

Abstract
All herpesviruses encode a complex of two proteins, referred to as the nuclear egress complex (NEC), which together facilitate the exit of assembled capsids from the nucleus. Previously, we showed that the Kaposi's sarcoma-associated herpesvirus (KSHV) NEC specified by the ORF67 and ORF69 genes when expressed in insect cells using baculoviruses for protein expression forms a complex at the nuclear membrane and remodels these membranes to generate nuclear membrane-derived vesicles. In this study, we have analyzed the functional domains of the KSHV NEC proteins and their interactions. Site-directed mutagenesis of gammaherpesvirus conserved residues revealed functional domains of these two proteins, which in many cases abolish the formation of the NEC and remodeling of nuclear membranes. Small in-frame deletions within ORF67 in all cases result in loss of the ability of the mutant protein to induce cellular membrane proliferation as well as to interact with ORF69. Truncation of the C terminus of ORF67 that resides in the perinuclear space does not impair the functions of ORF67; however, deletion of the transmembrane domain of ORF67 produces a protein that cannot induce membrane proliferation but can still interact with ORF69 in the nucleus and can be tethered to the nuclear membrane by virtue of its interaction with the wild-type-membrane-anchored ORF67. In-frame deletions in ORF69 have varied effects on NEC formation, but all abolish remodeling of nuclear membranes into circular structures. One mutant interacts with ORF67 as well as the wild-type protein but cannot function in membrane curvature and fission events that generate circular vesicles. These studies genetically confirm that ORF67 is required for cellular membrane proliferation and that ORF69 is the factor required to remodel these duplicated membranes into circular-virion-size vesicles. Furthermore, we also investigated the NEC encoded by Epstein-Barr virus (EBV). The EBV complex comprised of BFRF1 and BFLF2 was visualized at the nuclear membrane using autofluorescent protein fusions. BFRF1 is a potent inducer of membrane proliferation; however, BFLF2 cannot remodel these membranes into circular structures. What was evident is the superior remodeling activity of ORF69, which could convert the host membrane proliferations induced by BFRF1 into circular structures.
INTRODUCTION
Viruses have evolved methods to subvert cellular pathways and to utilize cellular processes for their replication, spread, and maintenance in the host cell. One such pathway that herpesviruses have clearly manipulated is that which is used to facilitate their egress from the site of assembly, the nucleus. For large protein assemblies, such as the 120-nm icosahedral herpesvirus capsid, a transport system out of the nucleus has to be used for subsequent maturation in the cytoplasm and exit of the infectious particle from the cell surface. All herpesviruses encode two proteins that play a key role in facilitating the exit of capsids from the nucleus (1–3). The proteins are commonly referred to as the nuclear egress complex (NEC). For the alphaherpesviruses (herpes simplex virus 1 [HSV-1] and pseudorabies virus [PRV]), these proteins, UL34 and UL31, participate in a molecular interaction that is required for nuclear membrane remodeling, “softening” of the nuclear lamina, and envelopment of the capsid at the inner nuclear membrane (INM) (4–10). UL34 is a type II membrane protein (11), and UL31 is a nuclear phosphoprotein (12), which in the presence of UL34 relocalizes to the nuclear membrane (8, 9, 13). The interaction between these two proteins may also stabilize the localization of UL34 in nuclear membranes (14). The herpesvirus NECs have been shown to facilitate INM envelopment by recruiting cellular or viral kinases to phosphorylate the nuclear lamina, a structure that is a physical barrier for capsid envelopment. This phosphorylation results in the dissolution of the lamina, thus allowing capsids to gain access to the INM (15–20). Interactions between the capsid and capsid-associated proteins are required to link these structures to the NEC during the budding process, also referred to as primary envelopment (21). The budding process at the INM is completed by membrane fission events that deposit the primary enveloped virion in the space between the inner and outer nuclear membranes (1, 3).
The Kaposi's sarcoma-associated herpesvirus (KSHV) NEC proteins are encoded by open reading frames (ORFs) ORF67, the functional ortholog of UL34, and ORF69, the functional ortholog of UL31. Previously, ORF69 was shown by Santarelli et al. (22) to localize to the nucleus and the nuclear periphery in KSHV lytically induced cells and colocalize with ORF67 in cotransfected cells. The Epstein-Barr virus (EBV) NEC is encoded by BFRF1 (UL34 ortholog) and BFLF2 (UL31 ortholog) genes, whose products have been shown to participate in physical interactions at the nuclear membrane (23, 24) and are required for nuclear egress of EBV (25, 26). EBV BFRF1 has been shown to cause proliferation of cellular membranes (22, 23) and also colocalizes with ORF69, suggesting trans complementation of the gammaherpesvirus proteins (22). The coexpression of these two proteins of PRV alone is sufficient to alter nuclear membranes, resulting in vesicle formation in transfected mammalian cells (13). Recently, we discovered, by using baculoviruses for gene expression, that the coexpression of the KSHV NEC proteins ORF67 and ORF69 could induce the same phenotype in insect cells (27). In our observations, we discovered that ORF67 is a potent inducer of host membrane proliferation. ORF67, by virtue of its location in nuclear, perinuclear, and cytoplasmic structures, as judged by confocal microscopy of fluorescent protein fusions in living cells, is able to mediate proliferation of these membranes. These membrane proliferations are structurally akin to stacks of nuclear envelopes or tubules which, depending on the sectional nature of ultrastructural analyses, appear as regular interconnected tubules, some of which have a honeycomb appearance. However, when ORF69 is present, which also causes significant relocalization of ORF67 to the nuclear margins, there is a conversion of these membrane proliferations into very distinct circular structures, which in most cases are intranuclear, adjacent to the nuclear membrane (27). Thus, our conclusion was that ORF69 was required for remodeling host membranes into circular vesicles. In the cells expressing the PRV NEC, it was possible to image intranuclear vesicles as well as vesicles within membrane invaginations of the inner nuclear membrane as well as fusion with the outer nuclear membrane (13).
Functional domains of the alphaherpesvirus and betaherpesvirus NEC proteins have been identified and used to determine the physical associations and interactions of these proteins; in many cases, the interaction domains reside within the conserved regions (3, 28–31). For both the PRV and HSV-1 NEC, the interaction domains that are important for mediating complex formation have been mapped and the roles of the transmembrane (TM) region in UL34 and the UL34 C terminus have been determined (28, 32, 33, 60). It appears that the luminal region of UL34, which is very short, is not required either for virus replication or for NEC formation. The TM region, although important for membrane anchorage, can be changed to any of a number of sequences from other inner nuclear membrane-localized proteins and still retain functional activity (33). The human cytomegalovirus (HCMV) NEC proteins encoded by genes UL50 (UL34) and UL53 (UL31) have been purified, and their hydrodynamic properties have been examined in vitro (30). These studies showed that UL53 exists as a dimer in solution whereas UL50 lacking the TM region behaves as a monomer. Furthermore, an alpha-helix in UL53 is important for the bimolecular interaction between these proteins. Extensive mutagenesis of the murine CMV proteins has determined the importance of the conserved regions for interactions, the location of a nuclear localization signal in M53, and the effect of dominant negative mutants of the NEC (34–36).
Although the alphaherpesvirus and betaherpesvirus NEC proteins have been extensively studied, the KSHV complex is not completely characterized. To that end, in this study we have uncovered essential functional and interaction domains of ORF67 and ORF69 by using a combination of colocalization assays of autofluorescent protein fusions, reconstitution of Venus protein fluorescence, and ultrastructural analyses of nuclear membrane remodeling events. We have shown using genetic methods that the loss of interaction between these two proteins results in the loss of circular vesicle formation. Finally we show that the KSHV NEC proteins are potent inducers of cellular membrane proliferation and remodeling and that ORF69 is a potent factor for remodeling nuclear membrane proliferations, which form as a consequence of EBV BFRF1 expression.
MATERIALS AND METHODS
Cells and viruses.
Spodoptera frugiperda (Sf9 and Sf21) cells were grown in Grace's insect cell medium, supplemented with 10% fetal calf serum (Gibco-Invitrogen). Sf9 and Sf21 cells were propagated as described by Perkins et al. (37) and Okoye et al. (38). Baculoviruses were made using the BAC-to-BAC system from Invitrogen. The procedures for this as well as for the transfection and amplification of baculoviruses are described in detail in the work of Perkins et al. (37) and Okoye et al. (38). Human embryonic kidney 293T (HEK-293T) cells were grown in alpha-minimum essential medium (α-MEM) supplemented with 10% fetal calf serum (Gibco-Invitrogen). These cells were passaged every 2 days, after being seeded at a density of 8 × 106 cells per T150 flask.
