

Abstract
Entry of enveloped viruses requires fusion of viral and cellular membranes. Fusion requires the formation of an intermediate stalk structure, in which only the outer leaflets are fused. The stalk structure, in turn, requires the lipid bilayer of the envelope to bend into negative curvature. This process is inhibited by enrichment in the outer leaflet of lipids with larger polar headgroups, which favor positive curvature. Accordingly, phospholipids with such shape inhibit viral fusion. We previously identified a compound, 5-(perylen-3-yl)ethynyl-2′-deoxy-uridine (dUY11), with overall shape and amphipathicity similar to those of these phospholipids. dUY11 inhibited the formation of the negative curvature necessary for stalk formation and the fusion of a model enveloped virus, vesicular stomatitis virus (VSV). We proposed that dUY11 acted by biophysical mechanisms as a result of its shape and amphipathicity. To test this model, we have now characterized the mechanisms against influenza virus and HCV of 5-(perylen-3-yl)ethynyl-arabino-uridine (aUY11), which has shape and amphipathicity similar to those of dUY11 but contains an arabino-nucleoside. aUY11 interacted with envelope lipids to inhibit the infectivity of influenza virus, hepatitis C virus (HCV), herpes simplex virus 1 and 2 (HSV-1/2), and other enveloped viruses. It specifically inhibited the fusion of influenza virus, HCV, VSV, and even protein-free liposomes to cells. Furthermore, aUY11 inhibited the formation of negative curvature in model lipid bilayers. In summary, the arabino-derived aUY11 and the deoxy-derived dUY11 act by the same antiviral mechanisms against several enveloped but otherwise unrelated viruses. Therefore, chemically unrelated compounds of appropriate shape and amphipathicity target virion envelope lipids to inhibit formation of the negative curvature required for fusion, inhibiting infectivity by biophysical, not biochemical, mechanisms.
INTRODUCTION
Viral entry is an attractive antiviral target. Clinical entry inhibitors target viral glycoproteins to inhibit their attachment to cellular receptors (e.g., maraviroc) (1) or to prevent the conformational changes of virion glycoproteins needed to drive fusion between viral and cellular membranes (e.g., enfuvirtide) (2). These approaches, unfortunately, have the same limitations as targeting any other viral protein, such as selection for resistance and narrow specificity (3). Experimental compounds have been proposed to inhibit the infectivity of unrelated enveloped viruses by disruption of virion envelopes (e.g., PD 404182 [42]), inhibition of fusion by poorly understood mechanisms (e.g., arbidol [48]), or by irreparable damage to the virion lipid envelopes (e.g., LJ-001 [49]). From these and related studies, it is clear that antiviral approaches targeting conserved structural elements of virions, such as the virion envelope, can result in broad-spectrum activity against unrelated enveloped viruses.
All enveloped viruses require the fusion of viral and cellular membranes to enter the cell (4). The energy required for this process is provided by the attachment, binding, and conformational changes of viral fusion proteins, which undergo major structural rearrangements during fusion from the pre- to the postfusion states. The three different classes of viral fusion proteins differ structurally but mediate fusion in an overall similar manner (5). The insertion of the viral fusion peptides disrupts the target membranes, resulting in the formation of a fusion intermediate known as the hemifusion stalk, in which only the outer leaflets of the two membranes are fused (6). The inner leaflets subsequently fuse, resulting in a small pore that then enlarges to allow full fusion.
Formation of the hemifusion stalk requires the lipid bilayer to form local negative curvature, an energetically demanding rearrangement. Fusion is consequently promoted by enrichment in the outer leaflet of lipids that induce negative curvature, such as oleic acid (6). Conversely, enrichment in the outer leaflet of lipids that favor positive curvature, specifically those with hydrophilic headgroups larger than their hydrophobic tails (such as lysophosphatidylcholine [LPC]), impairs fusion (6). Such phospholipids prevent the fusion of enveloped viruses, such as influenza virus (7, 8), rabies virus (9), and Sendai virus (10). However, such phospholipids also tend to be disruptive to all membranes (including cellular ones), to have signaling activities, to be toxic, and to be too rapidly metabolized to be useful as drugs.
We previously identified a synthetic compound, 5-(perylen-3-yl)ethynyl-2′-deoxy-uridine (dUY11), with the same overall shape and amphipathicity as such lipids (11). dUY11 inhibited the formation of the negative curvature necessary to form the hemifusion stalk. Furthermore, dUY11 prevented the fusion of a model enveloped virus, vesicular stomatitis virus (VSV). Consistently, dUY11 inhibited the infectivity of several enveloped viruses at nanomolar concentrations but did not inhibit the infectivity of nonenveloped viruses. Although dUY11 is a nucleoside derivative, it does not act by classical nucleosidic mechanisms (11).
We had proposed that dUY11 was the first member of a family of compounds that inhibit infectivity by biophysical (not biochemical) mechanisms, according to their shapes and amphipathicities, not to chemical interactions. We called these compounds rigid amphipathic fusion inhibitors (RAFIs). According to our model, chemically distinct molecules of the same overall shape and amphipathicity should also target virion envelope lipids to prevent fusion of viral and cellular membranes. Furthermore, all of these compounds should act by the same mechanisms against all enveloped viruses, including important human pathogens.
We had shown that an arabino-configured nucleoside, 5-(perylen-3-yl)ethynyl-arabino-uridine (aUY11), also inhibited the infectivity of herpes simplex virus 1 (HSV-1) (11). However, the antiviral mechanism of aUY11 remained uncharacterized. We have now tested our model using the arabino-derived aUY11 and clinically important viruses, including influenza virus and hepatitis C virus (HCV). The arabino-derived aUY11 differs from the deoxy-derived dUY11 in that it has a 2′ hydroxyl group in the nucleoside moiety. If the compounds acted by traditional biochemical mechanisms, then this modification should result in different biological activities. If these compounds acted by biophysical mechanisms, however, then the arabino-configured aUY11 and the deoxyuridine-configured dUY11 should act by the same mechanisms. Moreover, the antiviral mechanisms should be the same against several enveloped viruses, regardless of their particular fusion proteins.
Here, we report that aUY11 interacts with the envelope lipids of influenza virus, HCV, HSV-1, and HSV-2 and inhibits the infectivity of these and other enveloped viruses. aUY11 inhibits lipid mixing in fluorescence-dequenching fusion assays for influenza virus, HCV, and VSV, which represent the three classes of fusion proteins. Furthermore, aUY11 localizes to the hydrophobic core of the lipid membrane of protein-free liposomes and inhibits their acid-induced fusion to cells in the absence of any viral protein. aUY11 did not affect membrane fluidity but instead inhibited the transition of model lipid bilayers from lamellar to hexagonal phases, a transition that requires the formation of negative curvature. Such activities are consistent with the inhibition of the negative curvature necessary for virion envelopes to fuse to cell membranes as the main antiviral mechanism of 5-perylene-ethynyl uracil deoxy- or arabino-derived nucleosides. We conclude that aUY11 and dUY11 act by the same biophysical mechanisms, targeting the virion envelope lipids to prevent the formation of the negative curvature necessary for fusion, thereby inhibiting the infectivity of enveloped viruses. Our findings show that RAFIs are indeed a novel family of antiviral molecules that act by biophysical mechanisms against unrelated enveloped viruses, including clinically important human pathogens.
MATERIALS AND METHODS
Compounds.
The synthesis of aUY11 and other RAFIs has been described previously (11–13). The compounds were dissolved in dimethyl sulfoxide (DMSO) as 10 mM stocks, stored at −20°C, and resuspended just before use to the indicated concentrations in warm Dulbecco's modified Eagle's medium (DMEM). Equivalent concentrations of DMSO were used in the controls.
Cells and viruses.
African green monkey Vero fibroblasts, NIH 3T3 cells, and Madin-Darby canine kidney (MDCK) cells were cultured in DMEM supplemented with 5% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in 5% CO2. Huh7.5 cells (14) and HEK293 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in 5% CO2.
To prepare HSV-1 and HSV-2 stocks, Vero cells were infected at a multiplicity of infection (MOI) of 0.05 PFU/cell of HSV-1 strain KOS or HSV-2 strain 186 (from P. Schaffer, Harvard Medical School). Viral stocks were prepared according to standard procedures (15).
To prepare VSV or Sindbis virus stocks, Vero cells were infected with VSV or Sindbis virus (MOI, 0.02 PFU/cell) at 37°C in 5% CO2. The inoculum was removed, and the infected cells were overlaid with DMEM-5% FBS. Culture medium was collected after 48 h and centrifuged at 3,200 × g for 30 min at 4°C to pellet cell debris. The supernatant was then centrifuged at 10,000 × g for 2 h at 4°C, and the resulting virion pellet was resuspended in DMEM, aliquoted, and stored at −80°C.
HCV strain JFH-1 (16) was originally obtained from Takaji Wakita (Tokyo Metropolitan Institute for Neuroscience, Tokyo, Japan) through Lorne Tyrrell (University of Alberta, Edmonton, Canada). To prepare virus stock, Huh7.5 cells were infected with 0.2 focus-forming units (FFU)/cell of HCV JFH-1 and passaged as necessary. After 7 days, culture medium was collected and centrifuged at 800 × g for 5 min at 4°C to pellet cell debris. The supernatant was filtered through a 0.22-μm filter and concentrated using Amicon 100,000-molecular-weight cutoff filters (Millipore, Billerica, MA). The resulting virus stocks were titrated by focus-forming assays and stored at −80°C.
