

Abstract
Replication of the human papillomavirus (HPV) DNA genome relies on viral factors E1 and E2 and the cellular replication machinery. Bromodomain-containing protein 4 (Brd4) interacts with viral E2 protein to mediate papillomavirus (PV) genome maintenance and viral transcription. However, the functional role of Brd4 in the HPV life cycle remains to be clearly defined. In this study, we provide the first look into the E2-Brd4 interaction in the presence of other important viral factors, such as the HPV16 E1 protein and the viral genome. We show that Brd4 is recruited to actively replicating HPV16 origin foci together with HPV16 E1, E2, and a number of the cellular replication factors: replication protein A70 (RPA70), replication factor C1 (RFC1), and DNA polymerase δ. Mutagenesis disrupting the E2-Brd4 interaction abolishes the formation of the HPV16 replication complex and impairs HPV16 DNA replication in cells. Brd4 was further demonstrated to be necessary for HPV16 viral DNA replication using a cell-free replication system in which depletion of Brd4 by small interfering RNA (siRNA) silencing leads to impaired HPV16 viral DNA replication and recombinant Brd4 protein is able to rescue viral DNA replication. In addition, releasing endogenous Brd4 from cellular chromatin by using the bromodomain inhibitor JQ1(+) enhances HPV16 DNA replication, demonstrating that the role of Brd4 in HPV DNA replication could be uncoupled from its function in chromatin-associated transcriptional regulation and cell cycle control. Our study reveals a new role for Brd4 in HPV genome replication, providing novel insights into understanding the life cycle of this oncogenic DNA virus.
INTRODUCTION
Human papillomaviruses (HPVs) are small, double-stranded DNA viruses that replicate in differentiating cutaneous and mucosal epithelia (1). They are one of the most prevalent sexually transmitted pathogens in the world. High-risk HPVs are known etiological agents of cervical, anogenital, and head and neck cancers (2), with HPV16 being responsible for over 50% of cervical cancer cases worldwide (3–5).
HPVs specifically infect basal epithelial cells. HPV genome replication occurs during two different stages of the viral life cycle. In the infected basal epithelial cells, the viral genomes replicate an average of once per cell cycle during S phase, in synchrony with the host DNA replication (6). This allows the viral genome to be maintained as stable episomes at 50 to 100 copies per cell. This stage of DNA replication ensures a persistent infection in the basal layer of the epidermis. Terminal differentiation of infected cells triggers vegetative viral DNA replication, producing viral genomes, which can then be assembled into virions and be released from the surface of differentiated epithelium (7).
Replication of the HPV genome is carried out by viral E1 and E2 proteins in combination with various components of the cellular DNA replication machinery (7). E2 binds to several consensus E2 binding sites near the HPV origin of replication (Ori) and recruits E1 to the viral Ori (8, 9). The cooperative binding of E1 and E2 proteins to the viral Ori forms an E1/E2/Ori complex, in which E1 builds a hexameric ring around the viral DNA and functions as the helicase to unwind the HPV Ori for initiation of viral DNA replication (10). For successful completion of the viral DNA replication, many components of the cellular replication machinery are recruited by E1 and E2 to the viral origin of replication. For example, E1 has been shown to recruit the cellular DNA polymerase alpha/primase subunits to the viral replication origin (11–13). E1 interaction with the chaperone protein hsp40 and the single-stranded DNA-binding protein replication protein A (RPA) has also been shown to enhance E1 binding to the Ori and to facilitate processing of the replication fork, respectively (14, 15). Furthermore, interaction of E1 and hSNF5 proteins has been shown to stimulate HPV DNA replication (16).
Bromodomain-containing protein 4 (Brd4) is a critical host interacting partner for the PV E2 protein (17). Brd4 binds to both interphase chromatin and mitotic chromosomes through its double bromodomains, which specifically recognize acetylated histones. It interacts with the N terminus of E2 proteins from most PVs through its C-terminal domain (CTD) (18). During cell division, the interaction between Brd4 and bovine papillomavirus type 1 (BPV1) E2 tethers the E2/viral genome complexes to host mitotic chromosomes to ensure faithful partitioning of replicated viral episomes to the nuclei of both daughter cells (17). This function contributes to BPV1 episome maintenance during latent infection (17). The E2-Brd4 interaction also plays an important role in E2-mediated viral oncogene transcription (18–20).
In host cells, Brd4 functions in cellular transcription by recruiting P-TEFb (21, 22). Dysfunction of Brd4 has been linked to several cancers, including acute myeloid leukemia and breast cancer (23, 24). Other reports have shown that Brd4 interacts with the DNA damage response protein, ATAD5, and replication factor C (RFC), suggesting that Brd4 plays a role in cellular DNA replication and DNA damage repair (25–27). In accordance with Brd4's role in DNA replication, a recent study from our laboratory discovered that Brd4 functions in Merkel cell polyomavirus (MCV) genome replication through interaction with MCV large T antigen (28). HPV and MCV share similar properties in many aspects of the viral life cycles. For example, the viral genomes of both HPV and MCV are maintained as circular double-stranded DNA episomes during latent infection. These viral genomes replicate poorly in monolayer cell cultures, and both types of virions are commonly shed from healthy human skin surfaces. In addition, MCV large T antigen and the HPV E1/E2 viral replication proteins are functionally conserved in binding the viral replication origin and interacting with Brd4. Together, these observations suggest that Brd4 may also be important for HPV DNA replication. In addition, Ilves et al. demonstrated that abrogation of the E2-Brd4 interaction reduces BPV1 genome replication in some cell lines (29), although the underlying mechanism was not fully investigated. These observations prompted us to investigate the role of Brd4 in HPV replication.
In this study, we show that Brd4 is recruited to actively replicating HPV type 16 (HPV16) Ori foci together with HPV16 E1, E2, and a number of the cellular replication factors, including RPA70, RFC1, and DNA polymerase δ. The Brd4 function in HPV16 DNA replication was further demonstrated by a number of different approaches, including E2 mutagenesis, Brd4 silencing, rescue of HPV16 replication in a cell-free replication system with recombinant Brd4 protein, and a chemical compound that promotes release of Brd4 from chromatin. Our study reveals a new role of Brd4 in HPV DNA replication that is distinct from its chromatin-associated transcriptional regulation function, providing novel insights into understanding the life cycle of this oncogenic DNA virus.
MATERIALS AND METHODS
Cell culture, cell lines, and transfection.
Cells of the human papillomavirus-negative cervical cancer cell line C33A were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) with 10% fetal bovine serum (HyClone) and 1% penicillin–streptomycin (Invitrogen). Escherichia coli strain BL21 Rosetta-gami was cultured in 2× YT medium (1.6% tryptone, 1% yeast extract, 0.5% NaCl).
For immunofluorescence (IF) staining, immunofluorescence-fluorescence in situ hybridization (immuno-FISH) analysis, and Southern blotting, C33A cells were transfected with Fugene 6 (Promega) following the manufacturer's protocol. For the in vitro replication assay and immunoprecipitation (IP) experiments, cells were transfected using the calcium phosphate method at 30 to 40% confluence in 15-cm dishes. Briefly, 25 μg DNA was mixed with 163 μl 2 M CaCl2 in a 1.3-ml final volume. The DNA-CaCl2 mixture was slowly dropped into 1.3 ml 2× HBS (55 mM HEPES, 0.4 M NaCl, 1.5 mM Na2HPO4 [pH 7.0]) while vortexing. Then, DNA mixture was overlaid onto culture medium. Small interfering RNA (siRNA) transfection was performed using DharmaFECT II siRNA transfection reagent (Thermo Scientific Dharmacon) following the manufacturer's protocol. In some cases, siRNA transfection was performed using the calcium phosphate method as described above.
For bromodeoxyuridine (BrdU) labeling, C33A cells were transfected with Fugene HD (Promega) following the manufacturer's protocol. At 42 h posttransfection (p.t.), the cells were treated with 10 μM BrdU for 20 min, washed 2 times with medium, and cultured for an additional hour before being fixed with acetone.
Reagents.
Antibodies used for IF, immuno-FISH, and immunoblotting are listed as follows: anti-human β-actin (Chemicon), anti-hemagglutinin-horseradish peroxidase (anti-HA-HRP) (Roche), anti-Xpress (Invitrogen), anti-Flag M2 (Sigma), anti-RFC1 (H-300) (Santa Cruz), anti-RPA70 (Cell Signaling), anti-BrdU (Invitrogen), anti-HPV16 E2 (Millipore), anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) (United States Biological), and anti-DNA polymerase δ (Santa Cruz). The polyclonal rabbit anti-Brd4 antibodies and mouse anti-HA were made in the laboratory. The Brd4-specific inhibitor, JQ1(+), and its isomer compound, JQ1(−), were dissolved in dimethyl sulfoxide (DMSO) as 1,000× stocks as previously described (30). BrdU (Sigma) was dissolved in DMSO to 10 mM. siGENOME siRNAs targeting human BRD4 and control nontargeting siRNA 1 were purchased from Thermo Scientific Dharmacon.
Recombinant plasmid construction.