Plasmids.
Previously, the genes encoding the ORF67 proteins and ORF69 were amplified from the KSHV BAC36 genome (39). ORF67 and ORF69 were cloned in the baculovirus transfer vector pFastBac 1 (Invitrogen; pFB1), which had been modified to encode C-terminal fusions with V5, hemagglutinin (HA), enhanced green fluorescent protein (EGFP), and mCherry (27). The pFB1 transfer vector was modified to make N-terminal fusions with the Split-Venus genes. VenusN (amino acids 1 to 154) and VenusC (amino acids 155 to 238) were amplified using Phusion polymerase (NEB) and pVenus-VSVG (Addgene plasmid 11914) (40) as a template and cloned into pFB1 using the BamHI and EcoRI sites. A five-glycine flexible linker sequence was incorporated at the C terminus of each Split-Venus gene upstream of the EcoRI site. These plasmids after sequencing were designated pFB-NVenusN and pFB-NVenusC. Both ORF67 and ORF69 were transferred from pFB-ORF67CV5 and pFB-ORF69CV5, using EcoRI and HindIII digestion of these plasmids. The C-terminal V5 tag was also transferred into the Venus vectors. ORF67 and ORF69 sequences were also transferred from the plasmids carrying C-terminal V5, HA, GFP, and mCherry tags into pcDNA 3.1(−) (Invitrogen) using EcoRI and HindIII digestion. The EBV genes BFRF1 and BFLF2 were amplified using PCR assays (Table 1) and the EBV Akata bacterial artificial chromosome (BAC) as a template (41). The genes were cloned as EcoRI-SpeI fragments into pFB1-CV5 and sequenced, and the correct genes were moved into pFB1-CGFP and pFB1-CmCherry as EcoRI-SpeI fragments and into pFB1-NVenusN and pFB1-NVenusC as EcoRI-HindIII fragments.
Table 1.
Sequences of PCR primers used
| Primer name | Primer sequence (5′ to 3′)a |
|---|---|
| ORF67-F-EcoRI | GGAATTCACCATGAGTGTCGTTGGTAAGCGTGTAG |
| ORF67-R-SpeI | GGACTAGTGCTGGGCCTCATCCAAACCAAACC |
| ORF69-F-EcoRI | GGAATTCACCATGCCGAAATCAGTGTCCAGCCAC |
| ORF69-R-SpeI | GGACTAGTTAGGGCGTTGACAAGTGCGGTTCC |
| ORF67Δ61-65-F | GTGTACCACTTATGGGCGCACCTCCTGGAGGAGATGCACTACCCG |
| ORF67Δ61-65-R | GAGGTGCGCCCATAAGTGGTACACGCAACCGTGCTGCATTCTAAC |
| ORF67Δ88-93-F | ATATCGAATAATAAGTGCTTTCTGTGCCGACCAAGCGACGCCGTG |
| ORF67Δ88-93-R | CAGAAAGCACTTATTATTCGATATAACAAAGCAGGGAGAGAACGG |
| ORF67Δ128-132-F | TCTGTCGTGCTGACAAAGTTCAAGGCGAGCCTGGTGTTCTCAAAG |
| ORF67Δ128-132-R | CTTGAACTTTGTCAGCACGACAGACGTATTTTTGGCAAGGTATAC |
| ORF67Δ166-170-F | AGGCAGGTTTTGACGCCCAAAAGTCACAAGAGACTGAAGTCGCTG |
| ORF67Δ166-170-R | ACTTTTGGGCGTCAAAACCTGCCTGTAGTCCGTGAAGTAGGTACG |
| ORF69Δ90-94-F | AAAACAGTAATGTCTACCGTCGCACCCGGCCGCTGCGTCTCCCTC |
| ORF69Δ90-94-R | TGCGACGGTAGACATTACTGTTTTGGAGGTGCATATCGGGGTCTC |
| ORF69Δ99-102-F | TCTACCGTCGCAGTCTCCCTCTCTCCGTTTGGACACTCCTCAAAC |
| ORF69Δ99-102-R | AGAGAGGGAGACTGCGACGGTAGACAGGTCTAGGGGCAGCATTAC |
| ORF69Δ187-192-F | CAGAGCTTCCTGGCTAACCTGGTCCTAAAAACCTTTGGGCACGAT |
| ORF69Δ187-192-R | GACCAGGTTAGCCAGGAAGCTCTGCCGAACCACATCCATGGAGTG |
| ORF67-T264-R | GGACTAGTAAGACACAAGAAAGCGATCCCAGC |
| ORF67-T240-R | GGACTAGTGCTCCACTGAAAGGCGCCCAGAATC |
| ORF69-R17-F (Nar1) | GCGACCTCAACCGGCGCCAGTGGACCTGCAGAC |
| ORF69-R17-R | GTCTGCAGGTCCACTGGCGCCGGTTGAGGTCGC |
| ORF69-R21-25-F (PstI) | GGTCGCAGTGGACCTGCAGACATCGCAGCATGCTTGTCATCCCGC |
| ORF69-R21-25-R | GCGGGATGACAAGCATGCTGCGATGTCTGCAGGTCCACTGCGACC |
| ORF69-R21-39-F (KpnI) | GCATGCTTGTCATCCGCCCTGGCGTCGGTACCGCCCGGGGCGGCAAGCGCCAGCGTCAGC |
| ORF69-R21-39-R | GCTGACGCTGGCGCTTGCCGCCCCGGGCGGTACCGACGCCAGGGCGGATGACAAGCATGC |
| ORF69-R17-25-F (NarI) | GCGACCTCAACCGGCGCCAGTGGACCTGCAGAC |
| ORF69-R17-25-R | GTCTGCAGGTCCACTGGCGCCGGTTGAGGTCGC |
| BFLF2-F-EcoRI | GGAATTCACCATGCTACTGTTTATTTTCCAAAATGAG |
| BFLF2-R-SpeI | CCACTAGTATGGCCCCGGTCACCCCAGATGCC |
| BFRF1-F-EcoRI | GGAATTCACCATGGCGAGCCCGGAAGAGAGGCTC |
| BFRF1-R-SpeI | CCACTAGTGGTCCACCTCAGAAACATCAGGAG |
Restriction sites are in italics.
Mutagenesis.
In-frame deletions were made by overlap PCR procedures. The sequences of the overlap primers for each deletion are shown in Table 1. The template for each gene was pFB-ORF67CV5 and pFB-ORF69CV5. Each ORF67 and ORF69 gene carrying the deletion was sequenced to confirm the introduction of the correct mutation. Then, each mutant sequence was moved as an EcoRI-SpeI fragment into pFB-CGFP and pFB-CmCherry and as an EcoRI-HindIII fragment into pFB-NVenusN and pFB-NVenusC; the latter clones also transferred the C-terminal V5 sequence. Polypeptide chain termination mutants were made by standard PCR assays (Table 1). The QuikChange mutagenesis protocol (Stratagene) was used to make site-directed mutations in ORF69 using the primers in Table 1. The use of this method is described in detail in the work of Walters et al. (42). The template used was pcDNA-ORF69GFP (see above). Mutants ORF69 R21-39 and ORF69 R17-25 were amplified using the ORF69 R21-25 plasmid DNA as a template. Positive DNA clones carrying the mutation were confirmed by restriction enzyme and sequence analysis.
Gel electrophoresis and Western blotting.
Protein samples were prepared routinely from 1 × 106 Sf21 cells harvested 48 h postinfection. The cells were solubilized in RIPA buffer (27), clarified by centrifugation, and resolved in NuPage gels using morpholineethanesulfonic acid (MES) running buffer (Invitrogen). Protein samples were transferred to nitrocellulose membranes using the iBLOT apparatus (Invitrogen), and Western blots were processed using the enhanced chemiluminescence (ECL) method according to the manufacturer's protocol (GE Healthcare). Primary as well as secondary (horseradish peroxidase-conjugated) antibody dilutions ranged from 1:5,000 to 1:10,000. The mouse V5 antibody was obtained from Invitrogen, the rabbit GFP antibody was from Molecular Probes or Abcam, and the rabbit DsRed polyclonal antibody was purchased from BD Biosciences.
