

Abstract
Kaposi's Sarcoma-associated herpesvirus (KSHV) is maintained as a stable episome in latently infected pleural effusion lymphoma (PEL) cells. Episome maintenance is conferred by the binding of the KSHV-encoded LANA protein to the viral terminal repeats (TR). Here, we show that DNA replication in the KSHV TR is coupled with DNA recombination and mediated in part through the cellular replication fork protection factors Timeless (Tim) and Tipin. We show by two-dimensional (2D) agarose gel electrophoresis that replication forks naturally stall and form recombination-like structures at the TR during an unperturbed cell cycle. Chromatin immunoprecipitation (ChIP) assays revealed that Tim and Tipin are selectively enriched at the KSHV TR during S phase and in a LANA-dependent manner. Tim depletion inhibited LANA-dependent TR DNA replication and caused the loss of KSHV episomes from latently infected PEL cells. Tim depletion resulted in the aberrant accumulation of recombination structures and arrested MCM helicase at TR. Tim depletion did not induce the KSHV lytic cycle or apoptotic cell death. We propose that KSHV episome maintenance requires Tim-assisted replication fork protection at the viral terminal repeats and that Tim-dependent recombination-like structures form at TR to promote DNA repeat stability and viral genome maintenance.
INTRODUCTION
Nearly 20% of human cancers can be attributed to persistent viral infection (1). Human DNA and RNA viruses, such as human papillomavirus (HPV), Epstein-Barr virus (EBV), Kaposi's sarcoma-associated herpesvirus (KSHV), hepatitis B virus, hepatitis C virus, and human T-cell lymphotropic virus, have been associated with a broad spectrum of human cancers. These tumor-associated viruses maintain their genomes through multiple molecular mechanisms, some of which may lead to host genome instability, which is a commonly recognized intrinsic property of all cancers (2–4). Given the importance of genomic stability, it is imperative to understand how these viruses utilize host cell mechanisms to ensure their own survival.
KSHV, also known as human herpesvirus 8 (HHV8), was identified as the causative viral agent of Kaposi's sarcoma (KS), one of the most common cancers in human immunodeficiency virus-infected patients (5). KSHV is also associated with primary effusion lymphoma (PEL) and multicentric Castleman's disease, in addition to endemic forms of KS (6–8). Like all members of the herpesvirus family, KSHV is a double-stranded DNA virus that can persist for the life of the host. The virus can establish a stable latent infection in B lymphocytes similar to that of Epstein-Barr virus (EBV). During latency, the viral genome exists as a multicopy circular episome that replicates by recruiting host cellular replication machinery (9). The virus-encoded protein latency-associated nuclear antigen (LANA) plays a pivotal role in recruiting host cellular factors required for viral genome replication, partitioning, and maintenance during latent infection (10–12). LANA binds two LANA-binding sites (LBS1/2) within each terminal repeat (TR) sequence and is indispensable for viral genome replication and episome stability during latency (13–16).
The KSHV TR region is a complicated viral genome maintenance element. It consists of approximately 40 to 50 tandem repeats with approximately 800 bp of GC-rich, highly homologous DNA arranged in a head-to-tail orientation. Each repeat contains the same two LBS elements, each of which is capable of binding LANA and associated host factors involved in DNA replication origin formation. Despite the essential role of LANA and multiple TRs in viral genome maintenance during latency and the efficient DNA replication activity of plasmids containing multiple TRs bound to LANA, DNA replication can initiate outside the TRs in single-molecule replication assays (17). A similar observation has been made for EBV replication initiating outside OriP (18–21). Nevertheless, genetic evidence indicates that LANA binding to TR is essential for KSHV episome maintenance and genome stability (16). It is therefore important to understand how this genetic element confers viral genome stability and what host factors and processes it employs to achieve this essential viral function.
The mechanisms responsible for the DNA replication and stability of the terminal repeats remain unclear. In both prokaryotes and eukaryotes, replication forks often arrest or slip at repetitive GC-rich elements. It has been previously proposed that replication fork slippage or prolonged stalling induces rearrangements in repeated DNA sequences in all organisms (22–24). In eukaryotes, a stalled replication fork is protected by a specialized complex referred to as the “fork protection complex,” which in humans consists of the proteins Timeless-1 (Tim), Tipin, and Claspin (25–28). The Tim-Tipin complex is thought to function in response to DNA replication fork arrests, including those caused by complex DNA structures or protein-DNA complexes (29, 30). Whether DNA replication forks stall at LANA-binding sites or fork slippage occurs at the KSHV TR is largely unknown, yet it is likely to be critical for understanding how KSHV genome integrity is maintained and, potentially, whether this is linked to the mechanism of episome maintenance (31–33). Here, we investigate the potential role of the fork protection protein Tim at the KSHV TR and in LANA-dependent DNA replication and episome maintenance. Our findings suggest that LANA-dependent DNA replication fork pausing directs the formation of recombination-like structures at the TR and that this process is integrally linked to KSHV genome stability and episome maintenance.
MATERIALS AND METHODS
Cells, plasmids, shRNA, and antibodies.
KSHV-positive PEL cells (BCBL1, JSC-1, BC-1, and BC-3) were grown in RPMI medium (Gibco BRL) containing 15% fetal bovine serum and the antibiotics penicillin and streptomycin (50 U/ml). BJAB cells stably expressing N-terminal FLAG epitope-tagged LANA (FLAG-LANA) and containing the p8xTR plasmid were maintained in RPMI medium containing 10% fetal bovine serum, antibiotics, and 0.6 mg/ml G418. 293T cells were cultured in Dulbecco's modified Eagle's medium with 10% fetal bovine serum and antibiotics. All the cells were cultured at 37°C and 5% CO2. The plasmids containing 2×TR, 8×TR, and FLAG-LANA have been described previously (9, 14). Small hairpin RNAs (shRNAs) for Timeless (shTim) and the control (shCtrl) were obtained from the Sigma/TRC collection library of targeted shRNA plasmids (TRC no. 153090 and 157211), and validated clones were used to generate lentivirus particles in 293T-derived packaging cells. In some experiments (transient DNA replication in HEK293 cells), shRNA vectors were delivered by plasmid DNA transfection. Guinea pig anti-Timeless (a gift from A. Gotter) mouse monoclonal anti-actin (Sigma A3854), and rat anti-KSHV LANA (Advanced Biotechnologies Inc.; 13-210-100) antibodies were used for Western blotting. Anti-KSHV ORF45 antibody was provided by Yan Yuan, University of Pennsylvania, and anti-KSHV ORF50 antibody was provided by Erle Robertson, University of Pennsylvania.
Analysis of recombinational DNA structures.