Influenza virus strains A/Puerto Rico/8/1934 (H1N1), A/USSR/90/77 (H1N1), A/Aichi/2/68 (H3N2), and A/Port Chalmers/1/73 (H3N2) were obtained from Veronika von Messling (INRS-Institut Armand-Frappier, Research Centre, Quebec, Canada). To prepare virus stocks, MDCK cells were infected with 0.01 PFU/cell of influenza virus. The infected cells were then incubated in DMEM supplemented with 0.2% bovine serum albumin (BSA) and 2 μg/ml tosylphenylalanyl chloromethyl ketone (TPCK) trypsin (Sigma-Aldrich, Oakville, ON, Canada) in 5% CO2 at 33°C for approximately 2 days, until at least 90% of the cells displayed cytopathic effects (cell rounding with minimal detachment). Virions were harvested as described for VSV.
Murine cytomegalovirus (mCMV) strain RM427+ (17), containing a lacZ insertion in the nonessential immediate-early 2 gene, was originally obtained from Edward Mocarski (Emory University, Atlanta, GA) through Denise Hemmings (University of Alberta, Edmonton, Alberta, Canada). To prepare virus stocks, NIH 3T3 cells were infected with 0.01 PFU/cell of mCMV RM427+. The infected cells were incubated at 33°C in 5% CO2 for approximately 4 days, until at least 90% of the cells displayed cytopathic effects (cell rounding with minimal detachment). Two days after 90% of the cells displayed cytopathic effects, virions were harvested as described for HSV-1 and HSV-2.
Poliovirus (PV) strain Mahoney was obtained from Jiang Yin and Michael N. James (University of Alberta, Edmonton, Alberta, Canada) and propagated in Vero cells as described for VSV. Adenovirus (AdV) expressing green fluorescent protein (GFP) was obtained from Rene Jacobs and Dennis Vance (University of Alberta, Edmonton, Alberta, Canada) and was titrated in HEK293 cells maintained with DMEM-10% FBS. Mammalian orthoreovirus type 3 was kindly provided by Maya Shmulevitz (University of Alberta, Edmonton, Alberta, Canada).
Infectivity assays.
Vero cells (5 × 105 cells/well in 6-well plates) were infected with a 200-μl inoculum containing approximately 200 PFU of HSV-2, VSV, Sindbis virus, poliovirus, or reovirus preexposed to aUY11, dUY11, or DMSO vehicle for 10 min at 37°C. The inocula were removed after 1 h, and the monolayers were washed and overlaid with 2% methylcellulose or 1% agarose (for reovirus) containing 5% FBS. The infected cells were incubated in 5% CO2 at 37°C until well-defined plaques developed (typically, 1 to 2 days postinfection). The cells were then fixed and stained with crystal violet in methanol (0.5% [wt/vol] crystal violet, 17% [vol/vol] methanol in H2O).
For influenza virus, MDCK cells (6 × 105 cells/well in 6-well plates) were infected with a 200-μl inoculum containing approximately 200 PFU of influenza virus A/Puerto Rico/8/1934, A/USSR/70/99, A/Aichi/2/68, or A/Port Chalmers/1/73 preexposed to compound or DMSO vehicle for 10 min at 37°C. The inocula were removed after 1 h, and the monolayers were washed and overlaid with 0.8% agarose (containing 0.1 μg/ml TPCK-trypsin). Plaques were visualized using neutral red or crystal violet staining.
Huh7.5 cells (9 × 104 cells/well in 24-well plates) were infected with 150 μl of HCV strain JFH-1 (containing ∼100 FFU) preexposed to aUY11, dUY11, or DMSO vehicle for 10 min at 37°C. The inocula were removed 4 h later, and the monolayers were washed and overlaid with fresh DMEM-10% FBS containing no drug. The infected cells were fixed at 72 h postinfection with cold methanol-acetone (1:1) and processed for immunocytochemistry for HCV core protein. Cells were incubated with primary mouse IgG anti-core antibody MA1-080 (Thermo Scientific, Rockford, IL) diluted 1:300 for 2 h at room temperature, followed by addition of secondary biotinylated horse anti-mouse IgG antibody (Vector Laboratories, Burlingame, CA) for 30 min at room temperature. The fixed cells were then treated with the avidin-biotin-peroxidase complex (Vectastain ABC kit; Vector Laboratories) for 30 min, followed by addition of ImmPact SG peroxidase substrate (Vector Laboratories). Foci were counted under the microscope.
For mCMV, NIH 3T3 cells (3 × 105 cells/well in 12-well plates) were infected with a 150-μl inoculum containing approximately 200 PFU of mCMV RM427+ preexposed for 10 min at 37°C to compound or DMSO vehicle. The inocula were removed after 1 h, and the monolayers were washed and overlaid with DMEM-10% FBS. Foci of infected cells were detected after 24 h using a LacZ cell detection kit (InvivoGen, San Diego, CA).
For AdV, virions were exposed to aUY11, dUY11, or DMSO for 10 min at 37°C prior to infecting HEK293 cell monolayers (5 × 105 cells/well in 12-well plates). The inocula were removed after 1 h, and the monolayers were washed and overlaid with fresh DMEM-10% FBS. Infected cells expressing GFP were visualized after 24 h using a fluorescence microscope with a UV light source (Leica DM IRB, Itzlar, Germany).
Time-of-addition assays.
For pretreatment experiments, MDCK, Huh7.5, or Vero cells were treated with aUY11 or dUY11 for 1 h at 37°C. The cells were then washed three times with DMEM warmed to 37°C and infected with influenza virus A/PR/34/8, HCV JFH-1, or HSV-1, respectively, in the absence of any drug. Infectivity was assessed by plaque or focus formation. To test whether aUY11 or dUY11 was effective when added to already-infected cells, MDCK, Huh7.5, or Vero cells were infected with 5 or 0.5 PFU/cell of influenza virus A/PR/34/8, HCV JFH-1, or HSV-1 KOS. The inocula were removed after 1 h of adsorption. The infected cells were washed and then overlaid with DMEM supplemented with 10% FBS (HCV) or 5% FBS (influenza virus and HSV-1) and aUY11 for 24 (influenza virus and HSV-1) or 48 (HCV) hours. The supernatants and cell lysates were then harvested as described previously. Influenza virus and HSV-1 virions were pelleted by centrifugation at 10,000 × g and resuspended in drug-free DMEM. HCV virions were concentrated using Amicon centrifugal filters with a 100-kDa molecular-mass cutoff. Titrations were performed using logarithmic dilutions of the viruses (in drug-free medium) and MDCK, Vero, or Huh7.5 cells. Harvested virions were examined for the presence of aUY11 by obtaining its fluorescence spectra.
Liposome preparation.
Cholesterol, β-oleoyl-γ-palmitoyl-l-α-phosphatidylcholine (POPC), and 1,2-dioleoyl-l-α-phosphatidylcholine (DOPC) were obtained from Sigma-Aldrich (Oakville, ON, Canada). POPC liposomes were prepared by hydrating 2 μmol dry lipid with 1 ml of 180 mM Na2HPO4, 10 mM citric acid (pH 7.4), followed by vortexing. DOPC-cholesterol (1.7:1 molar ratio) liposomes were prepared by the hydration method. DOPC and cholesterol were dissolved and mixed in 500 μl chloroform, which was then evaporated; 1 ml of 180 mM Na2HPO4, 10 mM citric acid (pH 7.4) was added to the resulting lipid film, and the mixture was vortexed to form large multilamellar liposomes. Octadecyl rhodamine B chloride (R18; Invitrogen, Grand Island, NY) was added (to a final concentration of 5 mol%) as required by first mixing R18 and lipids as ethanol and chloroform solutions, respectively.
R18 labeling of VSV, HCV, and influenza virus virions.
Virions were labeled with 0.59 μM (VSV and HCV) or 1.8 μM (influenza virus) R18 by incubating VSV, influenza virus (108 PFU), or HCV JFH-1 (106 FFU) with 1.97 μl (VSV and HCV) or 5.91 μl (influenza virus) of 300 μM R18 in ethanol in 1 ml of 180 mM Na2HPO4, 10 mM citric acid (pH 7.4) for 1 h at room temperature on a rotary shaker in the dark. The labeled virions were purified through a column containing 4 ml of Sephadex G-100 resin. The column was equilibrated by washing it twice with approximately 12 ml of 180 mM Na2HPO4, 10 mM citric acid (pH 7.4) (henceforth referred to as fusion buffer). Labeled virions (1 ml) were then added to the column and eluted with fusion buffer. Fractions of approximately 500 μl were collected every minute for 15 min. The viral-protein concentration in each fraction was determined using a Bradford assay (Bio-Rad). Fractions of labeled VSV, HCV, or influenza virus virions were titrated on Vero, Huh7.5, or MDCK cells. For HCV and influenza virus, fractions containing infectious virus were then pooled and concentrated using Amicon 100,000-molecular-weight cutoff filters (Millipore, Billerica, MA). The pooled fractions were tested for R18 incorporation by R18 dequenching after addition of Triton X-100 to a final concentration of 0.1% (VSV and influenza virus) or 0.01% (HCV), using a QuantaMaster 40 scanning spectrofluorometer (Photon Technology International, Birmingham, NJ) equipped with a 75-W xenon lamp. R18 was excited at 560 nm. Fluorescence was detected at 590 nm using a model 814 switchable photon-counting/analog photomultiplier detection unit with an R1527 photomultiplier tube. Data were collected using FeliX32 software (Photon Technology International, Birmingham, NJ).
Spectra.