Plasmid pUC19-HPV16 containing the HPV16 genome inserted into the BamHI site of pUC19 is a generous gift from Paul F. Lambert. CMV4-Flag-16E2 has been described previously (17). HA-16E1 was kindly provided by Mart Ustav. The cDNA for full-length human Brd4 was amplified by PCR and subcloned into pcDNA4/HisMax C vector (pcDNA4C; Invitrogen) using BamHI and NotI digestion to generate pcDNA4C-hBrd4, in which human Brd4 was fused with an Xpress tag on the N terminus. Brd4 coding sequence was also inserted into BamHI/XhoI sites of pGEX-6P-1 (GE Healthcare) to obtain pGEX-6P-1-hBrd4 bearing a glutathione S-transferase (GST)-tagged hBrd4. pOZN-16E2, containing recoded HPV16 E2 fused with Flag and HA tags on its N terminus, has been described previously (31). HPV16 E2 was also amplified by PCR and inserted into BamHI/XhoI sites of pEGFPC1 (Clontech) to obtain green fluorescent protein (GFP)-16E2. HPV16 E2 mutant plasmids, GFP-16E2 E39A, GFP-16E2 R37A I73A, GFP-16E2 R304K, cytomegalovirus 4 (CMV4)-Flag-16E2 E39A, CMV4-Flag-16E2 R37A I73A, CMV4-Flag-16E2 R37A, and CMV4-Flag-16E2 I73A, were constructed using the QuikChange site-directed mutagenesis kit (Agilent Technologies) following the manufacturer's protocol. To obtain HPVOri, the HPV16 replication Ori (nucleotides [nt] 7855 to 96 in the HPV16 genome) was amplified using primers 5′-GCGGATCCCAAACCGTTTTGGGTT-3′ and 5′-GCGGATCCCTCTTTTGGTGCATAAAATG-3′ and inserted into the BamHI site of pcDNA4/HisMax C vector. The HA-16E1 open reading frame, including its upstream promoter and downstream polyadenylation signals, was cut by EcoRI/XhoI and inserted into the EcoRI/XhoI sites of HPVOri and pcDNA4/HisMax C to obtain HPVOri/E1 and HA-16E1, respectively. To generate a construct for expression of Brd4 in insect cells, two IgG binding sites and one tobacco etch virus (TEV) protease cutting site were fused to full-length hBrd4 to obtain IIT-hBrd4, which was amplified by PCR and inserted into KpnI/Xbal site of pFastBac1 (Invitrogen). pFast-IIT-hBrd4 was used as the donor plasmid to construct recombinant baculovirus Ac-IIT-hBrd4 using the Bac-to-Bac baculovirus expression system (Invitrogen). The HPV16 genome was isolated from pUC19-HPV16 by BamHI digestion, and religation was performed under a very diluted condition (3 to 4 μg/ml). Unligated DNA was removed using Plasmid-Safety ATP-dependent DNase (Epicentre) following the manufacturer's protocol. All constructs were verified by enzyme digestion, PCR, and DNA sequencing.
Protein expression and purification.
Full-length recombinant Brd4 (rBrd4) was expressed in E. coli. GST-Brd4 was induced using 0.8 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) in E. coli overnight at 18°C. Cells were then lysed by sonication in buffer containing 50 mM Tris, 0.1 mM EDTA, 2 mM dithiothreitol (DTT), and 0.2 mM phenylmethylsulfonyl fluoride (PMSF) (pH 7.6). GST-Brd4 was purified using glutathione agarose (Sigma) according to the manufacturer's protocol and washed 5 times with 50 mM Tris, 0.1 mM EDTA, 2 mM DTT, and 0.2 mM PMSF (pH 7.6) supplemented with 0.1% Tween 20. Brd4 was released from beads by PreScission protease digestion overnight at 4°C following the manufacturer's protocol. Released rBrd4 was passed through SP Sepharose (GE Lifescience) and washed with wash buffer WA150 and WA200, which includes 50 mM Tris, 0.1 mM EDTA, and 0.5 mM DTT, supplemented with 150 mM or 200 mM NaCl and protease inhibitors (pH 8.0). Purified rBrd4 was eluted with wash buffer containing 600 mM NaCl, dialyzed in 50 mM Tris, 0.1 mM EDTA, 0.5 mM DTT, 150 mM NaCl, and 0.1 mM PMSF (pH 7.5), and then concentrated on a Microcon column (Millipore).
To express rBrd4 in insect cells, either Ac-IIT-hBrd4 or wild-type baculovirus was used to infect Sf9 cells. Cells were lysed in IPP400 (20 mM HEPES, 0.4 M NaCl, 1 mM EDTA supplemented with protease inhibitors) by passing through a 20-gauge needle 15 times. Supernatant was isolated by centrifugation at 14,000 rpm for 30 min at 4°C, and the salt concentration was adjusted to 0.15 M. Recombinant IIT-Brd4 was purified using IgG-Sepharose 6 Fast Flow (GE Healthcare) following the manufacturer's protocol. The protein-bound Sepharose was washed with IPP150 (20 mM HEPES, 0.15 M NaCl, 1 mM EDTA, 0.1% Nonidet P-40 supplemented with protease inhibitors) four times and IPP400 supplemented with 0.1% Nonidet P-40 twice. rBrd4 was released from the Sepharose using TEV protease (Sigma) following the manufacturer's protocol. Expressed proteins were analyzed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), Coomassie brilliant blue staining, and/or immunoblotting.
Immunoblotting.
Cells were harvested at 48 h p.t. Cytoplasm was removed with a mixture containing 10 mM HEPES, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 0.2 mM PMSF, and 1 mM DTT, supplemented with protease inhibitors (pH 7.9). Nuclei were pelleted at 5,000 rpm for 5 min at 4°C. Nuclear proteins were extracted in a mixture containing10 mM HEPES, 200 mM NaCl, 3 mM MgCl2, 0.2 mM PMSF, and 0.1 mM DTT, supplemented with protease inhibitors (pH 7.9), and DNA was shredded by passing through a 22-gauge needle 10 times. Proteins were separated by SDS-PAGE, transferred to an Immobilon-P membrane (Millipore), and blotted with specific antibodies to detect proteins of interest (ECL enhanced chemiluminescence detection).
IF staining and immuno-FISH.
IF analysis was performed using a protocol described previously (32). Briefly, cells were fixed with 3% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 20 min at 48 to 64 h p.t., incubated in blocking/permeabilization buffer (3% bovine serum albumin [BSA] and 0.5% Triton X-100 in PBS), and then incubated with primary antibodies for 1 h at room temperature. After incubation, cells were washed 3 times using blocking/permeabilization buffer and incubated with fluorescently labeled secondary antibodies for another 1 h. Cells were counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) if necessary.
For immuno-FISH, cells were fixed with 1% PFA for 10 min at room temperature and washed with cold PBS twice on ice. Proteins were stained following the IF protocol described above, and RNA was digested with 25 μg/ml RNase A in PBS for 1 h at 37°C. Cells were fixed with 4% PFA for 10 min at room temperature, washed with cold PBS twice, 2× SSC (300 mM NaCl plus 30 mM trisodium citrate [pH 7.0]) twice, and finally once each with 70%, 80%, and 100% ethanol. Probes were labeled with biotin-dUTP (AppliChem) using a nick translation assay and incubated with cells for 5 min at 95°C. Cells were then incubated in a moist chamber at 37°C overnight. In situ hybridization signal was developed using the Trypticase soy agar (TSA)-biotin system (PerkinElmer) following the manufacturer's protocol.
IP.
C33A cells were pelleted at 48 h posttransfection and resuspended in buffer A (10 mM HEPES [pH 7.9], 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM dithiothreitol [DTT] supplemented with protease inhibitors [Roche Applied Science]). The resuspended cells were incubated on ice for 10 min, and Nonidet P-40 was added to a final concentration of 0.6%. After vortexing and centrifugation at 5,000 rpm for 5 min, the nuclear pellet was resuspended in ice-cold buffer B (20 mM HEPES [pH 7.9], 0.4 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, supplemented with protease inhibitors). To extract nuclear proteins, nuclei were passed through a 21-gauge needle 10 times and extracted at 4°C for 1 h. Nuclear proteins were isolated by centrifugation at 14,000 rpm for 15 min and diluted in buffer A. For Brd4 immunoprecipitation (IP), nuclear proteins were precleared with 3 μg normal rabbit (NR) IgG and 10 μl protein A-Sepharose 4 Fast Flow beads (GE Healthcare) at 4°C for 1 h. Precleared lysates were then divided equally and immunoprecipitated with 1.8 μg of either NR IgG antibody or Brd4 N-terminal antibody (Brd4NA) and 10 μl protein A beads (preblocked with 1% bovine serum albumin [BSA] in PBS at 4°C for 2 h) at 4°C for 7 h. The beads were then washed 3 times with 60 mM KCl base buffer (20 mM Tris [pH 8.0], 10% glycerol, 5 mM MgCl2, 60 mM KCl, and protease inhibitors [Roche Applied Science]) and eluted with SDS sample buffer. Input and IP samples were resolved on an SDS-PAGE gel and immunoblotted as described above. For Flag-tagged M2 IP, 10 μl of anti-Flag M2 affinity gel beads (Sigma) was used.
Southern blot analysis and in vitro replication assay.
For Southern blot analysis, episomal DNA was isolated by the Hirt extraction method. Less than 1 million cells were resuspended in 250 μl buffer I (50 mM Tris and 10 mM EDTA, supplemented with 50 μg/ml RNase A [pH 7.5]) and frozen at −80°C for more than 20 min. Cells were mixed with 250 μl 1.2% SDS and incubated for 5 min at room temperature. After incubation in 350 μl buffer III (3 M CsCl, 1 M KAc, 0.67 M HAc) for 10 min at room temperature, the samples were centrifuged at 16,000 × g for 10 min. Supernatant was loaded onto a Miniprep column (Qiagen) and washed twice with a mixture containing 10 mM Tris, 50 μM EDTA, 80 mM KAc, and 60% ethanol (pH 7.5). DNA was eluted with water or elution buffer (Qiagen). To linearize DNA and remove transfected DNA, 600 to 800 ng DNA from Hirt extraction was digested with XhoI/DpnI for 2 h at 37°C. To detect transfected DNA, episomal DNA was extracted at 6 h p.t. and digested with XhoI. Probes targeting the HPV16 E1 gene were labeled with [α-32P]dCTP (3,000 Ci/mmol) using Prime-It II random primer labeling kit (Agilent Technologies) following the manufacturer's instructions. The hybridization was performed at 65°C overnight.