Confocal microscopy.
Sf21 cells seeded in Lab-Tek 4-well chamber slides (Nunc) at a density of 2 × 106 cells per chamber were infected with 100 μl (per well) of the baculovirus mixture. The infected cells were analyzed in a Zeiss LSM 510 confocal microscope 48 to 72 h after infection as described by Henson et al. (43). 293T cells plated in Lab-Tek 2-well chamber slides at a density of 8 × 105 cells per chamber were transfected with a mixture of 3 μl of polyethyleneimine (PEI) reagent (1 mg/ml) and 1 μg of plasmid DNA. The transfected cells were imaged by confocal microscopy 24 h posttransfection as described above.
Bimolecular fluorescence complementation (BiFC) assay.
Sf21 cells in 12-well trays (1 × 106 cells per well) were coinfected or mock infected with recombinant baculoviruses expressing VenusN and VenusC fusions. Viruses were mixed in equal volumes and 100 μl was added to the cells. The infected cells were harvested 48 h postinfection in the growth medium, pelleted in an Eppendorf tube, resuspended in 100 μl of phosphate-buffered saline (PBS), and transferred to a 96-well tray for analysis. All assays were read in a Glomax (Promega) plate reader using the blue filter (excitation at 460 nm and emission at 515 to 575 nm).
TEM.
Sf21 cells (5 × 106) were used for all transmission electron microscopy (TEM) experiments. Infected cells were processed for EM 68 to 72 h after infection. For the 293T cell line, 3.5 × 106 cells in 60-mm petri dishes were transfected with 3 μg of each plasmid DNA (pcDNA-ORF69V5 plus pcDNA-ORF67GFP) and 18 μl of PEI reagent (1 mg/ml). Cells were processed for TEM at 24 or 48 h posttransfection. Infected and transfected cells were processed for conventional thin-section TEM as described by Huang et al. (44) and Perkins and McCaffery (45). Samples were examined using either a Philips EM 420 or an FEI Tecnai 12 electron microscope; images were obtained with an SIS Megaview III camera (Olympus).
Data and figure preparation.
Scanned autoradiographs and digital electron micrographs or confocal images were converted to TIFF files and imported into Adobe Photoshop for figure compilation.
RESULTS
Functional regions of ORF67 and ORF69.
After aligning the gammaherpesvirus sequences for each gene, we chose to generate small-in frame deletions in a number of conserved regions of the ORF67 and ORF69 protein coding sequences (Fig. 1). For ORF67, they included Δ61–65, Δ88–93, Δ128–132, and Δ166–170, and for ORF69, they were Δ90–94, Δ99–102, and Δ187–192. We also made two truncation mutations in the ORF67 gene that specify polypeptide termination after the predicted transmembrane region (T264) and before the transmembrane domain (T240). These mutations were created in the baculovirus transfer vector, pFB1, which also specified a C-terminal V5 epitope sequence (27). Following sequence analysis, the mutants were moved into different pFB1 vectors that specified C-terminal GFP and mCherry fusions (27). The genes were transferred into the baculovirus genome using recombineering methods, and the viruses isolated were used to infect Sf21 cells to examine the expression of the mutant polypeptides, using the V5 antibody (Fig. 2). All the mutants accumulated stable amounts of protein, although ORF67Δ128–132 accumulated slightly reduced amounts of protein.
Fig 1.

Polypeptide sequences of ORF67 and ORF69 and mutations made in these proteins. Shown are the amino acid sequences of KSHV ORF67 and ORF69 (strain BCBL-1 BAC36) (59). The in-frame deletions made are highlighted by black boxes, and the positions of the polypeptide truncation mutations are indicated by the arrows (T240 and T264; truncations after amino acids 239 and 263, respectively). The transmembrane domain residues of ORF67 predicted using MacVector are shown in bold. Underlined residues are those that are conserved in the gammaherpesviruses (KSHV, EBV, rhesus rhadinovirus, murine herpesvirus 68, and herpesvirus saimiri). Residues that are completely conserved in all human herpesvirus proteins that are located close to or within the in-frame deletions are in red. The amphipathic alpha-helix in ORF67 predicted by Milbradt et al. (29) is marked by a purple box, as is an alpha-helix in the N terminus of ORF69 that was determined using MacVector software. The arginine-rich region in the N terminus of ORF69, which may contain a nuclear localization signal (NLS), is shown in a green box.
Fig 2.
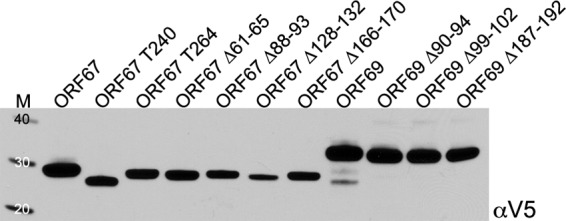
Biochemical analysis of ORF67 and ORF69 mutant polypeptides. Infected Sf21 cells were harvested at 48 h postinfection, and protein lysates were prepared. Polypeptides were analyzed by NuPage gel electrophoresis (4 to 12% acrylamide), and the membrane following protein transfer was probed with a mouse anti-V5 antibody. Protein standards (kDa) are in lane M.
NEC formation monitored by fluorescent protein fusions.
Previously, using autofluorescent protein fusions, we could observe using live-cell analysis the relocalization of ORF69 to the nuclear margins in the presence of ORF67 and alterations of the nuclear membrane as judged by the distribution of fluorescence during complex formation (27). The localization of ORF67 and ORF69 mutants to the correct cellular structure was confirmed by single infections with viruses expressing either GFP or mCherry fusions (data not shown). We then coinfected cells with wild-type ORF67 or ORF69 and examined the localization of the mutant polypeptides using confocal microscopy (Fig. 3). In coinfections with the virus expressing wild-type ORF69mCherry, all the ORF67 in-frame deletion mutants failed to interact with this protein, as judged by the absence of relocalization of red fluorescence to the nuclear margins (Fig. 3A). This was also observed when the fluorescent fusions were switched, that is, wild-type ORF69GFP and ORF67mCherry mutants. In the wild-type panel, there are still some cells that display diffuse ORF69mCherry fluorescence partly because of the asynchronous nature of the expression of the two proteins in coinfected cells as well as the fact that not all cells are coinfected with the two viruses. The images displayed in Fig. 3A were obtained at 48 h postinfection and were similar to results obtained when the cells were imaged at 68 to 72 h postinfection (data not shown). In the case of the ORF69 mutants, it was observed that Δ90–94 could associate somewhat with ORF67 and colocalize with this protein only after prolonged incubation (Fig. 3B). There was also a punctate distribution of colocalized fluorescence signal similar to that seen with the wild-type proteins; however, there was still not complete coincidence of the two fluorophores in some cells and the changes at the nuclear margins were not as pronounced. The other two ORF69 mutants, Δ99–102 and Δ187–192, did relocalize to the nuclear margins in many cells, but the pattern of fluorescence was different and there were significantly greater amounts of GFP signal that did not localize with the mCherry signal (see merged images). In addition, there was also more ORF67 fluorescence in the cytoplasmic membranes compared to the distribution in the presence of wild-type ORF69.
Fig 3.

Colocalization of ORF67 and ORF69 mutants at the nuclear margins. Sf21 cells were coinfected with baculoviruses expressing GFP and mCherry fusion proteins to discover whether the mutant proteins colocalize with the wild-type partner of the NEC by confocal microscopy. (A) Wild-type (WT) and mutant ORF67 polypeptides fused to GFP were examined in the presence of wild-type ORF69mCherry 48 h following infection. Magnification, ×63. (B) The distribution of wild-type and mutant ORF69 polypeptides fused to GFP was examined in the presence of wild-type ORF67mCherry, in this case 68 h after infection. Magnification, ×100. The scans from both the green and red channels are shown as well as the digitally merged images.