Emission spectra of aUY11 or dUY11 were collected using the QuantaMaster 40 spectrofluorometer. aUY11 or dUY11 was added to 2.5 ml of 180 mM Na2HPO4, 10 mM citric acid (pH 7.4) buffer, or to 2.5 ml of 1-octanol (Sigma), to a final concentration of 48 nM or 0.48 nM, respectively, in a polymethacrylate cuvette prewarmed to 37°C. Alternately, aUY11 or dUY11 was added to 107 PFU of VSV, HSV-1 or -2, or influenza virus; 106 FFU of HCV; or 2 nmol POPC liposomes in 2.5 ml of fusion buffer at 37°C to a final concentration of 48 nM. For other experiments, approximately 104 influenza virus, HCV, or HSV-1 virions produced by cells treated with vehicle, or equivalent volumes from cells treated with aUY11, were diluted to 2.5 ml in 180 mM Na2HPO4, 10 mM citric acid (pH 7.4). Emission spectra were obtained at the maximum excitation wavelength, 455 nm, and examined from 475 to 575 nm. Spectra were normalized to the highest fluorescence signal intensity obtained for all conditions.
Confocal microscopy.
Near-confluent Vero cells seeded onto 22- by 22-mm coverslips (Fisher) placed in 6-well plates were cultured in DMEM-5% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin in 5% CO2 for ∼16 h at 37°C. The cells were first incubated with 250 nM PKH26-GL fluorescent dye (Sigma) for 10 min at 37°C and were then washed twice with 2 ml/well of room temperature DMEM. The washed cells were exposed to 2 μM aUY11 or dUY11 for 1, 5, 15, 40, or 120 min at 37°C. The RAFIs were removed, and the cells were washed twice with 2 ml/well of DMEM at room temperature. The cells were then fixed with 10% formalin for 30 min at room temperature, washed once with 1× PBS at room temperature, and mounted onto glass slides using Mowiol mounting medium (10% Mowiol in 25% glycerol in 0.2 M phosphate buffer, pH 7.4).
Confocal microscopy was performed using a Leica SP5 laser scanning confocal microscope with a 100× oil immersion (numerical aperture, 1.44) lens. Fluorescent images were obtained using a 5-mW argon laser (458 nm) to excite RAFIs and a 1-mW HeNe laser (543 nm) to excite PKH26, with band-pass filters of 470 to 535 nm (RAFIs) and 560 to 650 nm (PKH26). The pinhole aperture was set to 1.0 Airy unit for each channel. The images were collected as 8-bit images using Leica Application Suite (LAS) microscope software and adjusted for contrast and brightness in Microsoft PowerPoint. Scale bars were added using LAS or Fiji ImageJ (NIH, Bethesda, MD) software.
Differential scanning calorimetry.
Mixtures of dielaidoylphosphatidylethanolamine (DEPE) (Avanti Polar Lipids, Alabaster, AL) and aUY11 were made by dissolving the components in chloroform-methanol (2:1). The solvent was then evaporated under nitrogen gas and placed in a vacuum desiccator for 3 h. The dried films were hydrated with 0.8 ml of 20 mM PIPES, 0.14 M NaCl, 1 mM EDTA, pH 7.4, by vortexing; degassed; and placed in the sample cell of a Nano II calorimeter (Calorimeter Sciences Corp., UT), and buffer was placed in the reference cell. The lamellar-to-inverted-hexagonal phase transition temperature was evaluated at a heating scan rate of 1°C/min. The cell volume was 0.34 ml, and the total lipid concentration was 2.5 mg/ml. The results were plotted in ORIGIN 7.0 and analyzed with DA-2 (Microcal, Inc.)
Fusion assays.
Vero, MDCK, or Huh7.5 cells were cultured in DMEM-5% FBS (Vero and MDCK) or DMEM-10% FBS (Huh7.5) as described previously. The cells were washed with phosphate-buffered saline (PBS) (1 mM KH2PO4, 150 mM NaCl, 3 mM Na2HPO4 [pH 7.4]) and incubated for less than 5 min at 37°C in 3 ml of 1× trypsin in PBS to generate single-cell suspensions. The single-cell suspensions were then washed twice by centrifugation in fusion buffer at 800 × g for 5 min at 4°C in an A-4-62 rotor in an Eppendorf 5810R centrifuge.
R18-labeled VSV fusion experiments were performed as previously described (11). R18-labeled influenza virus or HCV (1 × 104 PFU) was exposed to 0.1% DMSO or RAFI in 100 μl for 10 min at 37°C and then incubated on ice for 3 min. The virions thus exposed were then diluted 3.5-fold by mixing with 1 × 106 prechilled MDCK (influenza virus) or Huh7.5 or Vero (HCV) cells in 250 μl of fusion buffer and incubated on ice for 30 min (influenza virus) or 60 min (HCV) to allow binding but not fusion. The virus-cell complexes were then washed with fusion buffer by centrifugation at 300 × g for 5 min at 4°C. The virus-cell pellets were resuspended in 2.5 ml of the same buffer prechilled to 4°C. Fusion was triggered by increasing the temperature to 37°C and lowering the pH to 5 (influenza virus) or 5.5 (HCV) by adding 500 mM citric acid. For HCV fusion, we also tested pH 4, 4.5, 5, and 6. An identical sample was kept at pH 7.4 by adding fusion buffer, and an additional nonfusion control was maintained at 4°C (pH 7.4). Equivalent aliquots were removed at discrete time points, fixed with 10% formalin, and transferred to polymethacrylate cuvettes (Sigma, Oakville, ON, Canada). Fluorescence was excited at 560 nm and detected at 590 nm, using a QuantaMaster 40 scanning spectrofluorometer and FeliX32 software. Percent fusion was calculated from changes in fluorescence according to the following equation: percent fusion = [(F5.5/Fmax) − (Finitial/Fmax)] × 100, where F5.5 is the fluorescence at pH 5.5 at each time point, Finitial is the initial fluorescence of the complex, and Fmax is the total fluorescence after Triton X-100 lysis to a final concentration of 0.01% (HCV) or 0.1% (influenza virus). F7.4, the fluorescence at pH 7.4 for each time point, is also shown on the graphs.
R18-labeled DOPC-cholesterol liposomes (2 nmol) were exposed to 0.1% DMSO or 2 μM aUY11 in a minimal volume for 10 min at 37°C and then incubated on ice for 3 min. The liposomes thus exposed were then mixed with 1 × 106 prechilled Vero cells in fusion buffer and incubated on ice for 10 min. The liposome-cell mixtures were then diluted to 2.38 ml in fusion buffer and warmed to 37°C prior to transfer to polymethacrylate cuvettes. Fusion was triggered by increasing the temperature to 37°C and lowering the pH to 5.5 by addition of 500 mM citric acid. An identical sample was kept at pH 7.4 by adding fusion buffer. Fluorescence was excited at 560 nm and detected at 590 nm, using a QuantaMaster 40 scanning spectrofluorometer and FeliX32 software. Percent fusion of liposomes was calculated from changes in fluorescence according to the following equation: percent fusion = {[(F5.5 − Finitial)/(Fmax − Finitial)] − [(F7.4 − Finitial)/(Fmax − Finitial)]} × 100, where F5.5 is the fluorescence at pH 5.5 at each time point, F7.4 is the fluorescence at pH 7.4 at each time point, Finitial is the fluorescence of the complex after incubation for 10 min at pH 7.4 at 37°C, and Fmax is the total fluorescence after lysis with Triton X-100 (at a final concentration of 0.1%).
Membrane fluidity.
1,6-Diphenyl-1,3,5-hexatriene (DPH) (Sigma) (18) was dissolved in tetrahydrofuran and then added to DOPC-cholesterol liposomes (20 nmol) to a final concentration of 2 μM. To allow insertion of DPH into the hydrophobic core of the lipid membrane, the liposome-DPH mixture was incubated for 10 min at 37°C. The DPH-labeled liposomes were then incubated with aUY11 or cholesterol for 10 min at 37°C and transferred to cuvettes prewarmed to 37°C. DPH fluorescence was excited at 350 nm, and emission was measured at 450 nm using a QuantaMaster 40 scanning spectrofluorometer and FeliX32 software. Fluorescence polarization (P) was calculated according to the following equation: P = (IVV − IVH)/(IVV + IVH), where IVV and IVH are the intensities obtained with polarizers aligned parallel and perpendicular to the polarized excitation beam, respectively.
RESULTS
aUY11 inhibits the infectivity of otherwise unrelated enveloped viruses, including important human pathogens.
We first tested the effects of the arabino-configured aUY11 (Fig. 1) on the infectivity of unrelated DNA or RNA enveloped viruses, including important human pathogens (influenza virus, HCV, HSV-2, and Sindbis virus) and model viruses (mCMV and VSV). These viruses are internalized by different mechanisms, fuse to different cell membranes, and have fusion proteins of all three classes. However, they all require the formation of negative membrane curvature for the fusion of their envelopes with the cellular membranes.
Fig 1.

Chemical and three-dimensional (3D) structures of the RAFIs aUY11, dUY11, aUY12, dUY5, and dUY1. The 3D structures (11) are displayed in three orthogonal perspectives (gray, carbon; blue, hydrogen; red, oxygen; dark blue, nitrogen).