For the in vitro replication assay, C33A cells were transfected with siRNA targeting Brd4 (siBrd4) or nontarget siRNA. At 36 h p.t., cells were retransfected with HA-16E1 and pOZN-16E2 plasmids. After 48 h, cells were swollen in a mixture containing 20 mM HEPES, 5 mM KCl, 1.5 mM MgCl2, 1 mM DTT, and 1 mM PMSF (pH 8.0) and lysed with a Dounce homogenizer on ice. Supernatant was frozen at −80°C for the in vitro replication assay. The reaction mixture contained 40 mM creatine phosphate (pH 7.7; di-Tris salt), 7 mM MgCl2, 100 μg/ml creatine kinase, 0.5 mM DTT, 3.3 μM [α-32P]dCTP (3,000 Ci/mmol), 200 ng religated HPV16 genome, and 150 to 200 μg cellular extracts, supplemented with 4 mM ATP, 200 μM (each) CTP, UTP, and GTP, and 80 μM (each) dATP, dTTP, and dGTP in a 50-μl volume. Mixtures were incubated for 2 h at 37°C. DNA was extracted with phenol, precipitated with ethanol, and linearized with BamHI. Purified recombinant Brd4 was added to the Brd4 rescue reaction mixture. The band intensities of radiographs were analyzed using a PhosphorImager (Typhoon 9400; GE Healthcare).
Flow cytometry analysis and image analysis.
To perform flow cytometry, cells were harvested by trypsinization, resuspended and fixed with 70% ethanol and stained with 25 μg/ml propidium iodide. RNA was digested with 100 μg/ml RNase A. Flow cytometry results were analyzed using FlowJo software.
IF and immuno-FISH results were observed under an Olympus IX81 inverted fluorescence microscope and analyzed using Slidebook 5.0 software. The percentage of cells showing different foci or a different localization pattern was quantified from approximately 50 to 200 positively transfected cells. Means and standard deviations (SD) were calculated from at least three independent experiments. Images were cropped using ImageJ. Relative intensities were analyzed using ImageJ software.
RESULTS
Brd4 colocalizes with E2 in large nuclear foci only in the presence of HPV16 genomes.
Most of the previous studies have examined the E2 and Brd4 interaction in the absence of other viral components, such as the HPV genome and viral proteins. In this study, we set out to investigate the interaction of Brd4 and HPV16 E2 in cells carrying the HPV16 genome and E1 protein. Interestingly, when C33A cells are transfected with HA-16E1, GFP-16E2, and the HPV16 genome, Brd4 is found colocalized in large, punctate dots with E2 (Fig. 1). In contrast, in the absence of the HPV16 genome, exogenously expressed GFP-16E2 is diffuse, as reported previously (33, 34), while Brd4 staining gives a characteristic speckled pattern in the nucleus (Fig. 1). This suggests that the HPV16 genome can enrich the interaction/colocalization of Brd4 and HPV16 E2 into subnuclear domains and/or promote the recruitment of the HPV16 E2/Brd4 complex to the HPV16 replication centers. This result, together with the observation that Brd4 similarly colocalizes with MCV viral replication foci shown in our recent study (28), prompted us to further investigate the potential function of the E2-Brd4 interaction in HPV16 replication.
Fig 1.
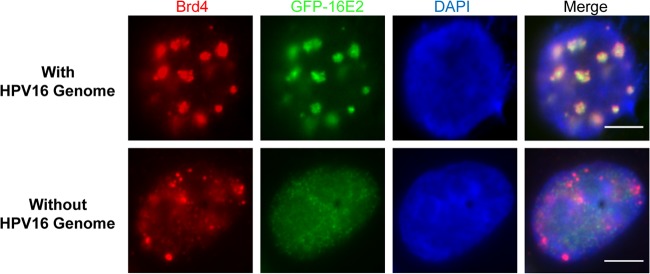
Brd4 and HPV16 E2 colocalize in punctate nuclear foci only in the presence of HPV16 genome. C33A cells were cotransfected with pUC19-HPV16, GFP-16E2, and HA-16E1. The pUC19-HPV16 plasmid was replaced by pUC19 in the “without-genome” transfection control. At 48-h p.t., cells were fixed and stained using anti-Brd4 antibody and counterstained with DAPI. This experiment was repeated at least 3 times. Bars, 5 μm.
HPV16 E1, E2, and Brd4 are recruited to the viral Ori replication foci.
The HPV16 genome contains the viral upstream regulatory region (URR), which includes transcription regulatory elements and the viral DNA replication origin. HPV E2 initiates viral gene transcription by recruiting many transcription factors, including Brd4, to the URR (21, 22, 35–39). The data shown in Fig. 1 suggest that Brd4 may be recruited by E2 to the viral URR to contribute to E2 viral transcription and/or replication function. Additionally, Brd4 may simply be enriched at the HPV16 genome through the binding of acetylated histones on HPV minichromosomes (40). To set up a system that would allow us to specifically examine the function of Brd4 in HPV16 replication instead of viral transcription and minichromosome association, we constructed an artificial episome that contains the 145-bp minimum HPV replication Ori (Fig. 2A), which we termed “HPVOri.” This episome excludes most of the URR transcription regulatory elements, such as the binding sites for C/EBP, NF1, OCT-1, and the TATA box of the viral early promoter, retaining only three E2 binding sites (E2BS), one E1 binding site (E1BS), a GT-1 binding site, and an SP1 binding site (Fig. 2A). According to mutagenesis studies performed in HPV11 URR, neither GT1 site nor SP1 site deletion dramatically affects viral early promoter activities (41), suggesting that these sites may not contribute significantly to the early promoter transcription. Therefore, this plasmid with the minimum HPV16 Ori allows us to specifically examine the recruitment of Brd4 and its impact on HPV16 replication.
Fig 2.

Brd4 and HPV16 E2 colocalization in nuclear foci is dependent on both HPV E1 and viral origin. (A) Schematic diagrams of the artificial HPVOri/E1 episome. E1BS, E1 binding site; E2BS, E2 binding site; P97, TATA box of viral early promoter; GT-1, transcription factor GT-1; Sp1, specificity protein 1. (B) Brd4 and HPV16 E2 colocalize in foci with E1-Ori. C33A cells were cotransfected with GFP-16E2 and either HPVOri/E1, HA-16E1, or HPVOri. At 60 h p.t., cells were fixed and costained using anti-Brd4 (red) and anti-HA (blue) antibodies. Bars, 5 μm. This experiment was repeated more than 3 times. (C) Quantification of the size of E1 foci at different time points posttransfection. C33A cells were cotransfected with GFP-16E2 and HPVOri/E1 and costained as in panel B at 12, 18, 24, 36, and 48 h p.t. The area of E1 foci was quantified from about 30 cells at each time point using ImageJ. The red dashed line represents the mean area of E1 foci at 48 h p.t. Foci with an area larger than this mean value are considered big foci. The mean area of these big foci (numbers in black) and the percentage of big foci in total foci (numbers in red) were calculated for each time point and are shown at the top of each column. (D) Brd4, 16E2, and 16E1 coimmunoprecipitate as a complex. C33A cells were cotransfected with CMV4-Flag-16E2 and either empty vector (V) or HA-16E1 (E1). At 48 h p.t., nuclear proteins were harvested and immunoprecipitated with the indicated antibodies. The precipitates were immunoblotted with specific antibodies as indicated. This experiment was repeated 3 times.
Using the HPVOri plasmid, we observed a similar phenomenon to that seen in Fig. 1 with E2 and Brd4 colocalized in large foci only in the cells cotransfected with HPVOri, E1, and E2 constructs (data not shown). To visualize cells transfected with the HPV16 Ori construct and to also study the focus formation in the presence of HPV16 E1 protein, we inserted an HA-16E1 expression cassette, which includes HA-tagged 16E1 under the control of a CMV promoter, in HPVOri to generate the HPVOri/E1 plasmid (Fig. 2A). With this plasmid, HPV16 Ori-positive cells can be identified as those stained positive for HA-16E1.
During the HPV life cycle, E2 binds to its binding sites in HPV16 Ori. Its interaction with E1 enhances E1 binding to the viral Ori and recruits cellular replication factors to the viral DNA replication complex (42–45). To monitor the localization pattern of Brd4, E1, and E2 in cells carrying HPV16 Ori, cells transfected with GFP-16E2 and HPVOri/E1 were stained by IF in three colors to show Brd4 in red, E1 in blue, and E2 displaying GFP in green fluorescence. In these cells, we detected a clear colocalization pattern of Brd4, E2, and E1 assembled into large foci in 55.1% ± 3.4% of E1 and E2 double-positive cells (Fig. 2B; n ≥ 3). (n represents the number of times the experiment was repeated.) The formation of Brd4-E1-E2 foci requires the HPV16 Ori because in cells transfected with GFP-16E2 and HA-16E1, which does not carry the HPV16 Ori, both E1 staining and E2 staining are diffuse in the nucleus of most cells and in only 19.8% ± 1.5% of E1 and E2 double-positive cells do the viral proteins colocalize as tiny dots (Fig. 2B; n ≥ 3). This phenomenon was also reported in a previous study (33). Interestingly, in these cells, Brd4 staining is markedly altered from its normal punctate pattern to a more diffuse pattern that only partially colocalizes with the tiny E1/E2 dots (Fig. 2B). In cells transfected with GFP-16E2 and HPVOri that does not carry the E1 expression cassette, E2 staining is diffuse, despite the presence of HPVOri, while Brd4 maintains its usual speckled pattern (Fig. 2B). This result is surprising because we expected E2 to bind the E2BSs within the Ori plasmid and to recruit Brd4. However, Brd4 was recruited to the large foci only when E1, E2, and HPV16 Ori were all present (Fig. 2B). This observation suggests that E1 plays an important role in building these foci and recruiting Brd4. Together, these results indicate that recruitment of Brd4 into the large nuclear foci is dependent on the formation of the intact E1/E2/Ori replication complex. We also performed a time course experiment to look at the kinetics of viral replication focus formation over time (Fig. 2C). Smaller Brd4, E1, and E2 replication foci were observed at earlier times posttransfection. The size of the replication foci as detected by E1 staining continued to increase after 12 h posttransfection and reached a plateau at 36 h posttransfection. The Brd4/E2/E1 complex was further investigated by Brd4 immunoprecipitation using cells transfected with CMV-Flag-16E2 and either empty vector or HA-16E1. Consistent with previous findings, Brd4 is able to pull down E2 (Fig. 2D). Interestingly, E1 was also pulled down with E2 and Brd4, providing biochemical evidence that these proteins assemble into a functional complex.
