

Abstract
Human papillomavirus 16 (HPV16) enters its host cells by a process that most closely resembles macropinocytosis. Uncoating occurs during passage through the endosomal compartment, and the low pH encountered in this environment is essential for infection. Furin cleavage of the minor capsid protein, L2, and cyclophilin B-mediated separation of L2 and the viral genome from the major capsid protein, L1, are necessary for escape from the late endosome (LE). Following this exodus, L2 and the genome are found colocalized at the ND10 nuclear subdomain, which is essential for efficient pseudogenome expression. However, the route by which L2 and the genome traverse the intervening cytoplasm between these two subcellular compartments has not been determined. This study extends our understanding of this phase in PV entry in demonstrating the involvement of the Golgi complex. With confocal microscopic analyses involving 5-ethynyl-2′-deoxyuridine (EdU)-labeled pseudogenomes and antibodies to virion and cellular proteins, we found that the viral pseudogenome and L2 travel to the trans-Golgi network (TGN) following exit from the LE, while L1 is retained. This transit is dependent upon furin cleavage of L2 and can be prevented pharmacologically with either brefeldin A or golgicide A, inhibitors of anterograde and retrograde Golgi trafficking. Additionally, Rab9a and Rab7b were determined to be mediators of this transit, as expression of dominant negative versions of these proteins, but not Rab7a, significantly inhibited HPV16 pseudovirus infection.
INTRODUCTION
Papillomaviruses (PV) comprise a large family of nonenveloped viruses that infect epithelial sites in many species. The high-risk human papillomaviruses (HPV) are the causative agents of some common malignancies, most notably cervical cancer. The virus capsid, which encloses the 8-kb double-stranded circular DNA genome, is composed of 72 pentamers of the major capsid protein L1 and up to 72 copies of L2, the minor capsid protein. The L1 protein possesses the ability to self-assemble in the absence of L2. In the virion, each molecule of L2 is associated with an L1 pentamer, but it is largely buried within the intact, mature virus capsid and exerts its influence largely after the entry and uncoating process, rather than during initial host engagement (1).
Elucidation of the infectious entry by PV has been established mainly through the in vitro study of HPV pseudovirions, which are composed of L1-L2 capsids surrounding a reporter plasmid. These particles, structurally indistinguishable from native virions, are considered to be relevant tools to study PV infection and entry (2). Following the initial interaction of HPV pseudovirions with the host cell (in vitro) or the basement membrane (in vivo), a conformational change in the capsid leads to exposure of the N terminus of L2, which allows access of furin to a consensus furin/proprotein convertase site at the N terminus that is conserved across all known L2 species (3). This cleavage is necessary for infectious entry of the virus, although it is dispensable for endocytic entry. Infection in the presence of a furin inhibitor or with particles containing L2 furin cleavage mutants results in the accumulation of uncoated capsids, L2, and DNA in a late endosomal compartment. The carboxy terminus of L2 has been shown to mediate the exit of L2 and the viral genome from the endosomal compartment (4). Additionally, the interaction of L2 with the chaperone cyclophilin B helps to segregate the L2-genome complex away from the residual L1 shell, which remains in the late endosome/lysosome (5). Due to the lack of evidence to the contrary, it has been assumed that an L2-viral genome complex escapes from the endosomal compartment and traverses the cytoplasm, possibly mediated by an interaction between L2 and dynein, to reach the nucleus (6, 7). Entry of the L2-genome complex into the nucleus likely depends on the breakdown of the nuclear envelope, as infection requires mitotic division (8). L2 and the genome are then targeted to the transcriptionally active nuclear subdomain, ND10 (9).
In this study, we have examined the postendosomal trafficking of the PV pseudogenome and L2 by utilizing HPV16 pseudovirions containing 5-ethynyl-2′-deoxyuridine (EdU)-labeled DNA, in contrast to our earlier studies, which had used 5-bromo-2′-deoxyuridine (BrdU)-labeled DNA (9). The detection method for EdU is sufficiently more sensitive that it has allowed us to identify the Golgi complex, specifically the trans-Golgi network (TGN), as playing an important role in HPV16 pseudovirus infectious entry. L2 and the pseudogenome, but not L1, appear to travel to this compartment following uncoating in the late endosome (LE). The importance of this trafficking for infectious entry was confirmed by biochemical inhibition of Golgi function, and the late endosome-to-TGN transfer was shown to depend on specific Rab GTPases. This report represents the first description of PV dependence on the Golgi for its infectious entry.
MATERIALS AND METHODS
Pseudovirus preparation.
