

Abstract
The varicella-zoster virus (VZV) ORF61 protein is necessary for normal replication in vitro and virulence in human skin xenografts in the severe combined immunodeficiency mouse model in vivo. These experiments identify a hydrophobic domain that mediates ORF61 self-interaction. While not needed to inhibit host cell defenses, disruption of this domain (residues 250 to 320) severely impairs VZV growth, transactivation of the immediate early 63 and glycoprotein E genes, and the pathogenesis of VZV skin infection in vivo.
TEXT
Varicella-zoster virus (VZV) is a human alphaherpesvirus that causes varicella (chicken pox) during primary infection, establishes lifelong latency in the host, and reactivates to cause zoster (shingles) (1). VZV has a double-stranded DNA genome of approximately 125 kbp which encodes at least 70 unique viral proteins. Although not present in the virion tegument, ORF61 is a 467-amino-acid protein expressed along with IE62 shortly after virus entry (2). ORF61 truncation and promoter mutations that severely limit ORF61 synthesis are associated with little or no VZV replication in cultured cells (3, 4). Our investigations of the ORF61 promoter mutant virus revealed that this protein is critical for VZV pathogenesis in skin in vivo (4). ORF61 acts as a weak transactivator for a few viral promoters in transient-transfection experiments and regulates expression of the major VZV glycoprotein gE in infected cells (5). ORF61 also has important functions in counteracting intrinsic antiviral cellular responses. ORF61 potently suppresses the tumor necrosis factor alpha (TNF-α)-activated NF-κB response in a dose-dependent manner (6). In addition, both VZV infection and ORF61 expression disrupt promyelocytic leukemia protein (PML) and Sp100 nuclear bodies (NBs) (7, 8). Our laboratory has demonstrated that ORF61 causes a dramatic reduction in PML NB frequency in VZV-infected cells in vitro and in skin cells infected within their tissue microenvironment in vivo (6, 8). This process depends upon three functional SUMO-interacting motifs (SIMs) in ORF61, which are essential for dispersal of PML NBs in infected cells in vitro and in human skin cells in xenografts in SCID mice infected in vivo (6). The disruption of ORF61 SIMs resulted in severely impaired virulence of VZV in skin xenografts in vivo (6). Incomplete dispersal of PML NBs by VZV inhibits egress of VZV capsids from the infected cell nucleus, impairing the synthesis of infectious virus progeny (8). This ORF61-dependent antiviral mechanism has biological significance, since skin cells and neurons and satellite cells in human dorsal root ganglia (DRG) infected with VZV in vivo contain large PML NBs that sequester newly assembled nucleocapsids (8).
ORF61 is functionally related to ICP0 encoded by herpes simplex viruses 1 and 2 (HSV-1 and HSV-2) and can functionally complement an HSV-1 ICP0 deletion mutant (9). ORF61 has a RING finger domain that is highly conserved among all alphaherpesvirus orthologs. Since putative SIMs are present in ORF61 orthologs, we proposed that SIM-dependent functions are likely to be conserved within this family proteins. However, except for the RING domain and the SIMs, these orthologs share very limited sequence identity.
This study was designed to identify other functional domains in ORF61 that are important for VZV replication and pathogenesis. First, an analysis of the ORF61 amino acid sequence was performed with the Eukaryotic Linear Motif (ELM) resource and revealed a globular domain located between amino acids 250 to 320, suggesting that a structural domain might be present within this region (10). Analysis using CLC Main Workbench software showed that this region is hydrophobic, with a local hydrophobicity count of 52% compared with 42% for the full-length ORF61 protein. In addition, the Kyte-Doolittle plot indicated that this domain displays distinctively higher hydrophobicity than the other regions except for the zinc finger domain (amino acids 1 to 60) (Fig. 1A, bottom panel). Therefore, we designated this domain the ORF61 hydrophobic domain (HPD) (Fig. 1A, top panel) and hypothesized that the HPD might mediate a hydrophobic interaction between ORF61 molecules. We investigated whether ORF61 self-interacts in transfected cells (Fig. 1B, lanes 1 to 3). Two ORF61 expression constructs, pFlag-ORF61, in which ORF61 is fused with a Flag tag in the N terminus, and pHis-ORF61, in which ORF61 is fused with a 6×His tag in the C terminus, were made by inserting Flag- or His-tagged fragments into pcDNA3.1. Melanoma cells were transfected with pHis-ORF61 alone or together with pFlag-ORF61 and lysed with coimmunoprecipitation buffer (20 mM HEPES [pH 7.2], 90 