schuerenberg M, Luebbert C, Deininger S, Mueller R, Suckau D. J. Biomol. Tech. 2007;18:38. [Google Scholar]
- (112).Nye GJ, Norris JL, Nickerson S. 54th ASMS Conference on Mass Spectrometry and Allied Topics; Seattle, WA. 2006. [Google Scholar]
- (113).Hankin JA, Barkley RM, Murphy RC. J. Am. Soc. Mass Spectrom. 2007;18:1646. doi: 10.1016/j.jasms.2007.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Dekker LJM, van KJJA, Reedijk ML, Burgers PC, Gruters RA, Osterhaus ADME, Luider TM. Rapid Commun. Mass Spectrom. 2009;23:1183. doi: 10.1002/rcm.3981. [DOI] [PubMed] [Google Scholar]
- (115).Baluya DL, Garrett TJ, Yost RA. Anal. Chem. 2007;79:6862. doi: 10.1021/ac070958d. [DOI] [PubMed] [Google Scholar]
- (116).Sugiura Y, Shimma S, Setou M. Anal. Chem. 2006;78:8227. doi: 10.1021/ac060974v. [DOI] [PubMed] [Google Scholar]
- (117).Delvolve AM, Woods AS. J. Mass Spectrom. 2011;46:1046. doi: 10.1002/jms.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Gustafsson JOR, McColl SR, Hoffmann P. J. Proteomics Bioinf. 2008;1:458. [Google Scholar]
- (119).Schwartz SA, Weil RJ, Johnson MD, Toms SA, Caprioli RM. Clin. Cancer Res. 2004;10:981. doi: 10.1158/1078-0432.ccr-0927-3. [DOI] [PubMed] [Google Scholar]
- (120).Pierson J, Norris JL, Aerni HR, Svenningsson P, Caprioli RM, Andren PE. J. Proteome Res. 2004;3:289. doi: 10.1021/pr0499747. [DOI] [PubMed] [Google Scholar]
- (121).Meistermann H, Norris JL, Aerni H-R, Cornett DS, Friedlein A, Erskine AR, Augustin A, De VMMC, Ruepp S, Suter L, Langen H, Caprioli RM, Ducret A. Mol. Cell. Proteomics. 2006;5:1876. doi: 10.1074/mcp.M500399-MCP200. [DOI] [PubMed] [Google Scholar]
- (122).Zavalin A, Todd EM, Rawhouser PD, Yang J, Norris JL, Caprioli RM. J. Mass Spectrom. 2012;47:1473. doi: 10.1002/jms.3108. [DOI] [PubMed] [Google Scholar]
- (123).Holle A, Haase A, Kayser M, Hohndorf J. J. Mass Spectrom. 2006;41:705. doi: 10.1002/jms.1041. [DOI] [PubMed] [Google Scholar]
- (124).Anderson NL, Anderson NG. Mol. Cell. Proteom. 2002;1:845. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- (125).Kutz KK, Schmidt JJ, Li L. Anal. Chem. 2004;76:5630. doi: 10.1021/ac049255b. [DOI] [PubMed] [Google Scholar]
- (126).Spraggins JM, Grove KJ, Cornett DS, Caprioli RM. 60th Annual Conference on Mass Spectrometry and Allied Topics; Vancouver, Canada. 2012. [Google Scholar]
- (127).Spraggins JM, Cornett DS, Angel P, Caprioli RM. 59th ASMS Conference on Mass Spectrometry and Allied Topics; Denver, CO. 2011. [Google Scholar]
- (128).Cornett DS, Frappier SL, Caprioli RM. Anal. Chem. 2008;80:5648. doi: 10.1021/ac800617s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Greer T, Sturm R, Li L. J. Proteomics. 2011;74:2617. doi: 10.1016/j.jprot.2011.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Fehniger TE, Vegvari A, Rezeli M, Prikk K, Ross P, Dahlback M, Edula G, Sepper R, Marko-Varga G. Anal. Chem. 2011;83:8329. doi: 10.1021/ac2014349. [DOI] [PubMed] [Google Scholar]
