

Abstract
A highly reproducible quantitative PCR (Q-PCR) assay was used to study the stability of human papillomavirus (HPV) in undifferentiated keratinocytes that maintain viral episomes. The term “stability” refers to the ability of episomes to persist with little copy number variation in cells. In investigating the mechanism of action of PA25, a previously published compound that destabilizes HPV episomes, aphidicolin was also found to markedly decrease episome levels, but via a different pathway from that of PA25. Since aphidicolin is known to activate DNA damage response (DDR) pathways, effects of inhibitors and small interfering RNAs (siRNAs) acting within DDR pathways were investigated. Inhibitors of Chk1 and siRNA directed against ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia Rad3-related (ATR) pathways significantly reduced viral episomes, suggesting that these pathways play a role in maintaining HPV episome stability. Inhibitors of Chk2 and DNA-PK had no effect on episome levels. Pharmacological inhibition of ATM proteins had no effect on episome levels, but ATM knockdown by siRNA significantly reduced episome levels, suggesting that ATM proteins are playing an important role in HPV episome stability that does not require kinase activity. These results outline two pathways that trigger episome loss from cells and suggest the existence of a little-understood mechanism that mediates viral DNA elimination. Together, our results also indicate that HPV episomes have a stability profile that is remarkably similar to that of fragile sites; these similarities are outlined and discussed. This close correspondence may influence the preference of HPV for integration into fragile sites.
INTRODUCTION
Human papillomaviruses (HPVs) are a large family of DNA viruses that infect stratified squamous epithelia of the skin, oropharynx, urogenital tract, and anus. A subset of high-risk viruses are essential, although not sufficient, to cause cancer (1). The small, approximately 8-kb viral genome is maintained as a circular extrachromosomal element, or episome, which encodes a limited number of proteins that are required for its establishment, replication, and maintenance. These proteins include E1, which, in cooperation with E2, binds and licenses the ori for DNA replication by forming a hexameric helicase to unwind viral DNA and initiate the loading of host cell replication factors (2). E2 is a protein with multiple functions that include transcriptional repression of the viral oncogenes E6 and E7 (3). E6 and E7 are required for maintenance of the viral genome, including assisting unscheduled rounds of DNA synthesis in differentiating keratinocytes in support of the productive phase of viral replication (4, 5).
A major risk factor for carcinogenic progression following infection by high-risk HPV is viral persistence (6). Episomal DNA persistence is thought to lead to viral DNA integration. Almost all cervical cancers exhibit integration at a single chromosomal locus, lending support for both the idea of a clonal origin of cervical cancer and the view that integration is key to carcinogenic progression (7). Integration occurs widely throughout the host genome but with a preference for fragile sites (8, 9). It has long been recognized that most HPV-associated cancers have integrated HPV sequences that do not retain the E1/E2 genes. The loss of E2 leads to upregulation of E6 and E7, genomic instability, and carcinogenic progression (10).
Cells maintaining HPV have been isolated from low-grade HPV lesions (11, 12) and created in the laboratory from primary cells transfected with HPV genomes (13–15). In general, high-risk HPV episomes are necessary for establishment of stable, episome-maintaining cell lines. This is most likely due to the ability of high-risk HPVs to influence keratinocyte phenotypes such as extension of cell life span, immortalization, circumvention of innate immunity, and promotion of DNA synthesis, which are required for both episome maintenance and the prolonged life span of the host cell line (4, 16, 17).
Keratinocytes maintaining high-risk HPV genomes in vitro, whether derived from clinical samples or engineered in the laboratory, generally maintain a constant number of episomes per cell (15, 18–20). The E2 protein and E8Ê2C have been implicated as playing an important role in HPV episome copy number control via their role in modulating both E1-dependent replication and transcriptional repression (21). Specific viral genes have been shown to be required for episome maintenance (6, 13, 22, 23). Control of viral episome levels has also been studied with respect to long-term cell population dynamics following viral DNA integration, and with respect to the productive replication phase of the viral life cycle during keratinocyte differentiation (15, 24, 25). It is known that significant episome loss follows interferon treatment in the HPV16 episome-maintaining cell line W12, and that the effect is not dependent upon the cytolytic loss of cells (25, 26). However, the cellular pathways responsible for episome elimination in response to interferon are unknown.
DNA viruses have a close association with DNA damage response (DDR) pathways, which are increasingly recognized as a first line of cellular antiviral defense (27, 28). Viruses represent a threat to genomic stability, and as such, the host cell acts to minimize genome damage and eliminate the foreign DNA. The virus employs strategies to circumvent detection and elimination, often redirecting the host DDR machinery to achieve these goals. The ataxia-telangiectasia mutated (ATM) and ATM and Rad3-related (ATR) serine/threonine protein kinases are primary sensors of DNA damage (29). ATM is chiefly involved with sensing and triggering the response to double-stranded DNA breaks. ATR has a wider function in organizing the response to a variety of DNA insults, including stalled replication forks and exposure of single-stranded regions of DNA. Acting downstream of ATM and ATR are the Chk2 and Chk1 kinases, respectively, which phosphorylate a host of substrates to coordinate the DDR (30). Elements of both the ATM and ATR pathways are activated in HPV-positive cells, and a role for ATM activation has been implicated in productive HPV DNA replication (31–34).
Stable maintenance, defined as the ability of cells to maintain a constant copy number of viral episomes over many cell passages in vitro, is well studied. However, little is known of the factors that contribute to episome stability over short periods of time in culture. We employ a quantitative PCR (Q-PCR) assay for HPV episomes that permits the rapid and accurate examination of changes in episome levels over short periods of time in culture (15, 19). Two DNA binding compounds, polyamide 1 (PA1) and polyamide 25 (PA25), targeting HPV were recently reported to cause a large, rapid loss of episomes in monolayer and organotypic cultures (19). In investigating the mechanism by which PA25 acts to destabilize episomes, we discovered that aphidicolin by itself also elicits episome loss from cells. We further found that ATR and Chk1, but not Chk2, play a role in stabilizing the HPV episome. ATM small interfering RNA (siRNA), but not ATM kinase inhibitor, also causes significant episome loss. The results reported here begin to reveal a role for DDR pathways in episome stability and suggest close parallels between HPV episomes and fragile sites. These results may help to provide insight into HPV persistence and integration.
MATERIALS AND METHODS
Cells and cell culture.
Tissue culture of normal foreskin keratinocytes (Ker-4) and keratinocytes maintaining HPV16 (W12E cells), HPV18 (Ker4-18 cells), or HPV31 was previously described (15, 19). Cells were cultured on mitomycin C-treated J2 3T3 cells in media containing three parts Dulbecco's modified Eagle medium (DMEM) and one part F12 medium. Culture media were supplemented with 0.4 μg/ml hydrocortisone, 10 ng/ml cholera toxin, 5 μg/ml insulin, 24 μg/ml adenine, 5 μg/ml transferrin, 20 pM 3,3′,5-triiodo-thyronine (T3), 5 ng/ml epidermal growth factor (EGF), 100 U/ml penicillin,100 μg/ml streptomycin, and 5% fetal bovine serum (FBS). Cells were passaged at 70% confluence using a split ratio of 1:10 (Ker-4, W12E, and HPV31 cells) or 1:20 (Ker4-18).
Inhibitors and siRNAs.