The ORF67 polypeptide truncation mutants were also examined by confocal microscopy (Fig. 4A and B). ORF67 T240 when expressed alone accumulated in what appeared to be large cytoplasmic aggregates, possibly in the cisternae of the endoplasmic reticulum (ER) (Fig. 4A). When the virus expressing ORF69mCherry was also present, the ORF67 T240 GFP signal changed and relocalized to the nucleus in a diffuse pattern overlapping with the ORF69mCherry fluorescence. One cell that does not express ORF69mCherry still contains ORF67 T240 in cytoplasmic aggregates (Fig. 4A, bottom). This indicates that the truncation mutant T240 polypeptide can still interact with ORF69. We also tested whether ORF67 T240 could be tethered to the membrane by interaction with wild-type ORF67. Cells were coinfected with ORF67 T240 GFP and ORF67V5 and imaged by confocal microscopy. Judging by the redistribution of the GFP signal to the nuclear margins and perinuclear structures, the T240 mutant interacts with ORF67 and can be positioned at the nuclear membrane. ORF67 T264 was present in a similar distribution in cellular membranes when expressed alone (data not shown). In the presence of ORF69GFP, there is relocalization of both proteins to nuclear margins with additional changes in the membrane-associated fluorescence (Fig. 4B).
Fig 4.

Localization and interactions of the polypeptide truncation mutants of ORF67. Infections similar to those described in the Fig. 3 legend were carried out using the ORF67 T240 and T264 mutants. (A) The distribution and interactions of ORF67 T240 fused to GFP were examined by confocal microscopy following infection for 48 h. (B) Colocalization of ORF67 T264mCherry and ORF69GFP at the nuclear margins. Magnification, ×100. For panel B and lower images of panel A, the scans from both the green and red channels are shown as well as the digitally merged images.
BiFC using the Split-Venus system.
Bimolecular fluorescence complementation (BiFC) has been used successfully to investigate protein-protein interactions in living cells and in real time. The first assays utilized yellow fluorescent proteins, but our approach was to use the Split-Venus method (46–48). In this method, the Venus gene is split into two fragments, an N-terminal fragment (residues 1 to 154) and a C-terminal fragment (residues 155 to 238). These fragments do not fluoresce on their own or when fused to a test protein. Fusions are made to a test protein at either the N or the C terminus separated by a linker sequence. If the two test proteins interact within the cell, the VenusN and VenusC are brought into close proximity with each other and fluorescence is observed. This method was used to show that ICP27 forms a head-to-tail intramolecular interaction (46). We decided to fuse the Split-Venus fragments to the N termini of both ORF67 and ORF69 for this assay as these are both located in the nucleoplasm. Sf21 cells infected with viruses expressing these fusions did not display any detectable fluorescence; however, when coinfections were performed using viruses expressing VenusN and VenusC, a significant fluorescence signal was observed which increased in intensity over time (Fig. 5). This was observed in cells coinfected with viruses expressing ORF67 fused to VenusN and ORF67 fused to VenusC, indicating a self-interaction between these molecules (Fig. 5A). The same was also seen with ORF69 fusions to the Split-Venus fragments. This self-interaction phenotype has been observed for other herpesvirus NEC proteins (30). The fluorescence observed within the cells from reconstituted Venus fluorescent protein or from the ORF67/ORF69 GFP-expressing viruses was the same or similar. Reconstitution of Venus fluorescence was also seen when cells were coinfected with viruses expressing ORF67 and ORF69 fused to Split-Venus, indicating bimolecular interactions between these proteins (Fig. 5B). The strength of this assay for visualizing interactions in living cells is further bolstered by the ability to quantitate the relative interaction between these proteins using a fluorescence reader. Similar infections were performed, and the relative Venus fluorescence was quantitated after 48 h postinfection (Fig. 5C). Judging from the fluorescent signal, it is apparent that the ORF69 self-interaction is stronger than the ORF67 self-interaction. The interaction between ORF67 and ORF69 results in greater amounts of fluorescence, indicating an even stronger interaction, although there is a difference depending on the Venus fragment fusion to each gene.
Fig 5.

Split-Venus fluorescence reconstitution by interactions of the KSHV NEC. Sf21 cells were infected with viruses expressing fusions with VenusN and VenusC. (A and B) A significant amplification of fluorescence was observed and visualized by confocal microscopy for both self-interactions (A) and interactions between ORF67 and ORF69 (B). The Venus fluorescence observed was similar to the distribution of GFP-tagged ORF67 and ORF69 (magnification, ×63). (C) These interactions were quantitated by measuring fluorescence as a consequence of Venus reconstitution following coinfection of Sf21 cells with viruses expressing VenusN and VenusC fusions. Fluorescence produced was measured after 48 h of infection, and the measurements of three independent infections were plotted.
To generate a more quantitative measurement of the interactions between the ORF67 and ORF69 mutant polypeptides, all the mutant ORF67 and ORF69 genes were moved into the baculovirus vector specifying the N-terminal VenusN and VenusC fusions. These mutants, once recombined into the virus, were tested for interaction with each other and with the NEC partner, and the results are shown in Fig. 6. All the ORF67 in-frame deletion mutants display a reduction in their ability to interact with each other (Fig. 6A) as well as a significant reduction in their ability to interact with ORF69 (Fig. 6B) as judged by the reduction in fluorescence emission. The level of fluorescence reduction for self-interaction ranges from approximately 4- to 8-fold; the reduction in fluorescence observed during the interaction with ORF69 was greater than 10-fold. Both the truncation polypeptide mutants retain the ability to self-interact, albeit at slightly reduced levels, and the reduction in self-interaction was more so for T240, probably either because of the loss of the membrane-anchoring region or as a consequence of aggregate formation seen by confocal microscopy. T240 and T264 interact strongly with ORF69 as judged by Venus fluorescence reconstitution, and this confirmed the confocal results. The ORF69 mutants, on the other hand, displayed varied phenotypes. The Δ187–192 mutant demonstrated significantly lower levels of fluorescence reconstitution both in the self-interaction and during interaction with ORF67. The Δ99–102 mutant still retained some activity in both the same two tests. The Δ90–94 mutant was still able to interact with itself and with ORF67 at almost the same level as that of the wild-type protein or better. All the mutants, because they express the C-terminal V5 tag, were examined for their ability to express and accumulate stable amounts of protein; as judged by Western blotting methods, this was the case for all the mutants (data not shown).
Fig 6.
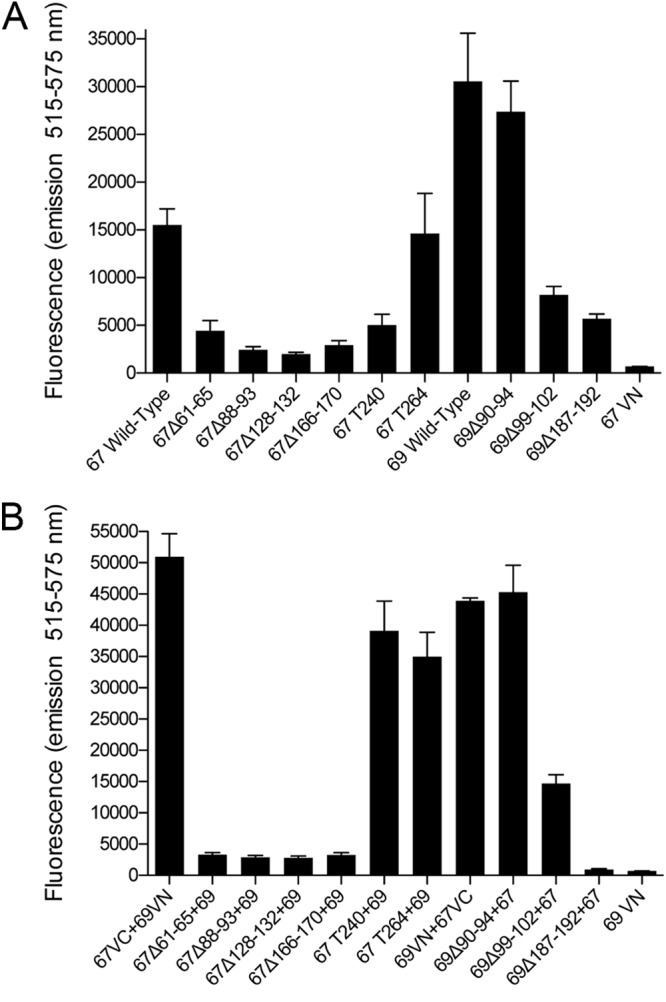
Use of BiFC to quantitate interactions between mutant polypeptides ORF67 and ORF69. Sf21 cells were coinfected with baculoviruses expressing fusions with VenusN and VenusC to quantify both self-interactions (A) and interactions between ORF67 and ORF69 (B). Cells were analyzed for reconstituted Venus fluorescence 48 h postinfection. The data for three independent experiments were plotted. In panel A, the two viruses encoded either VenusN or VenusC in the same wild-type or mutant background, whereas in panel B, the ORF67-expressing viruses used were VenusC fusions and the ORF69-expressing viruses were VenusN fusions. 67VN and 69VN are single infections.