Brd4 colocalizes with the HPV16 DNA replication complex.
Previous studies showed that the E1 and E2 proteins coexpressed in cells localize to defined nuclear foci and induce a cellular DNA damage response (33, 46). To rule out the possibility that the E1, E2, and Brd4 foci observed in this study are sites of host chromosomal DNA replication and to demonstrate that these foci contain the HPV Ori, immuno-FISH was performed. C33A cells cotransfected with E2 and HPVOri/E1 were examined by IF to detect the viral proteins and by FISH to detect the HPV16 Ori. FISH using HPV16 Ori-specific probes detected large foci of HPV16 Ori similar to those detected by IF, as shown in Fig. 2B, while no signal was detectable when nonspecific probes targeting the MCV genome were used (Fig. 3A and B). In addition, both E1 and E2 colocalized with the HPV16 Ori in large nuclear foci in nearly all cells carrying viral replication foci (Fig. 3A and B). Brd4 was also detected in these foci containing the HPV16 Ori (data not shown). These results further demonstrate that the large nuclear foci containing E1, E2, and Brd4 are centered on the HPV16 Ori.
Fig 3.

Brd4 colocalizes with HPV16 E1 and E2 proteins in foci harboring actively replicating HPV episomes. (A and B) HPV16 E1 and E2 colocalize with actively replicating HPV episome. C33A cells were cotransfected with HPVOri/E1 and CMV4-Flag-16E2. At 48 h p.t., biotin-labeled probes targeting HPVOri/E1 (Specific Probe) were used to detect the replicating DNA while probes targeting Merkel cell polyomavirus genome (Non-Specific Probe) were used as a nonspecific control. HPV16 E1 (A) and E2 (B) were stained using anti-HA and anti-Flag antibody, respectively. The cells were also counterstained with DAPI. Bars, 5 μm. (C) Brd4 colocalizes with E2 in HPV replicating foci. C33A cells were cotransfected with GFP-16E2 or pEGFPC1 and either HPVOri/E1, HA-16E1, or HPVOri. At 42 h p.t., the cells were labeled with BrdU for 20 min and grown for an additional hour prior to acetone fixation. Cells were immunostained for Brd4 (blue) and BrdU (red). Bars, 5 μm. Experiments were repeated at least 3 times.
Since the HPV16 foci we observed contain the viral replication proteins E1 and E2 and the HPV16 Ori, we next determined if these foci are centers of viral replication. To test this, C33A cells cotransfected with GFP-16E2 and various vectors carrying 16E1 and/or Ori were pulse-labeled with BrdU to detect its incorporation into newly synthesized DNA. As shown in Fig. 3C, in cells transfected with GFP-16E2 and HPV16 Ori, GFP-16E2 and BrdU staining was diffuse in the nucleus and Brd4 was in its normal nuclear speckled pattern. Cells transfected with HPVOri/E1 and the empty GFP vector, pEGFPC1, also displayed a background BrdU staining and the normal speckled Brd4 pattern, while most of the GFP was removed by the acetone fixation used in the BrdU staining (Fig. 3C). The diffuse BrdU staining in these cells cotransfected with HPV16 Ori and either E1 or E2 alone indicates that they do not have active HPV16 Ori replication (Fig. 3C). Since these cells also have an unaltered Brd4 localization pattern, the result suggests that both E1 and E2 are needed to support viral replication and Brd4 recruitment. In accordance with this notion, cells cotransfected with GFP-16E2 and HPVOri/E1 displayed clear colocalization of GFP-16E2 and BrdU in large HPV16 Ori foci in 82% ± 8.8% of transfected cells (Fig. 3C; n = 3). Interestingly, Brd4 and E1 were also clearly present in the BrdU-labeled foci with E2 in these cells (Fig. 3C) (data not shown). It is clear that formation of these large foci was dependent on HPV16 Ori, as in cells transfected with GFP-16E2 and HA-16E1, which does not carry HPV16 Ori, 54% ± 11.1% of cells had only small nuclear dots with GFP-16E2, Brd4, and BrdU colocalized, while the rest had diffuse GFP-16E2 staining in the nucleus (Fig. 3C; n = 3). This result is consistent with previous reports showing that E1 and E2 can form small foci that colocalize with BrdU and cellular replication factors on host chromatin (33, 34). The small foci formed in the absence of HPV16 Ori are likely sites of E1- and E2-mediated nonspecific unwinding/replication on the cellular genome. Our results demonstrate that Brd4 and the HPV replication proteins are present together at the HPV Ori in active viral replication compartments and suggest that Brd4 may be involved in HPV replication.
HPV genome replication requires the cellular replication machinery, and it has previously been shown that host replication factors are recruited to HPV replication compartments (33, 34). To further demonstrate that the large Brd4-E2-E1 foci are viral replication centers, we next determined if host replication proteins are recruited to these foci. For this, C33A cells cotransfected with Flag-16E2 or GFP-16E2 and various vectors carrying 16E1 and/or Ori were immunostained for the RPA component, RPA70, replication factor C1 (RFC1), or DNA polymerase δ. As shown in Fig. 4, cells transfected with both E2 and HPVOri/E1 showed large E2-positive nuclear foci, which colocalized with all three replication proteins tested. RPA70 was observed colocalized with large foci in 67% ± 7.8% of cells, while RFC1 and DNA polymerase δ colocalized with large foci in 51% ± 5.8% and 57.5% ± 2.7% of transfected cells, respectively. In the absence of HPV16 Ori, E2 and E1 formed small foci that only partially colocalized with RPA70 and RFC1 in 61% ± 0.8% and 32% ± 4.9% of transfected cells, respectively (Fig. 4A and B; n = 3). DNA polymerase δ did not localize to the small foci and was diffuse in the nucleus without HPV Ori (Fig. 4C; n = 3). Without E1, E2 cotransfected with the HPV16 Ori construct did not form nuclear foci, and all three replication factors remained diffuse (Fig. 4). These results further support that the large foci formed in cells transfected with both E2 and HPVOri/E1 are sites of HPV DNA replication. The fact that Brd4 colocalizes with these replication factors in the HPV16 Ori foci provide further support for its role in HPV16 replication.
Fig 4.
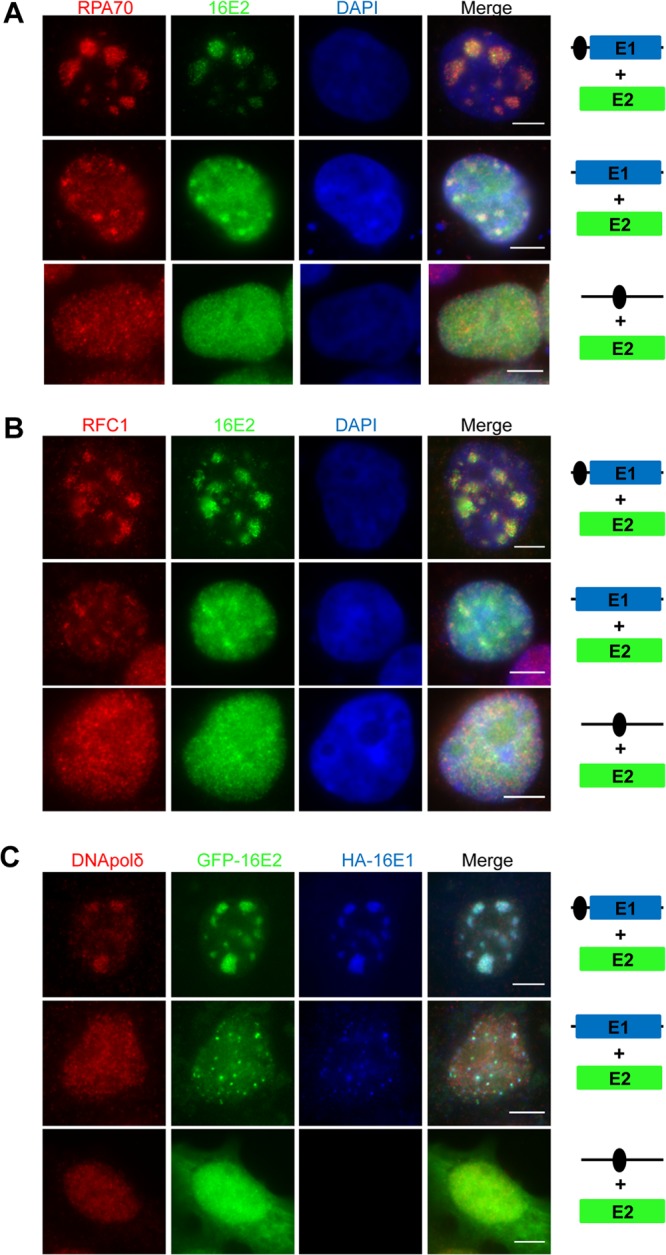
Host replication factors are recruited to the HPV16 E1/E2 foci in a HPV origin-dependent manner. (A and B) RPA70 and RFC1 are recruited to HPV16 Ori replication foci. C33A cells were cotransfected with Flag-16E2 expression vector and either HPVOri/E1, HA-16E1, or HPVOri. At 48 h p.t., the cells were fixed and immunostained for E2 (anti-Flag [α-Flag]; green) and RPA70 (red) (A) or RFC1 (red) (B). The cells were also counterstained with DAPI. (C) DNA polymerase δ is a component of the HPV16 replication complex. C33A cells were cotransfected with GFP-16E2 and either HPVOri/E1, HA-16E1, or HPVOri. At 64 h p.t., cells were fixed and costained with anti-DNA polymerase δ (red) and anti-HA (blue) antibodies. Bars, 5 μm. Experiments were repeated at least 3 times.