Pharmacological inhibitors were used at concentrations that had been previously reported to be effective in cells and against their enzymatic targets (35–39). Inhibitors were dissolved as stock solutions in 100% dimethyl sulfoxide (DMSO) and added to cell cultures at the indicated concentrations with a final vehicle concentration of 0.1% DMSO. Hydroxyurea was prepared in water. Chemical inhibitors (with sources and concentrations indicated in parentheses) used in the study included aphidicolin (Sigma catalog no. A0781; 0.1 μM to 25 μM), nocodazole (Sigma catalog no. M1404; 250 nM), hydroxyurea (Sigma catalog no. 8627; 2 mM), ATM inhibitor KU55933 (Tocris; 10 μM), Chk1 inhibitors PF477736 (Axon Medchem catalog no. 1379; 200 nM) and Chir124 (Axon Medchem catalog no. 1636; 200 nM), Chk2 inhibitor II hydrate (Sigma catalog no. C3742; 5 μM), and DNA-PK inhibitor NU7441 (Tocris; 1 μM).
All siRNAs were ON-TARGETplus SMARTpools purchased from Dharmacon, including Chk1 (catalog no. J-003255), Chk2 (catalog no. J-003256), ATR (catalog no. J-003202), ATM (catalog no. J-003201), and Nontargeting Control 2 (catalog no. D-001810). The siRNA was delivered 72 h prior to assay at a concentration of 100 nM using DharmaFect 1 transfection reagent (Dharmacon; catalog no. T-2001) according to the manufacturer's recommendations.
For ATM- or ATR-specific quantitative reverse transcription-PCR (Q-RT-PCR), RNA was extracted and reverse transcribed using a Maxima First-Strand cDNA synthesis kit from Fermentas (catalog no. K164125). cDNA (2.5 ng) was amplified using PCR primer sets obtained from Integrated DNA Technologies (IDT; San Jose, CA) at a final concentration of 300 nM. The ATR primer sequences were as follows: for the reverse primer, 5′-CCCAGACAAGCATGATCCAG-3′; and for the forward primer, 5′-GAAGATGATGACCACACTGAGA-3′. The ATM primers were as follows: for the reverse primer, 5′-CCTCAACACTTCTGACCATCT-3′; and for the forward primer, 5′-GTGCCTAAACAAAGCTCTCAG-3′. PCR conditions included 40 cycles of 95°C for 10 s, 60°C for 10 s, and 72°C for 10 s.
HPV episome stability assay.
HPV-maintaining cells were split into 24-well plates. The following day, inhibitors were added to replicate wells and cells cultured for 48 h. Total DNA was extracted by the use of DNAzol reagent (Invitrogen; catalog no. 10503-027), and quantitative PCR (Q-PCR) was performed using TaqMan probes on an ABI PRISM 7700 Sequence Detector (Applied Biosystems, Foster City, CA) as previously described (15, 19). The probes (IDT) were labeled with the 5′ reporter dye FAM (6-carboxyfluorescein) and the 3′ quencher dye TAMRA (6-carboxytetramethylrhodamine). Q-PCR mixtures contained 20 ng total DNA, a final concentration of 1× JumpStart Taq ReadyMix (Sigma; catalog no. D7440), 200 nM each primer (IDT), and 250 nM probe (IDT) in a reaction volume of 25 μl. Standard curves were generated using cloned HPV16, HPV18, and HPV31 DNA. The HPV episome copy number was calculated from the standard curve, and the effects of inhibitors were plotted as a percentage of the vehicle-treated control value.
Southern blotting.
Total cellular DNA was extracted by lysing the cells with 20 mM Tris (pH 8), 100 mM EDTA, 150 mM sodium chloride, and 1% sodium dodecyl sulfate (SDS). Proteinase K (Invitrogen; catalog no. 100005393) (50 μg/ml) was added, and lysates were incubated overnight at 37°C. Samples were extracted with phenol-chloroform-isoamyl alcohol (25:24:1 [vol/vol]) until the interphase was clear followed by 2 rounds of chloroform extraction. Total DNA was precipitated with 2 volumes of ethanol and incubated overnight at −20°C. Pellets were resuspended in 0.5 ml Tris-EDTA (TE) buffer (pH 8.0) and sheared by 10 passages through an 18-gauge needle, and 50 μg/ml RNase A (Sigma; catalog no. R4642) was added for 1 h at 37°C. DNA was again phenol-chloroform extracted and ethanol precipitated. DNA pellets were resuspended in TE buffer, and 5 μg was digested with BamHI or HindIII (New England BioLabs). DNA was subjected to electrophoresis at 5 V/cm for 18 h, transferred onto positively charged nylon membranes (Roche), and probed with HPV16 genomic DNA that had been gel purified and labeled with [32P]dCTP using a Random Primed DNA Labeling kit (Roche) according to the manufacturer's directions. Membranes were exposed to phosphor screens and imaged with a Molecular Dynamics Phosphorimager.
Western blotting.
Cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed in radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% NP-40, 2% sodium deoxycholate, 0.1% SDS) supplemented with 2× Halt Protease & Phosphatase Inhibitor Cocktail (ThermoScientific; catalog no. 1861284). Lysates were incubated with 150 U/ml DNase I (ThermoScientific; catalog no. 89835) for 30 min at room temperature with mixing. Protein concentrations were determined by a bicinchoninic acid (BCA) assay (ThermoScientific; catalog no. 23227), and 50 μg of protein was subjected to electrophoresis in Tris-glycine (4% to 20%) gels (NuPAGE). Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes using an iBlot system (Invitrogen), and the membranes were blocked with 5% nonfat milk–TBST buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1% Tween 20) overnight at 4°C. Membranes were then incubated at room temperature for 2 h with one of the following antibodies: ATR (Santa Cruz; catalog no. sc-28901), pATR (Santa Cruz; catalog no. sc-109912), ATM (Abcam; catalog no. ab78), pATM (Abcam; catalog no. ab81292), Chk1 (Cell Signaling; catalog no. 2G1D5), pChk1 (Cell Signaling; catalog no. 133D3), Chk2 (Cell Signaling; catalog no. 1C12), pChk2 (Cell Signaling; catalog no. C13C1), Rad9 (Santa Cruz; catalog no. sc-32489), pRad9 (Santa Cruz; catalog no. sc-130213), RPA32 (Abcam; catalog no. ab2175), or β-actin (Abcam; catalog no. ab6276). Primary antibodies were detected with goat anti-rabbit or goat anti-mouse poly-horseradish peroxidase (poly-HRP) (Pierce; catalog no. 32230 and 32260) (1:25,000) or donkey anti-goat IgG-HRP (Santa Cruz; catalog no. sc-2033) (1:5,000) secondary antibody.
Fluorescence-activated cell sorter (FACS) analysis.
W12E cells were treated for 24 h with aphidicolin, nocodazole, or hydroxyurea and harvested for cell cycle analysis by trypsinization. Cells were washed twice in 4°C phosphate-buffered saline (PBS); 1 ml of modified Videlov's propidium iodide staining solution was added, and each sample was subjected to a vigorous vortex procedure. Samples were stained for a minimum of 1 h at 4°C and analyzed using a Becton Dickinson FACSCalibur 4-color flow cytometer (Becton Dickinson). Data analysis was performed using BD CellQUEST Pro v6.0.1 software.