- (131).Perdian DC, Lee YJ. Anal. Chem. 2010;82:9393. doi: 10.1021/ac102017q. [DOI] [PubMed] [Google Scholar]
- (132).Schober Y, Guenther S, Spengler B, Roempp A. Rapid Commun. Mass Spectrom. 2012;26:1141. doi: 10.1002/rcm.6192. [DOI] [PubMed] [Google Scholar]
- (133).Vegvari A, Fehniger TE, Gustavsson L, Nilsson A, Andren PE, Kenne K, Nilsson J, Laurell T, Marko-Varga G. J. Proteomics. 2010;73:1270. doi: 10.1016/j.jprot.2010.02.021. [DOI] [PubMed] [Google Scholar]
- (134).Landgraf RR, Prieto-Conaway MC, Garrett TJ, Stacpoole PW, Yost RA. Anal. Chem. 2009;81:8488. doi: 10.1021/ac901387u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Deutsch E. Proteomics. 2008;8:2776. doi: 10.1002/pmic.200890049. [DOI] [PubMed] [Google Scholar]
- (136).Roempp A, Schramm T, Hester A, Klinkert I, Both J-P, Heeren RMA, Stoeckli M, Spengler B. Meth. Mol. Biol. 2011;696:205. doi: 10.1007/978-1-60761-987-1_12. [DOI] [PubMed] [Google Scholar]
- (137).Sanchez J-C, Corthals GL, Hochstrasser DF. Biomedical Applications of Proteomics. Wiley-VCH; Weinheim: 2004. [Google Scholar]
- (138).Jardin-Mathe O, Bonnel D, Franck J, Wisztorski M, Macagno E, Fournier I, Salzet M. J. Proteomics. 2008;71:332. doi: 10.1016/j.jprot.2008.07.004. [DOI] [PubMed] [Google Scholar]
- (139).Nilsson A, Fehniger TE, Gustavsson L, Andersson M, Kenne K, Marko-Varga G, Andren PE. PLoS One. 2010;5:e11411. doi: 10.1371/journal.pone.0011411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Stoeckli M, Staab D, Schweitzer A. Int. J. Mass Spectrom. 2007;260:195. [Google Scholar]
- (141).Kallback P, Shariatgorji M, Nilsson A, Andren PE. J. Proteomics. 2012;75:4941. doi: 10.1016/j.jprot.2012.07.034. [DOI] [PubMed] [Google Scholar]
- (142).Pirman DA, Yost RA. Analytical chemistry. 2011;83:8575. doi: 10.1021/ac201949b. [DOI] [PubMed] [Google Scholar]
- (143).Hamm G, Bonnel D, Legouffe R, Pamelard F, Delbos JM, Bouzom F, Stauber J. J. Proteomics. 2012;75:4952. doi: 10.1016/j.jprot.2012.07.035. [DOI] [PubMed] [Google Scholar]
- (144).Mantini D, Petrucci F, Pieragostino D, Del Boccio P, Sacchetta P, Candiano G, Ghiggeri GM, Lugaresi A, Federici G, Di Ilio C, Urbani A. J. Proteomics. 2010;73:562. doi: 10.1016/j.jprot.2009.11.004. [DOI] [PubMed] [Google Scholar]
- (145).Ndukum J, Atlas M, Datta S. Bioinformation. 2011;6:45. doi: 10.6026/97320630006045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Norris JL, Cornett DS, Mobley JA, Andersson M, Seeley EH, Chaurand P, Caprioli RM. Int. J. Mass Spectrom. 2007;260:212. doi: 10.1016/j.ijms.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Hussong R, Hildebrandt A. Methods Mol. Biol. 2010;604:145. doi: 10.1007/978-1-60761-444-9_11. [DOI] [PubMed] [Google Scholar]
- (148).Shin H, Sampat MP, Koomen JM, Markey MK. Omics : a journal of integrative biology. 2010;14:283. doi: 10.1089/omi.2009.0119. [DOI] [PubMed] [Google Scholar]