Brd4 is important for transient HPV16 replication in cells.
Since Brd4 is clearly recruited to the HPV replication Ori foci that are actively replicating, we further investigated the role of the Brd4-E2 interaction in HPV16 DNA replication using the HPV16 E2 R37A I73A mutant, which is unable to interact with Brd4. As observed before, in cells transfected with wild-type E2 and HPVOri/E1, large foci were found in 56.9% ± 5.1% of E2-positive cells (Fig. 5A; n = 3). Conversely, when the E2-Brd4 interaction was abolished with E2 R37A I73A mutations, large foci or tiny dots were found in only <10% of cells and most cells had diffuse staining of E2, E1, and Brd4 (Fig. 5A; n = 3) (data not shown). In the few cells with E1/E2 foci, Brd4 staining was more diffuse and only partially colocalized with the foci. Because large HPV replication foci cannot be efficiently formed in the E2 R37A I73A cells, this set of results suggested that the E2-Brd4 interaction is essential for recruitment of Brd4 and formation of the HPV16 Ori replication foci. We also studied the E2 E39A mutant, which does not interact with E1 and, therefore, does not support HPV replication. As predicted, cells transfected with both the E2 E39A mutant and HPVOri/E1 no longer support the large HPV replication focus formation as in the wild-type cells. This result confirms that the E1 and E2 interaction is important for the formation of HPV replication foci. Interestingly, this E2 E39A mutant did not completely eliminate E1/E2 colocalization since, in 26.3% ± 3.5% of the transfected cells, E1, E2, and Brd4 were observed to colocalize in tiny dots (Fig. 5A; n = 3). However, these foci are much smaller than those observed in wild-type E2 transfected cells. It is possible that while the E2 E39A mutant failed to recruit E1 to form the large HPV Ori replication complexes, E1 can still bind to the HPV Ori in an E2-independent manner as previously described (47) to form the small foci. Brd4 is recruited in these small foci presumably through binding to the E2 E39A mutant. We also examined the E2 R302K and R304K mutants, which do not interact with viral DNA but retain Brd4 binding (48), for their ability to form replication foci. Similar to the E39A mutant, in cells transfected with HPVOri/E1 and either E2 R302K or R304K, ∼35% had tiny dots, while the rest had diffuse staining of E2, E1, and Brd4 (Fig. 5A) (data not shown). Large replication foci were not found with these DNA binding-deficient mutants. From these results, it seems that the E2-Brd4 interaction, E2-E1 interaction, and E2-DNA binding are all necessary for the formation of large HPV replication foci, suggesting that these interactions are important for viral replication.
Fig 5.

The Brd4-E2 interaction is important for HPV16 replication. (A) Assembly of Brd4 into the HPV16 replication complex requires intact E1-E2 and E2-Brd4 interactions and E2-DNA binding. C33A cells were cotransfected with HPVOri/E1 and either GFP-16E2, GFP-16E2 E39A, GFP-16E2 R37A I73A, or GFP-16E2 R304K. At 48 h p.t., cells were fixed and costained with anti-Brd4 (red) and anti-HA (blue) antibodies. (B) The E2-Brd4 interaction is required for HPV16 DNA replication in vivo. C33A cells were cotransfected with HPVOri/E1 and either CMV4-Flag-16E2 (E2wt), CMV4-Flag-16E2 E39A, CMV4-Flag-16E2 R37A I73A, CMV4-Flag-16E2 R37A, CMV4-Flag-16E2 I73A, or CMV4 (Vector). To detect transfected DNA, episomal DNA was extracted and subjected to Southern blotting without DpnI treatment at 6 h p.t. At 48 h p.t., episomal DNA was extracted and transfected DNA was removed by DpnI treatment. Newly synthesized DNA was analyzed by Southern blotting. The intensity of each replicated DNA band was quantified by ImageJ and normalized to E2wt. (C) Western blot analysis of the HPV16 E1 and E2 proteins transfected in panel B. Cells were transfected as in panel B. At 48 h p.t., 25 μg total protein was analyzed by Western blotting using anti-16E2, anti-HA, and antiactin antibodies. Experiments were repeated at least 3 times. (D) HPV16 E2 R37A I73A interacts with 16E1 similarly to wild-type (WT) 16E2. C33A cells were cotransfected with HA-16E1 and either CMV4 (Vector), the CMV4-Flag-16E2 wild type, or CMV4-Flag-16E2 R37A I73A. Cell lysates were immunoprecipitated with Flag-tagged M2 antibody. The precipitates were immunoblotted with specific antibodies as indicated.
To further analyze the impact of the E2-Brd4 and E1-E2 interactions on HPV16 Ori DNA replication, we performed an in vivo replication assay comparing various E2 mutants with wild-type E2. C33A cells were transfected with HPVOri/E1 and the E2 constructs indicated in Fig. 5B. Episomal DNA was collected at 6 and 48 h p.t. for Southern blot analysis. The 6-h-p.t. DNA samples, which served as transfection controls, showed that transfection efficiencies were similar, and an equal amount of the initial viral DNA template was loaded for each lane (Fig. 5B). Western blot analysis showed that E1 and the various E2 constructs were also expressed in cells at comparable levels (Fig. 5C). For analysis of newly synthesized HPV16 Ori, the 48-h-p.t. DNA samples were digested with XhoI to linearize the plasmid and with DpnI to remove input plasmid. As shown in Fig. 5B, cells transfected with wild-type E2 supported efficient replication of HPVOri. This HPVOri replication was dependent on E2, as cells transfected with empty vector did not replicate the HPV16 Ori plasmid. As expected, the E2 E39A mutant with the abrogated E1-E2 interaction was nearly completely impaired in HPV16 Ori replication. This was not likely due to a change in the viral protein expression because E1 protein detected by Western blotting was not lower when the replication-defective E2 E39A mutant was used as opposed to the wild-type E2 (Fig. 5C). More importantly, the E2 R37A I73A double mutations, which disrupt the E2-Brd4 interaction, also dramatically reduced HPV16 Ori replication (Fig. 5B; P < 0.05, n = 3). Co-IP analysis showed that the E2 R37A I73A double mutant interacts with 16E1 similarly to wild-type 16E2 (Fig. 5D), suggesting that its reduced DNA replication activity is not caused by a defect in E1 binding. Notably, the E2 R37A and I73A single mutants could replicate HPV16 Ori slightly better than the R37A I73A double mutant. The partial inhibition of the HPV16 replication by these two single mutants, especially the R37A mutant, has been reported in a previous study using detailed structure-function analysis to separate the transcriptional activation and replication activities of HPV16 E2 (49). This previous study by Sakai et al. showed that E2 I73A only slightly inhibited HPV16 replication (49). Using the same condition as in Sakai et al. by cotransfecting cells with three separate plasmids for E1, E2, and Ori, respectively, we also observed a similar result to their study (data not shown). The more significant inhibition of the viral replication by the E2 I73A mutant observed in our study suggests that carrying the Ori and E1 on the same plasmid, HPVOri/E1, provides a more sensitive way to detect replication efficiency in transient replication. In both cases, the single mutants could support a little more HPVOri replication than the E2 R37A I73A mutant, probably because each of them still retains some interactions with Brd4. Together, these results suggest that the E2-Brd4 interaction is necessary for HPV16 Ori replication.
Brd4 is important for HPV DNA replication in vitro.
Brd4 plays an important role in regulating host gene transcription and cellular growth (50). Brd4 knockdown could therefore induce cell cycle arrest, which would indirectly inhibit HPV DNA replication. This prevented us from using Brd4 silencing as a fair approach to test the functional role of Brd4 in HPV DNA replication in cells. To overcome this problem, we set up a cell-free replication system as an alternative strategy. In the in vitro replication assay, protein lysates to be used as a source of replication factors were isolated from cells transfected with both HPV16 E1 and E2 proteins and either nontargeting control siRNA or Brd4-specific siRNA (Fig. 6). These lysates were mixed with the HPV16 genome template and in vitro replication components to set up the reaction (see details in Materials and Methods). As shown in Fig. 6A, lysates from cells expressing both E1 and E2 boosted HPV16 viral DNA synthesis by more than 9-fold compared to the cell lysates without E1 and E2 (compare “CO” lanes to the “No E1/E2” lane), even though a low level of E1/E2-independent viral DNA synthesis was detected in the no-E1/E2 lysate. Compared with reactions using control siRNA-transfected C33A cells, Brd4 knockdown cell lysates showed a more than 4-fold reduction in HPV16 DNA replication in vitro (Fig. 6A; P < 0.001, n = 3). This is not likely due to a change in the viral protein expression because E1 and E2 expression was not affected by Brd4 knockdown (Fig. 6D). This result suggests that Brd4 may directly contribute to HPV16 DNA replication.
Fig 6.