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, 20% glycerol) in the presence of Benzonase (25 units/ml) at 48 h after transfection. Immunoprecipitation was then performed with Flag (M2) monoclonal antibody (Sigma), and the precipitated complex was subjected to Western blotting with His antibody (Delta Biolabs). His-ORF61 was detected in the immunocomplex when it was coexpressed with Flag-ORF61 but was not detected when expressed alone (Fig. 1B, lanes 1 and 2), suggesting that His-ORF61 interacted with Flag-ORF61. An antibody heavy chain band was detected in all immunoprecipitation reactions with Flag antibody (Fig. 1B). No protein was detected in the antibody-free control (Fig. 1B, lane 3), which further confirmed the specificity of this interaction. The predicted HPD was then deleted in both pHis-ORF61 and pFlag-ORF61 constructs, resulting in pHis-ΔHPD. When pHis-ΔHPD was cotransfected with pFlag-ORF61, only a very limited amount of His-ΔHPD protein was detected in the Flag-precipitated complex (Fig. 1B, lane 4). This weak interaction was specific, as confirmed with both the antibody-free control and the His-ΔHPD-alone control (Fig. 1B, lanes 5 and 6). The strong interaction between Flag-ORF61 and His-ORF61 and the dramatically reduced interaction between Flag-ORF61 and His-ΔHPD were further confirmed in the reciprocal coimmunoprecipitation experiment using His antibody (Fig. 1C). To examine if ORF61 self-associates in VZV-infected cells, ORF61 in VZV-infected cells was evaluated by Western blotting. Both ORF61 monomers and oligomers were detected (Fig. 1D), which confirms the ORF61 oligomerization in VZV-infected cells. Taken together, these results demonstrated that ORF61 self-interacts and the interaction is mediated by ORF61 HPD.
Fig 1.

Identification of a hydrophobic domain (HPD) in ORF61 and its contribution to ORF61 self-interaction. (A) Schematic representation of the ORF61 motifs (SIM-N and SIM-C) and subdomains (RING finger and HPD). The numbers represent the amino acid positions of the motifs and domains. A hydrophobicity plot of the whole ORF61 was calculated by the Kyte-Doolittle method (14), and the HPD domain is highlighted with a dashed box. (B) Coimmunoprecipitation of Flag-tagged ORF61 with His-tagged ORF61 or the His-tagged ΔHPD mutant protein. Cells were transfected with ORF61 and the ΔHPD mutant as illustrated. The transfected cell lysate was subjected to immunoprecipitation with Flag monoclonal antibody and then immunoblotted with His polyclonal antibody. (C) Reciprocal coimmunoprecipitation. The transfected cell lysate was subjected to immunoprecipitation with His polyclonal antibody and then immunoblotted with Flag monoclonal antibody. (D) ORF61 oligomerization in VZV-infected cells. VZV-infected cell lysate was subjected to SDS-PAGE and then immunoblotted with ORF61 polyclonal antibody.
In order to determine whether this ORF61 HPD is conserved among alphaherpesviruses, sequence alignment between ORF61 HPD and other alphaherpesvirus orthologs was performed using ClustalW2. Similar regions were identified in the ORF61 orthologs in simian varicella virus (SVV), equine herpesvirus (EHV), and feline herpesvirus (FeHV) orthologs (Fig. 2). The identity of this region between VZV ORF61 and the SVV ortholog was ∼65% compared to ∼23 to 27% in EHV and FeHV (Fig. 2). Of interest, VZV and SVV belong to the Varicellovirus genus within the Alphaherpesvirinae subfamily. The sequence alignment showed that some residues within this domain are conserved across these viruses and, more importantly, the most conserved residues are hydrophobic amino acids, suggesting that regional hydrophobicity is shared by these viruses and it is probably a conserved mechanism of self-associations of the ORF61 homologs in these viruses. As noted, VZV ORF61 has functions similar to HSV ICP0 in transient transfection and in infected cells (5, 9). ICP0 expressed from baculovirus or adenovirus vectors also self-interacts; the C-terminal residues 633 and 775 are necessary for its multimerization (11). However, no region in ICP0 was found to have a significant level of identity to the ORF61 HPD and a hydrophobicity analysis showed that the hydrophobic residue count of the ICP0 self-association domain was similar to that of the full-length ICP0 protein (both are ∼55%). This information suggests that ICP0 self-interacts via a mechanism that differs from the ORF61 hydrophobic interaction. The relatively high degree of conservation of the ORF61 HPD within the varicelloviruses but not other orthologs suggests that HDP-dependent self-association might make a unique contribution to the replication and pathogenesis of this subfamily, which differs from the other alphaherpesviruses.