RESULTS
W12E cells maintaining approximately 300 to 500 HPV16 episome copies per cell and exhibiting no evidence of viral DNA integration were first studied. Viral DNA in Southern blots was associated predominantly with episomal bands, and no additional bands indicative of integration were detected (Fig. 1). Initial experiments queried whether cell cycle progression was required for the antiviral effect of PA25. Three agents that activate cell cycle checkpoints were initially tested for their ability to block PA25-mediated loss of HPV16 episomes. Cell cycle status had no measurable effect upon the antiviral activity of PA25. Aphidicolin, nocodazole, and hydroxyurea all acted to block cells within the cell cycle as anticipated (Fig. 2). Arrest of W12E cells in G2/M by pretreatment of cells with nocodazole (Fig. 3A) or at the G1/S-phase boundary by treatment with hydroxyurea (Fig. 3B) or aphidicolin (Fig. 3C) did not prevent the loss of episomes following treatment with PA25. Nocodazole by itself had no effect on episome levels as measured by Q-PCR (Fig. 3A), while hydroxyurea treatment caused a small apparent decrease in episome levels that was not statistically significant (Fig. 3B). In contrast, aphidicolin alone, at a concentration of 4 μM, elicited a robust decrease in HPV16 episome levels in W12E cells (Fig. 3C).
Fig 1.

Figure demonstrating maintenance of HPV16 as episomes in W12E cells. (A) The HPV16 episome copy number is derived using a standard curve. Representative Q-PCR amplification curves show the relationship between log increases in HPV16 plasmid DNA level and threshold cycle (Ct) value. The inset shows a linear regression plot of the cloned HPV16 genome standard. Efficiency of PCR amplification is calculated as E = −1 + 10(−1/slope). Twelve Q-PCRs with 20 ng input DNA from W12E cells are shown. W12E cells are maintained at early passage with HPV16 copy numbers ranging from 300 to 500 copies/cell. Clustered curves (block arrow) represent unknown W12E cell DNA sample replicates. (B) Southern blot analysis of HPV16 episome-containing W12E cells. A 5-μg mass of total DNA from W12E cells was digested with BamHI or HindIII, and episomes were detected by probing with full-length [32P]dCTP Random Primed HPV16 DNA. Two exposures of the same Southern blot are shown.
Fig 2.

FACS analysis reveals effects of inhibitors used to activate cell cycle check points. (A) Effects of increasing doses of aphidicolin on arresting cells in the S-phase and at the G1/S boundary. (B) Nocodazole causes accumulation of cells in the G2/M phase. (C) Hydroxyurea arrests cells at the G1/S boundary.
Fig 3.

Inhibitors of cell cycle progression were tested for effects on the antiviral activity of compound PA25. (A) W12E cells were treated with 250 nM nocodazole (Noc; Sigma catalog no. M1404) with or without 1 μM PA25 (n = 6). (B) W12E cells were treated for 24 h with 2 mM hydroxyurea (HU) with or without 1 μM PA25. HU had a small effect on HPV16 episome levels that was not statistically significant. HU did not block PA25 activity. (C) Effects of 4 μM aphidicolin (Aph) on W12E cell episomes in the presence of increasing amounts of PA25 or 0.1% DMSO control for 48 h. **, aphidicolin versus DMSO control (n = 12 samples from 3 independent experiments); *, aphidicolin plus PA25 versus PA25 alone (n = 6); P = <0.01. (D) HPV18 and HPV31 genomes are also lost following aphidicolin treatment. HPV31 or HPV18 episome-maintaining cell lines were treated for 48 h with 4 μM aphidicolin and episomes quantified by real-time PCR (n = 6; *, P = <0.01).
These results were surprising because a variety of pharmaceutically active and toxic compounds, including etoposide and podophyllotoxin (data not shown), in addition to hydroxyurea and nocodazole (Fig. 3), have little or no significant effect on HPV episome levels. Furthermore, aphidicolin contributed to HPV episome loss in an additive manner when it was provided prior to treatment with multiple, increasing PA25 doses (Fig. 3C). This observation indicated that aphidicolin and PA25 were causing episome loss via different cellular pathways.
The effects of aphidicolin on two additional HPV genotypes, HPV18 and HPV31, were then also tested. Aphidicolin, at a concentration of 4 μM, resulted in a statistically significant loss of approximately 40% of HPV31 episomes and 50% of HPV18 episomes after 48 h (Fig. 3D), showing that the effect was not limited to a single HPV genotype or cell type.
An aphidicolin dose-response analysis conducted with W12E cells showed that increasing amounts of the inhibitor to 10 μM resulted in a greater loss of episomes, with 80% of the viral DNA eliminated after 48 h at the highest dose (Fig. 4). The loss of episomes appeared to correlate with aphidicolin effects on the cell cycle (Fig. 2). The lowest doses (0.1 and 0.4 μM), which had smaller effects on HPV episome copy number, allowed cells to enter S phase, where they stalled and accumulated. On the other hand, the higher doses, which caused greater episome loss, blocked S-phase entry, resulting in congregation of cells at the G1/S boundary (Fig. 2).
Fig 4.
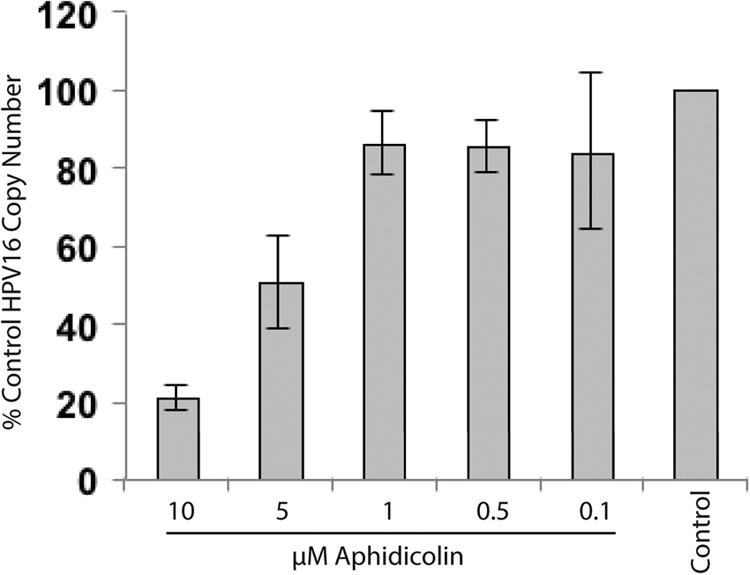
Aphidicolin decreases HPV16 episome copy numbers in a dose-dependent manner. W12E cells were treated for 48 h with increasing aphidicolin concentrations (n = 6).
Aphidicolin is known to activate a DDR in the ATR/Chk1 pathway due to stalling of replication forks. It can also activate the ATM/Chk2 pathway due to collapse of replication forks and generation of double-stranded DNA (dsDNA) breaks. The presence of HPV episomes can also cause an elevation of cellular DDR signaling, and in fact our own experiments found that a variety of DDR elements were activated in cells that maintain HPV episomes relative to normal keratinocytes (Fig. 5A). In addition, we found that aphidicolin activates both the ATM and ATR pathways in HPV16-positive cells (Fig. 5B). For this reason, we set out to test the hypothesis that DDR elements contribute to HPV episome stability in undifferentiated keratinocytes.
Fig 5.