DNA for the two-dimensional (2D) gels was extracted by the cetyltrimethylammonium bromide (CTAB) method, as described by Allers and Lichten (34). Two-dimensional gel electrophoresis was performed essentially as described previously (35). For 2D gels, DNA was cut with DraI to generate an 8.6-kb fragment containing 8×TR. For one-dimensional gel analysis, DNA was isolated using the CTAB method, as described above, and then digested with Bfa1, which generates ∼800-bp fragments for unit length TRs. In some cases, the fragments were further digested with either mung bean nuclease (MBN) or T7 Endo-1 nucleases (T7endo) (New England Biolabs) for 15 min. at 37°C in restriction enzyme buffer. The DNA was then subjected to Southern transfer to a nylon membrane and hybridized with PCR-generated probes specific for TR or the control 95000 region, which recognizes DNA fragments between positions 95000 and 96500.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed as described previously (36). Rabbit polyclonal antibodies for ChIP were either raised or purchased for anti-IgG (Santa Cruz Biotechnology), anti-FLAG (Sigma), anti-Timeless (a gift from A. Gotter), anti-Tipin (Bethyl), anti-DNA polymerase δ (Polδ) (Bethyl), and mouse monoclonal anti-mcm3 (Abcam). Primers for the KSHV genome included the KSHV TR (GGGCGCCCTCTCTCTACTG; CCCAAACAGGCTCACACACA), KSHV 50000 region (CGGCAGTGCATCCTTTTTAATAA; CCTCTTTTGTGTCTTTCCGTGTCT), and cellular β-actin (GCCATGGTTGTGCCATTACA; GGCCAGGTTCTCTTTTTATTTCTG).
DNA replication assays.
DNA replication assays have been described previously (37). For transient-replication assays in HEK293 cells, p8×TR was used as a replication template and digested with HinDIII or DpnI plus HinDIII. Bromodeoxyuridine (BrdU) incorporation was measured as described previously (38). BrdU combined with propidium iodide (PI) staining for fluorescence-activated cell sorter (FACS) analysis was described previously (39).
KSHV episome maintenance by pulsed-field gel electrophoresis (PFGE).
BCBL1, BC-1 and BC-3, and JSC-1 cells were infected with lentivirus expressing shTim or shCtrl shRNA. Seventy-two hours postinfection, cells were resuspended in agarose plugs and incubated for 48 h at 50°C in lysis buffer (0.2 M EDTA [pH 8.0], 1% sodium sarcosyl, 1 mg/ml proteinase K). The agarose plugs were washed twice in TE buffer (10 mM Tris [pH 7.5] and 1 mM EDTA). Pulsed-field gel electrophoresis was performed as described previously for 23 h at 14°C with a linear ramping pulse of 60 to 120 s through 120°C (Bio-Rad CHEF Mapper) (40). DNA was transferred to nylon membranes by established methods for Southern blotting (41). The DNA was then detected by hybridization with a 32P-labeled probe specific for the KSHV TR region or cellular α-satellite repeat DNA control and visualized with a Molecular Dynamics PhosphorImager.
Establishing p8×TR stable cell lines.
FLAG-LANA-expressing BJAB cells were transfected with p8×TR plasmid. After 48 h, 0.6 mg/ml G418 was added to the medium to select episomal p8×TR cells. Four to 6 weeks after selection, we confirmed the episomal maintenance of p8×TR by Southern blotting.
Centrifugal elutriation.
Cell cycle fractionation using centrifugal elutriation was performed with a modified Beckman JE 5.0 using counterflow rates for KSHV-positive BCBL1 and p8×TR BJAB cells as described previously (38, 42).
BrdU IP and FACS.
Asynchronously growing cells were pulse-labeled with 50 μM BrdU for 30 min and collected by centrifugation, and BrdU immunoprecipitation (IP) was performed essentially as described previously (43). For FACS analysis, asynchronously growing cells were pulse-labeled with 50 μM BrdU for 30 min and collected by centrifugation. The cells were then fixed with 4% paraformaldehyde in vitro for 30 min at 4°C. Following fixation, the cells were washed in 1× phosphate-buffered saline (PBS) (three times for 5 min each), and the BrdU FACS was performed as described previously (43).
Cell cycle profile analysis.
To determine the cell cycle profiles of cells, cultures were either treated or left untreated and fixed in ice-cold 70% ethanol for at least 30 min. After fixation, the cells were stained with staining solution (0.5 mg/ml propidium iodide, 100 mg/ml RNase A) for 30 min in the dark. Samples were analyzed using an EPICS XL (Beckman-Coulter, Inc., Miami, FL), and 50,000 events were recorded. For all flow cytometry experiments, the WINMDI software program (Scripps Institute) was used to analyze the data.
TUNEL assay.
The terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end-labeling (TUNEL) assay to detect apoptotic cells was performed with the Apo-BrdU TUNEL assay kit (A23210; Invitrogen) according to the manufacturer's instructions. The positive (camptothecin-treated) and negative (untreated) control cells were the fixed human lymphoma (HL60) cells provided in the kit.
Genome maintenance assays.
BCBL1 cells were collected at the indicated time points with or without shRNA treatment, and the genomic DNA was isolated using the Promega DNA purification kit (catalog number A1120). After a brief sonication, genomic DNA was then assayed by real-time PCR using primers for the TR and 50000 regions of KSHV and normalized by the cellular DNA signal at the actin gene locus. For Southern analysis, genomic DNA was digested with Bfa1 and hybridized with probes for the KSHV TR, the KSHV 95000 region, and cellular α-satellite DNA.
RESULTS
Replication fork pausing and recombination structure formation at the KSHV TR.
The DNA structures formed during replication and recombination can be analyzed using two-dimensional neutral agarose gel (2D gel) electrophoresis (44–46). Initial attempts to analyze the complete 40 copies of the KSHV TR found in BCBL1 cells were unsuccessful, presumably due to the large number of repeats and the sizes of the DNA fragments. We therefore examined a plasmid containing 8×TRs that is maintained stably in BJAB cells that constitutively express LANA (Fig. 1A). The migration properties of prominent DNA structures have been well established, including the Y arcs formed by replication fork progression, replication pause sites that appear as bulges in the Y arc, origin bubble arcs, and various recombination-like structures that form at sites of collapsed replication forks or replication termination (Fig. 1B) (46). To stabilize recombination-like structures, DNA was isolated with the cationic detergent CTAB, which inhibits branch migration during DNA isolation and manipulation (34). To enrich for any transient structures that occur for short periods of the cell cycle, we isolated cells from different stages of the cell cycle using centrifugal elutriation (Fig. 1C). 2D gel analysis of 8×TR DNA revealed various cell cycle-dependent DNA structures (Fig. 1D). As cells progress through S phase (fractions 22 to 28), Y structures accumulate, along with the formation of a replication fork pause site (Fig. 1D, purple arrow, fraction 24). A weak bubble arc indicative of replication initiation appeared in mid-S phase (red arrow, fraction 28). Remarkably, a prominent double Y and recombination-like structure formed in mid-S phase (yellow arrow, fraction 26).
Fig 1.

Analysis of replication and recombination intermediates of TRs. (A) Schematic of the 8.3-kb DNA fragment containing the 8 TR sequences used for 2D gel analysis. (B) Cartoon interpretation of the major TR DNA replication and recombination structures observed in the 2D gels shown in panel D. (C) BJAB cells containing a p8×TR plasmid were fractionated by centrifugal elutriation and then assayed by FACS analysis of PI-stained cells. The x axis is the PI intensity (DNA content), and the y axis is the cell number (events). (D) Two-dimensional neutral agarose gel analysis of 8×TRs isolated using the CTAB method from asynchronous (Asyn) BJAB cells after cell cycle fractionation. Replication fork pausing (purple arrows), recombination structures (yellow arrows), and replication bubbles (red arrows) are indicated.