Fig 2.

Sequence alignment of ORF61 HPD with orthologs in other alphaherpesviruses by ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/). An asterisk represents positions which have a single, fully conserved residue, a colon represents conservation between groups of strongly similar properties (scoring >0.5 in the Gonnet PAM 250 matrix), and a period represents conservation between groups of weakly similar properties (scoring <0.5 in the Gonnet PAM 250 matrix).
To evaluate the role of the ORF61 HPD during VZV replication, we generated a VZV recombinant virus in which the ORF61 gene sequence encoding residues 250 to 320 was deleted. Mutagenesis was done using our pOka cosmid system (12). First, ORF61 HPD was removed from pvSpe23, which includes ORF54 to ORF71. The resultant cosmid was designated pvSpe23-ORF61ΔHPD. Melanoma cells were then transfected with the intact pvSpe23 or pvSpe23-ORF61ΔHPD along with the other three pOka cosmids, pvSpe14, pvFsp73, and pvPme2. Recombinant pOka virus was recovered from transfections done with intact pvSpe23 at 7 days posttransfection, while plaques only became visible in cells transfected with pvSpe23-ORF61ΔHPD at 14 days posttransfection. The recombinant virus was designated pOka-ORF61ΔHPD. The absence of the ORF61 HPD coding sequence from the mutant virus genome was confirmed by sequencing the PCR fragment, which spans the deleted region (data not shown). Next, the growth kinetics of the ORF61 HPD deletion virus was compared with pOka in melanoma cells as described previously (4). Melanoma cells were inoculated with the two viruses at 103 PFU per 106 cells, and virus yields from day 1 to 6 after inoculation were quantitated. Virus yields from pOka-ORF61ΔHPD-infected cells were significantly lower (∼0.5 to 1 log less) than pOka at each time point over 6 days (P < 0.001) (Fig. 3A). Plaques formed by pOka-ORF61ΔHPD were also much smaller than pOka plaques (Fig. 3B). These data demonstrated that the ORF61 HPD, which mediates ORF61 self-interaction, was required for efficient VZV replication and cell-to-cell spread in cultured cells. The kinetics of the expression of ORF61 itself, an immediate early protein, IE63, and a VZV glycoprotein, gE, were analyzed in melanoma cells infected with pOka and pOka-ORF61ΔHPD at 103 PFU per 106 cells for 24, 48, and 72 h. Like ORF61, at least two species of the ORF61 ΔHPD mutant protein were detected, and as expected, they migrated slightly faster than ORF61 (Fig. 3C). Both IE63 and gE expression were substantially reduced in pOka-ORF61ΔHPD-infected cells compared with pOka-infected cells, with a greater effect on gE (Fig. 3C).
Fig 3.
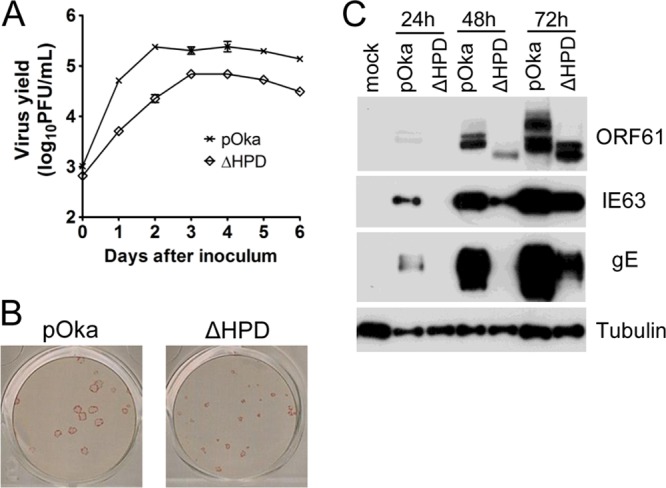
ORF61 HPD contributes to VZV replication in vitro and viral protein expression. (A) Replication of pOka and ORF61ΔHPD mutant viruses in melanoma cells over 6 days. Each point represents the mean ± standard error of the mean (SEM) of virus titers from three replicates. (B) Plaques formed by pOka and pOka-ORF61ΔHPD infection in melanoma cells for 5 days were visualized by immunostaining with anti-VZV serum. (C) VZV protein expression in melanoma cells infected with pOka and pOka-ORF61ΔHPD for 24, 48, and 72 h. Western blots of ORF61, IE63, gE, and tubulin are shown.