- (149).Liland KH, Almoy T, Mevik BH. Appl. Spectrosc. 2010;64:1007. doi: 10.1366/000370210792434350. [DOI] [PubMed] [Google Scholar]
- (150).Trede D, Kobarg JH, Oetjen J, Thiele H, Maass P, Alexandrov T. J. Integr. Bioinform. 2012;9:189. doi: 10.2390/biecoll-jib-2012-189. [DOI] [PubMed] [Google Scholar]
- (151).Fonville JM, Carter C, Cloarec O, Nicholson JK, Lindon JC, Bunch J, Holmes E. Anal. Chem. 2012;84:1310. doi: 10.1021/ac201767g. [DOI] [PubMed] [Google Scholar]
- (152).Tracy MB, Cooke WE, Gatlin CL, Cazares LH, Weaver DM, Semmes OJ, Tracy ER, Manos DM, Malyarenko DI. Proteomics Clin. Appl. 2011;5:440. doi: 10.1002/prca.201000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Lange E, Gropl C, Reinert K, Kohlbacher O, Hildebrandt A. Pac. Symp. Biocomput. 2006:243. [PubMed] [Google Scholar]
- (154).Trim PJ, Djidja MC, Muharib T, Cole LM, Flinders B, Carolan VA, Francese S, Clench MR. J. Proteomics. 2012;75:4931. doi: 10.1016/j.jprot.2012.07.016. [DOI] [PubMed] [Google Scholar]
- (155).Deininger SO, Becker M, Suckau D. Meth. Mol. Biol. 2010;656:385. doi: 10.1007/978-1-60761-746-4_22. [DOI] [PubMed] [Google Scholar]
- (156).Deininger S, Schuerenberg M, Luebbert C, Fuetterer A, Gerhard M, Suckau D, Roecken C, Ebert M, Walch A. J. Biomol. Tech. 2007;18:34. [Google Scholar]
- (157).Jones EA, van Remoortere A, van Zeijl RJM, Hogendoorn PCW, Bovee JVMG, Deelder AM, McDonnell LA. PLoS One. 2011;6:e24913. doi: 10.1371/journal.pone.0024913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Hsieh SY, Tseng CL, Lee YS, Kuo AJ, Sun CF, Lin YH, Chen JK. Mol. Cell. Proteomics. 2008;7:448. doi: 10.1074/mcp.M700339-MCP200. [DOI] [PubMed] [Google Scholar]
- (159).Trim PJ, Atkinson SJ, Princivalle AP, Marshall PS, West A, Clench MR. Rapid Commun. Mass Spectrom. 2008;22:1503. doi: 10.1002/rcm.3498. [DOI] [PubMed] [Google Scholar]
- (160).Goncalves A, Charafe-Jauffret E, Bertucci F, Audebert S, Toiron Y, Esterni B, Monville F, Tarpin C, Jacquemier J, Houvenaeghel G, Chabannon C, Extra JM, Viens P, Borg JP, Birnbaum D. Mol. Cell. Proteomics. 2008;7:1420. doi: 10.1074/mcp.M700487-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).He J, Shen D, Chung DU, Saxton RE, Whitelegge JP, Faull KF, Chang HR. Int. J. Oncol. 2009;35:683. doi: 10.3892/ijo_00000380. [DOI] [PubMed] [Google Scholar]
- (162).Lasch P, Beyer W, Nattermann H, Stammler M, Siegbrecht E, Grunow R, Naumann D. Appl Environ Microbiol. 2009;75:7229. doi: 10.1128/AEM.00857-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Liao CC, Ward N, Marsh S, Arulampalam T, Norton JD. BMC Cancer. 2010;10:410. doi: 10.1186/1471-2407-10-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Lasch P, Drevinek M, Nattermann H, Grunow R, Stammler M, Dieckmann R, Schwecke T, Naumann D. Anal. Chem. 2010;82:8464. doi: 10.1021/ac101036s. [DOI] [PubMed] [Google Scholar]