To determine if replication intermediates could also be observed in latently infected PEL cells, we examined the structure of TR DNA replication intermediates by 1D agarose gel electrophoresis and Southern blotting. Latently infected BCBL1 cells were subjected to cell cycle fractionation, and DNA from elutriation fractions 22 (G1), 26 (early S), 28 (mid-S), and 36 (G2/M) were isolated and digested with Bfa1, which has one restriction site in the TR and generates an ∼800-bp fragment. We observed that DNA molecules with greater than 1×TR mobility were enriched in elutriation fraction 26 (Fig. 2A, lower panel), the mid-S-phase fraction that is also enriched with recombination structures in 2D gels (Fig. 1D). The slower multimeric TR molecules did not form at other stages of the cell cycle and were dependent on isolation with CTAB (Fig. 2). These structures did not form on a control Bfa1 fragment of comparable size isolated from KSHV genome positions 95000 to 95600 (Fig. 2B). We did observe that CTAB-isolated TR DNA formed doublets at all stages of the cell cycle examined (Fig. 2A, bottom). We further analyzed the DNA structures by enzymatic digestion with T7endo, specific for Holliday junctions, or with MBN, specific for single-strand gaps in double-stranded replication intermediates, such as replication bubble or hemicatenane structures (Fig. 2C). We found that MBN, and to a lesser extent T7endo, showed only partial digestion of TR DNA isolated from fraction 26, suggesting that these structures are complex mixtures of DNA replication and recombination intermediates (Fig. 2C).
Fig 2.
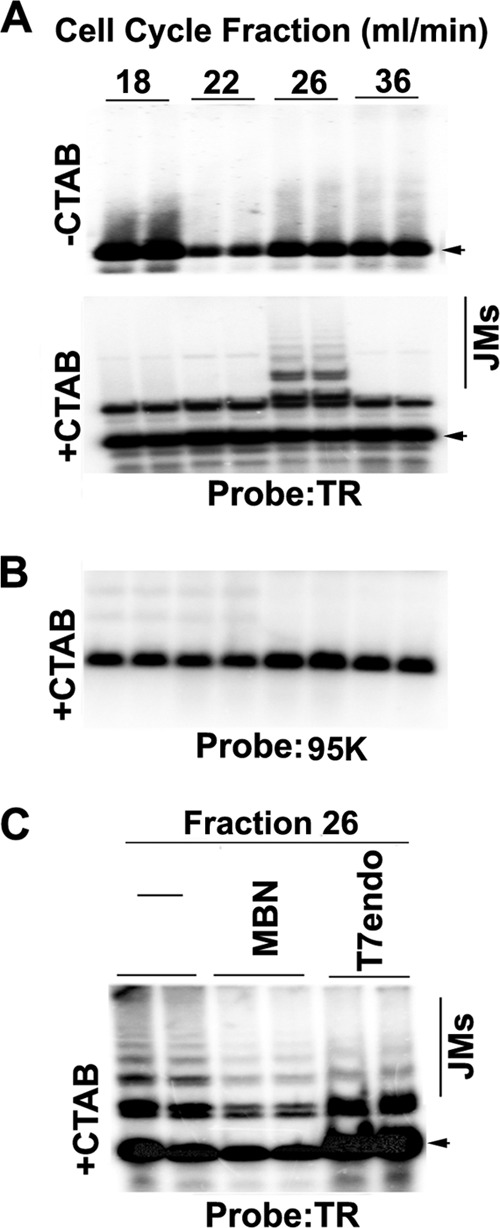
Evidence for recombination junctions formed at TRs. (A and B) BCBL1 cells were cell cycle fractionated by centrifugal elutriation as in Fig. 1C. DNA was extracted from fractions 22 (G1/S), 26 (early S), 28 (mid-S), and 36 (G2/M) with (+) or without (−) CTAB to stabilize recombination-like structures. DNA was isolated and cleaved by BfaI and then analyzed by Southern blotting from a one-dimensional agarose gel with probes for the TR (A) or control probe from the KSHV 95000 region (B). The positions of the expected 1×TR products are indicated by arrowheads. Recombination-generated joint molecules (JMs) of >1-TR size are indicated. (C) DNA was extracted from BCBL1 cells collected in elutriation fraction 26 (mid-S phase) with CTAB, digested with Bfa1, and then mock treated (lanes 1 and 2 from left; −) or treated with MBN (lanes 3 and 4) or with T7endo (lanes 5 and 6). DNA fragments were visualized by Southern blotting with a probe for the TR. Arrowheads indicate a 1× 801-bp TR fragment.
Replication fork pausing factor Tim is enriched at the TR.
To determine if known replication fork protection factors or DNA polymerase subunits accumulate in the TR region of the KSHV genome, we used ChIP assays with antibodies to human Tim, Tipin, Polδ, KSHV LANA, or control IgG. As expected, LANA was highly enriched in the KSHV TR region (Fig. 3A). We found that Tipin was also highly enriched and that Tim and Polδ were enriched relative to the IgG control and relative to a control region of the KSHV genome derived from the 50000 locus (Fig. 3B). We also tested whether Tim association with TR was dependent on LANA binding (Fig. 3C). A TR-containing plasmid (p2xTR) was transfected into HEK293 cells with or without a FLAG-LANA expression vector and then assayed by ChIP for binding of Tim or FLAG-LANA. We found that Tim bound to TR only in cells expressing FLAG-LANA (Fig. 3C). These results indicate that the replisome pausing and protection factor Tim associates with TRs in a LANA-dependent manner.
Fig 3.

Replication fork pausing factor TIM associates with the TR region of KSHV. (A) Schematic of KSHV TR organization and LANA-binding sites within each TR. (B) ChIP assay of BCBL1 cells with antibodies to LANA, Tim, Tipin, Polδ, or control IgG. ChIP DNA was analyzed by quantitative PCR (qPCR) with primers specific for the TR or the 50000 region. (C) 293 cells were transfected with p2xTR with either pFLAG-LANA or the pFLAG vector control and then assayed by ChIP at 72 h posttransfection. ChIP was performed with antibodies against FLAG, Tim, or control IgG and assayed for TR DNA by real-time PCR. (D) BCBL1 cells were fractionated by centrifugal elutriation and then assayed by PI staining and FACS. (E) ChIP assay with anti-LANA or control IgG at the TR region or the control 50000 region for each stage of the cell cycle, as indicated. (F) ChIP assays with antibody to Tim, Polδ, or control IgG were performed with primers for TR or the control 50000 region for each stage of the cell cycle, as indicated. The error bars represent standard deviations from the mean for at least three experimental replicates. P values were calculated by two-tailed t tests comparing the TR to control regions for relevant elutriation fractions. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We next tested whether the interaction of LANA, Tim, or Polδ with KSHV TRs is cell cycle dependent (Fig. 3D to F). BCBL1 cells were fractionated by centrifugal elutriation for separation of cell cycle stages, which were confirmed by PI staining followed by FACS analysis (Fig. 3D). Cell cycle-fractionated cells were then subjected to ChIP with antibodies to LANA and control IgG (Fig. 3E) or Tim, Polδ, and IgG (Fig. 3F). We found that LANA was equally enriched in the TR relative to the 50000 region at all stages of the cell cycle (Fig. 3E). In contrast, both Tim and Polδ were enriched in the TR in a cell cycle-dependent manner, with the predominant peaks of enrichment occurring at early to mid-S phase (fractions 22 and 24) for Tim (Fig. 3F). Neither Tim or Polδ was enriched in the KSHV 50000 region, indicating that there is selective enrichment of these factors at the TR.