Nuclear membrane remodeling by the NEC and the role of ORF69.
Previously, we had demonstrated the induction of nuclear membrane proliferation/duplication by the expression of ORF67 and the remodeling of these membranes into circular vesicles by the coexpression of the two proteins. This implied that ORF69 may be required for membrane fission events that give rise to these circular structures (27). To prove this genetically, we performed the same experiments with the mutants that fail to interact with each other. Sf21 cells were coinfected with baculoviruses expressing wild-type or mutant ORF67 and ORF69 proteins tagged with V5, and the cells were examined by TEM following 68 to 72 h of infection (Fig. 7). In cells expressing ORF67, large areas of membrane proliferations were observed, and in cells coinfected with baculoviruses expressing wild-type ORF67 and ORF69, we again saw accumulations of circular virion-sized vesicles in the nuclei of infected cells. We examined all the ORF67 mutants expressed by themselves for changes in cellular membranes. All the mutants with the exception of T264 failed to induce membrane proliferations; data are shown only for Δ61–65, T240, and T264. In cells coexpressing T264 and ORF69, circular vesicles were evident. When cells were coinfected with viruses expressing ORF69 mutants that either fail to interact (Δ187–192) or only weakly interact with ORF67 (Δ99–102), although cellular membrane proliferations were evident, they were not remodeled into circular structures. The ORF69 mutant Δ90–94, which can best interact with ORF67, as judged by the BiFC assay, was not able to remodel host cell membranes induced by ORF67 into circular structures. We determined quantitatively the location of membrane proliferations induced by ORF67 when expressed alone in insect cells. In 25 out of 26 cells that display this phenotype, the membrane proliferations were perinuclear with some continuity with the nuclear membrane; only one cell had intranuclear membrane proliferation. The vesicle structures that form in the presence of both ORF67 and ORF69 were intranuclear in 67 out of 70 cells displaying this phenotype, and only 3 cells had vesicles in a perinuclear/cytoplasmic site.
Fig 7.

Ultrastructural analysis of infected cells to demonstrate the effects of mutations on nuclear membrane remodeling. Infected Sf21 cells were processed for TEM analysis 68 to 72 h after infection. Bars, 400 nm. The nucleus (n) and cytoplasm (c) are indicated when clearly visible.
Nuclear membrane remodeling in mammalian cells by coexpression of ORF67 and ORF69.
To confirm the phenotypes observed in insect cells by the expression of ORF67 and ORF69 in a mammalian cell system, we used plasmid transfection methods and HEK-293T cells. Similar fluorescent protein fusions were made and expressed transiently using a CMV promoter-based expression vector. Confocal analysis of transfected cells demonstrated nuclear membrane and cytoplasmic localization of ORF67GFP and nuclear fluorescence of ORF69mCherry (Fig. 8A). When the two plasmids were cotransfected into the same cell cultures, relocalization to the nuclear margins of both fluorescent proteins was observed, and in some cells, the fluorescence observed suggested significant changes at the nuclear membrane (Fig. 8B). Similar transfected cells were also examined by ultrastructural methods; in this case, we used proteins that were tagged with the V5 epitope. However, we observed that ORF67V5 was poorly expressed in HEK-293T cells, and so we used ORF67GFP for these experiments and ORF69V5. In the cotransfected cells, both at the nuclear margins (Fig. 8C, middle and bottom) and in cytoplasmic sites (Fig. 8C, top) membrane proliferation was observed, and in some cases the membranes were remodeled into circular structures (Fig. 8C, middle and bottom). These remodeling events were observed but not to the same extent and efficiency as those of events detected in the arthropod cells. This is probably because of the lower levels of expression in these cells as well as having to use ORF67GFP for this experiment rather than a polypeptide with a smaller tag. Nuclear localization signals (NLS) have been predicted as well as mapped in the ORF69 orthologs encoded by the many herpesviruses (33, 35, 49, 50). Sequence analysis of ORF69 did not reveal a consensus NLS; however, there is present an arginine-rich region at the amino terminus surrounded by proline/glycine residues (Fig. 1). To test quickly whether these sequences mediate nuclear localization of ORF69, we made alanine substitution mutations in the arginine residues. These changes were made in ORF69 fused to GFP and the effect of those changes was examined by confocal microscopy (Fig. 8D and E). A sequence that resides between R17 and R25 clearly has an appreciable effect on the nuclear localization of ORF69GFP in HEK-293T cells.
Fig 8.

The KSHV NEC proteins colocalize at nuclear margins of mammalian cells and alter these membranes. (A) HEK-293T cells were transfected with plasmids expressing ORF67GFP or ORF69mCherry. (B) Cells were cotransfected with a plasmid expressing ORF67GFP and a plasmid expressing ORF69mCherry. The cells were imaged by confocal microscopy at 24 h posttransfection. (C) Similar cultures cotransfected with ORF67GFP- and ORF69V5-expressing plasmids demonstrate cellular membrane remodeling events. (D) HEK-293T cells were transfected with ORF69GFP-expressing plasmids and imaged by confocal microscopy 24 h posttransfection. A potential nuclear localization sequence within ORF69 is influenced by changes in arginines at positions R17, R21, R24, and R25. (E) Sequence of the N-terminal 50 amino acids of ORF69 showing the arginines that were sequentially changed to alanine. For example, R21-39 has alanine substitutions at all arginines between residues 21 and 39. Magnifications, ×63 (A and D) and ×100 (B). Bars in panel C are 200 nm, except for that in the center image, which is 100 nm. The nuclear (n) and cytoplasmic (c) compartments are indicated.
The EBV NEC and its interactions.
We next examined the effects of the gamma-1 herpesvirus NEC, which is encoded by genes BFLF2 and BFRF1 of EBV, on remodeling of host cell membranes. Previous studies showed membrane duplication in the vicinity of the nucleus by expression of BFRF1 in HEK-293T cells (22, 23). The EBV NEC genes were cloned into the same baculovirus transfer vector; C-terminally tagged V5, GFP, and mCherry and the recombinant viruses generated were examined for protein expression following infection of Sf21 cells. All the correct polypeptides were produced in infected cells and were of the right molecular weight as judged by Western blotting methods using antisera specific for the tag (Fig. 9A). Confocal analysis was also undertaken to confirm the cellular localization and colocalization of these two proteins. The distributions of the EBV protein fused to GFP and that fused to mCherry were similar in Sf21-infected cells (Fig. 9B). BFRF1 and BFLF2 proteins also colocalized at the nuclear margins, giving rise to characteristic fluorescent puncta around the nucleus (Fig. 9C). We also checked whether the EBV and KSHV NEC proteins interact with each other. This was done using the Split-Venus-expressing proteins. As shown in Fig. 10, the EBV and KSHV proteins can interact with each other but at slightly reduced levels, as judged by fluorescence output compared to that when the interaction was measured between each EBV or each KSHV protein. This was also confirmed by confocal analysis of coinfected cells showing relocalization to the nuclear margins (data not shown).
Fig 9.

Expression and cellular localization of the EBV NEC proteins BFRF1 and BFLF2. (A) Western blot analysis of Sf21 cells infected with viruses expressing V5-, GFP-, and mCherry-tagged BFRF1 and BFLF2. Protein standards (kDa) are in lane M. (B) Confocal analysis of the cellular localization of BFRF1 and BFLF2, fused to either GFP or mCherry, in infected Sf21 cells. (C) Colocalization of BFRF1 and BFLF2 at the nuclear margins of coinfected Sf21 cells imaged using confocal microscopy. Magnification for all images, ×100.
Fig 10.

Interactions between the EBV and KSHV NEC proteins. Coinfected Sf21 cells were processed for measurement of Split-Venus fluorescence as described in the legend to Fig. 6. The data derived from two independent infections demonstrate that interactions do occur between the EBV and KSHV NEC proteins.