Brd4 directly stimulates HPV16 replication in vitro. (A) Brd4 knockdown inhibits viral DNA synthesis in vitro, and this inhibition can be rescued by rBrd4. C33A cells were transfected with either an siRNA targeting Brd4 (KD) or nontargeting control siRNA (CO). At 36 h p.t., cells were transfected with pOZN-16E2 and HA-16E1. Cell extracts were prepared at 82 h p.t. for the in vitro replication assay using religated HPV16 genomes as the template. Cells transfected with the nontargeting siRNA but not the E1/E2 constructs were used as a negative control (no E1/E2). In the “rBrd4” reaction, 2 μg rBrd4 expressed and purified from E. coli was added to the replication assays with Brd4 knockdown extract to restore HPV16 replication. In the “Control” condition, an equal amount of nonspecific proteins isolated from the vector control E. coli were used. All reactions were performed in triplicate. The intensity of each replicated DNA band was quantified by ImageJ and normalized to the no-E1/E2 control. (B) Dose-dependent rBrd4 rescue of HPV16 in vitro replication. Cellular extracts and rBrd4 were prepared as in panel A. Increasing amounts of rBrd4 were added to the replication assays using Brd4 knockdown extracts. All reactions were performed in triplicate. The intensity of each replicated DNA band was quantified by ImageJ and normalized to one of the control replicates. (C) Western blotting of purified rBrd4. One microgram of rBrd4 purified from E. coli and an equal amount of nonspecific proteins isolated from the vector control cells were analyzed using anti-Brd4 immunoblotting. (D) Western blot analysis of the HPV16 E1, E2, and Brd4 proteins in cellular extracts used for in vitro replication. Cellular extracts used in panels A and B were analyzed by Western blotting using anti-Brd4, anti-16E2, anti-HA, and antiactin antibodies. Experiments were repeated at least 3 times. (E) Coomassie brilliant blue staining of rBrd4 purified from insect cells. Two micrograms of rBrd4 purified from Sf9 cells or an equal amount of nonspecific proteins isolated from wild-type baculovirus-infected Sf9 cells was analyzed using Coomassie brilliant blue staining. (F) Western blot analysis of the HPV16 E1 and Brd4 proteins in cellular extracts used in panel I. (G) Extra recombinant Brd4 does not further increase in vitro HPV16 replication when Brd4 is not depleted. Cellular extracts were prepared and in vitro replication was performed as in panel A. In the “rBrd4” reactions, 2 μg rBrd4 expressed and purified from Sf9 insect cells as shown in panel E was added. In the “Control” conditions, an equal amount of nonspecific proteins isolated from the wild-type baculovirus-infected Sf9 cells was used. All reactions were performed in triplicate. (H). Dose-dependent rescue of HPV16 in vitro replication using rBrd4 purified from insect cells. Cellular extracts were prepared and in vitro replication was performed as in panel B. rBrd4 purified from insect cells was used at 0.5, 1 and 2 μg. (I) rBrd4 partially restores HPV16 in vitro replication using cell lysates that express only the E1 protein. Cellular extracts were prepared as in panel A, except that, at 36 h p.t., cells were transfected with only HA-16E1. Cells transfected with the nontargeting siRNA cells but not the E1 construct were used as a negative control (no E1). HPV16 in vitro replication was performed as in panel B. Increasing amounts of rBrd4 purified from insect cells were added to the replication assays using Brd4 knockdown extracts. All reactions were performed in triplicate. The intensity of each replicated DNA band was quantified by ImageJ and normalized to one of the control replicates.
To demonstrate that this reduction of HPV16 DNA replication in Brd4 knockdown cell lysates was not due to cell cycle arrest caused by Brd4 knockdown, rBrd4 expressed and purified from E. coli was added to replication reaction mixtures to restore Brd4 levels (Fig. 6A). Compared to the control, addition of recombinant Brd4 increased the HPV16 genome replication by ∼2.5-fold (Fig. 6A, compare rBrd4 lanes with control lanes; P < 0.001, n = 3). The recombinant Brd4 also rescued in vitro HPV16 genome replication in Brd4 knockdown cell lysates in a dose-dependent manner, causing up to 276% increased replication of HPV16 DNA (Fig. 6B, compare 2-μg lanes to “KD” lanes; P < 0.001, n = 3). Notably, the recombinant Brd4 purified from E. coli contains both full-length Brd4 as well as shorter Brd4 fragments, which were likely caused by proteolytic cleavage (Fig. 6C). These shorter fragments may function as dominant-negative inhibitors to block the E2-Brd4 interaction, thus reducing the activity of full-length Brd4 in the rescue reactions. In fact, the shorter fragments of Brd4 have been shown to inhibit Brd4's activity in MCV DNA replication (28).
In addition to the Brd4 purified from E. coli, rBrd4 purified from insect cells also restored HPV16 replication in Brd4 knockdown lysates (Fig. 6E, G, and H). In contrast, rBrd4 did not increase HPV16 replication when cellular Brd4 was not depleted (Fig. 6G). This result provides additional support for the role of Brd4 in HPV16 replication. Since E1 protein of HPV1a is sufficient to initiate viral DNA replication (51) and the Brd4 CTD, which can block the E2-Brd4 interaction, does not significantly inhibit HPV16 replication in C33A cells (19, 29), we investigated if the Brd4 activity in HPV16 replication is solely dependent on its interaction with E2. In the in vitro replication assays using cellular extracts containing 16E1 but not 16E2, HPV16 replication could be detected in the control siRNA-treated samples and was also inhibited upon Brd4 knockdown (Fig. 6F and I). rBrd4 could partially restore the 16E1-mediated HPV16 replication (Fig. 6I). However, rBrd4 was able to more efficiently stimulate the viral replication in the reaction mixtures containing both E1 and E2 (compare the quantification data in Fig. 6H and I), suggesting that the E2-Brd4 interaction is important for the viral DNA replication in vitro. In addition, neither Brd4 knockdown nor recombinant Brd4 significantly affects in vitro DNA replication mediated by simian virus 40 (SV40) large T antigen (data not shown), which only interacts weakly with Brd4 (28). Together, the results shown in Fig. 6 demonstrate that Brd4 stimulates HPV16 DNA replication in the cell extract and is directly associated with HPV16 DNA replication.
Release of endogenous Brd4 from chromatin stimulates HPV16 DNA replication.
Most of Brd4's cellular functions are associated with its chromatin localization through a dynamic “on and off” interaction with acetylated histones (52). Since Brd4 can relocate from cellular chromatin to the HPV16 Ori replication complex (Fig. 1 to 3 and 5), we postulated that releasing Brd4 from chromatin may facilitate HPV16 DNA replication. The newly developed chemical compound JQ1(+) provides an excellent tool to test this possibility because it can prevent the binding of Brd4 to tetra-acetylated histone H4 peptides (30). We decided to test whether release of Brd4 from chromatin using JQ1(+) affects HPV16 Ori DNA replication. Brd4 binds acetylated chromatin and is normally observed in a nuclear speckled pattern in C33A cells. Treatment of C33A cells with 300 nM JQ1(+) could efficiently alter Brd4 localization from punctate foci on chromatin to a dimmer and more diffuse pattern throughout the nucleus, indicating the release of endogenous Brd4 from chromatin (Fig. 7A). Lower concentrations of JQ1(+) only slightly released Brd4 from chromatin, while the inactive stereoisomer JQ1(−) or DMSO vehicle control did not noticeably affect Brd4 staining in C33A cells. To determine if JQ1(+) could affect HPV16 DNA replication, HPVOri/E1 and E2 cotransfected cells were split into five dishes and each was treated with 100, 200, and 300 nM JQ1(+), 300 nM JQ1(−), or an equal volume of DMSO. The extracted episomal DNA samples were digested with both DpnI and XhoI and used in Southern blotting to analyze the HPV16 Ori replication product, whereas the samples without DpnI digestion were used as a loading control. As shown in Fig. 7B, 300 nM JQ1(+) treatment, which efficiently releases Brd4 from chromatin, increased HPV16 Ori replication, while the other drug concentrations had little effect. Treatment with 300 nM JQ1(+) did not affect E2 expression but slightly increased E1 expression, likely due to stimulation of HPVOri/E1 DNA replication in these cells (Fig. 7B). JQ1(+) also stimulated HPV replication in cells carrying Ori and E1 encoded by two separate plasmids, HPVOri and HA-16E1, cotransfected with CMV4-Flag-16E2 (data not shown), further supporting a direct role of JQ1(+) in stimulating the viral DNA replication. Using flow cytometry analysis, we confirmed that 300 nM JQ1(+) treatment does not cause cell cycle arrest (Fig. 7C; n = 3), suggesting that the induced HPV16 Ori DNA replication was not caused by a cell cycle effect associated with JQ1(+) treatment. We also examined how JQ1(+) affects the kinetics of HPV16 replication focus formation by monitoring BrdU incorporation over time. Compared to DMSO-treated cells, JQ1(+) treatment leads to increased BrdU focus size during the early time points and the percentage of large BrdU replication foci throughout the experiment (Fig. 7D). These results suggest that the release of Brd4 from cellular chromatin stimulates HPV16 DNA replication, likely by relocating Brd4 to the HPV16 Ori replication complex. The data also demonstrate that the role of Brd4 in HPV DNA replication could be uncoupled from its cell cycle and transcriptional regulation function.
Fig 7.