Tim depletion inhibits TR DNA replication.
To determine if Tim has a functional role in TR replication, we tested the effect of Tim depletion on the transient DNA replication of TR-containing plasmids (Fig. 4). Plasmids containing 8×TRs are competent for transient replication in HEK293 cells that coexpress LANA (47, 48). We therefore assayed the effects of shRNA-mediated Tim depletion on transient replication of 8×TR plasmids in HEK293 cells that were cotransfected with either FLAG-LANA or control FLAG vector (FV). We used two different shRNA-containing vectors that target different regions of the Tim mRNA. Transient shRNA transfection led to partial depletion of Tim but had no significant effect on the expression of FLAG-LANA (Fig. 4A). Transient DNA replication of 8×TRs was measured by comparing the DpnI-resistant DNA (Fig. 4B, arrow) relative to total HinDIII-digested DNA in Southern blot analysis. We found that Tim depletion led to an ∼4- to 5-fold reduction in LANA-dependent DNA replication of p8×TR at 72 h posttransfection (Fig. 4B and C). The inhibition of 8×TR replication was not a result of an indirect cell cycle arrest, since FACS profiling showed no major cell cycle changes relative to shCtrl-treated cells (Fig. 4D).
Fig 4.
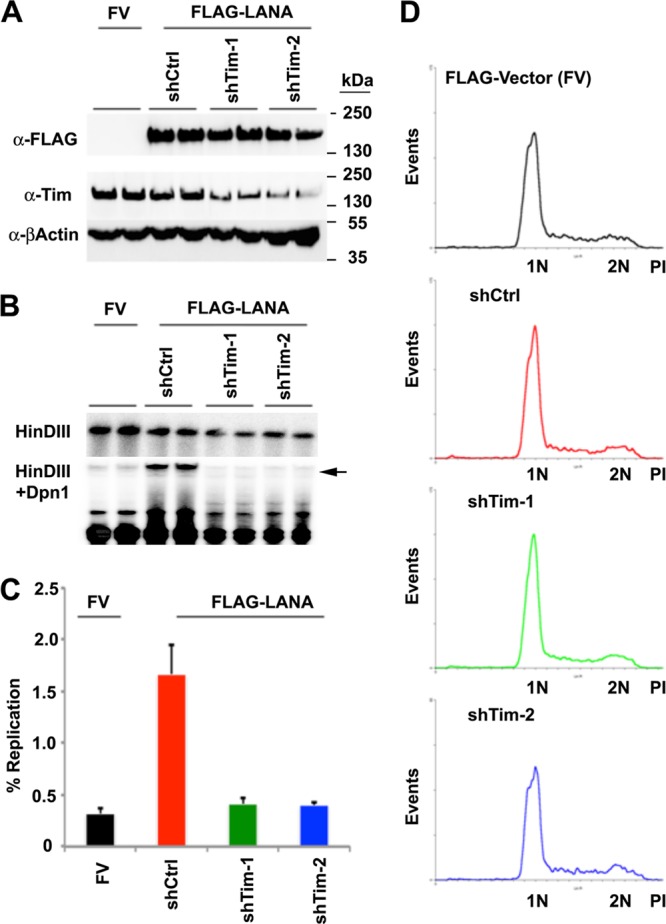
Inhibition of TR DNA replication by Tim depletion. (A) Western blot of cell extracts from 293 cells transfected with FV or FLAG-LANA expression vectors and with plasmid expression vectors for shCtrl, shTim-1, or shTim-2, as indicated above each lane. Western blots were probed with antibodies to FLAG (α-FLAG) (top), Tim (middle), or β-actin (bottom). (B) Transient DNA replication assay for p8xTR plasmid in 293 cells cotransfected with FV or FLAG-LANA with or without shTim plasmid cotransfection. Plasmid DNA was isolated 72 h posttransfection and subjected to restriction enzyme digestion with DpnI-HinDIII (bottom) or HinDIII alone (top). The arrow indicates DpnI-resistant replicated 8×TR DNA. (C) Quantification of the ratio between DpnI-resistant and total HinDIII-cut DNA to measure the percentage of 8-TR DNA replication. The error bars indicate standard deviations for three experimental replicates. (D) FACS analysis of PI-stained cells for cell cycle profile analysis of shCtrl- and shTim-transfected cells used for replication assays shown in panel B.
Tim depletion causes loss of episomal KSHV genomes from latently infected cells.
To determine if Tim plays a role in KSHV episomal maintenance, we assayed the effect of Tim depletion on KSHV genomes in latently infected PEL cells (Fig. 5). BCBL1 cells were infected with lentivirus expressing Tim shRNAs (shTim-1 or shTim-2) or control shRNA. We found Tim protein was efficiently depleted (>85%) with no detectable loss of β-actin protein expression and only a small decrease in LANA protein expression (Fig. 5A). To monitor KSHV episomes, we assayed KSHV-positive cells by PFGE, followed by Southern blotting (Fig. 5B to D). Ethidium bromide staining of PFGE gels indicated that Tim depletion did not lead to a gross degradation of chromosomal DNA, and equal amounts of total DNA were loaded for each sample (Fig. 5B). Hybridization with a probe for the cellular α-satellite repeat DNA also indicated that equal amounts of cellular DNA were loaded and intact (Fig. 5C, top). Hybridization of PFGE blots with KSHV TR DNA detected both circular (slower-migrating form) and linear (faster-migrating forms) genomes (Fig. 5C, bottom). Quantification of several independent PFGE experiments revealed that shTim depletion with either shTim-1 or shTim-2 led to an ∼3- to 4-fold loss of KSHV circular episomes relative to cellular DNA (Fig. 5D). Linear forms of the KSHV genome were also reduced, indicating that Tim depletion does not trigger KSHV lytic replication. These findings suggest that Tim depletion leads to a loss of KSHV genomes from latently infected BCBL1 cells. To rule out the possibility that this is a BCBL1 cell line-specific effect, we repeated the experiment with three different KSHV-positive PEL cell lines, JSC-1, BC-1, and BC-3 (Fig. 5E to I). In each cell line, Tim depletion was monitored (Fig. 5E) and KSHV episome maintenance was measured by PFGE and Southern blotting (Fig. 5F and G). We found that Tim depletion leads to a significant loss of KSHV episomes in various PEL cell lines (Fig. 5H).
Fig 5.