To examine whether these proteins can induce similar nuclear membrane proliferations in insect cells, we carried out ultrastructural analyses of cells infected with the virus expressing BFRF1 alone and also coinfected with viruses expressing BFRF1 and BFLF2 (Fig. 11). We first determined that the EBV and KSHV proteins are expressed in cells that are coinfected with the viruses expressing V5-tagged proteins. In each case, both proteins were detected to accumulate in abundant amounts by Western blotting methods. Infected cells that express BFRF1 displayed significant nuclear membrane proliferation. These membranes have a tubular structure as well as interconnected stacks of tubules, some of which had a circular appearance depending on the sectional plane of the cell. Within the cells infected with the BFRF1-expressing virus and displaying membrane proliferation, the majority, 44 out of 50, contained perinuclear/cytoplasmic region-localized proliferations; only in 4 cells were they located within the nucleus. In the presence of BFLF2, these membranes were not converted to circular structures but were now localized primarily within the nucleus because of the relocalization of BFRF1 to the nuclear margins. This was also seen in cells coinfected with the viruses expressing ORF67 and BFLF2. The ORF67-induced membrane proliferations were not converted into discrete circular structures but were localized to the nucleus. However, if cells were coinfected with viruses expressing BFRF1 and KSHV ORF69, the cellular membranes were remodeled into circular form, demonstrating the superior activity of ORF69 in remodeling cellular membranes. Cells coinfected with ORF67 and ORF69 contained a number of vesicles that reside within inner nuclear membrane invaginations. This last result shows that the KSHV NEC can recapitulate nuclear egress in this ex vivo system.
Fig 11.

Remodeling of cellular membranes by the EBV and KSHV NEC. Sf21 cells were infected with the baculovirus expressing the EBV BFRF1 or coinfected with baculoviruses expressing the EBV and KSHV NEC proteins. Infected cells were processed for electron microscopy 72 h after infection. ORF69 was able to remodel nuclear membrane proliferations induced by either BFRF1 or ORF67; however, EBV BFLF2 did not display the same activity. Bars, all 400 nm, except for that in the ORF67+ORF69 panel, which is 200 nm. The nucleus (n) and cytoplasm (c) are indicated. The bottom right panel shows the results of Western blot analysis of Sf21 cells infected with combinations of the KSHV and EBV NEC-expressing viruses. The blot was probed with anti-V5 antibody. Protein standards (kDa) are shown in lane M.
DISCUSSION
Previously, Klupp et al. (13) demonstrated a remarkable visual phenotype: formation of nuclear membrane-derived vesicles by coexpression in mammalian cells of the PRV NEC. Thus, these two proteins alone could remodel cellular membranes into virion-sized envelope structures in the absence of a budding capsid or the conserved protein kinase. These vesicles were present within the nucleus and in nuclear membrane invaginations, and some were visualized in the process of fusing with the outer nuclear membrane. Subsequently, during a study of the expression of KSHV structural proteins in insect cells, using baculoviruses for expression, we discovered that we could achieve this phenotype by coexpression of the KSHV NEC proteins ORF67 and ORF69 (27). In our study, we also were able to differentiate the roles of the individual proteins in this remodeling phenotype. Thus, we could show significant nuclear/perinuclear membrane proliferation in the presence of OR67 alone and the remodeling of these membranes into circular structures when ORF69 was also present. ORF69 on its own did not have any effect on host-derived membranes. We proposed that ORF69 acts to recruit the cellular fission machinery to modify the tubular nuclear membrane structures, a process which would occur during primary envelopment. Direct genetic evidence for this proposition was obtained in this study, where we now can show that the formation of these vesicles is dependent on the interaction between these two proteins.
The remarkable activity of the KSHV NEC proteins in altering host cellular membranes was visualized in the insect cell infected with recombinant baculoviruses. This may have been a fortuitous choice because of the higher and more stable expression levels of ORF67 and ORF69, the likelihood that many of the cells are infected or coinfected, and possibly the cellular microenvironment that allowed us to visualize quite easily and consistently these remodeling events by ultrastructural methods and analyze the effects of mutations in these proteins using similar methods. This was not the case when using plasmid transfection experiments in a mammalian cell line that is efficiently transfected. Thus, although our system uses an arthropod cell line that grows at a lower temperature, it is a valid and tractable method to discover mechanistic pathways of the herpesvirus NEC. For our experiments, we used epitope- or fluorescently tagged proteins, which allowed us to follow protein expression and localization in the absence of reagents for these proteins. These tags, we believe, do not disrupt the functions and interactions of ORF67 and ORF69 in this system, but the tags should be tested for activity in KSHV-infected cells in future. Studies for the first time have also shown in Drosophila melanogaster cells the presence of a cellular pathway that utilizes nuclear egress/export of large assemblies that does not occur through the small nuclear pore but by the same mode as that of herpesvirus egress (51). This investigation revealed that the exit of ribonucleoprotein (RNP) granules out of the nucleus likely occurs in a similar fashion. This pathway may have been coopted by pathogens that assemble in the nucleus, such as herpesviruses, for their spread in infected cells (51, 52). It has also been suggested that this transport pathway may be used to transport nuclear protein aggregates out into the cytosol for processing by the autophagosome (53). The information gained from studying KSHV NEC-mediated nuclear membrane remodeling events could be used to discover the mechanism of this newly discovered cellular process that involves vesicle-mediated transfer of large assemblies through the nuclear envelope.
EBV BFRF1, when overexpressed in mammalian cells, can induce proliferation of cellular membranes (22, 23). We showed in our system that ORF67 has the same property and in this study confirm that activity for BFRF1 in insect cells. Both are potent inducers of membrane proliferation as seen in insect cells, where large areas within the cell contain these membrane structures. This hyperproliferation represents a significant change within the cell that is driven by the overexpression of these viral proteins. All the mutations in the ectodomain of ORF67 abolish this activity, implying that there are many domains responsible for this activity within ORF67. These mutations also eliminate the ability of the ORF67 polypeptides to self-interact with each other, something that may be a prerequisite for induction of membrane proliferation. Mechanistically, how ORF67 and BFRF1 function in membrane proliferation is still unclear, although recent data demonstrate interactions between BFRF1 and the cellular ESCRT pathway (54). Evident in the literature are also reports that demonstrate examples of cellular INM proteins that display this activity when overexpressed. This has been established for lamins and other nuclear membrane-associated proteins (55). Overexpression of these proteins results in formation of intranuclear membrane arrays, and this activity can be transferred to GFP by the incorporation of an NLS and the CaaX motif from N-ras (56). The CaaX motif is generally present at the C termini of many lamins, and posttranslational modification of this motif is required for their membrane association (57). The overexpression of the GFP chimeric protein results in both intranuclear membrane arrays and endoplasmic reticulum (ER)-like cisternae adjacent to the nucleus (56). In our studies, we have observed membrane proliferation primarily in the perinuclear regions and within the nucleus when the interacting protein (ORF69 or BFLF2) is present. ORF67 is present in both nuclear and perinuclear membranes as judged by confocal analysis. The circular vesicles that form as a consequence of ORF69 coexpression have been observed in the nucleus with continuity with the nuclear membranes. Additional studies, including TEM examination of serial thin sections through infected cells and correlation of light and electron microscopy data, should reveal more information on the organization of these vesicles.
ORF69 is a potent factor that can remodel nuclear membrane proliferations into circular structures. Again, how it does this is still unclear and has yet to be determined, although it may interact with and recruit cellular factors for membrane curvature and/or fission to facilitate this activity. We now have also discovered that the KSHV NEC pair, particularly ORF69, are superior to the other human herpesvirus NEC proteins in membrane remodeling events. Expression of the HSV-1 and HCMV NEC proteins in insect cells has produced different phenotypes. Although the UL34 and UL50 proteins induce membrane proliferation, it was difficult to discern reproducible vesicle formation when the virus expressing UL31 or UL53 was included (data not shown). For the KSHV NEC, 88% of the cells displayed vesicles derived from the nuclear membrane (27). In this study, we demonstrate that ORF69 can remodel membrane structures induced by the EBV BFRF1 into vesicular structures, something which the EBV homologue BFLF2 could not accomplish. In transfected cells, Santarelli et al. (22) showed that coexpression of BFRF1 and ORF69 increased membrane duplication but conversion to vesicles was not observed. Hence, the superior activity of the KSHV NEC could potentially be harnessed and used to gain more insight into the functions of this complex and how it recruits cellular factors for these activities. Genetic data are now provided in this study showing that in this ex vivo system the interaction between these proteins is required for vesicle formation, which confirms observations that were demonstrated for the HSV-1 NEC in infected cells (10).