- (165).Wittwer M, Heim J, Schar M, Dewarrat G, Schurch N. Syst. Appl. Microbiol. 2011;34:12. doi: 10.1016/j.syapm.2010.11.006. [DOI] [PubMed] [Google Scholar]
- (166).Roesch-Ely M, Nees M, Karsai S, Ruess A, Bogumil R, Warnken U, Schnolzer M, Dietz A, Plinkert PK, Hofele C, Bosch FX. Oncogene. 2007;26:54. doi: 10.1038/sj.onc.1209770. [DOI] [PubMed] [Google Scholar]
- (167).Agar NY, Malcolm JG, Mohan V, Yang HW, Johnson MD, Tannenbaum A, Agar JN, Black PM. Anal. Chem. 2010;82:2621. doi: 10.1021/ac100113w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Cazares LH, Troyer DA, Wang B, Drake RR, Semmes OJ. Anal. Bioanal. Chem. 2011;401:17. doi: 10.1007/s00216-011-5003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Seeley EH, Caprioli RM. Anal. Chem. 2012;84:2105. doi: 10.1021/ac2032707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Li H, Hummon AB. Anal. Chem. 2011;83:8794. doi: 10.1021/ac202356g. [DOI] [PubMed] [Google Scholar]
- (171).Xiong X, Xu W, Eberlin LS, Wiseman JM, Fang X, Jiang Y, Huang Z, Zhang Y, Cooks RG, Ouyang Z. J. Am. Soc. Mass Spectrom. 2012;23:1147. doi: 10.1007/s13361-012-0361-7. [DOI] [PubMed] [Google Scholar]
- (172).Ye H, Greer T, Li L. Bioanal. 2011;3:313. doi: 10.4155/bio.10.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Chen R, Hui L, Sturm RM, Li L. J. Am. Soc. Mass Spectrom. 2009;20:1068. doi: 10.1016/j.jasms.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Sinha TK, Khatib-Shahidi S, Yankeelov TE, Mapara K, Ehtesham M, Cornett DS, Dawant BM, Caprioli RM, Gore JC. Nat. Methods. 2008;5:57. doi: 10.1038/nmeth1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Crecelius AC, Cornett DS, Caprioli RM, Williams B, Dawant BM, Bodenheimer B. J. Am. Soc. Mass Spectrom. 2005;16:1093. doi: 10.1016/j.jasms.2005.02.026. [DOI] [PubMed] [Google Scholar]
- (176).Chughtai K, Jiang L, Greenwood TR, Klinkert I, Amstalden van Hove ER, Heeren RM, Glunde K. Anal. Chem. 2012;84:1817. doi: 10.1021/ac203373h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Attia AS, Schroeder KA, Seeley EH, Wilson KJ, Hammer ND, Colvin DC, Manier ML, Nicklay JJ, Rose KL, Gore JC, Caprioli RM, Skaar EP. Cell host & microbe. 2012;11:664. doi: 10.1016/j.chom.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Trede D, Schiffler S, Becker M, Wirtz S, Steinhorst K, Strehlow J, Aichler M, Kobarg JH, Oetjen J, Dyatlov A, Heldmann S, Walch A, Thiele H, Maass P, Alexandrov T. Anal. Chem. 2012;84:6079. doi: 10.1021/ac300673y. [DOI] [PubMed] [Google Scholar]
- (179).Han J, Schey KL. Invest. Ophthalmol. Vis. Sci. 2006;47:2990. doi: 10.1167/iovs.05-1529. [DOI] [PubMed] [Google Scholar]
- (180).Grey AC, Schey KL. Mol. Vis. 2008;14:171. [PMC free article] [PubMed] [Google Scholar]