Loss of KSHV episomal form in PEL cells after Tim depletion. (A) BCBL1 cells were infected with shCtrl, shTim-1, or shTim-2 and then assayed by Western blotting for Tim (top), LANA (middle), or β-actin (bottom). (B) Ethidium bromide (EtBr) staining of PFGE gels of BCBL1 cells after infection with shCtrl or shTim lentiviruses for 72 h. (C) A Southern blot of PFGE gels was probed in cellular α-satellite DNA (top) or the KSHV TR region (bottom). Episomes and linear forms of KSHV genomes are indicated. (D) Quantification of the percentage of episomal DNA loss between shCtrl- and shTim-treated cells relative to α-satellite DNA as determined by PhosphorImager analysis. P values were determined by a chi-square test. (E) KSHV-positive PEL cell lines JSC-1, BC-1, and BC-3 were infected with shCtrl or shTim for 72 h and then assayed by Western blotting for Tim (top) or β-actin (bottom). (F) EtBr staining of PFGE for JSC-1, BC-1, and BC-3 cells at 72 h after lentivirus infection. (G) A Southern blot of PFGE gels was probed in cellular α-satellite DNA (top) or the KSHV TR region (bottom). KSHV episomal genomes are indicated. (H) Quantification of the percentage of KSHV episomal DNA loss between shCtrl and shTim relative to cellular α-satellite as determined by PhosphorImager analysis of PFGE. P values were determined by a chi-square test. The error bars indicate standard deviations.
Tim depletion leads to a selective loss of KSHV DNA.
The loss of KSHV episomes from PEL cells could be due to a failure of defectively replicated genomes to enter PFGE gels. To better assess the mechanism of KSHV genome loss from BCBL1 cells, we performed Southern blot analysis of total genomic DNA without addition of CTAB at 48 and 72 h postinfection with shTim lentivirus (Fig. 6). Ethidium bromide staining of total cellular DNA did not reveal a global loss of DNA integrity at these times after Tim depletion (Fig. 6A, right). Southern blot hybridization with a TR probe revealed the formation of doublets at 48 h and a reduction of signal intensity at 72 h. A second probe for KSHV DNA (95000 region) revealed a loss of signal intensity at 48 and 72 h relative to cellular α-satellite repeat DNA (Fig. 6B). Real-time PCR analysis of KSHV genome relative copy numbers also indicated a loss of KSHV DNA in the TR and 50000 region at 72 h, with a slight increase in TR copy numbers occurring at the 48-h time point (Fig. 6C). This is consistent with Southern blot analysis showing stable doublet forms of TR at the 48-h time point.
Fig 6.
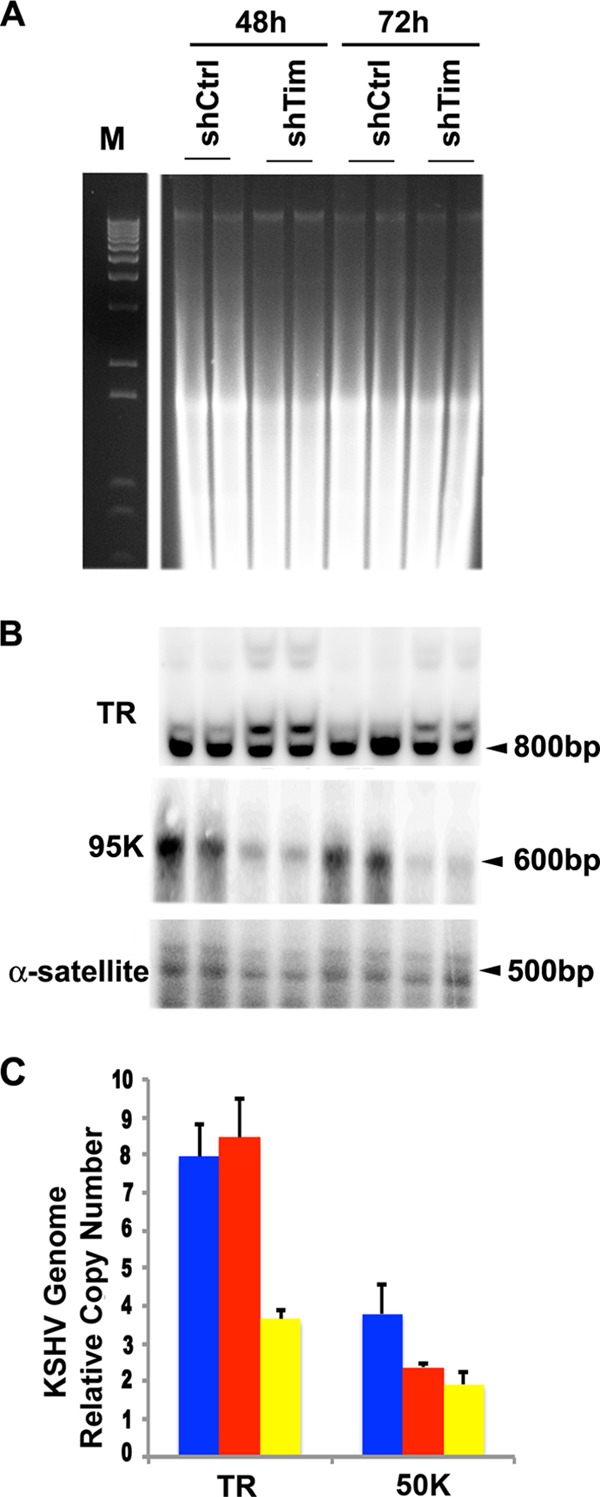
Selective loss of KSHV DNA from BCLB1 cells. (A) DNA from BCLB1 cells treated with shCtrl or shTim for 48 or 72 h was isolated by a total genomic DNA isolation procedure, followed by digestion with BfaI, and then analyzed by ethidium bromide staining after agarose gel electrophoresis (right). M, molecular weight markers. (B) A Southern blot of the same gel as in panel A was probed with TR (top), KSHV 95000 (middle), or cellular α-satellite repeat (bottom) DNA. (C) The KSHV genome copy number was determined from DNA isolated from BCBL1 cells after infection with shCtrl or shTim for 48 or 72 h and assayed by real-time PCR with primers specific for the TR or 95000 region relative to β-actin DNA using the ΔCt method. The error bars indicate standard deviations.
Tim depletion does not block total cellular DNA synthesis or induce KSHV reactivation or apoptosis.
To determine if Tim depletion in PEL cells had additional effects on the host cells, we examined the cell cycle profile after FACS analysis using PI staining at 48 h after lentivirus infection (Fig. 7A). We found that Tim depletion led to an increase in G2/M cells relative to cells in S and G1 phases. This is consistent with previous findings that Tim depletion leads to an elongated G2/M phase but only modest reduction in global DNA synthesis, as measured by BrdU incorporation (49). We also examined whether Tim depletion resulted in activation of KSHV lytic-cycle gene expression (Fig. 7B). Western blot analysis of BCBL1 cells infected with shTim-1 or shTim-2 lentivirus showed an efficient depletion of Tim protein but no apparent increase in the KSHV immediate-early proteins ORF50 and ORF45 (Fig. 7B). This indicates that Tim depletion does not cause KSHV lytic reactivation. We next examined the effects of shTim on the replication rates of the KSHV TR region by measuring site-specific BrdU incorporation at 48 h postinfection (Fig. 7C). BCBL1 cells were pulse-labeled with BrdU, followed by BrdU IP assays with PCR analysis at the positions to the left of the TR (TR-L), within the TR (TR), or to the right of the TR (TR-R). Tim depletion produced a small increase in BrdU incorporation at the TR but no consistent or significant change at positions to the left or right of the TR (Fig. 7C). This result is consistent with a role for Tim in regulating replication fork stalling in the TR region. We also tested whether global BrdU incorporation was altered by Tim depletion (Fig. 7D). We found that Tim depletion led to a small decrease in global BrdU incorporation (from 49% to 42% or 44%), consistent with the observed reduction in S-phase cells (Fig. 7A). Finally, we tested whether Tim depletion induced cellular apoptosis (Fig. 7E). BCBL1 cells infected with shCtrl or shTim were analyzed by Apo-BrdU TUNEL assay (Fig. 7E). While positive-control samples for apoptosis scored highly positive for BrdU, essentially no TUNEL-positive apoptotic cells were detected in Tim-depleted or control shRNA-infected cells. Thus, Tim depletion does not cause apoptosis in BCBL1 cells.