Influenza virus, HSV-2, or Sindbis virus virions (∼200 PFU) were exposed to aUY11, dUY11, or DMSO vehicle control at 37°C in DMEM for 10 min prior to infecting Vero (HSV-2 and Sindbis virus) or MDCK (influenza virus) cell monolayers. Infectivity was assessed by plaquing efficiency. For HCV infections, JFH-1 virions (∼200 FFU) were exposed to aUY11, dUY11, or DMSO vehicle control in serum-free medium for 10 min at 37°C prior to infecting Huh7.5 cell monolayers. Foci of infected cells were detected by immunocytochemistry and counted under the microscope. Infectivity was expressed as a percentage of the infectivity of virions treated with vehicle control.
aUY11 inhibited the infectivity of several strains of influenza virus (50% inhibitory concentration [IC50], 0.078 to 0.283 μM), HCV JFH-1 (IC50, 0.187 μM), HSV-2 (IC50, 0.031 μM), and Sindbis virus (IC50, 0.006 μM) (Fig. 2A). Similarly, dUY11 inhibited the infectivity of several strains of influenza virus (IC50, 0.097 to 0.187 μM), HCV JFH-1 (IC50, 0.107 μM), and HSV-2 (IC50, 0.052 μM) (Fig. 2A). The IC50s against each virus were similar for aUY11 and dUY11. We had previously shown that aUY11 and dUY11 both inhibit the infectivity of HSV-1 at similar concentrations (IC50, 0.131 μM and 0.048 μM, respectively) (11).
Fig 2.

aUY11 is active against enveloped but otherwise unrelated viruses. (A) Infectivity of HSV-2, HCV, influenza virus, or Sindbis virus exposed to aUY11 (open squares) or dUY11 (filled circles), tested by plaquing efficiency or focus-forming efficiency (average ± standard deviation [SD]; n ≥ 3). (B) Infectivity of model DNA and RNA enveloped viruses exposed to aUY11 or dUY11, tested by plaquing efficiency or focus-forming efficiency (average ± SD; n ≥ 3). (C) Infectivity of nonenveloped poliovirus (open triangles), adenovirus (filled inverted triangles), or reovirus (filled diamonds) exposed to aUY11 (average ± range; n = 2).
aUY11 also inhibited the infectivity of model DNA and RNA viruses, mCMV and VSV, respectively. mCMV RM427+ virions (∼200 PFU) were exposed to aUY11, dUY11, or DMSO vehicle control in DMEM for 10 min at 37°C prior to infecting NIH 3T3 cell monolayers. Foci of infected cells were identified 24 h later by LacZ expression. Infectivity was expressed as focus-forming efficiency relative to that of vehicle-treated virions. VSV virions (∼200 PFU) were exposed to aUY11, dUY11, or DMSO vehicle control in DMEM for 10 min at 37°C prior to infecting Vero cell monolayers. Infected cells were incubated until plaques developed. Infectivity was assessed by plaquing efficiency and is expressed relative to that of vehicle-treated virions. aUY11 and dUY11 inhibited the infectivity of mCMV (IC50, 0.037 μM and 0.013 μM, respectively) and VSV (IC50, 0.005 μM and 0.002 μM, respectively) (Fig. 2B).
aUY11 inhibited the infectivity of unrelated enveloped viruses, including important human pathogens, in the nanomolar range (Table 1). In contrast, and consistent with the proposed mechanism of action, aUY11 did not inhibit the infectivity of three nonenveloped viruses, poliovirus, adenovirus, and reovirus (Fig. 2C). Taken together, our results suggest that aUY11 likely targets virion envelopes.
Table 1.
Inhibition of infectivity of enveloped but otherwise unrelated viruses or nonenveloped viruses
| Virus family | Virus | IC50 (μM)a |
|
|---|---|---|---|
| aUY11 | dUY11 | ||
| Orthomyxoviridae | Influenza virus A/PR/8/34 (H1N1) | 0.283 ± 0.130 | 0.187 ± 0.051 |
| Influenza virus A/USSR/90/77 (H1N1) | 0.078 ± 0.019 | 0.097 ± 0.021 | |
| Influenza virus A/Aichi/2/68 (H3N2) | 0.078 ± 0.007 | 0.143 ± 0.045 | |
| Influenza virus A/PC/1/73 (H3N2) | 0.090 ± 0.026 | 0.117 ± 0.032 | |
| Flaviviridae | HCV JFH-1 | 0.187 ± 0.060 | 0.107 ± 0.041 |
| Togaviridae | Sindbis | 0.006 ± 0.001 | 0.011 ± 0.005b |
| Rhabdoviridae | VSV | 0.005 ± 0.001 | 0.002 ± 0.001 |
| Herpesviridae | HSV-1 KOS | 0.131 ± 0.034b | 0.048 ± 0.012b |
| HSV-2 186 | 0.031 ± 0.008 | 0.052 ± 0.003 | |
| mCMV | 0.037 ± 0.016 | 0.013 ± 0.004 | |
| Picornaviridae | Polio | >200 | >200b |
| Adenoviridae | Adenovirus | >20 | >20b |
| Reoviridae | Reovirus | >20 | ND |
Average ± SD (n = 3). ND, not done.
Data from reference 11.
aUY11 localizes to virion envelope lipids.
Fluorescence spectra depend on the polarity of the fluorochrome environment. To test the localization of aUY11 in virions, we analyzed its fluorescence spectra in different environments. aUY11 was added to 2.5 ml of aqueous fusion buffer or to 2.5 ml of 1-octanol to a final concentration of 48 nM or 0.48 nM, respectively, in a polymethacrylate cuvette prewarmed to 37°C. Alternatively, aUY11 was added to 107 PFU of VSV, 107 PFU of HSV-1, 106 PFU of influenza virus, 106 FFU of HCV, or 2 nmol protein-free POPC liposomes in the same aqueous buffer to a final concentration of 48 nM. We also analyzed the fluorescence spectra of dUY11 in the same environments. Emission spectra were obtained at the maximum excitation wavelength, 455 nm, and were normalized to the highest fluorescence signal intensity obtained for all conditions (set as 1).
As we had observed for dUY11 in the presence of a model enveloped virus, VSV (11), the fluorescence spectra of aUY11 were most similar when in the presence of HSV-1, HSV-2, influenza virus, or HCV virions or protein-free liposomes (Fig. 3A). These viruses have different glycoproteins and different glycoprotein contents but share a hydrophobic environment in the lipid cores of their envelopes. These spectra were most similar to the spectrum of aUY11 in octanol, which mimics the hydrophobic core of the membrane, and clearly distinct from its spectrum in aqueous buffer without virions or liposomes. These results are consistent with aUY11 localizing to a hydrophobic environment in virions, such as the hydrophobic lipid core of the virion envelope, as we had proposed based on our previous studies with dUY11 and a model enveloped virus (11). More interestingly, we show here that aUY11 localizes to a similar hydrophobic environment in clinically important viruses, such as influenza virus, HCV, and herpesviruses.
Fig 3.

aUY11 localizes to lipid membranes. (A) The emission spectra of aUY11 and dUY11 in virions or protein-free liposomes are most similar, and closely resemble their spectra in hydrophobic environments. aUY11 or dUY11 was added to aqueous buffer (gray) or to 1-octanol (black) to a final concentration of 48 nM or 0.48 nM, respectively. aUY11 or dUY11 (48 nM final concentration) was also added to 106 PFU of influenza virus A (blue), 106 FFU of HCV (orange), 107 PFU of HSV-1 (green), 107 PFU of HSV-2 (teal), or 107 PFU of VSV (pink) or to 2 nmol liposomes (red) in 2.5 ml of aqueous buffer. Fluorescence was excited at 455 nm. Emission spectra for dUY11 in VSV virions, liposomes, aqueous buffer, and octanol were determined previously (11). (B) The RAFI aUY11 localizes to cellular lipid membranes. Vero cell monolayers exposed to PKH26 general membrane dye for 10 min at 37°C were washed and then exposed to aUY11 or dUY11 for 1, 5, 15, 40, or 120 min at 37°C. Shown are confocal microscopy images; scale bars, 25 μm.
We also took advantage of the intrinsic fluorescence of aUY11 to evaluate its potential localization to other lipid membranes. Near-confluent Vero cell monolayers seeded onto coverslips were treated with 250 nM PKH26-GL fluorescent cell dye (a general membrane dye) for 10 min at 37°C and then exposed to 2 μM aUY11 or dUY11 for 1 to 120 min, also at 37°C. The cells were washed, fixed with 10% formalin, and mounted onto glass slides. aUY11 localized to plasma and intracellular membranes (Fig. 3B; see Movies S1A and B in the supplemental material). In summary, aUY11 localizes to cellular lipid membranes.
aUY11 protects cells from infection with influenza virus.
Since aUY11 localizes to cellular membranes, we next tested the effects of aUY11 on virion infectivity to cells treated before infection. Cells were exposed to aUY11 for 1 h at 37°C. Following three washes, the cells were infected with influenza virus, HCV, or HSV-1 and evaluated for plaque or focus formation. Infectivity was expressed as a percentage of the infectivity in cells pretreated with vehicle control. Under these conditions, aUY11 protected the cells from infection by influenza virus and, less efficiently, HCV and HSV-1 (IC50s, 0.55, 3.5, and 4 μM, respectively) (Fig. 4A). For influenza virus, aUY11 acted at concentrations similar to those when virions were exposed (the IC50 was only ∼1.5-fold higher than when virions were preexposed). Similarly, dUY11 protected cells from infection by influenza virus and, less efficiently, by HCV and HSV-1 (Fig. 4B). In summary, aUY11 and dUY11 protect cells from infection by influenza virus at concentrations similar to those at which they inhibit the infectivity of influenza virus virions.
Fig 4.