- (181).Thibault DB, Gillam CJ, Grey AC, Han J, Schey KL. J. Am. Soc. Mass Spectrom. 2008;19:814. doi: 10.1016/j.jasms.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (182).Grey AC, Schey KL. Invest. Ophthalmol. Vis. Sci. 2009;50:4319. doi: 10.1167/iovs.09-3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Su SP, McArthur JD, Aquilina JA. Exp. Eye Res. 2010;91:97. doi: 10.1016/j.exer.2010.04.010. [DOI] [PubMed] [Google Scholar]
- (184).Stella DR, Floyd KA, Grey AC, Renfrow MB, Schey KL, Barnes S. Invest. Ophthalmol. Vis. Sci. 2010;51:5153. doi: 10.1167/iovs.10-5302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Ronci M, Sharma S, Chataway T, Burdon KP, Martin S, Craig JE, Voelcker NH. J. Proteome Res. 2011;10:3522. doi: 10.1021/pr200148k. [DOI] [PubMed] [Google Scholar]
- (186).Hayasaka T, Goto-Inoue N, Sugiura Y, Zaima N, Nakanishi H, Ohishi K, Nakanishi S, Naito T, Taguchi R, Setou M. Rapid Commun. Mass Spectrom. 2008;22:3415. doi: 10.1002/rcm.3751. [DOI] [PubMed] [Google Scholar]
- (187).Garrett TJ, Dawson WW. Methods Mol. Biol. 2009;579:247. doi: 10.1007/978-1-60761-322-0_12. [DOI] [PubMed] [Google Scholar]
- (188).Vidova V, Pol J, Volny M, Novak P, Havlicek V, Wiedmer SK, Holopainen JM. J. Lipid Res. 2010;51:2295. doi: 10.1194/jlr.M005488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Hayasaka T, Goto-Inoue N, Zaima N, Shrivas K, Kashiwagi Y, Yamamoto M, Nakamoto M, Setou M. J. Am. Soc. Mass Spectrom. 2010;21:1446. doi: 10.1016/j.jasms.2010.04.005. [DOI] [PubMed] [Google Scholar]
- (190).Deeley JM, Hankin JA, Friedrich MG, Murphy RC, Truscott RJ, Mitchell TW, Blanksby SJ. J. Lipid Res. 2010;51:2753. doi: 10.1194/jlr.M007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Roy MC, Nakanishi H, Takahashi K, Nakanishi S, Kajihara S, Hayasaka T, Setou M, Ogawa K, Taguchi R, Naito T. J. Lipid Res. 2011;52:463. doi: 10.1194/jlr.M010546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Pol J, Vidova V, Hyotylainen T, Volny M, Novak P, Strohalm M, Kostiainen R, Havlicek V, Wiedmer SK, Holopainen JM. PLoS One. 2011;6:e19441. doi: 10.1371/journal.pone.0019441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Palmer AD, Griffiths R, Styles I, Claridge E, Calcagni A, Bunch J. J. Mass Spectrom. 2012;47:237. doi: 10.1002/jms.2049. [DOI] [PubMed] [Google Scholar]
- (194).Grey AC, Crouch RK, Koutalos Y, Schey KL, Ablonczy Z. Invest. Ophthalmol. Vis. Sci. 2011;52:3926. doi: 10.1167/iovs.10-7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Ablonczy Z, Gutierrez DB, Grey AC, Schey KL, Crouch RK. Adv. Exp. Med. Biol. 2012;723:75. doi: 10.1007/978-1-4614-0631-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Drexler DM, Tannehill-Gregg SH, Wang L, Brock BJ. J. Pharmacol. Toxicol. Methods. 2011;63:205. doi: 10.1016/j.vascn.2010.10.003. [DOI] [PubMed] [Google Scholar]
- (197).Yamada Y, Hidefumi K, Shion H, Oshikata M, Haramaki Y. Rapid Commun. Mass Spectrom. 2011;25:1600. doi: 10.1002/rcm.5021. [DOI] [PubMed] [Google Scholar]