Releasing Brd4 from chromatin by JQ1(+) increases HPV16 replication. (A) IF analysis of JQ1(+)-induced Brd4 release from host chromatin. C33A cells were treated with JQ1(+), JQ1(−), or DMSO at the indicated concentrations for 24 h. Cells were then fixed and immunostained with anti-Brd4 antibody and counterstained with DAPI. Bars, 5 μm. (B) JQ1(+) treatment stimulates HPV16 replication. C33A cells transfected with HPVOri/E1 and CMV4-Flag-16E2 were split equally into 6 dishes. Cells were treated with 100, 200, 300 nM JQ1(+), 300 nM JQ1(−), or an equal volume of DMSO. At 48 h p.t., episomal DNA was extracted and the transfected DNA was removed by DpnI treatment. DpnI- and XhoI-digested DNA (600 ng) was used to analyze the replication product, and 2.5 ng XhoI-treated DNA was used as loading control. Treated DNA was subjected to Southern blotting. Cellular extracts were analyzed by Western blotting using anti-16E2, anti-HA, and antiactin antibodies. NTC, nontransfection control. (C) Quantification of the size of BrdU foci at different time points posttransfection. C33A cells were transfected as in panel B and treated with either DMSO or 300 nM JQ1(+). Cells were labeled using BrdU as in Fig. 3C and costained with anti-HA E1 and anti-BrdU antibodies at 12, 18, 24, 36, and 48 h p.t. The area of BrdU foci was quantified for about 30 cells from each sample using ImageJ. The red dashed line represents the mean area of BrdU foci in the DMSO-treated sample at 48 h p.t. Foci with an area larger than the mean value are considered big foci. The mean area of these big foci (numbers in black) and the percentage of big foci in total foci (numbers in red) were calculated for each time point and are shown at the top of each column. (D) JQ1(+) treatment does not affect the cell cycle of C33A cells. C33A cells were treated as described in panel A. Flow cytometry analysis was performed to evaluate the impact of JQ1(+) treatments on the cell cycle. Experiments were repeated at least 3 times. The values for the cells treated with 300 nM JQ1(+), JQ1(−), or DMSO are the same as those shown in reference 28.
DISCUSSION
Brd4 functions as an episomal tether to allow hitchhiking of BPV 1 E2/episomes on mitotic chromosomes through mitosis (17). Inhibition of the Brd4 interaction with HPV E2s has been shown to displace the HPV16 or 31 genome from mitotic chromosomes (53), suggesting that Brd4 may play a similar role to support HPV episome maintenance. Brd4 also contributes to PV viral transcription regulation (18–20). A previous study has suggested a possible role of Brd4 in BPV1 genome replication (29). However, the functional role of Brd4 in the HPV viral life cycle remains to be clearly defined.
In this study, we show that Brd4 is recruited to the actively replicating HPV16 Ori along with HPV16 E1, E2, and a number of the cellular replication factors—RPA70, RFC1, and DNA polymerase δ. Mutagenesis disrupting either the E2-Brd4 interaction or the E1-E2 interaction abolished the formation of HPV16 replication complex and impaired HPV16 Ori DNA replication in cells. Brd4 was further demonstrated to be necessary for HPV16 viral DNA replication, since protein lysates from Brd4 knockdown cells were defective in a cell-free HPV16 DNA replication reaction and recombinant Brd4 protein was able to restore viral replication. Additionally, releasing endogenous Brd4 from chromatin using a chemical compound, JQ1(+), enhances HPV16 Ori DNA replication without affecting host cell cycle. This result shows that the role of Brd4 in HPV DNA replication could be uncoupled from its function in transcriptional regulation and cell cycle control. Our studies demonstrate a novel function of Brd4 in HPV replication, providing new insight into understanding the differentiation-dependent HPV life cycle.
In our study, Brd4 was first observed to form large foci with HPV16 E2 and the viral genome in cells cotransfected with E1 plasmid. Since Brd4 has been implicated in HPV transcription (18–20), to specifically examine Brd4's role in HPV16 replication, we constructed an artificial episome that contains the 145-bp minimum HPV16 replication Ori, excluding most of the URR transcription regulatory elements. This plasmid allowed us to specifically examine the recruitment of Brd4 and its impact on HPV16 replication in cells. Using this system, we discovered that E1, E2, and HPV16 Ori are all required for the formation of HPV16 Ori replication foci; omission of any one of the viral components could abolish the replication complex formation (Fig. 2 and 3). Immuno-FISH and BrdU incorporation further demonstrated that these foci contain actively replicating HPV16 Ori DNA. In addition, Brd4 was recruited to the large HPV16 Ori foci only when E1, E2, and HPV16 Ori were all present (Fig. 2B). These results indicate that the recruitment of Brd4 into the large HPV16 replication foci is dependent on the formation of the intact E1/E2/Ori complex. The conclusion was further supported by a mutagenesis study showing that the E1-E2 and E2-Brd4 interactions are critical for the HPV16 replication complex formation and Brd4 recruitment (Fig. 5). More importantly, these functional interactions are also critical for successful replication of viral DNA in cells (Fig. 5B).
Brd4's role in the cell cycle regulation complicates the analysis of its function in HPV replication using siRNA-mediated silencing in cells (54). We therefore reconstituted a cell-free replication system to directly assess the impact of Brd4 on HPV16 replication and control for the Brd4 knockdown effect on the cell cycle. We observed that Brd4 knockdown reduces viral replication, and more importantly, addition of recombinant Brd4 restores HPV replication in a dose-dependent manner (Fig. 6). This study provides direct evidence to support Brd4's function in HPV16 replication.
Brd4 dynamically associates with acetylated histone, and it is known to bind to cellular chromatin with a rapid “on and off” mode (52). The chromatin-bound Brd4 could be readily released upon signal-triggered histone deacetylation (55). Using the chemical compound JQ1(+) to dissociate Brd4 from chromatin leads to a stimulation of HPV16 Ori replication, suggesting that releasing Brd4 from chromatin may make it more accessible to be incorporated into the E1/E2/Ori replication complex. Together with previous studies, our data support a model in which chromatin-bound Brd4 tethers the E2 and viral episome complex to support the PV episome maintenance and viral transcription, whereas Brd4 released from chromatin may be recruited to the viral replication complex to support HPV viral replication.
A number of Brd4 features support its role in HPV replication. Brd4 has been shown to interact with the RFC complex (26). This interaction may promote the recruitment of cellular replication factories to the viral replication complex. This notion is supported by the observation that Brd4 colocalizes with replication factors RPA70, RFC1, and DNA polymerase δ in the actively replicating HPV16 Ori foci (Fig. 4). The HPV16 genome contains four E2 binding sites. It is possible that multiple E2 molecules binding to these sites can recruit an array of Brd4 molecules and cellular replication factors to form large multiprotein complexes on the HPV16 genome to support viral replication. In addition, recent reports show that HPV E1 and E2 recruit DNA damage response proteins to viral replication centers to support viral genome amplification in differentiated cells (46, 56). Sakakibara et al. observed that the E2 R37A I73A mutant was defective in recruiting DNA damage response proteins to host chromatin foci (33). These studies suggest that another possible role of Brd4 could be to recruit DNA damage response proteins to the HPV16 Ori complex to support viral replication.
Wild-type E2 supports the formation of the E1/E2/Brd4/Ori replication complex (Fig. 5), whereas large HPV replication foci cannot be efficiently formed in the E2 R37A I73A cells. This set of results suggested that the E2-Brd4 interaction is essential for recruitment of Brd4 and formation of the large HPV Ori replication foci. However, as described above, in a small portion of the cells, the E2 R37A I73A mutant defective in Brd4 binding can still form large nuclear foci with E1 and Brd4 (Fig. 5A) (data not shown). This observation suggests that the E2-Brd4 interaction is only partially required for Brd4 recruitment and that some other factor(s) may also be involved. As shown previously, E1 and E2 proteins coexpressed in cells localize to defined host chromatin foci (33, 34). It is possible that Brd4 may be recruited to these chromatin foci through the binding of acetylated histones, which is independent of E2 binding. We also observed an E2-independent interaction between E1 and Brd4 (data not shown), which could facilitate Brd4 recruitment to these foci when the E2-Brd4 interaction is abolished.
In summary, this study provides a first look into the E2-Brd4 interaction in the presence of other important viral factors, such as E1 and HPV16 Ori. This system better resembles HPV natural infection and allows us to observe an important function of Brd4 in HPV DNA replication, which was not detectable in the absence of E1 and Ori. This study provides new insights into the E2-Brd4 interaction in the HPV viral life cycle. These findings show that E1, E2, and Brd4 form a complex to support HPV viral DNA replication, identifying all components of this complex as possible targets to block viral replication. The interesting results obtained from the JQ1(+) study suggest that this chemical compound could be a valuable tool to eliminate HPV infection as it can release Brd4 from chromatin and abrogate the viral episome maintenance in latently infected cells. More importantly, since HPV genome amplification occurs exclusively in terminally differentiated cells to avoid detection by host immune surveillance, an impromptu viral genome amplification induced by JQ1(+) in the infected basal cells could trigger activation of the immune response to clear the viral latent infection before cancer development. This small molecule therefore offers a promising lead for development of antiviral inhibitors to treat HPV latent infection.
ACKNOWLEDGMENTS
We thank Paul F. Lambert (University of Wisconsin—Madison) for pUC19-HPV16, Mart Ustav (University of Tartu) for HA-16E1, and James E. Bradner (Dana-Farber Cancer Institute) for the JQ1 compounds. We thank the members of our laboratory for helpful discussions and critical reviews of the manuscript.
This work was supported by the HIV-Associated Malignancies Pilot Project Award (National Cancer Institute) and by National Institutes of Health (NIH) grants R01CA148768 and R01CA142723.