Stocks of matured HPV16 pseudovirus were produced and purified as previously described (10). Pseudovirus was produced with the p16Llw plasmid, which encodes codon-optimized versions of HPV16 L1 and L2. For some experiments p16LlCHA, which encodes HPV16 L1 and a carboxy-terminal HA-tagged L2 protein, or p16LlR12S, which encodes HPV16 L1 and an L2 containing a mutation within its furin cleavage site, was utilized as noted. Infection analyses were performed with pseudovirions with packaged green fluorescent protein (GFP) (pfwB) or red fluorescent protein (RFP) (prwB) expression plasmids. Microscopic analysis was performed with pseudovirions containing PYSEAP plasmid. All plasmids are described on our laboratory website (http://home.ccr.cancer.gov/lco/plasmids.asp). Labeling of the pseudogenome was performed by supplementing the growth media with 50 μM EdU at 6 h posttransfection during pseudovirus production.
Cell lines and antibodies.
HaCaT, HeLa, and 293TT cells were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS). The CHO-derived Δfurin cell line was grown in this medium with additional inclusion of 20 mM proline (11). MCF10A cells were grown as previously described (12). The polyclonal antiserum raised in rabbits against HPV16 capsids was previously described (13). The monoclonal antibody 33L1-7 (14) was a kind gift from Martin Sapp. The antihemagglutinin (anti-HA) antibody (clone HA.11) and antigiantin antibody (PRB-114C) were obtained from Covance Research Products. Antibodies to gm130 (610822) and p230 (611280) were from BD Transduction Laboratories. Promyelocytic leukemia protein (PML) was detected with an antiserum from MBL (PM001) and calnexin (C5C9) with an antiserum from Cell Signaling Technologies. EdU-labeled DNA was detected with a Click-iT imaging kit (C10337) manufactured by Invitrogen.
Immunofluorescent staining.
Cells were seeded onto glass no. 01 coverslips in a 24-well plate at a density of 8 × 104/well and cultured overnight. Twenty nanograms of pseudovirions was added to each well and allowed to bind and internalize for the time indicated in the figure legends for each experiment. Following this incubation, cells were fixed in ice-cold ethanol containing 15 mM glycine. EdU detection was performed according to the manufacturer's instructions, with the exception of the utilization of ethanol fixation. Following virus internalization, coverslips were inverted onto 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting solution (Prolong Gold; Molecular Probes). All images were acquired with a Zeiss LSM 510 confocal system interfaced with a Zeiss Axiovert 100 M microscope. Images were collated with Adobe Photoshop software.
Inhibitor analysis.
Brefeldin A (BFA) and golgicide A (GCA) were purchased from Calbiochem. For 50% inhibitory concentration (IC50) determination, HeLa or HaCaT cells were seeded at a density of 7 × 103/well in a 96-well plate. Following overnight adherence, inhibitors were added to the wells. The dilution series of BFA was started at 1.5 μM, and 3-fold dilutions were performed. Twofold dilutions of GCA were analyzed beginning from a starting concentration of 20 μM. Pseudovirions containing a GFP expression plasmid were then added to the wells. Incubation was allowed to continue for 48 h, and GFP expression was determined by flow cytometric analysis on a BD FACSCanto II system. For microscopic analysis in the presence of BFA and GCA, drug was added to preplated HeLa cells at concentrations of 150 nM and 5 μM, respectively. Pseudovirus at 10 ng/well was added and incubated for 24 h, at which time cells were fixed and processed as described above.
Analysis of Rab protein requirement.
The Rab7a and Rab9b wild-type (WT) and dominant negative (DN) expression constructs were a gift from Richard Pagano and obtained from AddGene (plasmids 12661, 12662, 12677, and 12676) (15). All constructs encode a dsRed RFP fusion protein. The Rab7b wild-type and dominant negative GFP fusion expression plasmids were a kind gift of Cecilia Bucci (16). These constructs have previously been characterized extensively regarding their subcellular localization and relocalization of relevant cellular proteins. They behaved as expected in our system in both regards (data not shown). For the experimental analysis, 293TT cells were transfected in a 6-well plate with 4 μg of plasmid. The following day, cells were reseeded to a 96-well plate format, and HPV16 pseudovirus containing either an RFP or GFP expression plasmid was added. Infection was allowed to continue for 48 h. RFP and GFP expression was determined by flow cytometric analysis. FlowJo software was utilized to determine the percentage of fluorescent transfected cells that were coincidentally infected with the fluorescent-protein-encoding HPV16 pseudovirus. GraphPad Prism software was used to determine significance using a two-tailed, unpaired t test.