- (198).Schwartz SA, Weil RJ, Thompson RC, Shyr Y, Moore JH, Toms SA, Johnson MD, Caprioli RM. Cancer Res. 2005;65:7674. doi: 10.1158/0008-5472.CAN-04-3016. [DOI] [PubMed] [Google Scholar]
- (199).Rauser S, Marquardt C, Balluff B, Deininger S-O, Albers C, Belau E, Hartmer R, Suckau D, Specht K, Ebert MP, Schmitt M, Aubele M, Hoefler H, Walch A. J. Proteome Res. 2010;9:1854. doi: 10.1021/pr901008d. [DOI] [PubMed] [Google Scholar]
- (200).Balluff B, Elsner M, Kowarsch A, Rauser S, Meding S, Schuhmacher C, Feith M, Herrmann K, Rocken C, Schmid RM, Hofler H, Walch A, Ebert MP. J. Proteome Res. 2010;9:6317. doi: 10.1021/pr100573s. [DOI] [PubMed] [Google Scholar]
- (201).Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, Dowsett M, Fitzgibbons PL, Hanna WM, Langer A, McShane LM, Paik S, Pegram MD, Perez EA, Press MF, Rhodes A, Sturgeon C, Taube SE, Tubbs R, Vance GH, van de Vijver M, Wheeler TM, Hayes DF. J. Clin. Oncol. 2007;25:118. doi: 10.1200/JCO.2006.09.2775. [DOI] [PubMed] [Google Scholar]
- (202).Balluff B, Rauser S, Meding S, Elsner M, Schone C, Feuchtinger A, Schuhmacher C, Novotny A, Jutting U, Maccarrone G, Sarioglu H, Ueffing M, Braselmann H, Zitzelsberger H, Schmid RM, Hofler H, Ebert MP, Walch A. Am. J. Pathol. 2011;179:2720. doi: 10.1016/j.ajpath.2011.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Nipp M, Elsner M, Balluff B, Meding S, Sarioglu H, Ueffing M, Rauser S, Unger K, Hofler H, Walch A, Zitzelsberger H. J. Mol. Med. 2012;90:163. doi: 10.1007/s00109-011-0815-6. [DOI] [PubMed] [Google Scholar]
- (204).Oezdemir RF, Gaisa NT, Lindemann-Docter K, Gostek S, Weiskirchen R, Ahrens M, Schwamborn K, Stephan C, Pfister D, Heidenreich A, Knuechel R, Henkel C. Clin. Biochem. 2012;45:7. doi: 10.1016/j.clinbiochem.2011.09.013. [DOI] [PubMed] [Google Scholar]
- (205).Meding S, Nitsche U, Balluff B, Elsner M, Rauser S, Schone C, Nipp M, Maak M, Feith M, Ebert MP, Friess H, Langer R, Hofler H, Zitzelsberger H, Rosenberg R, Walch A. J. Proteome Res. 2012;11:1996. doi: 10.1021/pr200784p. [DOI] [PubMed] [Google Scholar]
- (206).Oppenheimer SR, Mi D, Sanders ME, Caprioli RM. J. Proteome Res. 2010;9:2182. doi: 10.1021/pr900936z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Shimma S, Furuta M, Ichimura K, Yoshida Y, Setou M. Surf. Interface Anal. 2006;38:1712. [Google Scholar]
- (208).Djidja M-C, Claude E, Snel MF, Francese S, Scriven P, Carolan V, Clench MR. Anal. Bioanal. Chem. 2010;397:587. doi: 10.1007/s00216-010-3554-6. [DOI] [PubMed] [Google Scholar]