The small deletions made in ORF67 and ORF69 were a preliminary examination of the functional domains of these two proteins (Table 2). Because the NEC proteins have been analyzed in great detail in the alphaherpesvirus and betaherpesvirus families, we chose to focus on those regions of these two proteins that are specific to the gammaherpesvirus family. Any in-frame deletion in the nucleoplasmic domain of ORF67 abolished its interactions and its functions. Although the mutants localized to the correct cellular compartment, they failed to induce nuclear membrane proliferation and similarly failed to interact with ORF69. They also in all cases cannot self-interact, as judged by the Venus reconstitution assay. Thus, ORF67 is particularly sensitive to mutations, especially to polypeptide in-frame deletions (Table 2). The consequences of these mutations for the biochemical and functional properties of this protein cannot be separated by these types of changes and may require more subtle alterations to separate out the different domains of ORF67. Analysis of charge cluster mutants of HSV-1 UL34 and chimeric molecules of PRV UL34 has been more informative for separating out the domains required for INM targeting, interaction with UL31, and functional activity for nuclear egress and virus replication (10, 33, 60). Two conserved regions (CRs), CR1 and CR2, have been assigned to the N-terminal portion of the CMV (UL50/M50) proteins in which reside the UL53 interaction domain, an alpha-helix important for protein stability and possibly interaction with UL53, and a globular domain in which reside important residues for NEC formation (29, 34, 58). The ORF67 mutant Δ61-65 includes two residues, E61 and F62 (Fig. 1), whose counterparts (E56 and Y57 in UL50) have been shown to be essential for interaction with UL53 (29). The results of the truncation mutants of ORF67 were more informative by confirming that the sequences residing in the perinuclear space are not important for induction of membrane proliferation or for interaction with ORF69. The ability of the ORF67 TM deletion to retain both the ability to interact with ORF69 and to be tethered to nuclear membranes by virtue of its interaction with a TM-containing ORF67 polypeptide demonstrates the modular nature of this protein.
Table 2.
Summary of the phenotypes and activities of the ORF67 and ORF69 mutantsb
| Mutant | Self-interaction | NEC interactiona | Membrane proliferation | Remodeling into circular structures |
|---|---|---|---|---|
| ORF67 WT | + | + | + | NA |
| T240 | +/− | + | − | NA |
| T264 | + | + | + | NA |
| Δ61–65 | − | − | − | NA |
| Δ88–93 | − | − | − | NA |
| Δ128–132 | − | − | − | NA |
| Δ166–170 | − | − | − | NA |
| ORF69 WT | + | + | NA | + |
| Δ90–94 | + | + | NA | − |
| Δ99–102 | +/− | +/− | NA | − |
| Δ187–192 | +/− | − | NA | − |
Interaction between the nuclear egress complex proteins.
Abbreviations and symbols: WT, wild type; NA, not applicable; +/−, intermediate.
The deletions in ORF69 displayed more varied phenotypes. There were those that failed to interact with ORF67 and thus failed to remodel cellular membranes into circular vesicles, confirming that ORF69 in concert with ORF67 is required for this function. However, there was one mutant, Δ90-94, that retained the ability to interact efficiently with ORF67 but did not give rise to circular structures (Table 2). This information potentially elucidates a domain that is important for recruiting cellular factors that carry out the actual fission of membranes. The designation of conserved regions CR1 to CR4 for UL31 homologues was determined by Lotzerich et al. (35) using 36-amino-acid sequences from herpesvirus, and these regions span the N-terminal portion of the polypeptide and include a significant portion of the C-terminal half. All the ORF69 mutations reside within these conserved regions; however, the Δ90-94 mutant contains a number of highly conserved residues (Fig. 1), and an insertion mutant in this sequence in the M53 gene resulted in a lethal phenotype (35), indicating again the importance of this region for ORF69 function. Within the CR sequences for CMV UL53 resides a globular domain as well as the alpha-helix required for interaction with UL50 (29, 30). The predicted NLS in CMV UL53 is between residues 13 to 26 (50) and corresponds closely to a similar arginine-rich sequence in ORF69 residing between amino acids 17 and 25 that we show has an influence on nuclear localization of ORF69 (Fig. 8).
The variety of activities performed by the herpesvirus NEC proteins and the modular nature of their polypeptides demonstrate the complexity and sophistication of the interplay between these proteins and between the NEC and host membranes. The nuclear egress process to transport large assemblies out of the nucleus and into the cytosol has now been shown to occur for a cellular activity and thus gives more impetus to investigating the nature of this complex. Hence, investigations of the NEC in herpesvirus-infected cells as well as in an ex vivo system that is amenable to manipulation are valid approaches to discover the functions of this nuclear export pathway.
ACKNOWLEDGMENTS
Research support was provided by Public Health Service grants from the National Institutes of Health (RO1 AI 063182) and American Recovery and Reinvestment Act (ARRA) challenge grant (RC2 CA148402). Additional funding for shared equipment was provided by NIH-NCRR 1S10RR023454-01 (shared instrumentation grant for the Tecnai 12 G2 Spirit TEM) and NIH-NCI 1U54CA143868-01 (Physical Sciences of Cancer grant) (J.M.M.). G.C. was supported by an REU grant, Visualizations of Macromolecules in Biological Research (1005027), from the NIH.
We thank S. J. Gao (University of Southern California, Los Angeles, CA) for providing BAC36 and Lindsey Hutt-Fletcher (Louisiana State University, Shreveport, LA) for the EBV BAC. The plasmid carrying the Venus sequence was made by Jennifer Lippincott-Schwartz (NIH, Bethesda, MD) and acquired from the Addgene collection. The plasmid containing the mCherry ORF was a kind gift from Venu Raman (JHU, Baltimore, MD). Finally, we thank Ainsley Starghill for critical proofreading of the manuscript.