aUY11 and dUY11 protect cells from infection with influenza virus, HCV, or HSV-1. MDCK, Huh7.5, or Vero cells were treated with aUY11 (A) or dUY11 (B) for 1 h prior to infection with influenza virus, HCV, or HSV-1, respectively (pretreatment). Alternately, virions were preexposed to aUY11 (A) or dUY11 (B) prior to infection of cells (virion treatment). In both cases, infectivity was evaluated by plaquing or focus-forming efficiency and is expressed as a percentage relative to a DMSO vehicle-treated control. The error bars represent ranges from two experiments, with the exception of HSV-1 (B), which shows one experiment representative of two.
aUY11 inhibits the infectivity of influenza virus, HCV, or HSV-1 virions produced by treated cells.
Viruses acquire their envelopes from the membranes of infected cells, and aUY11 localizes to cell membranes (Fig. 3B). Therefore, we tested the effects of aUY11 on the infectivity of virions produced by already infected cells. MDCK, Huh7.5, or Vero cells were first infected with 5 or 0.5 PFU/cell of influenza virus, HCV, or HSV-1. After 1 h, the inocula were removed, and the cells were washed and overlaid with medium containing aUY11 for 24 or 48 h (for influenza virus and HSV-1 or HCV, respectively). Supernatants and cell lysates were harvested, and influenza virus and HSV-1 virions were pelleted and resuspended in medium with no drugs, whereas HCV virions were concentrated on a 100-kDa molecular-mass cutoff and then diluted in medium with no drug to be titrated. Almost no infectious virus could be recovered from already infected cells incubated with 6 μM aUY11 (Fig. 5A). The concentrations at which aUY11 inhibited the production of infectious virus by cells treated after infection were very similar to the concentrations at which it inhibited the infectivity of exposed virions. In cells infected at an MOI of 5, the IC50 for influenza virus, HCV, or HSV-1 was 0.2, 0.4, or 0.35 μM, respectively. In cells infected at an MOI of 0.5, the IC50 for influenza virus, HCV, or HSV-1 was 0.06, 0.14, or 0.28 μM, respectively. We had previously shown that the RAFI dUY11 does not affect viral replication in previously infected cells and has no apparent effects on virion assembly or integrity, only minimal ones on virion budding, and no cytotoxic effects (11). We have now shown (Fig. 3B) that aUY11 localizes to the membranes of treated cells. Therefore, the effects on the virions produced by cells treated after infection are likely the result of inhibition of the infectivity of the progeny virions, which acquire their envelopes by budding from the cell membranes to which aUY11 localizes. In this model, aUY11 in the cellular membranes would become incorporated into the virion envelopes during budding. To test the model, we took advantage of the intrinsic fluorescence of aUY11 and examined its fluorescence spectra in 104 influenza virus, HCV, or HSV-1 virions produced by cells treated with vehicle or in equivalent volumes from cells treated with aUY11. The characteristic emission spectrum of aUY11 was detected, at decreasing intensities, for samples treated with 6, 2, or 0.6 μM aUY11. The spectrum was not detected at lower concentrations, most likely as a result of the limiting virions (100- or 1,000-fold fewer HCV and influenza virus or HSV-1 virions, respectively, than in Fig. 3A). As expected, aUY11 fluorescence was not detected in samples from the untreated cells (Fig. 5B). In summary, aUY11 added to already infected cells associated with the progeny virions produced.
Fig 5.

aUY11 inhibits the infectivity of influenza virus, HCV, and HSV-1 virions produced by infected cells. (A) Cells infected with 5 or 0.5 PFU or FFU per cell of influenza virus, HCV, or HSV-1 were incubated in the presence of aUY11 for 23 h (influenza virus and HSV-1) or 44 h (HCV). Supernatants and cell lysates were harvested and titrated for the presence of infectious virus. (B) Virions harvested in panel A were tested for the presence of aUY11 by examining its fluorescence spectra. aUY11 fluorescence was excited at 455 nm.
aUY11 does not perturb membrane fluidity.
Compounds that target membranes can modulate their fluidity, which in turn affects the infectivity of enveloped virions (20). For example, cholesterol increases the ordering of phospholipid acyl chain packing (21). Decreases in membrane fluidity reduce the infectivity of enveloped virions, such as HIV (22). We therefore tested the effects of aUY11 on membrane fluidity, using the DPH fluorescence polarization method (23). As membrane fluidity decreases (such as by addition of cholesterol), the polarization of DPH fluorescence increases. The addition of aUY11 up to 20 μM (approximately 100 times the IC50) to DOPC liposomes did not result in any such increase in DPH polarization (Fig. 6A), indicating that aUY11 does not affect membrane fluidity.
Fig 6.

The RAFI aUY11 does not perturb membrane fluidity but prevents the formation of negative curvature in lipid structures. (A) Membrane fluidity was assessed by fluorescence polarization. aUY11 did not affect the fluidity of liposomes, whereas cholesterol decreased their fluidity, as expected and as shown by an increase in polarization. (B) Schematic representation of the lipid phase transitions measured by DSC. The transition from lamellar gel to lamellar liquid crystalline occurs at melting temperature (Tm); the transition from lamellar liquid crystalline to hexagonal phase occurs at temperature TH. (C) DSC of aUY11 in DEPE. The heating scans are shown with positive values and the corresponding cooling scans with negative values. The gel-to-liquid-crystalline phase transition temperature remains largely unaltered, at ∼37°C, with the addition of increasing amounts of aUY11 to DEPE, while the transition temperature to the hexagonal phase increases. This progression is shown in the enlarged graphs.
aUY11 inhibits the formation of lipid structures with negative curvature.
We next used differential scanning calorimetry (DSC) to test whether aUY11 inhibits the formation of lipid structures with negative curvature. DEPE lamellar phases were reconstituted with increasing concentrations of aUY11. The transition from the flat morphology of the lamellar phase to the negative curvature of the inverted hexagonal phase was evaluated by DSC (Fig. 6B). Less than 3% aUY11 in DEPE increased the transition temperature between the lamellar and inverted hexagonal phases by ∼1°C (Fig. 6C). These effects on the transition temperature required for the formation of negative monolayer curvature, characterized by a regression of 29 ± 7, indicate that aUY11 disfavors the formation of negative membrane monolayer curvature.
The cooling scans, which evaluate the reciprocal inverted-hexagonal-to-lamellar phase transition, exhibit a characteristic hysteresis caused by kinetic factors. They also show that aUY11 raises the hexagonal-to-bilayer transition temperature, characterized by a regression of 49 ± 7 (Fig. 6C). The facts that a characteristic transition due to hysteresis is recovered on each cooling scan and that the cooling regression is similar to that obtained in the heating scans demonstrate that aUY11 did not affect the integrity of the multilamellar membranes. They were not disrupted or lysed by aUY11, as can also be ascertained by the constancy of the main transition, which appears at 37°C on reheating. The hysteresis of DSC transitions of phosphatidylethanolamines has been previously noted (24).
Taken together, the DSC shows that aUY11 raises the transition temperature necessary to form inverted hexagonal phases without disrupting the membranes or affecting their fluidity (Fig. 6A). This is consistent with inhibitors of fusion that prevent the formation of negative monolayer curvature (25).
aUY11 inhibits the fusion of viral and cellular membranes.
Since aUY11 and dUY11 inhibit the changes in membrane curvature required for fusion (Fig. 6C), we next tested whether aUY11 inhibited virion-envelope-to-cell-membrane fusion. We used fluorescence-dequenching fusion assays, which analyze lipid mixing between the outer leaflets of virions and target cells.
VSV virions labeled with self-quenching concentrations of R18 were exposed to aUY11 prior to mixing with Vero cells. Fusion was triggered by lowering the pH and analyzed by the dequenching of R18 fluorescence. Fluorescence was dequenched by ∼15% when VSV virions exposed to DMSO vehicle were induced to fuse to target cells, but by only 5% when VSV virions exposed to aUY11 were induced to fuse under the same conditions (Fig. 7A).
Fig 7.
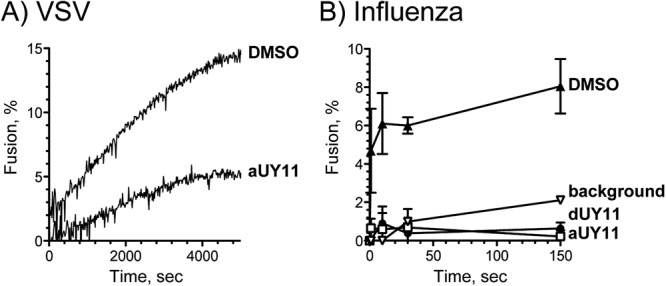
The RAFIs aUY11 and dUY11 inhibit fusion of VSV and influenza virus to cellular membranes. Shown are fluorescence dequenching of R18-labeled VSV virions preexposed to 60 nM aUY11 or DMSO vehicle during pH-dependent fusion to Vero cells (A) and influenza virus virions preexposed to 0.6 μM dUY11, aUY11, or DMSO vehicle during fusion to MDCK cells (average ± range; n = 2) (background, dequenching at neutral pH) (B).
We next tested the effects of aUY11 and dUY11 on fusion of a clinically important virus, influenza virus (A/Puerto Rico/8/34[H1N1]). Influenza virus virions labeled at self-quenching concentrations of R18 were exposed to aUY11 or dUY11 prior to mixing with MDCK cells. Fusion was triggered by increasing the temperature and decreasing the pH to 5. Under these conditions, fluorescence was dequenched by approximately 8% for influenza virus virions treated with DMSO vehicle but by less than 1% (background levels) for influenza virus virions treated with 600 nM dUY11 or aUY11, even after 2.5 h (influenza virus fuses in less than 10 min). The background dequenching at neutral pH was ∼2% after 2.5 h (Fig. 7B).