To determine the role of furin cleavage in the observed Rab inhibition, cells were treated as described above except that under one condition, pseudovirus was incubated with exogenous furin. Fifty microliters of supernatant from the furin-secreting CHO-Δfurin cells was added prior to pseudovirus addition. Infection was allowed to continue for 48 h, and RFP and GFP expression was determined by flow cytometric analysis. In the second method in which we examined furin dependence, we employed a variation of our recently described L2-based neutralization assay (12). In this assay, extracellular matrix (ECM) was prepared from MCF10A cells as previously described. Pseudovirus was added to the ECM and incubated in the presence of CHO-Δfurin cell supernatant or normal medium. Following an overnight incubation at 37°C, medium and unbound virus were removed by washing. At this point, the Rab-transfected cells were seeded into the ECM-virus-containing wells. Furin inhibitor (RYKR-cmk; Calbiochem) was added to a final concentration of 5 μM to the wells that were treated with exogenous furin in the previous step. Infection was allowed to continue for 48 h. RFP and GFP expression was determined by flow cytometric analysis.
RESULTS
Monitoring EdU-labeled pseudogenome trafficking.
We previously established a method for monitoring uncoating through the detection of 5-bromo-2′-deoxyuridine (BrdU)-labeled viral DNA (9). However, BrdU detection required nuclease treatment to expose the antigen, and the binding of the anti-BrdU antibody to the uncoated genome was variable and often resulted in a low fluorescent signal. The detection of EdU, which is an alternative to BrdU, may be more sensitive because it contains an alkyne that can be reacted with a small azide-containing detection reagent to form a stable triazole ring, thus eliminating the requirement for antibody-based detection. Although extensive antigen unmasking is not required for the small detection azide to access incorporated EdU, the detection protocol does result in some protein denaturation, as discussed below.
We first sought to determine the degree of uncoating required to detect the EdU-labeled pseudogenome. Using antibody-based detection, our previous work had demonstrated that the BrdU-labeled genome and an epitope tag at the carboxy terminus of L2 remained undetectable until 12 h postentry, with both of them showing a progressive increase in signal up to 48 h as more L2 and genome became accessible (9). Colocalization of DNA and L2 with ND10 bodies was not clearly detected until 24 h postentry. For the current study, we fixed and processed HeLa cells for simultaneous EdU and L1 protein detection at 2, 6, 12, 24, and 40 h postentry. As shown in Fig. 1, the pseudogenome was detectable at the earliest time point, 2 h postentry, at which time it completely colocalized with L1 (Fig. 1A and B; merged image in Fig. 1C). This localization was largely unaltered at the 6-h time point (Fig. 1D to F). As the infection progressed, a separation of L1 and the pseudogenome became evident by 12 h (Fig. 1G to I), with the separation being unequivocal by the 24-h time point (Fig. 1J to L). At 40 h, the majority of the pseudogenome signal was found in the nucleus, whereas L1 was retained in the cytoplasm (Fig. 1M to O). Figure 1P to R demonstrate the colocalization of the nuclear pseudogenome with PML, indicating localization at ND10, as previously described. We utilized the L1-7 antibody to detect L1 in this experiment. The epitope recognized by this antibody is typically inaccessible on the intact viral particle and not detectable by intracellular staining until 6 to 8 h postentry (17). The Sapp lab has recently shown that the detection conditions for EdU staining result in sufficient denaturation of the viral capsid that this L1 epitope can be detected prior to any cell-dependent uncoating events (5). Their study also demonstrated the segregation of capsid and genome by 18 h. In other experiments, we found that when cells were processed by the EdU detection protocol, an anti-L1 polyclonal antiserum detected L1 similarly to the L1-7 antibody while also permitting EdU detection (data not shown); however, the EdU processing procedure did not enable BrdU antibodies to detect BrdU-labeled pseudogenome (data not shown).
Fig 1.

Time course of L1 and pseudogenome colocalization. HeLa cells were incubated with HPV16 pseudovirus that contained EdU-labeled pseudogenome. Cells were fixed after various incubation times, as indicated, following pseudovirus addition. The antigens detected are also indicated within the individual panels. The merged images are in the rightmost panel of each row. The nuclei were detected with DAPI (blue) in all panels.
The viral genome localizes to the TGN.
In the above-described experiment, a perinuclear accumulation of EdU-labeled pseudogenome was evident at later time points, especially at 24 h. To identify this compartment, we costained for EdU and various cellular protein markers. We found partial colocalization with Lamp1, a marker of the late endosomal/lysosomal compartment, when we examined cells in which the EdU signal exhibited a more diffuse punctate pattern (data not shown), although the majority of anti-L1 staining colocalized with Lamp1 (Fig. 2A). However, we found minimal colocalization of EdU with Lamp1 in cells that exhibited a more defined accumulation of perinuclear pseudogenome (Fig. 2B). We also examined colocalization of EdU with markers of the trans-Golgi network (TGN) and Golgi complex: gm130, a cis-Golgi matrix protein (Fig. 2C); giantin, a component of the cis-Golgi and medial Golgi (Fig. 2D); p230, a peripheral membrane protein associated with the TGN (Fig. 2E); and calnexin, an endoplasmic reticulum (ER) resident (Fig. 2F). The EdU signal appeared to be immediately adjacent to both giantin and gm130. However, the fluorescent signals for EdU and anti-p230 were largely coincident, indicating localization of the viral genome to the TGN. No association with the ER was evident.