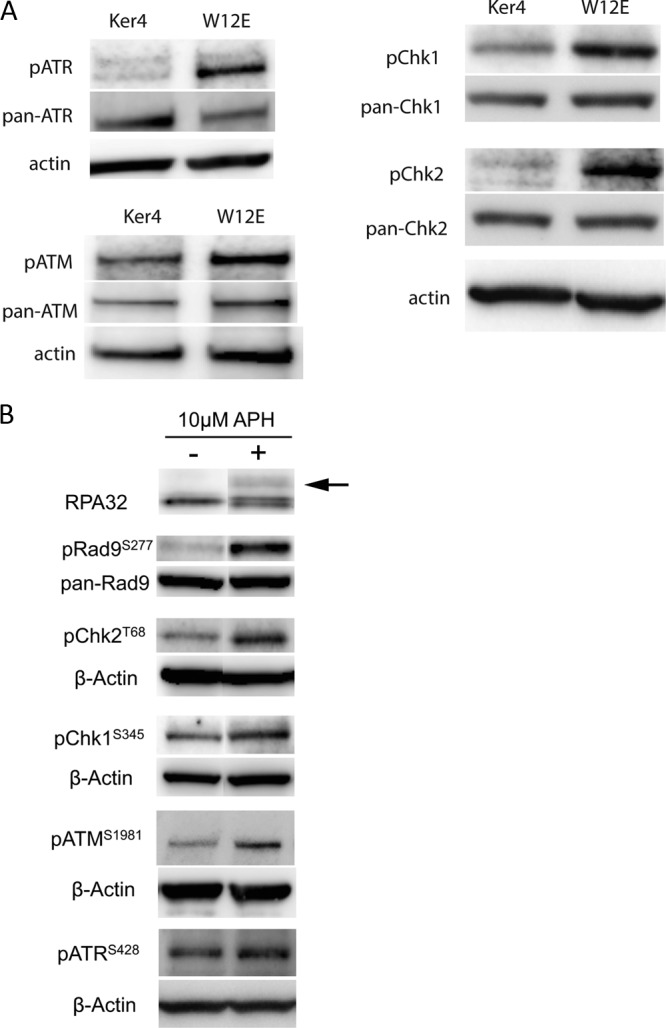
Western blots demonstrating activation of DNA damage repair elements in cells maintaining HPV episomes and following treatment of cells with aphidicolin. (A) Western blots showing activation of DNA damage repair elements in W12E cells relative to normal human keratinocytes (Ker4). ATM/ATR and Chk1/Chk2 are constitutively activated in HPV16 episome-maintaining cells. Western blots show basal levels of DDR elements recognized with antibodies directed against phosphorylated epitopes or recognizing full-length protein. (B) Western blots of extracts of W12E cells treated with aphidicolin (+) or of vehicle-treated controls (−). All DDR elements show various degrees of activation following aphidicolin treatment. The arrow indicates the position of migration of the phosphorylated form of RPA32.
We first tested inhibitors acting within the ATR/Chk1 pathway for their effects on HPV episome stability. Similar to the aphidicolin results, the Chk1 kinase inhibitor Chir124 produced a statistically significant 40% reduction in episome levels (Fig. 6A). Chir124 also reduced HPV episomes in an additive manner when coadministered with increasing doses of PA25 (Fig. 6B). This result suggested that, like aphidicolin (Fig. 3), Chir124 acts to destabilize episomes via a pathway different from that of PA25. PF477736, a second Chk1 inhibitor chemically distinct from Chir124, also caused a statistically significant 50% decline in HPV16 episome numbers (Fig. 6A). Additional inhibitors acting on other DDR pathway elements were also tested: KU55933, an ATM-specific kinase inhibitor, Chk2-specific inhibitor II, and the DNA-PK inhibitor NU7441 all had no effect on episome levels (Fig. 6A).
Fig 6.
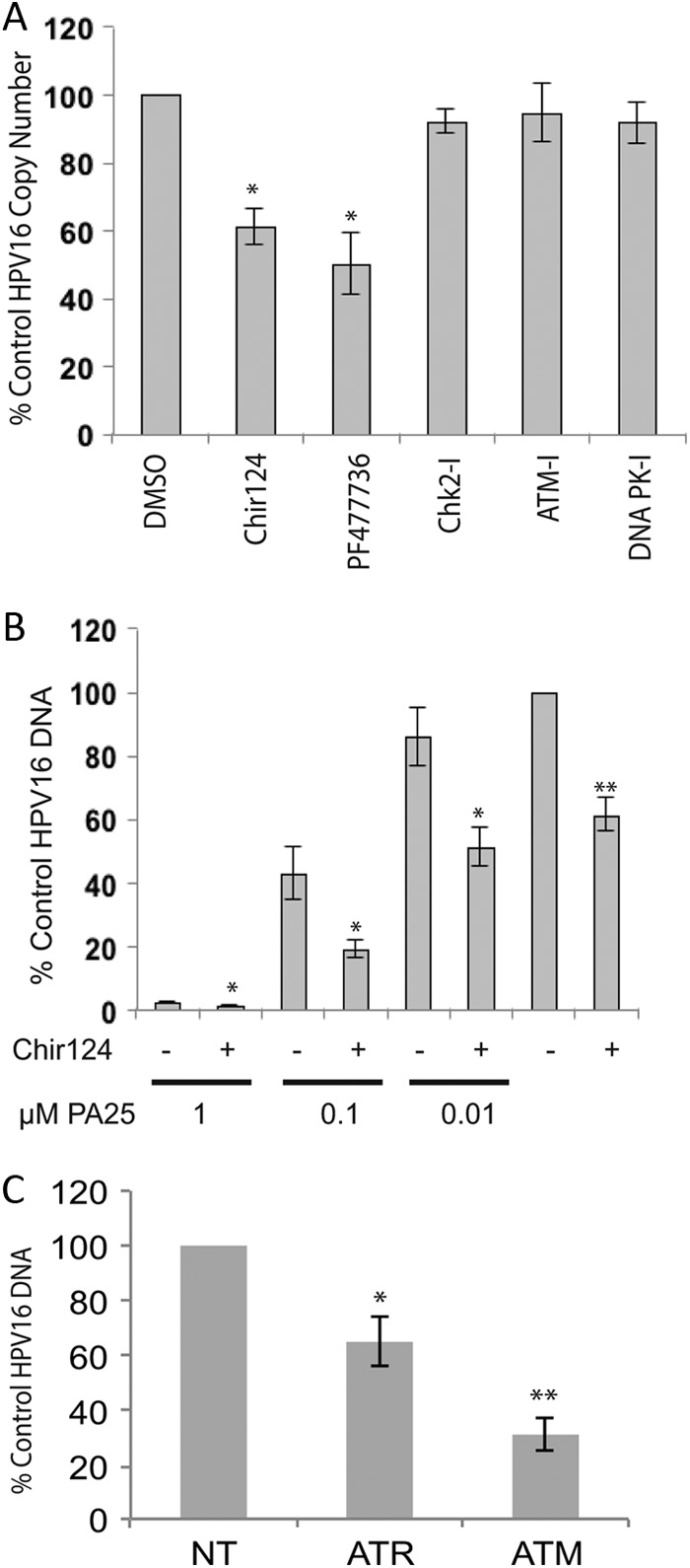
The effects of inhibitors or siRNA acting in the ATR/Chk1 or ATM/Chk2 pathways on W12E cell HPV episomal DNA. (A) W12E cells treated for 48 h with 10 μM ATM inhibitor (ATM-I; Tocris catalog no. KU55933), 5 μM Chk2 inhibitor II (Chk2-I; catalog no. Sigma C3742), or 1 μM DNA-PK inhibitor (DNA PK-I; Tocris catalog no. NU7441) displayed no changes in episome levels relative to control cells. In contrast, 200 nM Chk1 inhibitor PF477736 (Axon Medchem; catalog no. 1379) or Chir124 (Axon Medchem; catalog no. 1636) significantly reduced episome levels compared to vehicle-treated cell results (n = 6; *P < 0.01). (B) Chk1 inhibitor Chir124 significantly reduces HPV16 episomes in an additive manner with PA25 as measured by Q-PCR. W12E cells were treated with 200 nM Chir124 in the presence of increasing PA25 concentrations or 0.1% DMSO vehicle control for 48 h. Statistically significant Chir124 effects are indicated by asterisks: **, Chir124 versus DMSO control; *, aphidicolin plus PA25 versus PA25 alone; n = 6; P ≤ 0.01. (C) siRNA knockdown of ATR or ATM significantly reduces HPV16 episome levels. W12E cells were treated with 100 nM ATR, ATM, or nontargeting siRNA (NT) for 72 h and episomes quantified by Q-PCR. Experimental replicates were analyzed against NT-control siRNA replicates. *, n = 6, P = <0.01, error bars represent standard deviations; **, n = 3 independent experiments with 3 replicates each, P = <0.01, error bars represent standard errors of the means.