Footnotes
Published ahead of print 30 January 2013
REFERENCES
- 1. zur Hausen H, de Villiers EM. 1994. Human papillomaviruses. Annu. Rev. Microbiol. 48:427–447 [DOI] [PubMed] [Google Scholar]
- 2. zur Hausen H. 2009. Papillomaviruses in the causation of human cancers—a brief historical account. Virology 384:260–265 [DOI] [PubMed] [Google Scholar]
- 3. Munoz N, Bosch FX, de Sanjose S, Herrero R, Castellsague X, Shah KV, Snijders PJ, Meijer CJ. 2003. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348:518–527 [DOI] [PubMed] [Google Scholar]
- 4. Smith JS, Lindsay L, Hoots B, Keys J, Franceschi S, Winer R, Clifford GM. 2007. Human papillomavirus type distribution in invasive cervical cancer and high-grade cervical lesions: a meta-analysis update. Int. J. Cancer 121:621–632 [DOI] [PubMed] [Google Scholar]
- 5. Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 189:12–19 [DOI] [PubMed] [Google Scholar]
- 6. Gilbert DM, Cohen SN. 1987. Bovine papilloma virus plasmids replicate randomly in mouse fibroblasts throughout S phase of the cell cycle. Cell 50:59–68 [DOI] [PubMed] [Google Scholar]
- 7. Hebner CM, Laimins LA. 2006. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev. Med. Virol. 16:83–97 [DOI] [PubMed] [Google Scholar]
- 8. Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–1699 [DOI] [PubMed] [Google Scholar]
- 9. Yang L, Mohr I, Fouts E, Lim DA, Nohaile M, Botchan M. 1993. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc. Natl. Acad. Sci. U. S. A. 90:5086–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sedman J, Stenlund A. 1998. The papillomavirus E1 protein forms a DNA-dependent hexameric complex with ATPase and DNA helicase activities. J. Virol. 72:6893–6897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Conger KL, Liu JS, Kuo SR, Chow LT, Wang TS. 1999. Human papillomavirus DNA replication. Interactions between the viral E1 protein and two subunits of human DNA polymerase alpha/primase. J. Biol. Chem. 274:2696–2705 [DOI] [PubMed] [Google Scholar]
- 12. Masterson PJ, Stanley MA, Lewis AP, Romanos MA. 1998. A C-terminal helicase domain of the human papillomavirus E1 protein binds E2 and the DNA polymerase alpha-primase p68 subunit. J. Virol. 72:7407–7419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park P, Copeland W, Yang L, Wang T, Botchan MR, Mohr IJ. 1994. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc. Natl. Acad. Sci. U. S. A. 91:8700–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu JS, Kuo SR, Makhov AM, Cyr DM, Griffith JD, Broker TR, Chow LT. 1998. Human Hsp70 and Hsp40 chaperone proteins facilitate human papillomavirus-11 E1 protein binding to the origin and stimulate cell-free DNA replication. J. Biol. Chem. 273:30704–30712 [DOI] [PubMed] [Google Scholar]
- 15. Loo YM, Melendy T. 2004. Recruitment of replication protein A by the papillomavirus E1 protein and modulation by single-stranded DNA. J. Virol. 78:1605–1615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee D, Sohn H, Kalpana GV, Choe J. 1999. Interaction of E1 and hSNF5 proteins stimulates replication of human papillomavirus DNA. Nature 399:487–491 [DOI] [PubMed] [Google Scholar]
- 17. You J, Croyle JL, Nishimura A, Ozato K, Howley PM. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117:349–360 [DOI] [PubMed] [Google Scholar]
- 18. McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 80:9530–9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schweiger MR, You J, Howley PM. 2006. Bromodomain protein 4 mediates the papillomavirus E2 transcriptional activation function. J. Virol. 80:4276–4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu SY, Lee AY, Hou SY, Kemper JK, Erdjument-Bromage H, Tempst P, Chiang CM. 2006. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 20:2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19:523–534 [DOI] [PubMed] [Google Scholar]
- 22. Yang Z, Yik JH, Chen R, He N, Jang MK, Ozato K, Zhou Q. 2005. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19:535–545 [DOI] [PubMed] [Google Scholar]
- 23. Crawford NP, Alsarraj J, Lukes L, Walker RC, Officewala JS, Yang HH, Lee MP, Ozato K, Hunter KW. 2008. Bromodomain 4 activation predicts breast cancer survival. Proc. Natl. Acad. Sci. U. S. A. 105:6380–6385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. 2011. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478:524–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ishii H, Inageta T, Mimori K, Saito T, Sasaki H, Isobe M, Mori M, Croce CM, Huebner K, Ozawa K, Furukawa Y. 2005. Frag1, a homolog of alternative replication factor C subunits, links replication stress surveillance with apoptosis. Proc. Natl. Acad. Sci. U. S. A. 102:9655–9660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Maruyama T, Farina A, Dey A, Cheong J, Bermudez VP, Tamura T, Sciortino S, Shuman J, Hurwitz J, Ozato K. 2002. A mammalian bromodomain protein, Brd4, interacts with replication factor C and inhibits progression to S phase. Mol. Cell. Biol. 22:6509–6520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, Howley PM. 2011. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 31:2641–2652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang X, Li J, Schowalter RM, Jiao J, Buck CB, You J. 2012. Bromodomain protein Brd4 plays a key role in Merkel cell polyomavirus DNA replication. PLoS Pathog. 8:e1003021 doi:10.1371/journal.ppat.1003021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ilves I, Maemets K, Silla T, Janikson K, Ustav M. 2006. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 80:3660–3665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. 2010. Selective inhibition of BET bromodomains. Nature 468:1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zheng G, Schweiger MR, Martinez-Noel G, Zheng L, Smith JA, Harper JW, Howley PM. 2009. Brd4 regulation of papillomavirus protein E2 stability. J. Virol. 83:8683–8692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang R, Li Q, Helfer CM, Jiao J, You J. 2012. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 287:10738–10752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol. 85:8981–8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. 1999. Human papillomavirus DNA replication compartments in a transient DNA replication system. J. Virol. 73:1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim J, Lee D, Gwan Hwang S, Hwang ES, Choe J. 2003. BRCA1 associates with human papillomavirus type 18 E2 and stimulates E2-dependent transcription. Biochem. Biophys. Res. Commun. 305:1008–1016 [DOI] [PubMed] [Google Scholar]
- 36. Kumar RA, Naidu SR, Wang X, Imbalzano AN, Androphy EJ. 2007. Interaction of papillomavirus E2 protein with the Brm chromatin remodeling complex leads to enhanced transcriptional activation. J. Virol. 81:2213–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lee AY, Chiang CM. 2009. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem. 284:2778–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee D, Hwang SG, Kim J, Choe J. 2002. Functional interaction between p/CAF and human papillomavirus E2 protein. J. Biol. Chem. 277:6483–6489 [DOI] [PubMed] [Google Scholar]
- 39. Lee D, Lee B, Kim J, Kim DW, Choe J. 2000. cAMP response element-binding protein-binding protein binds to human papillomavirus E2 protein and activates E2-dependent transcription. J. Biol. Chem. 275:7045–7051 [DOI] [PubMed] [Google Scholar]
- 40. Moreno ML, Stein GS, Stein JL. 1987. Nucleosomal organization of a BPV minichromosome containing a human H4 histone gene. Mol. Cell. Biochem. 74:173–177 [DOI] [PubMed] [Google Scholar]
- 41. Zhao W, Chow LT, Broker TR. 1997. Transcription activities of human papillomavirus type 11 E6 promoter-proximal elements in raft and submerged cultures of foreskin keratinocytes. J. Virol. 71:8832–8840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiang CM, Ustav M, Stenlund A, Ho TF, Broker TR, Chow LT. 1992. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc. Natl. Acad. Sci. U. S. A. 89:5799–5803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ustav E, Ustav M, Szymanski P, Stenlund A. 1993. The bovine papillomavirus origin of replication requires a binding site for the E2 transcriptional activator. Proc. Natl. Acad. Sci. U. S. A. 90:898–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 10:449–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ustav M, Ustav E, Szymanski P, Stenlund A. 1991. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J. 10:4321–4329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5:e1000605 doi:10.1371/journal.ppat.1000605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dixon EP, Pahel GL, Rocque WJ, Barnes JA, Lobe DC, Hanlon MH, Alexander KA, Chao SF, Lindley K, Phelps WC. 2000. The E1 helicase of human papillomavirus type 11 binds to the origin of replication with low sequence specificity. Virology 270:345–357 [DOI] [PubMed] [Google Scholar]
- 48. Cardenas-Mora J, Spindler JE, Jang MK, McBride AA. 2008. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J. Virol. 82:7298–7305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sakai H, Yasugi T, Benson JD, Dowhanick JJ, Howley PM. 1996. Targeted mutagenesis of the human papillomavirus type 16 E2 transactivation domain reveals separable transcriptional activation and DNA replication functions. J. Virol. 70:1602–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang Z, He N, Zhou Q. 2008. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 28:967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gopalakrishnan V, Khan SA. 1994. E1 protein of human papillomavirus type 1a is sufficient for initiation of viral DNA replication. Proc. Natl. Acad. Sci. U. S. A. 91:9597–9601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. U. S. A. 100:8758–8763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Abbate EA, Voitenleitner C, Botchan MR. 2006. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol. Cell 24:877–889 [DOI] [PubMed] [Google Scholar]
- 54. Mochizuki K, Nishiyama A, Jang MK, Dey A, Ghosh A, Tamura T, Natsume H, Yao H, Ozato K. 2008. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 283:9040–9048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ai N, Hu X, Ding F, Yu B, Wang H, Lu X, Zhang K, Li Y, Han A, Lin W, Liu R, Chen R. 2011. Signal-induced Brd4 release from chromatin is essential for its role transition from chromatin targeting to transcriptional regulation. Nucleic Acids Res. 39:9592–9604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gillespie KA, Mehta KP, Laimins LA, Moody CA. 2012. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J. Virol. 86:9520–9526 [DOI] [PMC free article] [PubMed] [Google Scholar]