Fig 2.
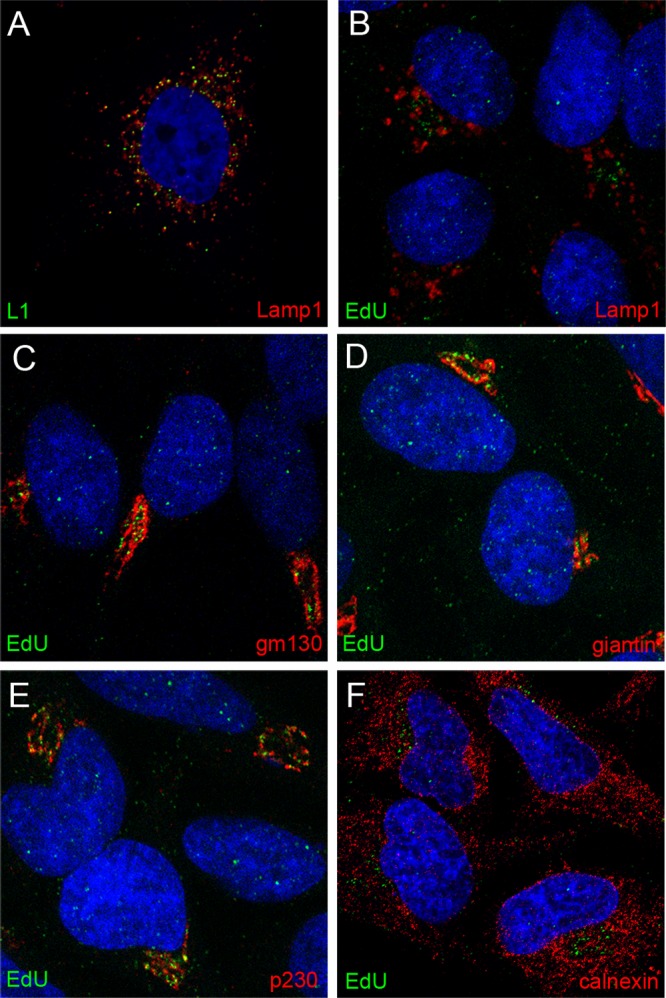
Localization of pseudogenome relative to subcellular markers. Cells were harvested at 24 h post-virus addition. Reagents used for detection are indicated within the individual panels. Please note that panel A shows HPV capsid staining, whereas all other panels show EdU detection, relative to subcellular markers. The nuclei were detected with DAPI (blue) in all panels.
Brefeldin A and golgicide A inhibit HPV16 infection.
Given the finding that the pseudogenome traffics to the TGN, we wanted to determine if TGN localization was required for HPV16 infection. We examined the effects of two pharmacological inhibitors, brefeldin A (BFA) and golgicide A (GCA), on pseudovirus infection. BFA inhibits the anterograde and retrograde transport of proteins between the ER and Golgi (18–20). However, BFA has also been shown to affect intraendosomal transport (21, 22). GCA is thought to be Golgi specific, as it selectively targets the ArfGEF GBF1, which is not required for intraendosomal trafficking but is required for retrograde trafficking to the TGN and Golgi complex (23). We analyzed the effects of both compounds on infection of HaCaT cells with an HPV16 pseudovirus carrying a GFP marker plasmid. As shown in Fig. 3, both effectively inhibit infection, with approximate IC50s of 14.84 nM for BFA and 0.62 μM for GCA. This result indicates that the microscopic detection of the genome in the TGN was likely a significant event for PV infection. Similar results were obtained with HeLa cells (data not shown). We also examined the effects of BFA and GCA treatment on the perinuclear accumulation of the viral pseudogenome. In this experiment, cells were left untreated or treated with either BFA or GCA for 24 h during pseudovirus entry and stained for EdU and Lamp1 (Fig. 4). The untreated cells show an asymmetrical perinuclear EdU pattern that is clearly distinct from Lamp1+ compartments (Fig. 4A to C). The cells treated with BFA show little Lamp1 colocalization but also no recognizable TGN pattern (Fig. 4D to F). This is consistent with BFA's pleiotropic effects on endosomal trafficking. The GCA-treated cells show retention of the pseudogenome in the Lamp1+ compartment (Fig. 4G to I). This is consistent with a block in the delivery of the pseudogenome from the late endosome (LE) to the TGN. As both inhibitors affect the localization of Golgi resident proteins, it was impossible to examine colocalization with the anti-p230 antibody. This observed redistribution was indicative of the drugs' functionality (data not shown).