We also investigated the effects of the HPD deletion on ORF61 protein stability and other ORF61 functions. Since ORF61 undergoes rapid proteasome-mediated degradation (13), ΔHPD mutant protein accumulation after proteasome inhibitor treatment was examined. Melanoma cells (106 cells) were inoculated with 103 PFU of pOka or 104 PFU of pOka-ORF61ΔHPD in order to achieve similar levels of expression of viral proteins. The purpose of using a 10-fold-higher inoculum of the ΔHPD mutant was to compare the protein accumulation within a certain amount of time from a more similar initial level. At 48 h postinfection, cells were treated with 10 µM MG132 for 6 h and then subjected to Western blot analysis. The accumulation of the mutated ΔHPD protein after MG132 treatment was much less than ORF61, although the expression levels of the two proteins were similar before treatment (Fig. 4A). Expression levels of IE63 and gE remained unchanged after MG132 treatment (Fig. 4A). This result suggested that ORF61 might associate with the ubiquitin proteasome complex via ORF61 HPD or ORF61 oligomer formation is a prerequisite for the subsequent recruitment of proteasome proteins.
Fig 4.

The contributions of ORF61 HPD to ORF61 functions and VZV virulence in vivo. (A) Melanoma cells infected with pOka or pOka-ORF61ΔHPD were treated with 10 μM MG132 for 6 h. Western blots with ORF61, IE63, gE, and tubulin antibodies are shown. (B) The effect of ORF61 HPD deletion on the gE promoter. Luciferase assays were performed on melanoma cells transfected with the gE promoter and pcDNA constructs expressing ORF61 or the ΔHPD mutant. (C) NF-κB suppression by ORF61 and the ΔHPD mutant. Luciferase assays were performed on melanoma cells that were cotransfected with NF-κB reporter plasmid and increasing amounts (10 to 250 ng) of pcDNA-ORF61ΔHPD and then treated with TNF-α. The graphs show the relative luciferase units (firefly luciferase units normalized with Renilla luciferase units). (D) (Upper panels) Confocal microscopy of melanoma cells infected with the ΔHPD mutant for 6 h and stained for ORF61 (red), PML (green), and nuclei (blue). The images show association of puncta (indicated by white arrows) formed by ΔHPD mutant proteins with PML NBs. Scale bar, 5 μm. (Lower panels) Confocal microscopy of melanoma cells infected with the ΔHPD mutant for 24 h and stained for ORF61 (red), PML (green), and nuclei (blue). The images show PML NB dispersal in pOKa-ORF61ΔHPD-infected cells. Infected nuclei with abundant expression of ΔHPD mutant proteins are outlined by dashed ovals. Scale bar, 10 μm. (E) Replication of pOka and ΔHPD mutant viruses in human skin xenografts in SCID mice. The graph shows the mean titers ± SEM for xenografts that yielded viruses at 10 and 21 days postinfection. **, P < 0.01; ***, P < 0.001; ΔHPD mutant viruses versus pOka (two-way analysis of variance).
With regard to effects on other functions, ORF61 is known to transactivate the gE promoter in transient luciferase assays (5). However, the ΔHPD mutant suppressed the gE promoter to 50% of the vector control (Fig. 4B). This observation suggests that ORF61 oligomerization via HPD or HPD-mediated ORF61 interaction with other transactivators is essential for ORF61 to function as a transactivator. This role of HDP might also have contributed to the low protein levels of the ΔHPD mutant after MG132 treatment. This mechanism most likely differs from that of ORF61 suppression of the NF-κB promoter, as the suppression of the TNF-α-activated NF-κB response by the ΔHPD mutant was indistinguishable from ORF61 in the NF-κB luciferase reporter assay described previously (Fig. 4C) (6). Since ORF61 plays a critical role in disrupting the intrinsic cell defenses that are mediated by PML NBs against VZV (6), the effects of HPD deletion on the ORF61 association with and dispersal of PML NBs were examined. The ΔHPD mutant colocalized with PML NBs in infected cells at 6 h postinfection and caused obvious PML NB dispersal by 24 h postinfection (Fig. 4D). This finding is consistent with the preservation of the ORF61 RING finger and SIM domains despite the HPD deletion.
In order to assess the effects on VZV pathogenesis, the capacity of pOka-ORF61ΔHPD and pOka to infect skin xenografts in SCID mice was compared as described previously (6). Similar titers of each virus were used to inoculate the xenografts (Fig. 4E). Infectious virus was recovered from 5 of 6 skin xenografts inoculated with pOka at day 10 and all 6 at day 21. In contrast, pOka-ORF61ΔHPD was recovered from only 2 of 6 skin xenografts at day 10 and 4 of 6 at day 21. The mean titer of pOka-ORF61ΔHPD was ∼1.9 log lower than pOka at day 10 (P < 0.01) and ∼2.1 log lower than pOka at day 21 (P < 0.001) (Fig. 4E). These data demonstrate that ORF61 HPD is required for robust VZV replication in the skin tissue microenvironment in vivo.