There were no available specific chemical ATR inhibitors of which we were aware, so we employed a siRNA strategy to target ATR for downregulation. ATR siRNA reduced ATR transcript levels by 78% relative to nontargeting control levels as measured by Q-RT-PCR. Q-PCR of the ATR siRNA-treated W12E cells demonstrated a 40% loss of HPV16 episomal DNA at 48 h (Fig. 6C). As a control for this experiment, we also tested the effects of siRNA directed against ATM. ATM siRNA caused a 64% reduction in ATM mRNA levels after 72 h of treatment. Surprisingly, ATM siRNA also caused a highly reproducible 70% decrease in HPV16 episome levels. The data shown represent a summary of three independent experiments, each having multiple replicates (Fig. 6C). The specificity of both ATR and ATM siRNAs was confirmed in a matrix of 22 siRNAs chosen from DDR pathways (data not shown).
The effects of aphidicolin and Chir124 on viral episome maintenance were also investigated by Southern blotting in order to confirm and extend the Q-PCR results (Fig. 7). Loss of HPV16 DNA following treatment with either inhibitor was time dependent. As expected, the samples digested with either BamHI or HindIII showed no evidence of viral DNA integration. Samples digested with BamHI, which linearizes HPV16 episomes, yielded the expected 7.9-kb HPV16 DNA that was progressively lost over 48 h in the presence of either aphidicolin or Chir124 (Fig. 7). Samples that had not been linearized (HindIII) also exhibited clear evidence of viral DNA loss over time. These samples did not display any noticeable changes in the ratios or electrophoretic migration of form I (supercoiled) DNA or form II (open circle) DNA, indicating that the two forms were lost at similar rates (Fig. 7).
Fig 7.
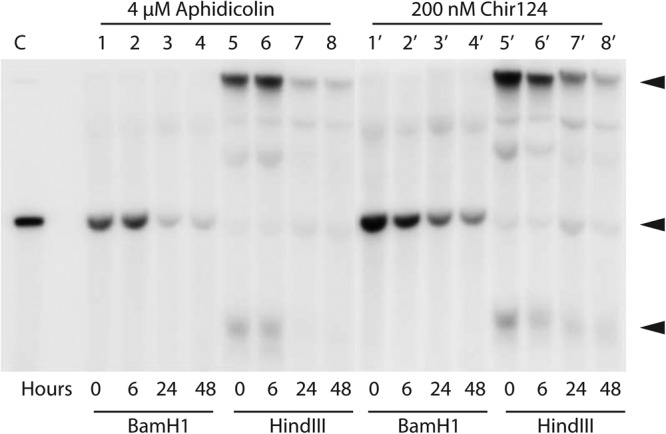
Southern blot of HPV16 DNA from W12E cells treated with 4 μM aphidicolin (lanes 1 to 8) or 200 nM Chir124 (lanes 1′ to 8′). Prior to loading, the DNA was digested either with BamHI (lanes 1 to 4 and 1′ to 4′), which linearizes the HPV16 episome, or with HindIII (lanes 5 to 8 and 5′ to 8′), which does not cut HPV16. Arrowheads indicate the positions of migration of open circle, linear, and supercoiled HPV forms (from top to bottom) in the uncut samples.
We next asked if siRNA knockdown of Chk1 or Chk2 would also affect HPV episome levels. Western blots of W12E cells transfected with siRNAs demonstrated the expected specificities and effects for the respective siRNAs. Chk1 siRNA reduced Chk1 protein levels while having no noticeable effect upon the related Chk2 kinase. Conversely, Chk2 siRNA specifically reduced Chk2 protein levels (Fig. 8A). The levels of HPV episomes in these cells were then quantified. Chk1 knockdown alone elicited a 60% to 70% reduction of HPV16. On the other hand, Chk2 knockdown alone had no significant effect upon HPV16 episomes relative to cells receiving nontargeting siRNA and untreated controls (Fig. 8B). A 24-h treatment with aphidicolin was also carried out following siRNA-mediated knockdown of Chk1 and Chk2. In the case of Chk1 siRNA, aphidicolin treatment caused no further reduction in episome levels. On the other hand, a 40% reduction in episome levels was seen following aphidicolin treatment of Chk2 siRNA-treated cells (Fig. 8B). Since Chk1 siRNA and aphidicolin do not exhibit an additive or additional effect on HPV episome levels, the data support the hypothesis that they decrease episome levels by a shared or closely related mechanisms.
Fig 8.
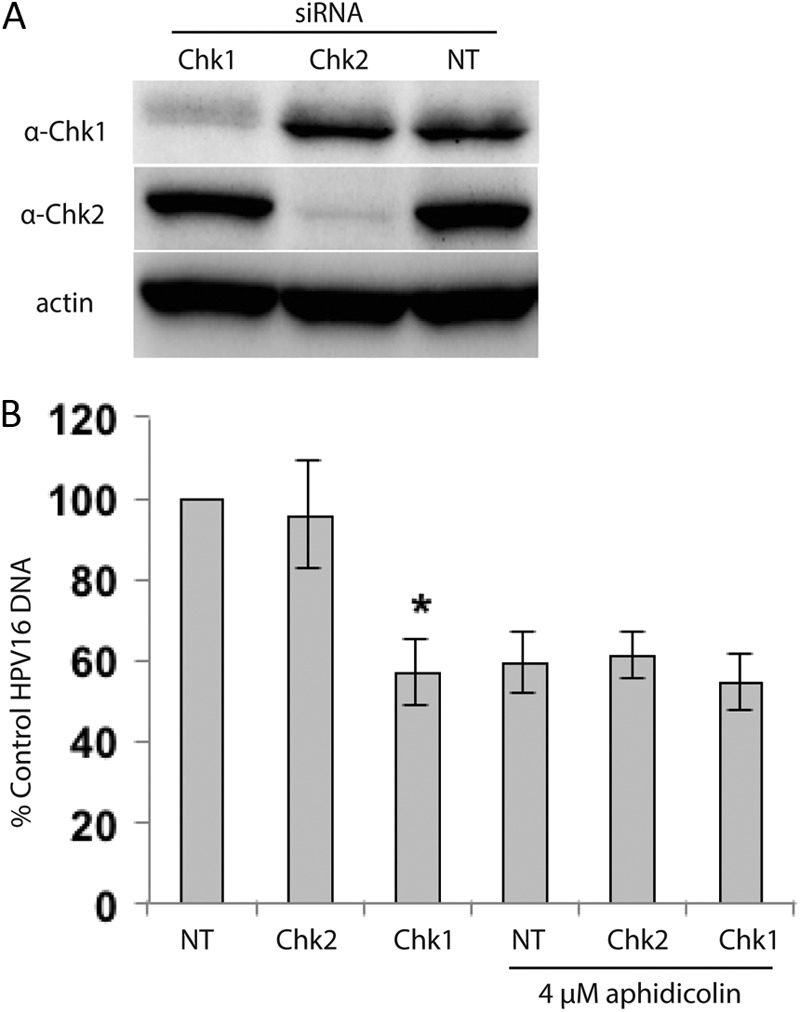
siRNA knockdown of Chk1, but not Chk2, significantly reduces HPV episomes in W12E cells, and aphidicolin treatment causes no additional episome loss. (A) Western blot showing specific knockdown of Chk1 and Chk2 (Cell Signaling 2G1D5 and 1C12 antibodies, respectively) following 72 h of incubation with 100 nM siRNA (Chk1 and Chk2 SMARTpool siRNA; Dharmacon). Control cells were treated with 100 nM nontargeting siRNA 2 (NT; Dharmacon). (B) Q-PCR analysis of HPV episomes following siRNA-mediated knockdown of Chk1 or Chk2. Following siRNA treatment for 72 h, W12E cells were incubated with 4 μM aphidicolin or 0.1% DMSO for 24 h. Reductions in HPV16 episome levels were observed following knockdown of Chk1 (Q-PCR, n = 6; *, P = <0.01). Aphidicolin treatment did not result in an additional decrease in the HPV copy number in cells receiving Chk1 siRNA.