- (209).Murphy AJ, Axt JR, de Caestecker C, Pierce J, Correa H, Seeley EH, Caprioli RM, Newton MW, de Caestecker MP, Lovvorn HN., 3rd Int. J. Cancer. 2012;131:E983. doi: 10.1002/ijc.27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Rahman SM, Gonzalez AL, Li M, Seeley EH, Zimmerman LJ, Zhang XJ, Manier ML, Olson SJ, Shah RN, Miller AN, Putnam JB, Miller YE, Franklin WA, Blot WJ, Carbone DP, Shyr Y, Caprioli RM, Massion PP. Cancer Res. 2011;71:3009. doi: 10.1158/0008-5472.CAN-10-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Morgan TM, Seeley EH, Fadare O, Caprioli RM, Clark PE. J. Urol. 2012;189:1097. doi: 10.1016/j.juro.2012.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (212).Bauer JA, Chakravarthy AB, Rosenbluth JM, Mi D, Seeley EH, De Matos Granja-Ingram N, Olivares MG, Kelley MC, Mayer IA, Meszoely IM, Means-Powell JA, Johnson KN, Tsai CJ, Ayers GD, Sanders ME, Schneider RJ, Formenti SC, Caprioli RM, Pietenpol JA. Clin. Cancer Res. 2010;16:681. doi: 10.1158/1078-0432.CCR-09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (213).Troendle F, Reddick C, Yost RA. J. Am. Soc. Mass Spectrom. 1999;10:1315. [Google Scholar]
- (214).Reyzer ML, Hsieh Y, Ng K, Korfmacher WA, Caprioli RM. J. Mass Spectrom. 2003;38:1081. doi: 10.1002/jms.525. [DOI] [PubMed] [Google Scholar]
- (215).Hsieh Y, Casale R, Fukuda E, Chen J, Knemeyer I, Wingate J, Morrison R, Korfmacher W. Rapid Commun. Mass Spectrom. 2006;20:965. doi: 10.1002/rcm.2397. [DOI] [PubMed] [Google Scholar]
- (216).Rohner TC, Staab D, Stoeckli M. Mech. Ageing Dev. 2005;126:177. doi: 10.1016/j.mad.2004.09.032. [DOI] [PubMed] [Google Scholar]
- (217).Shahidi-Latham SK, Dutta SM, Prieto-Conaway MC, Rudewicz PJ. Anal. Chem. 2012;84:7158. doi: 10.1021/ac3015142. [DOI] [PubMed] [Google Scholar]
- (218).Garrett TJ, Prieto-Conaway MC, Kovtoun V, Bui H, Izgarian N, Stafford G, Yost RA. Int. J. Mass Spectrom. 2007;260:166. [Google Scholar]
- (219).Acquadro E, Cabella C, Ghiani S, Miragoli L, Bucci EM, Corpillo D. Anal. Chem. 2009;81:2779. doi: 10.1021/ac900038y. [DOI] [PubMed] [Google Scholar]
- (220).Earnshaw CJ, Carolan VA, Richards DS, Clench MR. Rapid Commun. Mass Spectrom. 2010;24:1665. doi: 10.1002/rcm.4525. [DOI] [PubMed] [Google Scholar]
- (221).Kreye F, Hamm G, Karrout Y, Legouffe R, Bonnel D, Siepmann F, Siepmann J. J. Control. Release. 2012;161:98. doi: 10.1016/j.jconrel.2012.04.017. [DOI] [PubMed] [Google Scholar]
- (222).Huang JT, Hannah-Qiuhua L, Szyszka R, Veselov V, Reed G, Wang X, Price S, Alquier L, Vas G. J. Mass Spectrom. 2012;47:155. doi: 10.1002/jms.2046. [DOI] [PubMed] [Google Scholar]
- (223).Miki A, Katagi M, Kamata T, Zaitsu K, Tatsuno M, Nakanishi T, Tsuchihashi H, Takubo T, Suzuki K-I. J. Mass Spectrom. 2011;46:411. doi: 10.1002/jms.1908. [DOI] [PubMed] [Google Scholar]
- (224).Goodwin RJ, MacIntyre L, Watson DG, Scullion SP, Pitt AR. Rapid Commun. Mass Spectrom. 2010;24:1682. doi: 10.1002/rcm.4567. [DOI] [PubMed] [Google Scholar]
- (225).Goodwin RJA, Mackay CL, Nilsson A, Harrison DJ, Farde L, Andren PE, Iverson SL. Anal. Chem. 2011;83:9694. doi: 10.1021/ac202630t. [DOI] [PubMed] [Google Scholar]
- (226).Goodwin RJA, Scullion P, MacIntyre L, Watson DG, Pitt AR. Anal. Chem. 2010;82:3868. doi: 10.1021/ac100398y. [DOI] [PubMed] [Google Scholar]