Fig 7.

Tim depletion does not cause cell cycle arrest, apoptosis, or lytic reactivation of KSHV in BCBL1 cells. (A) BCBL1 cells were infected with shCtrl, shTim-1, or shTim-2 for 48 h and then stained with PI for FACS analysis. The cell cycle distribution is presented graphically as percentages of total cells in G1, S, and G2/M. (B) Western blot analysis of KSHV lytic proteins Rta and ORF45 and control actin for BCBL1 cells after infection with shCtrl or shTim or after treatment with tetradecanoyl phorbol acetate (TPA) and NaB for 48 h, as indicated. (C) BrdU-IP assay for BCBL1 cells after infection with shCtrl or shTim. BrdU incorporation was assayed by qPCR at KSHV locations for TRs, TR-L, and TR-R, as indicated. The error bars indicate standard deviations from the mean, and P values were determined by a chi-square test. (D) The percentages of cells undergoing DNA synthesis were measured by pulse-labeling with BrdU for 30 min, followed by staining with PI. BrdU intensity (Int) was monitored by anti-BrdU-conjugated fluorescein isothiocyanate (FITC) (y axis), and PI was monitored on the x axis. Shown are BCBL1 cells with no BrdU control (top left), shCtrl (top right), shTim-1 (bottom left), or shTim-2 (bottom right). (E) Apoptotic cells were quantified using the Apo-BrdU TUNEL assay. Control samples were camptothecin treated (+Con) or untreated (−Con) HL60 cells provided by the manufacturer (top). BCBL1 cells 48 h postinfection with shCtrl (bottom left) or shTim-1 (bottom right) are also shown. FACS analyses were performed for two independent experiments, and a representative experiment is shown.
Tim depletion causes accumulation of replication pause and recombination structures at the TR.
To further investigate the molecular basis for the loss of KSHV episomal maintenance caused by Tim depletion, we assayed the effect of Tim depletion on replication fork pausing structures at the TR using 2D neutral agarose gel analysis (Fig. 8). BJAB cells were infected with shCtrl or shTim lentivirus and then assayed 48 h postinfection (Fig. 8A and B). In asynchronous shCtrl cells, few replication fork pausing structures were observed (Fig. 8A, left), consistent with previous observations (Fig. 1). In contrast, replication fork pausing structures markedly accumulated in shTim-infected cells (Fig. 8A, right). shTim cells had ∼4-fold-increased replication fork and recombination structures, suggesting that replication was not being completed or that replication fork structures were inducing the formation of pathological recombination structures that destabilized the TR. We further characterized these structures by 1D gel electrophoresis (Fig. 8C). We found that Tim-depleted cells accumulated higher-order TR DNA structures, while these structures were absent in control shRNA. The structures were partially sensitive to both MBN and T7endo, suggesting that abnormal recombination structures were accumulating at the TR in shTim-depleted cells. We also tested the effect of Tim depletion on LANA, ORC2, and MCM3 binding by ChIP assay (Fig. 8D and E). We found that Tim depletion had no significant effect on LANA or ORC2 (Fig. 8D) but increased MCM3 occupancy ∼4-fold (Fig. 8E). These findings are consistent with a role for Tim in facilitating MCM helicase movement through or from the TR.
Fig 8.

Tim depletion causes accumulation of paused replication and recombination structures at the TR. (A) 2D neutral agarose gel electrophoresis and Southern blot analysis of p8xTR DNA in BJAB cells after infection with shCtrl (left) or shTim (right) lentivirus for 48 h. The arrow indicates the migration of recombination-like structures. (B) Western blot of cells used for pulsed-field analysis. Antibodies for Tim (top), LANA (middle), and β-actin (bottom) are indicated. (C) Scheme of 0.8-kb Bfa1-digested TR fragment (top) used for 1D gel analysis of replication and recombination structures (bottom). DNA was extracted from shCtrl- or shTim-infected cells with CTAB, digested with Bfa1, and then mock treated (lanes 1 to 4 from left; −) or treated with MBN (lanes 5 to 8) or with T7endo (lanes 9 to 12), as indicated. DNA fragments were visualized by Southern blotting with a probe for the TR. Recombination-generated JMs are indicated. (D) ChIP assays with anti-LANA, anti-ORC2, or control IgG were performed with primers for the TR region or the control 50000 region for shCtrl or shTim, as indicated. (E) ChiP assay as in panel D with anti-MCM3 or control IgG. The error bars represent standard deviations from the mean for at least three experimental replicates.
DISCUSSION
Eukaryotic DNA replication fork progression varies depending on DNA structure and nucleoprotein composition. There are genomic sites that inhibit replication fork progression, called replication fork blocking sites (RFBs), that are conserved from DNA viruses and bacteria to humans (30, 50). RFBs have been implicated in replication termination, DNA recombination, and genome maintenance and commonly function in association with the replication fork protection complex (51, 52). We and others have demonstrated that the EBV genome contains regions with different replication rates (18, 39) and that the EBV OriP can function as a site-specific RFB that requires Tim and Tipin for episome maintenance (43, 53). Like that of EBV, the KSHV episome maintenance element is a repetitive DNA structure that binds to the viral LANA and is responsible for faithful segregation of the viral genome during cell division. We show here that the KSHV episome maintenance element shares important common features in terms of replication fork-blocking activity, formation of recombination junctions, and requirement for the replication fork protection complex.
In this study, we provide evidence that the replication fork stalling and recombination-like structures occur in one or more TRs of KSHV (Fig. 1D), suggesting that recombination junctions are intermediates in the replication process of TR (Fig. 2C). Components of the replication fork protection complex, Tim and Tipin, are recruited to this region in a cell cycle- and LANA-dependent manner (Fig. 3). Tim depletion inhibited TR plasmid DNA replication (Fig. 4) and resulted in the loss of stable episomes in latently infected PEL cells (Fig. 5). Tim depletion caused the selective loss of KSHV DNA from BCBL1 cells (Fig. 6) but caused only moderate reduction in global cellular DNA synthesis, as measured by incorporation of BrdU (Fig. 7). Furthermore, Tim depletion caused aberrant replication and recombination of the TR (Fig. 8), which may activate deleterious rearrangement in this region, destabilizing the TR. These results indicate that Tim cooperates with LANA to facilitate faithful replication of the TRs and suggests that TR-associated recombination structures contribute directly to KSHV episome maintenance.