Fig 3.

Inhibition profiles of BFA and GCA. HaCaT cells were incubated with a dilution series of either BFA or GCA, and infection with a GFP-expressing pseudovirus was allowed to continue for 48 h. Following this, GFP expression was determined by flow cytometric analysis. The percent inhibition was determined by comparison to control infections. The results from triplicate infections are shown. The starting concentration of BFA was 150 nM, and 11 3-fold dilutions were examined. The starting concentration of GCA was 5 μM, and 11 2-fold dilutions were examined.
Fig 4.

Effects of BFA and GCA on pseudogenome trafficking. The localization of EdU-labeled pseudogenome was analyzed relative to the Lamp1 protein. Cells were left untreated (untx) or incubated in the presence of either BFA or GCA, as indicated. Merged images are shown in the rightmost panel of each row. Cells were harvested at 24 h post-virus addition. The nuclei were detected with DAPI (blue) in all panels.
Detection of TGN-localized L2.
We also evaluated the localization of L2 during entry. In our prior studies, we found that visualization of uncoated L2 required the use of an HA tag at the carboxy terminus of L2 and the detection of L2 with anti-HA reagents. Similar to the BrdU-labeled genome, in previous experiments, L2 was detected in the LE and the nucleus but not intermediate compartments. Since the EdU procedure was found to result in exposure of cryptic L1 epitopes, we evaluated whether that procedure might make it possible to also identify HA-L2 in the TGN. Under these conditions, it was possible to detect the accumulation of HA-L2 along with the pseudogenome in a perinuclear compartment (Fig. 5A). This was also shown to be adjacent to gm130 (Fig. 5B), indicating colocalization of L2 with the pseudogenome in the TGN. In our hands, murine antibodies provided the most sensitive detection of both the HA-tagged L2 and p230. Therefore, it is not possible to directly colocalize L2 with this marker of the TGN.
Fig 5.
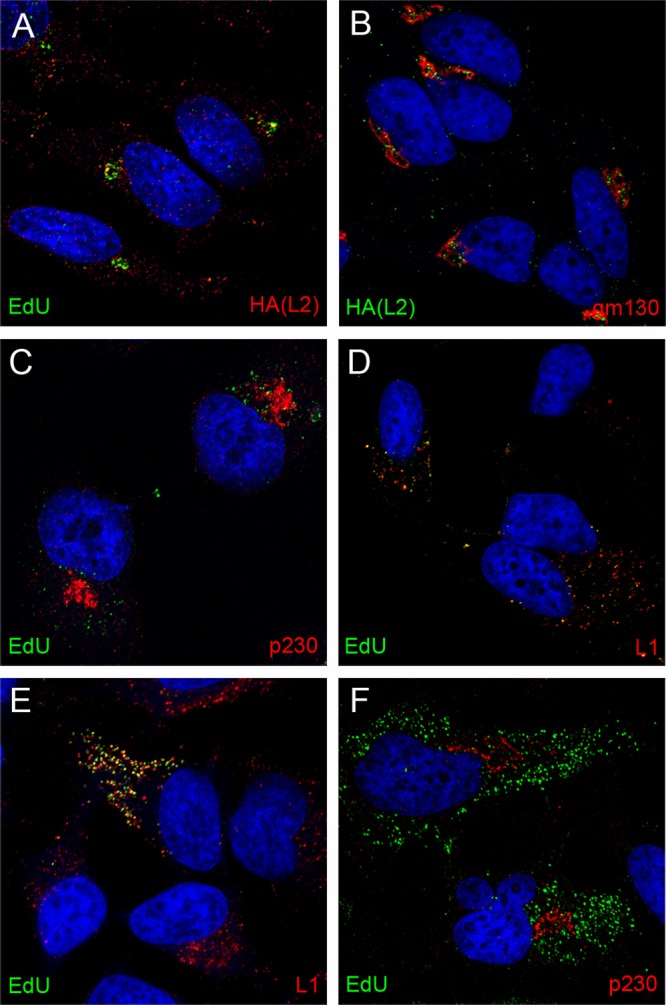
Dependence on furin-cleaved L2 for TGN localization. HA-tagged L2 was localized relative to the EdU-labeled genome (A) and gm130 (B) as indicated. EdU detection from L1-only VLPs is shown in panels C and D relative to p230 and L1 as indicated. EdU detection from pseudovirus containing an L2 furin cleavage mutant is shown in panels E and F relative to L1 and p230 as indicated. Cells were harvested at 24 h postentry. The nuclei were detected with DAPI (blue) in all panels.