In summary, we have identified a hydrophobic domain in ORF61 which is conserved among varicelloviruses within the alphaherpesvirus subfamily. We demonstrated that ORF61 HPD mediates ORF61 self-interaction and is required for normal VZV replication in cultured cells. Deletion of this domain also affected intracellular processing of ORF61 protein and its functions as a transactivator of other VZV genes. ORF61 self-interaction was not necessary for its capacity to inhibit intrinsic host cell responses to VZV. Importantly, ORF61 self-interaction through its HPD and the functions that depend on this interaction are needed for VZV virulence in skin in vivo.
ACKNOWLEDGMENT
This work was supported by the National Institute of Allergy and Infectious Diseases (grant AI053846).
Footnotes
Published ahead of print 23 January 2013
REFERENCES
- 1. Cohen JI, Straus SE, Arvin AM. 2007. Varicella-zoster virus replication, pathogenesis and management, p 2 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 5th ed Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Reichelt M, Brady J, Arvin AM. 2009. The replication cycle of varicella-zoster virus: analysis of the kinetics of viral protein expression, genome synthesis, and virion assembly at the single-cell level. J. Virol. 83:3904–3918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cohen JI, Nguyen H. 1998. Varicella-zoster ORF61 deletion mutants replicate in cell culture, but a mutant with stop codons in ORF61 reverts to wild-type virus. Virology 246:306–316 [DOI] [PubMed] [Google Scholar]
- 4. Wang L, Sommer M, Rajamani J, Arvin AM. 2009. Regulation of the ORF61 promoter and ORF61 functions in varicella-zoster virus replication and pathogenesis. J. Virol. 83:7560–7572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Moriuchi H, Moriuchi M, Straus SE, Cohen JI. 1993. Varicella-zoster virus (VZV) open reading frame 61 protein transactivates VZV gene promoters and enhances the infectivity of VZV DNA. J. Virol. 67:4290–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang L, Oliver SL, Sommer M, Rajamani J, Reichelt M, Arvin AM. 2011. Disruption of PML nuclear bodies is mediated by ORF61 SUMO-interacting motifs and required for varicella-zoster virus pathogenesis in skin. PLoS Pathog. 7:e1002157 doi:10.1371/journal.ppat.1002157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walters MS, Kyratsous CA, Silverstein SJ. 2010. The RING finger domain of varicella-zoster virus ORF61p has E3 ubiquitin ligase activity that is essential for efficient autoubiquitination and dispersion of Sp100-containing nuclear bodies. J. Virol. 84:6861–6865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, Sen N, Baiker A, Zerboni L, Arvin AM. 2011. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus. PLoS Pathog. 7:e1001266 doi:10.1371/journal.ppat.1001266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moriuchi H, Moriuchi M, Smith HA, Straus SE, Cohen JI. 1992. Varicella-zoster open reading frame 61 protein is functionally homologous to herpes simplex virus type 1 ICP0. J. Virol. 66:7303–7308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dinkel H, Michael S, Weatheritt RJ, Davey NE, Van Roey K, Altenberg B, Toedt G, Uyar B, Seiler M, Budd A, Jšdicke Dammert LMA, Schroeter C, Hammer M, Schmidt T, Jehl P, McGuigan C, Dymecka M, Chica C, Luck K, Via A, Chatr-Aryamontri A, Haslam N, Grebnev G, Edwards RJ, Steinmetz MO, Meiselbach H, Diella F, Gibson TJ. 2012. ELM-the database of eukaryotic linear motifs. Nucleic Acids Res. 40:D242–D251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ciufo DM, Mullen MA, Hayward GS. 1994. Identification of a dimerization domain in the C-terminal segment of the IE110 transactivator protein from herpes simplex virus. J. Virol. 68:3267–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Niizuma T, Zerboni L, Sommer MH, Ito H, Hinchliffe S, Arvin AM. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J. Virol. 77:6062–6065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Everett RD, Boutell C, McNair C, Grant L, Orr A. 2010. Comparison of the biological and biochemical activities of several members of the alphaherpesvirus ICP0 family of proteins. J. Virol. 84:3476–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kyte J, Doolittle R. 1982. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 157:105–132 [DOI] [PubMed] [Google Scholar]