According to our model, RAFIs should inhibit the infectivity of enveloped (but otherwise unrelated) viruses by the same mechanisms. We tested whether aUY11 also inhibited the fusion of another clinically relevant virus, HCV. To this end, we first developed a fusion assay using HCV JFH-1 virions and Huh7.5 cells. HCV JFH-1 virions labeled at self-quenching concentrations of R18 were mixed with Huh7.5 cells. Fusion was triggered by increasing the temperature to 37°C and decreasing the pH (Fig. 8A). As expected, HCV fused only to Huh7.5 cells, and not to Vero cells, at an optimum pH of 5.0 to 5.5, consistent with fusion to earlier endosomes (Fig. 8B). In summary, the requirements for pH and cell type in our fusion assays match the requirements for HCV infectivity.
Fig 8.

aUY11 inhibits fusion of HCV to Huh7.5 cells in a new HCV fluorescence dequenching fusion assay. (A) Fusion of HCV to Huh7.5 cells was tested at several pHs. The pH requirements in our fusion assay match those for HCV infectivity. The error bars indicate SD. (B) HCV fused to Huh7.5 cells but, as expected, not to Vero cells, which is consistent with the ability of HCV to infect the former but not the latter. (C) Fluorescence dequenching of R18-labeled HCV JFH-1 virions preexposed to 0.6 μM dUY11, aUY11, or DMSO vehicle during pH-dependent fusion to Huh7.5 cells (average ± SD; n = 3); background is the level of dequenching observed at neutral pH.
We then used the HCV fusion assays to test whether aUY11 or dUY11 inhibited the fusion of HCV to Huh7.5 cells. R18-labeled HCV JFH-1 virions were exposed to aUY11 or dUY11 and then mixed with Huh7.5 cells. Fluorescence was dequenched by approximately 12% for HCV virions treated with DMSO vehicle but by less than 5% for HCV virions treated with 600 nM dUY11 or aUY11 (IC99 in infectivity assays), in the range of the background dequenching at neutral pH in these assays (∼3%) (Fig. 8C).
aUY11 inhibits fusion by acting on lipids, not proteins.
Influenza virus has a class I fusion protein, whereas HCV is thought to have a class II and VSV a class III protein. Although these fusion proteins differ structurally and mechanistically, all fusions require curvature changes in the lipid envelopes. Therefore, RAFIs likely inhibit fusion by acting on the lipid membranes (to prevent formation of negative curvature), and not by targeting any viral protein. To test this model, we next analyzed the effects of aUY11 on the fusion of protein-free liposomes to cells, fusion that is induced under acidic conditions (26). We exposed R18-labeled protein-free DOPC liposomes to aUY11 and then added the liposomes thus exposed to cells. Fusion was triggered by decreasing the pH to 5.5 and monitored by fluorescence dequenching of R18. Under these conditions, fluorescence was dequenched by approximately 45% for liposomes treated with DMSO vehicle but by only approximately 20% for liposomes exposed to 2 μM aUY11 (Fig. 9). Therefore, aUY11 inhibits fusion that is not mediated by any viral protein.
Fig 9.
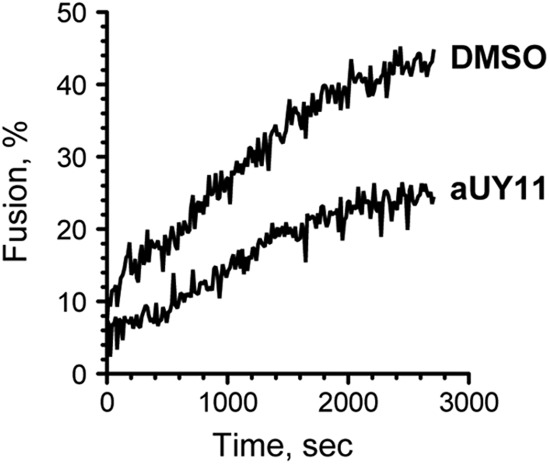
aUY11 inhibits acid-induced liposome fusion to cells. Shown is fluorescence dequenching of R18-labeled DOPC-cholesterol liposomes preexposed to 2 μM aUY11 or DMSO vehicle during acid-induced fusion to Vero cells.
aUY11 and other RAFIs inhibit infectivity and fusion at similar concentrations.
If RAFIs inhibited infectivity mainly by inhibiting fusion, then we expected the concentrations required to inhibit infectivity or fusion to be similar. To test whether such correlation existed, different concentrations of aUY11 and dUY11 were tested in plaquing efficiency and fusion assays, and the respective dose responses were analyzed. aUY11 and dUY11 inhibited fusion and plaquing at the most similar concentrations (Fig. 10). Consequently, their IC50s in fusion assays closely corresponded to their IC50s in plaquing efficiency assays (Table 2).
Fig 10.

RAFIs inhibit VSV plaquing efficiency and fusion to Vero cells at similar concentrations. Shown are comparisons of inhibition of fusion (open squares) or infectivity (filled circles) by RAFIs. Lipid-mixing assays were conducted as described in the legend to Fig. 7. For plaquing assays, approximately 200 VSV virions were exposed to increasing concentrations of aUY11, dUY11, aUY12, dUY5, or dUY1 for 10 min at 37°C. The virions thus exposed were then used to infect 5 × 106 Vero cells seeded in 6-well plates. Plaquing efficiency was calculated as a percentage of plaques formed by virions exposed to DMSO vehicle.
Table 2.
IC50 for fusion correlates with IC50 for plaquing efficiency
| RAFI | IC50 (μM) |
|
|---|---|---|
| Fusion | Plaquing | |
| dUY11 | 0.011 | 0.009 |
| aUY11 | 0.010 | 0.010 |
| dUY5 | 3.0 | 2.5 |
| aUY12 | 5.0 | 2.5 |
Structure-activity relationship studies had previously shown that amphipathicity, a hydrophilic head group larger than the hydrophobic group, and rigidity and planarity of the hydrophobic moiety were all necessary for inhibition of HSV-1 plaquing (11). Modifications that disrupt the amphipathicity and rigidity of RAFIs, therefore, disrupt their ability to inhibit infectivity. We then tested the effects of these modifications on fusion. dUY1 has a polar group in the hydrophobic moiety (and is less amphipathic than aUY11 or dUY11), whereas dUY5 has a nonplanar hydrophobic group (of a size similar to that of the hydrophilic moiety in aUY11 and dUY11), and aUY12 has a flexible and polar linker between the hydrophobic and hydrophilic groups (Fig. 1). Several concentrations of dUY1, dUY5, and aUY12 were tested in VSV fusion and plaquing efficiency assays. Although these RAFIs vary in their potencies by over 100-fold, each inhibited fusion and plaquing at similar concentrations (Fig. 10). Consequently, their IC50s in fusion closely corresponded to their IC50s in plaquing efficiency (Table 2).
DISCUSSION
We had reported that dUY11 inhibits the infectivity of otherwise unrelated enveloped viruses, and the fusion of VSV with cellular membranes, by targeting the envelope lipids to prevent the curvature changes required for fusion (11). We had proposed that other compounds with similar shapes should have similar activities against other enveloped viruses, including clinically important ones. aUY11, which has an arabinose moiety in place of the deoxyribose of dUY11, is biochemically and chemically distinct from dUY11. Nonetheless, the two compounds have the same overall three-dimensional shape, rigidity, and amphipathicity (Fig. 1). aUY11 inhibited the infectivity of HSV-1 (11), but its antiviral mechanisms or effects on other viruses remained unknown.
aUY11 inhibited, with similar potencies, the infectivity of clinically important viruses (influenza virus A, HCV, HSV-1, HSV-2, and Sindbis virus) with DNA or RNA genomes, which replicate in the nucleus or in the cytoplasm and which use different cellular receptors and enter cells by different fusion mechanisms. In contrast, it failed to inhibit the infectivity of nonenveloped viruses. The targets of aUY11 are therefore conserved only among enveloped viruses. We propose that aUY11 most likely targets the conserved lipid core of the envelopes. Consistently, aUY11 interacts with a hydrophobic component of enveloped virions (such as the envelope lipids) or protein-free liposomes and inhibits the fusion of these viruses and liposomes to target cells.
Influenza virus hemagglutinin (HA), like other class I fusion proteins, is predominantly composed of alpha helices containing an N-terminal hydrophobic fusion peptide (27) and forms trimers before and after fusion. The proposed mechanism for HA suggests the folding of the uncleaved protein to a metastable state (28), which is then activated by cleavage (27, 29). Fusion is triggered by low pH, resulting in irreversible conformational changes leading to a more stable postfusion conformation (30). HCV E2 is proposed to be a class II fusion protein, like those in other flaviviridae (31), which consist predominantly of beta sheets with the fusion peptide in internal loops (32). Class II fusion proteins are dimers parallel to the virion surface in their prefusion state but undergo conformational rearrangements to form postfusion trimers projecting perpendicularly from the virion envelope (32). VSV G protein is a class III fusion protein consisting of a mixed alpha helix and beta sheet structure (33). Class III fusion proteins undergo reversible conformational changes. Extended exposure to low pH inactivates the virions, but fusion is recovered when the pH is raised again (34).