DISCUSSION
A highly reproducible Q-PCR assay makes it possible to examine inhibitors and siRNAs for their ability to cause instability and loss of HPV episomes. Numerous studies have contributed to our understanding of HPV episome maintenance (for recent reviews, see references 6 and 40), but little work has focused on understanding conditions affecting short-term stability and loss of HPV episomes. The present studies show the feasibility of such an approach while outlining a role for DDR elements in HPV episome stability. These studies also help elucidate two apparently independent pathways that control HPV episome stability and loss. DNA binding compounds represented by PA1 and PA25 trigger one pathway, and aphidicolin and inhibitors of the ATR/Chk1 axis reveal the other. Both pathways lead to rapid loss of episomes by a process that we are seeking to understand. The process must ultimately involve the degradation or export of HPV episome DNA. If loss is occurring by degradation, the process is rapid since no evidence of viral DNA degradation intermediates is found by Southern blotting. In addition, there must be substantial selectivity, or discrimination of self DNA from non-self DNA, in that viral genomes are targeted for loss and the host DNA is spared. Understanding these features of viral DNA loss has the potential to contribute to our knowledge of such disparate processes as the interferon mechanism of action, and HPV persistence and integration.
PA25 is one of a series of synthetic, higher-order distamycin derivatives initially designed to target the ori of HPV18. The compound causes a dramatic and rapid loss of HPV episomes in vitro, verifying that the acute instability of HPV episomes can be achieved under certain conditions (19). Experiments initially showed that the action of PA25 was not affected by cell cycle checkpoint arrest enacted by a number of agents. In the process, it was discovered that aphidicolin alone was able to cause the loss of HPV episomes. Additionally, when HPV-maintaining cells were treated with both PA25 and aphidicolin, an additive effect on episome loss was observed. This interesting observation allowed us to conclude that aphidicolin and PA25 were acting to destabilize episomes via different cellular pathways. Two chemically distinct Chk1 inhibitors were also additive with PA25 in their ability to trigger HPV episome loss. This observation, and the fact that aphidicolin does not cause additional episome loss when added after Chk1 siRNA knockdown, is consistent with aphidicolin causing episome instability via an ATR/Chk1 pathway. These results also suggest that the ATR/Chk1 pathway is not playing a role in PA25-mediated episome loss.
It was previously shown using an infectious assay that aphidicolin and other agents that block cell cycle progression are able to effectively block HPV infection (41). Our results present a clear difference from those results by focusing on episome DNA levels and not infectivity. We demonstrate that agents activating cell cycle checkpoints, such as hydroxyurea and nocodazole, have little or no effect on HPV episome numbers, but that DNA polymerase inhibition by aphidicolin causes significant episome loss. Aphidicolin and hydroxyurea both cause replication stress, but hydroxyurea, which acts by depleting cells of deoxynucleoside triphosphates (dNTPs), had no significant effect on HPV episomes. Therefore, replication stress alone is not sufficient to cause episome instability.
Aphidicolin is a polymerase inhibitor that causes replication forks to stall and single-stranded DNA (ssDNA) to accumulate due to the continued activity of the MCM helicase (42). As such, it is a potent activator of the ATR/Chk1 pathway (43). It is well documented that viruses commonly activate host cellular DDR pathways, and that this activation is important for a number of viral processes such as establishment of infection, viral persistence, and completion of the viral life cycle. We found that treatment of HPV-maintaining cells with inhibitors or siRNAs directed against ATR or Chk1 resulted in significant loss of HPV episomal DNA. Hence, our studies suggest an important relationship between cellular signaling through ATR/Chk1 and the stability of HPV episomes and indicate that episomes are lost under conditions that compromise replication fork stability. Like the MCM helicase in replicating mammalian DNA, the E1 helicase in the HPV replication fork may remain active and become uncoupled from DNA polymerase in the presence of aphidicolin. In this way, ssDNA may be generated, stimulating activation of the ATR/Chk1 pathway. Interestingly, transfected papillomavirus E1 alone or with E2 is sufficient for activation of both ATR and ATM (32, 34). In light of the present results, it is conceivable that aphidicolin uncouples polα and E1, resulting in a stalled, unstable replication fork. However, multiple examples of E1-independent maintenance and replication of HPV episomes have been reported (44–47), making it possible only to speculate on a role for E1 in the episome loss reported here. For this reason, we are interested in examining the status of the viral genome under conditions of instability. However, we have been unsuccessful at colocalizing markers of stalled replication forks with the unamplified HPV genome in undifferentiated cells due to a lack of sensitivity of our in situ hybridization procedure. These efforts are continuing.
It is worthwhile to seek out examples of genomic DNA that exhibit signs of instability under similar conditions. We find a notable similarity between the stability of fragile sites and that of HPV episomes (48) (Table 1). Aphidicolin is widely used as an agent for inducing the expression of chromosomal fragile sites (49, 50). Fragile sites are typically visually measured as breakage points in mitotic chromosomes. Consequently, low levels of aphidicolin that inhibit DNA synthesis, and yet allow cell cycle progression into mitosis, are generally the standard used in such studies. Other notable similarities between the stability of fragile sites and that of the HPV episome also bear mentioning (Table 1). Aphidicolin was previously shown to promote the expression of fragile sites via the ATR/Chk1 pathway, and ATR/Chk1, but not ATM/Chk2, is required for maintaining the stability of fragile sites (51–53). ATM has also been shown to play a role in fragile site stability, but only in the absence of ATR (55).
Table 1.
A summary of similarities in stability between common and rare fragile sites and HPV episomes
| Treatment | Stability of fragile site in indicated categorya |
HPV stabilityb | Reference(s) | |
|---|---|---|---|---|
| Common | Rare | |||
| Aphidicolin | Yes | Yes | Yes | (49, 50) |
| ATM dependent | No | No | No, yesb | (51) |
| ATR dependent | Yes | Yes | Yes | (51, 52) |
| Chk1 dependent | Yes | — | Yes | (53) |
| Chk2 dependent | No | — | No | (53) |
| Distamycin sensitive | No | Yes | Yes | (19, 54) |
| AT-rich DNA | Yes | Yes | Yes | (48) |
Treatment/gene effect on loss of common and rare fragile site stability. —, not reported.
Treatment/gene effect causes loss of HPV episome stability.
cATM siRNA but not ATM inhibitor causes episome loss.