Footnotes
Published ahead of print 30 January 2013
REFERENCES
- 1. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 2. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 3. Mettenleiter TC, Muller F, Granzow H, Klupp BG. 7 November 2012. The way out: what we know and do not know about herpesvirus nuclear egress. Cell. Microbiol. [Epub ahead of print.] doi:10.1111/cmi.12044 [DOI] [PubMed] [Google Scholar]
- 4. Bjerke SL, Roller RJ. 2006. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 347:261–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J. Virol. 82:8094–8104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reynolds AE, Liang L, Baines JD. 2004. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J. Virol. 78:5564–5575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roller RJ, Bjerke SL, Haugo AC, Hanson S. 2010. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J. Virol. 84:3921–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J. Gen. Virol. 81:2397–2405 [DOI] [PubMed] [Google Scholar]
- 12. Chang YE, Roizman B. 1993. The product of the UL31 gene of herpes simplex virus 1 is a nuclear phosphoprotein which partitions with the nuclear matrix. J. Virol. 67:6348–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klupp BG, Granzow H, Fuchs W, Keil GM, Finke S, Mettenleiter TC. 2007. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. U. S. A. 104:7241–7246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yamauchi Y, Shiba C, Goshima F, Nawa A, Murata T, Nishiyama Y. 2001. Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. J. Gen. Virol. 82:1423–1428 [DOI] [PubMed] [Google Scholar]
- 15. Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5:e1000275 doi:10.1371/journal.ppat.1000275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Leach NR, Roller RJ. 2010. Significance of host cell kinases in herpes simplex virus type 1 egress and lamin-associated protein disassembly from the nuclear lamina. Virology 406:127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marschall M, Feichtinger S, Milbradt J. 2011. Regulatory roles of protein kinases in cytomegalovirus replication. Adv. Virus Res. 80:69–101 [DOI] [PubMed] [Google Scholar]
- 18. Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 81:6459–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857 [DOI] [PubMed] [Google Scholar]
- 20. Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 80:494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Santarelli R, Farina A, Granato M, Gonnella R, Raffa S, Leone L, Bei R, Modesti A, Frati L, Torrisi MR, Faggioni A. 2008. Identification and characterization of the product encoded by ORF69 of Kaposi's sarcoma-associated herpesvirus. J. Virol. 82:4562–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gonnella R, Farina A, Santarelli R, Raffa S, Feederle R, Bei R, Granato M, Modesti A, Frati L, Delecluse HJ, Torrisi MR, Angeloni A, Faggioni A. 2005. Characterization and intracellular localization of the Epstein-Barr virus protein BFLF2: interactions with BFRF1 and with the nuclear lamina. J. Virol. 79:3713–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lake CM, Hutt-Fletcher LM. 2004. The Epstein-Barr virus BFRF1 and BFLF2 proteins interact and coexpression alters their cellular localization. Virology 320:99–106 [DOI] [PubMed] [Google Scholar]
- 25. Farina A, Feederle R, Raffa S, Gonnella R, Santarelli R, Frati L, Angeloni A, Torrisi MR, Faggioni A, Delecluse HJ. 2005. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J. Virol. 79:3703–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Granato M, Feederle R, Farina A, Gonnella R, Santarelli R, Hub B, Faggioni A, Delecluse HJ. 2008. Deletion of Epstein-Barr virus BFLF2 leads to impaired viral DNA packaging and primary egress as well as to the production of defective viral particles. J. Virol. 82:4042–4051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Desai PJ, Pryce EN, Henson BW, Luitweiler EM, Cothran J. 2012. Reconstitution of the Kaposi's sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J. Virol. 86:594–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liang L, Baines JD. 2005. Identification of an essential domain in the herpes simplex virus 1 UL34 protein that is necessary and sufficient to interact with UL31 protein. J. Virol. 79:3797–3806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Milbradt J, Auerochs S, Sevvana M, Muller YA, Sticht H, Marschall M. 2012. Specific residues of a conserved domain in the N terminus of the human cytomegalovirus pUL50 protein determine its intranuclear interaction with pUL53. J. Biol. Chem. 287:24004–24016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sam MD, Evans BT, Coen DM, Hogle JM. 2009. Biochemical, biophysical, and mutational analyses of subunit interactions of the human cytomegalovirus nuclear egress complex. J. Virol. 83:2996–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schnee M, Ruzsics Z, Bubeck A, Koszinowski UH. 2006. Common and specific properties of herpesvirus UL34/UL31 protein family members revealed by protein complementation assay. J. Virol. 80:11658–11666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ott M, Tascher G, Hassdenteufel S, Zimmermann R, Haas J, Bailer SM. 2011. Functional characterization of the essential tail anchor of the herpes simplex virus type 1 nuclear egress protein pUL34. J. Gen. Virol. 92:2734–2745 [DOI] [PubMed] [Google Scholar]
- 33. Schuster F, Klupp BG, Granzow H, Mettenleiter TC. 2012. Structural determinants for nuclear envelope localization and function of pseudorabies virus pUL34. J. Virol. 86:2079–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bubeck A, Wagner M, Ruzsics Z, Lotzerich M, Iglesias M, Singh IR, Koszinowski UH. 2004. Comprehensive mutational analysis of a herpesvirus gene in the viral genome context reveals a region essential for virus replication. J. Virol. 78:8026–8035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lotzerich M, Ruzsics Z, Koszinowski UH. 2006. Functional domains of murine cytomegalovirus nuclear egress protein M53/p38. J. Virol. 80:73–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Popa M, Ruzsics Z, Lotzerich M, Dolken L, Buser C, Walther P, Koszinowski UH. 2010. Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J. Virol. 84:9035–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Perkins EM, Anacker D, Davis A, Sankar V, Ambinder RF, Desai P. 2008. Small capsid protein pORF65 is essential for assembly of Kaposi's sarcoma-associated herpesvirus capsids. J. Virol. 82:7201–7211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okoye ME, Sexton GL, Huang E, McCaffery JM, Desai P. 2006. Functional analysis of the triplex proteins (VP19C and VP23) of herpes simplex virus type 1. J. Virol. 80:929–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhou FC, Zhang YJ, Deng JH, Wang XP, Pan HY, Hettler E, Gao SJ. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J. 1997. ER-to-Golgi transport visualized in living cells. Nature 389:81–85 [DOI] [PubMed] [Google Scholar]
- 41. Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324–6332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Walters JN, Sexton GL, McCaffery JM, Desai P. 2003. Mutation of single hydrophobic residue I27, L35, F39, L58, L65, L67, or L71 in the N terminus of VP5 abolishes interaction with the scaffold protein and prevents closure of herpes simplex virus type 1 capsid shells. J. Virol. 77:4043–4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Henson BW, Perkins EM, Cothran JE, Desai P. 2009. Self-assembly of Epstein-Barr virus capsids. J. Virol. 83:3877–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huang E, Perkins EM, Desai P. 2007. Structural features of the scaffold interaction domain at the N terminus of the major capsid protein (VP5) of herpes simplex virus type 1. J. Virol. 81:9396–9407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Perkins EM, McCaffery JM. 2007. Conventional and immunoelectron microscopy of mitochondria. Methods Mol. Biol. 372:467–483 [DOI] [PubMed] [Google Scholar]
- 46. Hernandez FP, Sandri-Goldin RM. 2010. Herpes simplex virus 1 regulatory protein ICP27 undergoes a head-to-tail intramolecular interaction. J. Virol. 84:4124–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kerppola TK. 2006. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 1:1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kerppola TK. 2009. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: characteristics of protein fragment complementation. Chem. Soc. Rev. 38:2876–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kim S, Ahn BC, O'Callaghan DJ, Kim SK. 2012. The early UL31 gene of equine herpesvirus 1 encodes a single-stranded DNA-binding protein that has a nuclear localization signal sequence at the C-terminus. Virology 432:306–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Milbradt J, Auerochs S, Marschall M. 2007. Cytomegaloviral proteins pUL50 and pUL53 are associated with the nuclear lamina and interact with cellular protein kinase C. J. Gen. Virol. 88:2642–2650 [DOI] [PubMed] [Google Scholar]
- 51. Speese SD, Ashley J, Jokhi V, Nunnari J, Barria R, Li Y, Ataman B, Koon A, Chang YT, Li Q, Moore MJ, Budnik V. 2012. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 149:832–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hatch EM, Hetzer MW. 2012. RNP export by nuclear envelope budding. Cell 149:733–735 [DOI] [PubMed] [Google Scholar]
- 53. Rose A, Schlieker C. 2012. Alternative nuclear transport for cellular protein quality control. Trends Cell Biol. 22:509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lee CP, Liu PT, Kung HN, Su MT, Chua HH, Chang YH, Chang CW, Tsai CH, Liu FT, Chen MR. 2012. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog. 8:e1002904 doi:10.1371/journal.ppat.1002904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Linde N, Stick R. 2010. Intranuclear membranes induced by lipidated proteins are derived from the nuclear envelope. Nucleus 1:343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ralle T, Grund C, Franke WW, Stick R. 2004. Intranuclear membrane structure formations by CaaX-containing nuclear proteins. J. Cell Sci. 117:6095–6104 [DOI] [PubMed] [Google Scholar]
- 57. Prufert K, Vogel A, Krohne G. 2004. The lamin CxxM motif promotes nuclear membrane growth. J. Cell Sci. 117:6105–6116 [DOI] [PubMed] [Google Scholar]
- 58. Milbradt J, Auerochs S, Sticht H, Marschall M. 2009. Cytomegaloviral proteins that associate with the nuclear lamina: components of a postulated nuclear egress complex. J. Gen. Virol. 90:579–590 [DOI] [PubMed] [Google Scholar]
- 59. Yakushko Y, Hackmann C, Gunther T, Ruckert J, Henke M, Koste L, Alkharsah K, Bohne J, Grundhoff A, Schulz TF, Henke-Gendo C. 2011. Kaposi's sarcoma-associated herpesvirus bacterial artificial chromosome contains a duplication of a long unique-region fragment within the terminal repeat region. J. Virol. 85:4612–4617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bjerke SL, Cowan JM, Kerr JK, Reynolds AE, Baines JD, Roller RJ. 2003. Effects of charged-cluster mutations on the function of herpes simplex virus type 1 UL34 protein. J. Virol. 77:7601–7610 [DOI] [PMC free article] [PubMed] [Google Scholar]