L2 is required for TGN localization of the pseudogenome.
As noted in the introduction, L1 can self-assemble into virus-like particles (VLPs) that have many features of the intact virus particle, including their ability to interact with the cell surface and cellular endocytic machinery in a manner indistinguishable from that of L1-L2-containing infectious particles (24, 25). It has been shown that HPV16 L1 VLPs can package DNA similarly to L2-containing particles; however, these VLPs are not infectious (2). Previous studies have shown that L2 was required for endosome escape of the genome and that the packaged DNA in L1 VLPs does not get transferred to the nucleus (4). The increased sensitivity described above for detecting nonnuclear L2 made it possible to evaluate whether there is a role for L2 in delivering the pseudogenome to the TGN. As shown in Fig. 5, no coincident staining of L1-only-delivered pseudogenome was detected with p230 (Fig. 5C), indicating the dependence on L2 for TGN localization of the pseudogenome. Figure 5D shows that the majority of the EdU signal from the L1-only pseudovirions remains colocalized with L1 at later time points. We also examined the localization of EdU-labeled pseudogenome either with L2 furin cleavage mutants (Fig. 5E and F) or in the presence of a furin inhibitor (data not shown). These results mimicked that observed with the L1-only particles. The pseudogenome remained colocalized with L1 (Fig. 5E) and did not reach the TGN (Fig. 5F).
Rab GTPase requirement for TGN entry.
Taken together, the above results indicate that TGN localization is important for infection and requires L2, specifically furin-cleaved L2. To explore the mechanism by which the virus enters the TGN, we focused on its transit from the LE to the TGN, as low pH, which is a characteristic of cellular penetration via endosomes, is known to be essential for HPV16 infection, and the pseudogenome was localized in Lamp1+ compartments, which are predominantly the LE and lysosomes (9, 24, 26). Rab GTPases are required for the intracellular trafficking of protein cargo between various compartments (reviewed in reference 27). From the LE, cargo can traffic into the lysosome or the TGN via either Rab9a or Rab7b (16, 28, 29). To determine whether either of these Rab GTPases was required, we studied infection in the presence of GDP-locked dominant negative forms of Rab7a, Rab7b, and Rab9a (15). Rab7a represented a negative control, as it has been reported previously to be dispensable for HPV16 infection (30).
293TT cells were used for these experiments because they are highly susceptible to HPV16 pseudovirus infection and express high levels of transfected genes. The cells were transfected with either wild-type (WT) expression plasmids or dominant negative (DN) plasmids for Rab7a, Rab7b, and Rab9a. All constructs encoded a GFP- or RFP-fused protein for easy detection of the transfected subset. At 24 h posttransfection, cells were infected with either an RFP- or GFP-expressing HPV16 pseudovirus, selected to complement the fluorescence expressed by the transgene. At 48 h postinfection (72 h posttransfection), the percentage of transfected cells that were also infected was determined. Infection was compared between the WT protein and the DN version. All experiments were performed in triplicate and repeated on three occasions, with similar results. A representative experiment is shown in Fig. 6A. No effect was obvious with Rab7a deficiency. However, both the Rab7b and Rab9a DN proteins induced a significant decrease (P value = 0.0004 and P value = 0.0092, respectively) in infection.
Fig 6.

Effect of expression of DN Rab proteins on HPV16 pseudovirus infection. (A) 293TT cells were transfected with WT or DN Rab expression plasmids. At 24 h, these cells were infected with HPV16 pseudovirus containing a GFP expression plasmid (for Rab7a and Rab9a) or an RFP expression plasmid (for Rab7b). Following 48 h of infection, RFP and GFP coexpression was determined. The percentage of transfected cells that were also infected was determined. The plain bar indicates this percentage for the WT protein; the bar with the horizontal stripes represents the DN protein. Rab 7a is shown in white, Rab 9a in light gray, and Rab 7b in dark gray. (B) The transfected cells were either incubated with exogenous furin during the infection (left side of graph) or plated over furin-cleaved pseudovirus in the presence of furin inhibitor (right side of graph).