Using inhibitors and siRNA, we demonstrated additional parallels between HPV episomal DNA and fragile sites beyond aphidicolin sensitivity. HPV episomes and fragile sites are both sensitive to Chk1 inhibitors and siRNA, and to ATR siRNA, but not to ATM, Chk2, or DNA-PK inhibitors (Table 1). ATM appears to be an interesting, more complicated exception: while inhibition of ATM kinase activity had no effect on HPV episomes, in agreement with previous findings (33), ATM siRNA promoted a large loss of episomes. Previous ATM kinase inhibitor studies described DNA repair defects in cells that are not observed in ATM-null cells, suggesting that even the inactivated protein has roles in DNA repair (56, 57). This was supported by two recent publications showing that the presence of full-length, kinase-dead ATM results in early embryonic lethality in mouse models (58, 59). The enzymatically inactive ATM lacks dominant-negative interfering activity and yet interferes with homologous recombination to a greater extent than seen in ATM-null cells. Recent work has suggested a role for homologous recombination in HPV episome replication based upon colocalization of pathway members, including pATM, Chk2, and 53BP1, with HPV genomes in undifferentiated cells (60). Simian virus 40 (SV40) and Epstein-Barr virus (EBV) also both utilize homologous recombination for efficient replication (22, 37). Thus, while kinase inhibition of either ATM or Chk2 causes no loss of HPV episomes, the acute loss of episomes following ATM siRNA knockdown is consistent with an important role for homologous recombination in HPV episome stability in the undifferentiated keratinocyte.
Other parallels between fragile sites and HPV episomes are also evident (Table 1). Fragile sites are AT-rich islands with the potential to form stable secondary structures that contribute to instability during replication stress. The AT-rich nature of fragile sites also contributes to the sensitivity of a subset of rare fragile sites especially prone to damage by the natural product distamycin A, a drug that binds the minor groove of AT-rich DNA sequences (54). It is intriguing that HPV genomes are also AT rich, with the HPV16 genome having approximately 63% AT sequence (61). Some regions of HPV, especially within the long control region (LCR), are exceedingly AT rich. It is to these sequences that we targeted distamycin-like synthetic compounds such as PA25, which have a remarkable propensity to destabilize and eliminate HPV genomes (19). While we do not know of any effects that PA25 and related compounds have on fragile sites, we previously showed that high concentrations of distamycin A destabilize HPV episomes (19), demonstrating yet another connection with fragile sites. Any similarity in stability between fragile sites and HPV genomes is of potential importance because HPV integration is central to current models of HPV-related carcinogenesis (1) and because a preference for HPV integration at fragile sites is well established (8, 9). If HPV episomes and fragile sites share similar traits of instability, then it is likely that a single cellular event would result in destabilization of both. A simplified path to HPV integration is therefore more easily envisaged, predicted, and possibly prevented.
In summary, we report here that HPV episomal DNA is sensitive to aphidicolin, inhibitors of Chk1, and knockdown of ATR, ATM, and Chk1 by siRNA. We also show that ATR/Chk1 is likely in the pathway mediating the aphidicolin effect on episomal stability. Furthermore, we show evidence for at least two pathways mediating episomal stability since PA25 does not cause episome instability via the same pathway as aphidicolin, ATR, or Chk1. It is notable that most of these approaches and pathways have also been shown to elicit fragile site expression (49). We believe our results provide a framework for exploring HPV episome stability that has the potential to provide insights into HPV DNA persistence, integration, and oncogenesis.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI062182, AI068159, and AI083803.
We thank Thomas Glover, Martin Arlt, and Thomas Wilson (Department of Human Genetics, University of Michigan) for helpful discussions regarding fragile sites. Richard West (West Labs Scientific, LLC) carried out the FACS analysis. Lou Laimins (Northwestern University) provided the HPV31-containing cells used in this study, and Paul Lambert (University of Wisconsin) provided W12E cells.
C.F. and J.K.B. are cofounders of NanoVir and hold a significant ownership position in the company.
Footnotes
Published ahead of print 30 January 2013
REFERENCES
- 1. zur Hausen H. 2002. Papillomaviruses and cancer: from basic studies to clinical application. Nat. Rev. Cancer 2:342–350 [DOI] [PubMed] [Google Scholar]
- 2. Stenlund A. 2003. Initiation of DNA replication: lessons from viral initiator proteins. Nat. Rev. Mol. Cell Biol. 4:777–785 [DOI] [PubMed] [Google Scholar]
- 3. McBride AA, Romanczuk H, Howley PM. 1991. The papillomavirus E2 regulatory proteins. J. Biol. Chem. 266:18411–18414 [PubMed] [Google Scholar]
- 4. Garner-Hamrick PA, Fostel JM, Chien WM, Banerjee NS, Chow LT, Broker TR, Fisher C. 2004. Global effects of human papillomavirus type 18 E6/E7 in an organotypic keratinocyte culture system. J. Virol. 78:9041–9050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moody CA, Laimins LA. 2010. Human papillomavirus oncoproteins: pathways to transformation. Nat. Rev. Cancer 10:550–560 [DOI] [PubMed] [Google Scholar]
- 6. Bodily J, Laimins LA. 2011. Persistence of human papillomavirus infection: keys to malignant progression. Trends Microbiol. 19:33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woodman CB, Collins SI, Young LS. 2007. The natural history of cervical HPV infection: unresolved issues. Nat. Rev. Cancer 7:11–22 [DOI] [PubMed] [Google Scholar]
- 8. Thorland EC, Myers SL, Gostout BS, Smith DI. 2003. Common fragile sites are preferential targets for HPV16 integrations in cervical tumors. Oncogene 22:1225–1237 [DOI] [PubMed] [Google Scholar]
- 9. Thorland EC, Myers SL, Persing DH, Sarkar G, McGovern RM, Gostout BS, Smith DI. 2000. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res. 60:5916–5921 [PubMed] [Google Scholar]
- 10. zur Hausen H. 1991. Human papillomaviruses in the pathogenesis of anogenital cancer. Virology 184:9–13 [DOI] [PubMed] [Google Scholar]
- 11. Doorbar J, Parton A, Hartley K, Banks L, Crook T, Stanley M, Crawford L. 1990. Detection of novel splicing patterns in a HPV16-containing keratinocyte cell line. Virology 178:254–262 [DOI] [PubMed] [Google Scholar]
- 12. Meyers C, Frattini MG, Hudson JB, Laimins LA. 1992. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science 257:971–973 [DOI] [PubMed] [Google Scholar]
- 13. Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. 2000. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J. Virol. 74:6622–6631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frattini MG, Lim HB, Laimins LA. 1996. In vitro synthesis of oncogenic human papillomaviruses requires episomal genomes for differentiation-dependent late expression. Proc. Natl. Acad. Sci. U. S. A. 93:3062–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garner-Hamrick PA, Fisher C. 2002. HPV episomal copy number closely correlates with cell size in keratinocyte monolayer cultures. Virology 301:334–341 [DOI] [PubMed] [Google Scholar]
- 16. Hebner CM, Laimins LA. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 16:83–97 [DOI] [PubMed] [Google Scholar]
- 17. Münger K, Howley PM. 2002. Human papillomavirus immortalization and transformation functions. Virus Res. 89:213–228 [DOI] [PubMed] [Google Scholar]