Our findings are consistent with the established roles of TR and LANA in KSHV episome maintenance and DNA replication (10, 11). Our results are less consistent with single-molecule-fiber fluorescence in situ hybridization (FISH) studies that suggest that replication neither initiates nor pauses in TRs in latently infected BCBL1 cells (17). In contrast to single-molecule studies, we found that replication pausing and recombination occur and, to a lesser extent, origin bubbles form at KSHV TRs. These structures were readily detected in 8×TR plasmids that were maintained as stable episomes in LANA-expressing BJAB cells. For technical reasons, we were unable to resolve TR replication structures with DNA from full-length KSHV genomes due to the very large number of repeats. However, we could detect recombination-like structures in viral genomes derived from BCBL1 cells using 1D gel methods (Fig. 2). We also observed enrichment of Tim, Tipin, and Polδ, which further suggests that replication pausing occurs at TRs in latently infected PEL cells (Fig. 3). Numerous differences in the methods employed may account for these conflicting observations (17). First, replication initiation within TRs is likely to be more frequent on small plasmids lacking alternative zones of DNA replication, as was observed for EBV OriP-containing plasmids (18). We also found that replication pausing and origin bubbles could be detected only in cells highly enriched in S phase and that these structures represent less than 1% of the total population of DNA structures isolated from asynchronous cells. Therefore, it is possible that single-molecule analysis underrepresents these relatively rare replication events due to the limited number of molecules examined. Single-molecule analysis may also lack the resolution to observe short pausing and recombination events that occur within the TR DNA, which may be less than ∼1 kb. While we cannot exclude the possibility that 8×TR plasmid replication is different than that of full-length KSHV genomes, our findings support roles for Tim, Tipin, and Polδ accumulation in both 8×TR plasmids and viral episomes in latently infected cells. Therefore, we favor the interpretation that LANA-dependent replication initiation, pausing, and recombination do occur at some essential level within TRs in latently infected B cells.
Programmed pausing of DNA replication forks can have specialized chromosome functions. In prokaryotes, RFBs promote a form of sister chromatid interaction referred to as chromosome kissing, due to the formation of hemicatenated DNA replication intermediates (54). Persistence of these DNA junctions provides a mechanism of sister chromatid cohesion that may facilitate homologous recombination repair in G2 phase and amphitelic chromosome segregation in mitosis (55). In eukaryotes, different types of replication fork barriers may lead to different DNA structures and chromosome functions. Our studies suggest that while both EBV and KSHV maintenance elements have RFB activities, the DNA structures have significantly different properties. In particular, we found that upon Tim depletion, replication forks at EBV OriP collapse and induce the formation of DNA double-strand breaks (43). In contrast, Tim depletion at KSHV TRs prolongs replication fork pausing and induces pathological recombination (Fig. 8). It has been proposed that the recombination proteins mediate the spatial chromosome organization and pairing during meiotic cell division (56). Recombination precedes the dynamic chromosomal movement to biorient chromosomes for equal segregation. It would be interesting to know whether replication-coupled recombination at KSHV TRs may work as a homologue pairing center to spatially organize its episomes for faithful segregation. The Tim homologue in yeast, Swi1, differentially regulates recombination at RFBs based on their genomic context (57). At the mating type switch locus in Schizosaccharomyces pombe, Swi1 is required for an epigenetically stable imprinting signal (58, 59). Whether similar imprinting occurs at the KSHV and EBV maintenance elements is not known, but the establishment of stable episomal maintenance has been shown to depend on a rare epigenetic event (60). It is therefore tempting to speculate that programmed pausing by LANA at the TR and by EBNA1 at OriP generates critical epigenetic recombination-dependent structures essential for the establishment and maintenance of episomal viral genomes.
In conclusion, KSHV episome stability requires the fork protection complex protein Tim to regulate the formation of recombination-like structures that form during latent-cycle replication of the TRs. Further work is needed to understand and identify specific partners involved in this Tim-dependent replication pathway to ensure KSHV TR stability. Finally, it remains to be shown that programmed DNA replication fork barriers, induced by proteins like EBNA1 and LANA, and the formation of DNA recombination structures at these viral episome maintenance elements contribute directly to viral chromosome segregation, as has been proposed in other organisms.
ACKNOWLEDGMENTS
We thank Andreas Wiedmer for technical support and the Wistar Institute Cancer Center Core Facilities for genomics and flow cytometry.
This work was supported by NIH grant CA117830 to P.M.L. and NIH/NCI grant RO1 CA082036 to K.M.K. The Wistar Institute received NCI Cancer Center core grant P30 CA10815.
Footnotes
Published ahead of print 16 January 2013
REFERENCES
- 1. Parkin DM. 2006. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 118:3030–3044 [DOI] [PubMed] [Google Scholar]
- 2. Butel JS, Fan H. 2012. The diversity of human cancer viruses. Curr. Opin. Virol. 2:449–452 [DOI] [PubMed] [Google Scholar]
- 3. Martin D, Gutkind JS. 2008. Human tumor-associated viruses and new insights into the molecular mechanisms of cancer. Oncogene 27(Suppl. 2):S31–S42 [DOI] [PubMed] [Google Scholar]
- 4. Moore PS, Chang Y. 2010. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 10:878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869 [DOI] [PubMed] [Google Scholar]
- 6. Cesarman E. 2002. The role of Kaposi's sarcoma-associated herpesvirus (KSHV/HHV-8) in lymphoproliferative diseases. Recent Results Cancer Res. 159:27–37 [DOI] [PubMed] [Google Scholar]
- 7. Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 1:273–296 [DOI] [PubMed] [Google Scholar]
- 8. Mesri EA, Cesarman E, Boshoff C. 2010. Kaposi's sarcoma and its associated herpesvirus. Nat. Rev. Cancer. 10:707–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stedman W, Deng Z, Lu F, Lieberman PM. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi's sarcoma-associated herpesvirus latent replication origin. J. Virol. 78:12566–12575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ballestas ME, Kaye KM. 2011. The latency-associated nuclear antigen, a multifunctional protein central to Kaposi's sarcoma-associated herpesvirus latency. Future Microbiol. 6:1399–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lieberman PM, Hu J, Renne R. 2007. Chapter 24, Maintenance and replication during latency. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 12. Verma SC, Lan K, Robertson E. 2007. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 312:101–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644 [DOI] [PubMed] [Google Scholar]
- 14. Ballestas ME, Kaye KM. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 75:3250–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garber AC, Hu J, Renne R. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 277:27401–27411 [DOI] [PubMed] [Google Scholar]
- 16. Ye FC, Zhou FC, Yoo SM, Xie JP, Browning PJ, Gao SJ. 2004. Disruption of Kaposi's sarcoma-associated herpesvirus latent nuclear antigen leads to abortive episome persistence. J. Virol. 78:11121–11129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verma SC, Lu J, Cai Q, Kosiyatrakul S, McDowell ME, Schildkraut CL, Robertson ES. 2011. Single molecule analysis of replicated DNA reveals the usage of multiple KSHV genome regions for latent replication. PLoS Pathog. 7:e1002365 doi:10.1371/journal.ppat.1002365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Norio P, Schildkraut CL. 2004. Plasticity of DNA replication initiation in Epstein-Barr virus episomes. PLoS Biol. 2:e152 doi:10.1371/journal.pbio.0020152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Norio P, Schildkraut CL. 2001. Visualization of DNA replication on individual Epstein-Barr virus episomes. Science 294:2361–2364 [DOI] [PubMed] [Google Scholar]