The fusion proteins of influenza virus, HCV, and VSV represent all three different classes, with different structures and fusion mechanisms. However, aUY11 and dUY11 inhibited fusion of all three viruses at similar concentrations. Their targets are therefore not likely the fusion proteins but something even more conserved among enveloped viruses. aUY11 also inhibited acid-induced fusion of protein-free liposomes to cells (in the absence of any viral protein), strongly supporting the model in which RAFIs inhibit infectivity by acting on lipids. Although liposome fusion was inhibited only partially, RAFIs are the only small molecules known to inhibit this process, indicating that they directly target the lipids. The lipids in the envelopes of all enveloped viruses (or vesicles) must form a hemifusion stalk structure when fusing to target cell membranes (4). This process requires the formation of negative curvature by the outer leaflet of the envelope. aUY11 inhibits the transition from the lamellar phase (flat) to the hexagonal II phase (with negative curvature), indicating that it inhibits the formation of the negative curvature required for fusion. These results support the model in which RAFIs target virion envelope lipids to prevent fusion of viral and cellular membranes by biophysical mechanisms (i.e., inhibiting the formation of negative curvature in virion envelopes).
Virion fusion is affected by the lipid composition of the envelope (35). The molecular shape of lipids affects the formation of the negative curvature of the hemifusion stalk fusion intermediate. Enrichment of lipids with polar headgroups of larger diameter than their hydrophobic tails in the outer leaflet favors positive curvature, increasing the activation energy required to form the hemifusion stalk and thereby inhibiting viral fusion (6, 36). Accordingly, addition of exogenous lipids of the appropriate shape and polarity (such as LPC) prevents the fusion of many enveloped viruses (8–10, 37–39). Phospholipids, however, are not likely to be pharmacologically useful.
RAFIs are small synthetic compounds of appropriate shape and polarity to inhibit the formation of the negative curvature necessary for the hemifusion stalk without disrupting membranes. aUY11 has no obvious effects on cellular fusions, such as those required for cell replication (11) or endocytosis. aUY11 did not inhibit the infectivity of the nonenveloped poliovirus or reovirus, the entry of which requires internalization by endocytosis and fusion of endocytotic vesicles. aUY11 had no obvious effects on the morphology of intracellular membranous organelles or on exocytosis (data not shown). aUY11 has no cytotoxic or cytostatic effects, further supporting its lack of effect on cellular fusions (11). We speculate that the energy-consuming cellular processes that modulate the lipid composition and curvature of all cellular membranes overcome the increases in activation energy resulting from the effects of aUY11, whereas metabolically inert virions cannot overcome this increase in activation energy. However, this model has yet to be formally tested. Moreover, the activities of aUY11, or any other RAFI, in vivo are still unknown, as are their pharmacokinetics/pharmacodynamics. Studies in animal models are necessary to assess the actual potential of RAFIs as clinical antivirals.
Other membranotropic inhibitors of entry have been described. C5A inhibited the entry of several enveloped viruses, including HCV and HIV (40, 41), by virucidal mechanisms. Another inhibitor of HCV and HIV infectivity, PD 404182, was proposed to disrupt virions, perhaps by interfering with membrane fluidity or curvature (42). It might also act as a weak RAFI. Other compounds target virion envelopes and are nonvirucidal (43). Arbidol interacts with lipid membranes (44, 45) and might also target tryptophan residues in virion proteins (46, 47), inhibiting the fusion of enveloped viruses, such as influenza virus and HCV (47, 48). LJ-001 was proposed to damage lipid membranes (49), perhaps through generation of reactive oxygen species after exposure to light (F. Vigant, J. Lee, Z. Akyol-Ataman, L. Robinson, M. E. Jung, and B. Lee, presented at the 11th Annual Symposium on Antiviral Drug Resistance, Hershey, PA, 2010), damage that would be repaired by metabolically active cells, but not by inert virions.
Clinical entry inhibitors target viral glycoproteins, cellular receptors, or their interactions. The small molecule maraviroc inhibits the interactions between HIV gp120 and CCR5 (1), whereas the biomimetic peptide enfuvirtide inhibits the structural rearrangements of HIV gp41 (19, 50–52). Antibodies have also been explored as inhibitors of infectivity, mostly against respiratory syncytial virus (RSV) (53). Unfortunately, peptides and antibodies have poor oral bioavailability or stability (54). Furthermore, all these approaches suffer from the limitations of targeting any viral protein, such as selection for resistance (3, 55), which is particularly important for the viral surface proteins that commonly change rapidly to avoid immune responses (56). In contrast, RAFIs target viral envelope lipids, which are not encoded in the viral genome and are an essential structural component of all enveloped viruses. Therefore, they are expected to have a higher barrier to selection for resistance. Consistent with this expectation, we have not yet been able to select for dUY11-resistant HSV-1 mutants.
Importantly, RAFIs also protect cells from infection. aUY11 was particularly effective at preventing infection by influenza virus, which is internalized by endocytosis before fusion is triggered by the conformational changes in the virion glycoproteins at the low pH in the late endosomes (57). Pretreatment of cells was less effective at inhibiting the infectivity of HSV-1, which fuses to most cell types at neutral pH at the plasma membrane (58), or HCV, which follows a more complex entry pathway to fuse at a pH typical of earlier endosomes. aUY11 is internalized to many intracellular membranes. It may well accumulate in the late endosomes, which would result in a high-concentration reservoir to which the influenza virus, but not HSV-1 or HCV, virions would be exposed before fusion. Alternatively, the lack of inactivation of HSV-1 or HCV at low pH may allow these virions to make repeated attempts to fuse until one is successful.
aUY11 is also effective for treating cells already infected with influenza virus, HCV, or HSV-1. Although it does not protect the cells from virus-induced cytopathic effects, as expected from its mechanisms of action, the virions produced from the infected cells are not infectious. This effect is likely due to the incorporation of aUY11 in progeny virion envelopes, which are acquired during budding from the cellular membranes to which aUY11 localizes.
In summary, chemically distinct compounds with the same overall three-dimensional shape and amphipathicity inhibit viral infectivity by inhibiting fusion of viral and cellular membranes. aUY11 inhibits viral fusion due to its shape and amphipathicity through biophysical, not biochemical, mechanisms. Specifically, it inhibits the formation of the negative curvature necessary for the formation of the hemifusion stalk, a critical step in the fusion of enveloped viruses. aUY11 acts by the same biophysical mechanisms on several enveloped but otherwise unrelated viruses, including important human pathogens, such as influenza virus, HCV, and HSV-1/2. Here, we defined the mechanisms of RAFIs against such clinically important viruses. These mechanisms are fully consistent with those we had proposed based on dUY11 and a single model enveloped virus, VSV (11). We conclude that RAFIs are a novel family of antiviral compounds that act by biophysical mechanisms to prevent fusion of viral to cellular membranes.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by Canadian Institute of Health Research grants to L.M.S. (MOP 106452) and R.M.E. (MOP 86608) and the Burroughs Wellcome Fund (L.M.S.). L.M.S. is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease. V.A.K. was supported by the Molecular and Cellular Biology Program of the Russian Academy of Sciences; A.V.U. was supported by the Dynasty Foundation and a Russian President scholarship for young investigators (project SP-2494.2012.4). C.C.C. thanks the Natural Sciences and Engineering Research Council of Canada and Alberta Innovates-Health Solutions for graduate scholarships.
We are grateful to Takaji Wakita, Charles Rice, Veronika von Messling, Edward Mocarski, and Maya Shmulevitz for the kind gifts of HCV JFH-1, Huh7.5 cells, influenza virus strains, mCMV, and reovirus T3, respectively, and to Gary Eitzen for his assistance with the fusion assays.
Footnotes
Published ahead of print 2 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02882-12.