- 18.Bedell MA, Hudson JB, Golub TR, Turyk ME, Hosken M, Wilbanks GD, Laimins LA. 1991. Amplification of human papillomavirus genomes in vitro is dependent on epithelial differentiation. J. Virol. 65:2254–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Edwards TG, Koeller KJ, Slomczynska U, Fok K, Helmus M, Bashkin JK, Fisher C. 2011. HPV episome levels are potently decreased by pyrrole-imidazole polyamides. Antiviral Res. 91:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jeon S, Allen-Hoffmann BL, Lambert PF. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 69:2989–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8E2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol. 74:1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bodily JM, Mehta KP, Cruz L, Meyers C, Laimins LA. 2011. The E7 open reading frame acts in cis and in trans to mediate differentiation-dependent activities in the human papillomavirus type 16 life cycle. J. Virol. 85:8852–8862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wilson R, Fehrmann F, Laimins LA. 2005. Role of the E1–E4 protein in the differentiation-dependent life cycle of human papillomavirus type 31. J. Virol. 79:6732–6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang YE, Pena L, Sen GC, Park JK, Laimins LA. 2002. Long-term effect of interferon on keratinocytes that maintain human papillomavirus type 31. J. Virol. 76:8864–8874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herdman MT, Pett MR, Roberts I, Alazawi WO, Teschendorff AE, Zhang XY, Stanley MA, Coleman N. 2006. Interferon-beta treatment of cervical keratinocytes naturally infected with human papillomavirus 16 episomes promotes rapid reduction in episome numbers and emergence of latent integrants. Carcinogenesis 27:2341–2353 [DOI] [PubMed] [Google Scholar]
- 26. Chang YE, Laimins LA. 2000. Microarray analysis identifies interferon-inducible genes and Stat-1 as major transcriptional targets of human papillomavirus type 31. J. Virol. 74:4174–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Turnell AS, Grand RJ. 2012. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 93:2076–2097 [DOI] [PubMed] [Google Scholar]
- 28. Weitzman MD, Lilley CE, Chaurushiya MS. 2010. Genomes in conflict: maintaining genome integrity during virus infection. Annu. Rev. Microbiol. 64:61–81 [DOI] [PubMed] [Google Scholar]
- 29. Harper JW, Elledge SJ. 2007. The DNA damage response: ten years after. Mol. Cell 28:739–745 [DOI] [PubMed] [Google Scholar]
- 30. Stracker TH, Usui T, Petrini JH. 2009. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 8:1047–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banerjee NS, Wang HK, Broker TR, Chow LT. 2011. Human papillomavirus (HPV) E7 induces prolonged G2 following S phase reentry in differentiated human keratinocytes. J. Biol. Chem. 286:15473–15482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol. 85:8996–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5:e1000605 doi:10.1371/journal.ppat.1000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sakakibara N, Mitra R, McBride A. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 85:8981–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arienti KL, Brunmark A, Axe FU, McClure K, Lee A, Blevitt J, Neff DK, Huang L, Crawford S, Pandit CR, Karlsson L, Breitenbucher JG. 2005. Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J. Med. Chem. 48:1873–1885 [DOI] [PubMed] [Google Scholar]
- 36. Ashwell S, Zabludoff S. 2008. DNA damage detection and repair pathways—recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 14:4032–4037 [DOI] [PubMed] [Google Scholar]
- 37. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. 2004. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 64:9152–9159 [DOI] [PubMed] [Google Scholar]
- 38. Tavecchio M, Munik JM, Cano C, Newell DR, Curtin NJ. 2012. Further characterisation of the cellular activity of the DNA-PK inhibitor, NU7441, reveals potential cross-talk with homologous recombination. Cancer Chemother. Pharmacol. 69:155–164 [DOI] [PubMed] [Google Scholar]
- 39. Tse AN, Rendahl KG, Sheikh T, Cheema H, Aardalen K, Embry M, Ma S, Moler EJ, Ni ZJ, Lopes de Menezes DE, Hibner B, Gesner TG, Schwartz GK. 2007. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 13:591–602 [DOI] [PubMed] [Google Scholar]
- 40. McBride AA, Sakakibara N, Stepp WH, Jang MK. 2012. Hitchhiking on host chromatin: how papillomaviruses persist. Biochim. Biophys. Acta 1819:820–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 5:e1000318 doi:10.1371/journal.ppat.1000318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. 2005. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 19:1040–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith J, Tho LM, Xu N, Gillespie DA. 2010. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108:73–112 [DOI] [PubMed] [Google Scholar]
- 44. Angeletti PC, Kim K, Fernandes FJ, Lambert PF. 2002. Stable replication of papillomavirus genomes in Saccharomyces cerevisiae. J. Virol. 76:3350–3358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Egawa N, Nakahara T, Ohno S, Narisawa-Saito M, Yugawa T, Fujita M, Yamato K, Natori Y, Kiyono T. 2012. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol. 86:3276–3283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kim K, Lambert PF. 2002. E1 protein of bovine papillomavirus 1 is not required for the maintenance of viral plasmid DNA replication. Virology 293:10–14 [DOI] [PubMed] [Google Scholar]
- 47. Pittayakhajonwut D, Angeletti PC. 2010. Viral trans-factor independent replication of human papillomavirus genomes. Virol. J. 7:123 doi:10.1186/1743-422X-7-123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Durkin SG, Glover TW. 2007. Chromosome fragile sites. Annu. Rev. Genet. 41:169–192 [DOI] [PubMed] [Google Scholar]
- 49. Glover TW, Arlt MF, Casper AM, Durkin SG. 2005. Mechanisms of common fragile site instability. Hum. Mol. Genet. 14(Suppl 2):R197–R205 [DOI] [PubMed] [Google Scholar]
- 50. Mrasek K, Schoder C, Teichmann AC, Behr K, Franze B, Wilhelm K, Blaurock N, Claussen U, Liehr T, Weise A. 2010. Global screening and extended nomenclature for 230 aphidicolin-inducible fragile sites, including 61 yet unreported ones. Int. J. Oncol. 36:929–940 [DOI] [PubMed] [Google Scholar]
- 51. Casper AM, Nghiem P, Arlt MF, Glover TW. 2002. ATR regulates fragile site stability. Cell 111:779–789 [DOI] [PubMed] [Google Scholar]
- 52.Kumari D, Somma V, Nakamura AJ, Bonner WM, D'Ambrosio E, Usdin K. 2009. The role of DNA damage response pathways in chromosome fragility in Fragile X syndrome. Nucleic Acids Res. 37:4385–4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Durkin SG, Arlt MF, Howlett NG, Glover TW. 2006. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 25:4381–4388 [DOI] [PubMed] [Google Scholar]
- 54. Hori T, Takahashi E, Murata M. 1988. Nature of distamycin A-inducible fragile sites. Cancer Genet. Cytogenet. 34:189–194 [DOI] [PubMed] [Google Scholar]
- 55. Ozeri-Galai E, Schwartz M, Rahat A, Kerem B. 2008. Interplay between ATM and ATR in the regulation of common fragile site stability. Oncogene 27:2109–2117 [DOI] [PubMed] [Google Scholar]
- 56. Gamper AM, Choi S, Matsumoto Y, Banerjee D, Tomkinson AE, Bakkenist CJ. 2012. ATM protein physically and functionally interacts with proliferating cell nuclear antigen to regulate DNA synthesis. J. Biol. Chem. 287:12445–12454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. White JS, Choi S, Bakkenist CJ. 2010. Transient ATM kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci. Signal. 3:ra44 doi:10.1126/scisignal.2000758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Daniel JA, Pellegrini M, Lee BS, Guo Z, Filsuf D, Belkina NV, You Z, Paull TT, Sleckman BP, Feigenbaum L, Nussenzweig A. 2012. Loss of ATM kinase activity leads to embryonic lethality in mice. J. Cell Biol. 198:295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yamamoto K, Wang Y, Jiang W, Liu X, Dubois RL, Lin CS, Ludwig T, Bakkenist CJ, Zha S. 2012. Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice. J. Cell Biol. 198:305–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gillespie KA, Mehta KP, Laimins LA, Moody CA. 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 86:9520–9526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seedorf K, Krammer G, Durst M, Suhai S, Rowekamp WG. 1985. Human papillomavirus type 16 DNA sequence. Virology 145:181–185 [DOI] [PubMed] [Google Scholar]