- (227).Marko-Varga G, Fehniger TE, Rezeli M, Doeme B, Laurell T, Vegvari A. J. Proteomics. 2011;74:982. doi: 10.1016/j.jprot.2011.03.019. [DOI] [PubMed] [Google Scholar]
- (228).Ceuppens R, Dumont D, Van BL, Van d. P. B., Daniels R, Noben J-P, Verhaert P, Van d. G. E., Robben J, Clerens S, Arckens L. Int. J. Mass Spectrom. 2007;260:185. [Google Scholar]
- (229).DeKeyser SS, Kutz-Naber KK, Schmidt JJ, Barrett-Wilt GA, Li L. J. Proteome Res. 2007;6:1782. doi: 10.1021/pr060603v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Chansela P, Goto-Inoue N, Zaima N, Sroyraya M, Sobhon P, Setou M. Peptides. 2012;34:10. doi: 10.1016/j.peptides.2011.03.021. [DOI] [PubMed] [Google Scholar]
- (231).Hanrieder J, Ljungdahl A, Andersson M. J. Visualized Exp. 2012:3445/1. doi: 10.3791/3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Herbert Z, Rauser S, Williams L, Kapan N, Guentner M, Walch A, Boyan G. J. Morphol. 2010;271:1509. doi: 10.1002/jmor.10895. [DOI] [PubMed] [Google Scholar]
- (233).Hui L, Zhang Y, Wang J, Cook A, Ye H, Nusbaum MP, Li L. ACS Chem. Neurosci. 2011;2:711. doi: 10.1021/cn200042p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Ye H, Greer T, Li L. J Proteomics. 2012 doi: 10.1016/j.jprot.2012.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Zimmerman TA, Rubakhin SS, Sweedler JV. J. Am. Soc. Mass Spectrom. 2011;22:828. doi: 10.1007/s13361-011-0111-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Kruse R, Sweedler JV. J. Am. Soc. Mass Spectrom. 2003;14:752. doi: 10.1016/S1044-0305(03)00288-5. [DOI] [PubMed] [Google Scholar]
- (237).Rubakhin SS, Hatcher NG, Monroe EB, Heien ML, Sweedler JV. Curr. Pharm. Des. 2007;13:3325. doi: 10.2174/138161207782360708. [DOI] [PubMed] [Google Scholar]
- (238).Sparvero LJ, Amoscato AA, Kochanek PM, Pitt BR, Kagan VE, Bayir H. J. Neurochem. 2010;115:1322. doi: 10.1111/j.1471-4159.2010.07055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Koizumi S, Yamamoto S, Hayasaka T, Konishi Y, Yamaguchi-Okada M, Goto-Inoue N, Sugiura Y, Setou M, Namba H. Neuroscience. 2010;168:219. doi: 10.1016/j.neuroscience.2010.03.056. [DOI] [PubMed] [Google Scholar]
- (240).Hankin JA, Farias SE, Barkley RM, Heidenreich K, Frey LC, Hamazaki K, Kim H-Y, Murphy RC. J. Am. Soc. Mass Spectrom. 2011;22:1014. doi: 10.1007/s13361-011-0122-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Whitehead SN, Chan KHN, Gangaraju S, Slinn J, Li J, Hou ST. PLoS One. 2011;6:e20808. doi: 10.1371/journal.pone.0020808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Janfelt C, Wellner N, Leger P-L, Kokesch-Himmelreich J, Hansen SH, Charriaut-Marlangue C, Hansen HS. FASEB J. 2012 doi: 10.1096/fj.11-201152. [DOI] [PubMed] [Google Scholar]
- (243).Chaurand P, Schwartz SA, Caprioli RM. Anal. Chem. 2004;76:87A. [PubMed] [Google Scholar]