REFERENCES
- 1. Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. 2005. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49:4721–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wild CT, Shugars DC, Greenwell TK, McDanal CB, Matthews TJ. 1994. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. U. S. A. 91:9770–9774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 46:1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Earp LJ, Delos SE, Park HE, White JM. 2005. The many mechanisms of viral membrane fusion proteins. Curr. Top. Microbiol. Immunol. 285:25–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chernomordik LV, Kozlov MM. 2003. Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72:175–207 [DOI] [PubMed] [Google Scholar]
- 7. Chernomordik LV, Leikina E, Frolov VA, Bronk P, Zimmerberg J. 1997. An early stage of membrane fusion mediated by the low pH conformation of influenza hemagglutinin depends upon membrane lipids. J. Cell Biol. 136:81–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gunther-Ausborn S, Praetor A, Stegmann T. 1995. Inhibition of influenza-induced membrane fusion by lysophosphatidylcholine. J. Biol. Chem. 270:29279–29285 [DOI] [PubMed] [Google Scholar]
- 9. Gaudin Y. 2000. Rabies virus-induced membrane fusion pathway. J. Cell Biol. 150:601–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yeagle PL, Smith FT, Young JE, Flanagan TD. 1994. Inhibition of membrane fusion by lysophosphatidylcholine. Biochemistry 33:1820–1827 [DOI] [PubMed] [Google Scholar]
- 11. St Vincent MR, Colpitts CC, Ustinov AV, Muqadas M, Joyce MA, Barsby NL, Epand RF, Epand RM, Khramyshev SA, Valueva OA, Korshun VA, Tyrrell DL, Schang LM. 2010. Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc. Natl. Acad. Sci. U. S. A. 107:17339–17344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Andronova VL, Skorobogatyi MV, Manasova EV, Berlin I, Korshun VA, Galegov GA. 2003. Antiviral activity of some 5-arylethynyl 2′-deoxyuridine derivatives. Bioorg. Khim. 29:290–295 [DOI] [PubMed] [Google Scholar]
- 13. Skorobogatyi MV, Ustinov AV, Stepanova IA, Pchelintseva AA, Petrunina AL, Andronova VL, Galegov GA, Malakhov DA, Korshun VA. 2006. 5-Arylethynyl-2′-deoxyuridines, compounds active against HSV-1. Org. Biomol. Chem. 4:1091–1096 [DOI] [PubMed] [Google Scholar]
- 14. Blight KJ, McKeating JA, Rice CM. 2002. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 76:13001–13014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lacasse JJ, Schang LM. 2010. During lytic infections, herpes simplex virus type 1 DNA is in complexes with the properties of unstable nucleosomes. J. Virol. 84:1920–1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11:791–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stoddart CA, Cardin RD, Boname JM, Manning WC, Abenes GB, Mocarski ES. 1994. Peripheral blood mononuclear phagocytes mediate dissemination of murine cytomegalovirus. J. Virol. 68:6243–6253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lentz BR. 1989. Membrane “fluidity” as detected by diphenylhexatriene probes. Chem. Phys. Lipids 50:171–190 [Google Scholar]
- 19. Schmidt AG, Yang PL, Harrison SC. 2010. Peptide inhibitors of dengue-virus entry target a late-stage fusion intermediate. PLoS Pathog. 6:e1000851 doi:10.1371/journal.ppat.1000851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harada S. 2005. The broad anti-viral agent glycyrrhizin directly modulates the fluidity of plasma membrane and HIV-1 envelope. Biochem. J. 392:191–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lande MB, Donovan JM, Zeidel ML. 1995. The relationship between membrane fluidity and permeabilities to water, solutes, ammonia, and protons. J. Gen. Physiol. 106:67–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Harada S, Yusa K, Monde K, Akaike T, Maeda Y. 2005. Influence of membrane fluidity on human immunodeficiency virus type 1 entry. Biochem. Biophys. Res. Commun. 329:480–486 [DOI] [PubMed] [Google Scholar]
- 23. Shinitzky M, Barenholz Y. 1978. Fluidity parameters of lipid regions determined by fluorescence polarization. Biochim. Biophys. Acta 515:367–394 [DOI] [PubMed] [Google Scholar]
- 24. Epand RM, Epand RF. 1988. Kinetic effects in the differential scanning calorimetry cooling scans of phosphatidylethanolamines. Chem. Phys. Lipids 49:101–104 [Google Scholar]
- 25. Epand RM. 1986. Virus replication inhibitory peptide inhibits the conversion of phospholipid bilayers to the hexagonal phase. Biosci. Rep. 6:647–653 [DOI] [PubMed] [Google Scholar]
- 26. Connor J, Yatvin MB, Huang L. 1984. pH-sensitive liposomes: acid-induced liposome fusion. Proc. Natl. Acad. Sci. U. S. A. 81:1715–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wilson IA, Skehel JJ, Wiley DC. 1981. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3A resolution. Nature 289:366–373 [DOI] [PubMed] [Google Scholar]
- 28. Chen J, Lee KH, Steinhauer DA, Stevens DJ, Skehel JJ, Wiley DC. 1998. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 95:409–417 [DOI] [PubMed] [Google Scholar]
- 29. Wiley DC, Skehel JJ. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56:365–394 [DOI] [PubMed] [Google Scholar]
- 30. Bullough PA, Hughson FM, Skehel JJ, Wiley DC. 1994. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371:37–43 [DOI] [PubMed] [Google Scholar]
- 31. Krey T, d'Alayer J, Kikuti CM, Saulnier A, Damier-Piolle L, Petitpas I, Johansson DX, Tawar RG, Baron B, Robert B, England P, Persson MA, Martin A, Rey FA. 2010. The disulfide bonds in glycoprotein E2 of hepatitis C virus reveal the tertiary organization of the molecule. PLoS Pathog. 6:e1000762 doi:10.1371/journal.ppat.1000762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kielian M. 2006. Class II virus membrane fusion proteins. Virology 344:38–47 [DOI] [PubMed] [Google Scholar]
- 33. Roche S, Rey RA, Gaudin Y, Bressanelli S. 2007. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 315:843–848 [DOI] [PubMed] [Google Scholar]
- 34. Gaudin Y, Tuffereau C, Segretain D, Knossow M, Flamand A. 1991. Reversible conformational changes and fusion activity of rabies virus glycoprotein. J. Virol. 65:4853–4859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Teissier E, Pecheur EI. 2007. Lipids as modulators of membrane fusion mediated by viral fusion proteins. Eur. Biophys. J. 36:887–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chernomordik L. 1996. Non-bilayer lipids and biological fusion intermediates. Chem. Phys. Lipids 81:203–213 [DOI] [PubMed] [Google Scholar]
- 37. Chernomordik LV, Leikina E, Kozlov MM, Frolov VA, Zimmerberg J. 1999. Structural intermediates in influenza haemagglutinin-mediated fusion. Mol. Membr. Biol. 16:33–42 [DOI] [PubMed] [Google Scholar]
- 38. Stiasny K, Heinz FX. 2004. Effect of membrane curvature-modifying lipids on membrane fusion by tick-borne encephalitis virus. J. Virol. 78:8536–8542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vogel SS, Leikina EA, Chernomordik LV. 1993. Lysophosphatidylcholine reversibly arrests exocytosis and viral fusion at a stage between triggering and membrane merger. J. Biol. Chem. 268:25764–25768 [PubMed] [Google Scholar]
- 40. Bobardt MD, Cheng G, de Witte L, Selvarajah S, Chatterji U, Sanders-Beer BE, Geijtenbeek TB, Chisari FV, Gallay PA. 2008. Hepatitis C virus NS5A anchor peptide disrupts human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A. 105:5525–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cheng G, Montero A, Gastaminza P, Whitten-Bauer C, Wieland SF, Isogawa M, Fredericksen B, Selvarajah S, Gallay PA, Ghadiri MR, Chisari FV. 2008. A virocidal amphipathic alpha-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U. S. A. 105:3088–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chamoun AM, Chockalingam K, Bobardt M, Simeon R, Chang J, Gallay P, Chen Z. 2012. PD 404,182 is a virocidal small molecule that disrupts hepatitis C virus and human immunodeficiency virus. Antimicrob. Agents Chemother. 56:672–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Teissier E, Penin F, Pecheur EI. 2011. Targeting cell entry of enveloped viruses as an antiviral strategy. Molecules 16:221–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pecheur EI, Lavillette D, Alcaras F, Molle J, Boriskin YS, Roberts M, Cosset FL, Polyak SJ. 2007. Biochemical mechanism of hepatitis C virus inhibition by the broad-spectrum antiviral arbidol. Biochemistry 46:6050–6059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Villalain J. 2010. Membranotropic effects of arbidol, a broad anti-viral molecule, on phospholipid model membranes. J. Phys. Chem. B 114:8544–8554 [DOI] [PubMed] [Google Scholar]
- 46. Leneva IA, Russell RJ, Boriskin YS, Hay AJ. 2009. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antiviral Res. 81:132–140 [DOI] [PubMed] [Google Scholar]
- 47. Teissier E, Zandomeneghi G, Loquet A, Lavillette D, Lavergne JP, Montserret R, Cosset FL, Bockmann A, Meier BH, Penin F, Pecheur EI. 2011. Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS One 6:e15874 doi:10.1371/journal.pone.0015874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boriskin YS, Leneva IA, Pecheur EI, Polyak SJ. 2008. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 15:997–1005 [DOI] [PubMed] [Google Scholar]
- 49. Wolf MC, Freiberg AN, Zhang T, Akyol-Ataman Z, Grock A, Hong PW, Li J, Watson NF, Fang AQ, Aguilar HC, Porotto M, Honko AN, Damoiseaux R, Miller JP, Woodson SE, Chantasirivisal S, Fontanes V, Negrete OA, Krogstad P, Dasgupta A, Moscona A, Hensley LE, Whelan SP, Faull KF, Holbrook MR, Jung ME, Lee B. 2010. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. U. S. A. 107:3157–3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dwyer JJ, Wilson KL, Davison DK, Freel SA, Seedorff JE, Wring SA, Tvermoes NA, Matthews TJ, Greenberg ML, Delmedico MK. 2007. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. U. S. A. 104:12772–12777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, Alldredge L, Hunter E, Lambert D, Bolognesi D, Matthews T, Johnson MR, Nowak MA, Shaw GM, Saag MS. 1998. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4:1302–1307 [DOI] [PubMed] [Google Scholar]
- 52. Liu R, Tewari M, Kong R, Zhang R, Ingravallo P, Ralston R. 2010. A peptide derived from hepatitis C virus E2 envelope protein inhibits a post-binding step in HCV entry. Antiviral Res. 86:172–179 [DOI] [PubMed] [Google Scholar]
- 53. Huang K, Incognito L, Cheng X, Ulbrandt ND, Wu H. 2010. Respiratory syncytial virus-neutralizing monoclonal antibodies motavizumab and palivizumab inhibit fusion. J. Virol. 84:8132–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. 2010. Synthetic therapeutic peptides: science and market. Drug Discov. Today 15:40–56 [DOI] [PubMed] [Google Scholar]
- 55. Baba M, Miyake H, Wang X, Okamoto M, Takashima K. 2007. Isolation and characterization of human immunodeficiency virus type 1 resistant to the small-molecule CCR5 antagonist TAK-652. Antimicrob. Agents Chemother. 51:707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iannello A, Debbeche O, Martin E, Attalah LH, Samarani S, Ahmad A. 2006. Viral strategies for evading antiviral cellular immune responses of the host. J. Leukoc. Biol. 79:16–35 [DOI] [PubMed] [Google Scholar]
- 57. Yoshimura A, Kuroda K, Kawasaki K, Yamashina S, Maeda T, Ohnishi S. 1982. Infectious cell entry mechanism of influenza virus. J. Virol. 43:284–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wittels M, Spear PG. 1991. Penetration of cells by herpes simplex virus does not require a low pH-dependent endocytic pathway. Virus Res. 18:271–290 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.