Rab9a has been shown to be necessary for the recycling of furin from the late endosome to the TGN (31), and the cleavage of HPV L2 by furin is required for infection. Under the conditions of the previous experiment, L2 cleavage by furin takes place on the cell surface, although this cleavage can also occur on the extracellular matrix (ECM) if shed furin is provided. Although no effect on cell surface levels of furin has been reported with Rab9a inhibition, we wanted to ensure that the observed inhibition was not attributable to a putative decrease in accessible furin. The effect of Rab7b inhibition on furin trafficking has not been examined. We employed two methods to evaluate possible confounding results due to changes in furin trafficking, for Rab7b as well as Rab9a. In the first method, we simply provided exogenous furin to the transfected cells during virus infection, a procedure we have previously shown permits infection of furin-deficient cells (24). As shown in Fig. 6B, exogenous furin did not affect the inhibition by DN Rab7b (P value < 0.0038) or DN Rab9a (P value < 0.0001). In the second method, HPV16 virions were applied to an in vitro-generated ECM, and exogenous furin was provided under some conditions, whereas others were incubated in parallel without exogenous furin. Following this incubation, furin and unbound virions were removed by washing, and the transfected cells expressing the WT or DN Rab proteins were added. Cells added to the furin-cleaved subset were incubated in the presence of a furin inhibitor, to ensure that only furin-independent infection was evaluated. As with the first method, this stringent assay, in which the furin levels in the target cells do not affect infection, verified that HPV16 infection depended on functional Rab7b and Rab9a GTPases. P values for the inhibition with the DN proteins were 0.0005 for Rab7b and 0.0002 for Rab9a.
DISCUSSION
The advent of sensitive DNA detection techniques such as the EdU system has allowed us to recognize the TGN as an obligatory subcellular compartment through which L2 and the pseudogenome traverse during HPV pseudovirus infection. Our previous method for following viral DNA during entry had relied upon detection of BrdU-labeled pseudogenome with anti-BrdU antibodies, which was cumbersome and less sensitive. However, that approach did enable us to define the uncoating kinetics of incoming particles and the localization of the genome and L2 at the ND10 nuclear domain (9). The EdU detection procedure is more sensitive because its mild protein denaturing conditions enable the detection reagent to bind EdU at 2 h postentry, the earliest time point tested. The detection procedure also revealed cryptic L1 and L2-linked epitopes that are not exposed in mature virus particles. These results indicate that the EdU detection conditions can be used to follow pseudogenome trafficking, although the denaturing conditions of the procedure mean that it cannot be used to study uncoating per se, a physiologic process that requires exposure to cellular uncoating factors, rather than to the exogenous proteases supplied in the detection buffer. A caveat in any study that employs modified nucleoside analogs to trace viral DNA is the detrimental effect that this incorporation has on the infectivity of the labeled genomes (32). The dependence of the DNA localization on L2, specifically furin-cleaved L2, renders it unlikely that the localization that we describe is an artifact of the DNA-labeling and detection procedure. Additionally, EdU-labeled and BrdU-labeled pseudogenomes arrive at the same ND10 destination with similar kinetics despite the dramatically different chemistries of the labeling procedures.
Consistent with previous work, we observed segregation of L2 and the PV pseudogenome from the residual L1. At early time points, the DNA and L1 were coincident in punctate cytoplasmic vesicles that were Lamp1+ by 6 h (data not shown). At later times, L1 remained a resident of a Lamp1+ compartment, whereas the L2-genome complex was detected in a loose perinuclear vesicular aggregate, which corresponds to the TGN, as evidenced by its being adjacent to giantin and gm130, markers of the cis-medial Golgi and cis-Golgi, respectively, and its strong colocalization with the TGN marker p230.
The relevance of this Golgi localization to infectious entry was confirmed by inhibition studies with brefeldin A (BFA) and golgicide A (GCA), both of which reduced HPV16 pseudovirus infection. BFA and GCA prevent anterograde and retrograde movement of proteins through the Golgi stacks by different mechanisms. The molecular target of BFA is a subset of GTP exchange factors (GEFs) that activate the Arf1p small GTPase to recruit coat proteins (including COP1) that are integral to the formation of transport vesicles (33–35). Therefore, BFA has effects beyond those at the ER and Golgi. BFA was previously shown to inhibit HPV16 infection (36). It was reported that viral capsids localize to the ER at early time points (4 h), and this is disrupted following BFA treatment. The authors concluded that this indicated a COP1-dependent transport of capsids into the ER. However, the pleiotropic effects of BFA were not considered before drawing this conclusion, and the effect of this treatment on the localization of the viral genome and L2 was not examined. No other laboratories, including ours, have reported ER localization of PV capsids, and we have no explanation for this discrepancy. In our current study, we also considered the effect of GCA on HPV16 infection. GCA, a highly specific inhibitor of the cis-Golgi ArfGEF GBF1, induces disassembly of the Golgi and TGN and prevents Golgi-dependent bidirectional traffic (23). In contrast to BFA, GCA is thought to act solely at the Golgi.