- 20. Norio P, Schildkraut CL, Yates JL. 2000. Initiation of DNA replication within oriP is dispensable for stable replication of the latent Epstein-Barr virus chromosome after infection of established cell lines. J. Virol. 74:8563–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ott E, Norio P, Ritzi M, Schildkraut C, Schepers A. 2011. The dyad symmetry element of Epstein-Barr virus is a dominant but dispensable replication origin. PLoS One 6:e18609 doi:10.1371/journal.pone.0018609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldfless SJ, Morag AS, Belisle KA, Sutera VA, Jr, Lovett ST. 2006. DNA repeat rearrangements mediated by DnaK-dependent replication fork repair. Mol. Cell 21:595–604 [DOI] [PubMed] [Google Scholar]
- 23. Saveson CJ, Lovett ST. 1999. Tandem repeat recombination induced by replication fork defects in Escherichia coli requires a novel factor, RadC. Genetics 152:5–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Viguera E, Canceill D, Ehrlich SD. 2001. Replication slippage involves DNA polymerase pausing and dissociation. EMBO J. 20:2587–2595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chou DM, Elledge SJ. 2006. Tipin and Timeless form a mutually protective complex required for genotoxic stress resistance and checkpoint function. Proc. Natl. Acad. Sci. U. S. A. 103:18143–18147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gotter AL, Suppa C, Emanuel BS. 2007. Mammalian TIMELESS and Tipin are evolutionarily conserved replication fork-associated factors. J. Mol. Biol. 366:36–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noguchi E, Noguchi C, McDonald WH, Yates JR, III, Russell P. 2004. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol. Cell. Biol. 24:8342–8355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK. 2007. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 27:3131–3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lee JA, Carvalho CM, Lupski JR. 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131:1235–1247 [DOI] [PubMed] [Google Scholar]
- 30. Mirkin EV, Mirkin SM. 2007. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 71:13–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. 2005. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 19:1905–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Voineagu I, Freudenreich CH, Mirkin SM. 2009. Checkpoint responses to unusual structures formed by DNA repeats. Mol. Carcinog. 48:309–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Voineagu I, Narayanan V, Lobachev KS, Mirkin SM. 2008. Replication stalling at unstable inverted repeats: interplay between DNA hairpins and fork stabilizing proteins. Proc. Natl. Acad. Sci. U. S. A. 105:9936–9941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Allers T, Lichten M. 2000. A method for preparing genomic DNA that restrains branch migration of Holliday junctions. Nucleic Acids Res. 28:e6 doi:10.1093/nar/28.2.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brewer BJ, Fangman WL. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51:463–471 [DOI] [PubMed] [Google Scholar]
- 36. Chau CM, Lieberman PM. 2004. Dynamic chromatin boundaries delineate a latency control region of Epstein-Barr virus. J. Virol. 78:12308–12319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deng Z, Atanasiu C, Burg JS, Broccoli D, Lieberman PM. 2003. Telomere repeat binding factors TRF1, TRF2, and hRAP1 modulate replication of Epstein-Barr virus OriP. J. Virol. 77:11992–12001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhou J, Chau CM, Deng Z, Shiekhattar R, Spindler MP, Schepers A, Lieberman PM. 2005. Cell cycle regulation of chromatin at an origin of DNA replication. EMBO J. 24:1406–1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou J, Snyder A, Lieberman PM. 2009. Epstein-Barr virus episome stability is coupled to a delay in replication timing. J. Virol. 83:2154–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Johnson PG, Beerman TA. 1994. Damage induced in episomal EBV DNA in Raji cells by antitumor drugs as measured by pulsed field gel electrophoresis. Anal. Biochem. 220:103–114 [DOI] [PubMed] [Google Scholar]
- 41. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 42. Ritzi M, Tillack K, Gerhardt J, Ott E, Humme S, Kremmer E, Hammerschmidt W, Schepers A. 2003. Complex protein-DNA dynamics at the latent origin of DNA replication of Epstein-Barr virus. J. Cell Sci. 116:3971–3984 [DOI] [PubMed] [Google Scholar]
- 43. Dheekollu J, Lieberman PM. 2011. The replisome pausing factor Timeless is required for episomal maintenance of latent Epstein-Barr virus. J. Virol. 85:5853–5863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brewer BJ, Fangman WL. 1991. Mapping replication origins in yeast chromosomes. Bioessays 13:317–322 [DOI] [PubMed] [Google Scholar]
- 45. Gerbi SA. 2005. Mapping origins of DNA replication in eukaryotes. Methods Mol. Biol. 296:167–180 [DOI] [PubMed] [Google Scholar]
- 46. Liberi G, Cotta-Ramusino C, Lopes M, Sogo J, Conti C, Bensimon A, Foiani M. 2006. Methods to study replication fork collapse in budding yeast. Methods Enzymol. 409:442–462 [DOI] [PubMed] [Google Scholar]
- 47. Hu J, Garber AC, Renne R. 2002. Analysis of cis-and trans-requirements of LANA-dependent DNA replication in dividing cells. J. Virol. 76:11677–11687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hu J, Renne R. 2005. Characterization of the minimal replicator of Kaposi's sarcoma-associated herpesvirus latent origin. J. Virol. 79:2637–2642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dheekollu J, Wiedmer A, Hayden J, Speicher D, Gotter AL, Yen T, Lieberman PM. 2011. Timeless links replication termination to mitotic kinase activation. PLoS One 6:e19596 doi:10.1371/journal.pone.0019596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. White EJ, Emanuelsson O, Scalzo D, Royce T, Kosak S, Oakeley EJ, Weissman S, Gerstein M, Groudine M, Snyder M, Schubeler D. 2004. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc. Natl. Acad. Sci. U. S. A. 101:17771–17776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leman AR, Noguchi C, Lee CY, Noguchi E. 2010. Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 123:660–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leman AR, Noguchi E. 2012. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle 11:3945–3955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gahn TA, Schildkraut CL. 1989. The Epstein-Barr virus origin of plasmid replication, oriP, contains both the initiation and termination sites of DNA replication. Cell 58:527–535 [DOI] [PubMed] [Google Scholar]
- 54. Bastia D, Singh SK. 2011. “Chromosome kissing” and modulation of replication termination. Bioarchitecture 1:24–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maher RL, Branagan AM, Morrical SW. 2011. Coordination of DNA replication and recombination activities in the maintenance of genome stability. J. Cell. Biochem. 112:2672–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Storlazzi A, Gargano S, Ruprich-Robert G, Falque M, David M, Kleckner N, Zickler D. 2010. Recombination proteins mediate meiotic spatial chromosome organization and pairing. Cell 141:94–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pryce DW, Ramayah S, Jaendling A, McFarlane RJ. 2009. Recombination at DNA replication fork barriers is not universal and is differentially regulated by Swi1. Proc. Natl. Acad. Sci. U. S. A. 106:4770–4775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dalgaard JZ, Klar AJ. 2000. swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe. Cell 102:745–751 [DOI] [PubMed] [Google Scholar]
- 59. Vengrova S, Dalgaard JZ. 2004. RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching. Genes Dev. 18:794–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Leight ER, Sugden B. 2001. Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol. Cell. Biol. 21:4149–4161 [DOI] [PMC free article] [PubMed] [Google Scholar]