The HPV entry pathway, as exemplified by HPV16, is apparently unique. It is independent of clathrin, caveolin, flotillin, cholesterol, and dynamin, and although it shares many features with macropinocytotic uptake, including, among others, dependence on actin dynamics and receptor tyrosine kinases, some hallmarks of this mechanism are missing, such as cholesterol and myosin dependence (30). PV capsids pass through the early endosomes en route to late endosomes, and the low pH encountered during this passage is essential for infection, contributing to uncoating and/or endosome escape (24, 30, 37–39). It has been postulated that L2 participates in exodus of the virus from the endosomal compartment (4).
It is currently unclear how the virus traverses the cytosol en route to the nucleus. The bidirectional traffic of cargo in the cellular endosomal system is controlled by a series of membrane budding and fusion events mediated by various Rab family proteins, which are small Ras-like GTPases that cycle between their active, GTP-bound form and their inactive, GDP-bound state (reviewed in reference 40). Active, membrane-bound Rabs fulfill their role in vesicle trafficking by association with their specific effectors. Following hydrolysis, the GDP-bound Rab is solubilized from the membrane and can be recycled for another round of transport. Specific Rabs are physically associated with particular organelles and their associated transport vesicles. Rab5a is detected at the cytoplasmic surface of both the plasma membrane and early endosomes and mediates entry into the early endosome (41, 42), whereas Rab7a is associated with LEs and mediates LE-to-lysosome trafficking (41, 43, 44). Dominant negative (DN) versions of the Rabs have been used extensively to dissect the route that a cargo follows within the cell. Using expression of DN versions of Rab proteins, HPV16 and HPV31 infections have both been shown to be dependent on functional Rab5a and independent of Rab7a (30, 45). The Rab7a results, which we confirmed here, are consistent with viral escape from the late endosome prior to lysosome biogenesis.
Rab7b and Rab9a contribute to trafficking from the LE to the TGN, and DN versions of both genes inhibited infection. Rab7b has been shown to sort both CI-MPR and sortilin from the LE to the TGN (46), while Rab9a mediates LE-to-Golgi passage of a variety of cargoes (28, 29). These results further demonstrate the significance of Golgi trafficking in HPV16 infection. The observed localization of L2 and the viral pseudogenome likely indicates transit through this compartment en route to the nucleus. Apparently, the virus can enter the TGN by at least two routes, one that is Rab7b dependent and one that is Rab9a dependent. This diversity is not surprising, as some redundancy within the Rab pathways exists, and other cargoes, such as CI-MPR, are known to be capable of following more than one pathway (46).
It has been proposed that the endosomal system exists as an extensive tubular endosomal network (TEN) (27). Within this network, cargo is sorted to different destinations through selection by Rabs. Cargo that remains in the endosomal system continues into the lysosomes, whereas cargo that enters the TEN avoids this degradative route and is actively transported elsewhere. This idea is consistent with the observed separation of L1 from L2 and the pseudogenome. It is appealing to imagine that L1 represents a “nonselected” cargo and continues passage through the default pathway into lysosomes, whereas L2 and the associated pseudogenome are recruited into the TEN and sorted to the TGN. Sorting nexins (SNX) have been extensively linked to endosomal sorting events (47). In this regard, SNX17 may have a role in this selection of L2, as SNX17 has been reported to interact with HPV16 L2 protein during infectious entry (37), and inhibition of this interaction induces increased degradation of L2, which is associated with the increased delivery of L2 to the lysosomal compartment. SNX17 can also protect integrins from degradation by shunting them into a recycling pathway (48, 49) and can also serve as a cargo-specific adaptor (50). Although many cellular cargoes are known that route from endosomes to the TGN (reviewed in reference 27), this is the first report to describe the dependence of viral entry on an LE-to-TGN pathway.
L2 has also been shown to interact directly with the dynein light-chain DYNLT1, also known as Tctex1, which predominantly localizes to the Golgi apparatus (51), and small interfering RNA (siRNA)-mediated reduction of Tctex1 expression can prevent infection (7). This effect could be explainable by the effect of dynein depletion on intra-Golgi transport (52). Interestingly, the distribution of L2 in the dynein light-chain knockdown cells strongly resembles the Golgi-localized L2 and pseudogenome described in this study. Intriguingly, the dynein light-chain rp3, which shares 55% homology with Tctex1, has been shown to associate with the nuclear matrix, where a subset localized to ND10 (53). Tctex1 has also been found in interphase nuclei (54). Therefore, it is important to consider the possibility that the direct interaction of L2 with Tctex1 could indicate a role in the delivery mechanism of the L2-genome complex from the Golgi to the kinetochore in the mitotic cell. Although we have followed the virus one step closer to the nucleus, the final steps remain to be elucidated, perhaps by evaluating this and other possible post-Golgi pathways.
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
We gratefully acknowledge the gifts of the Δfurin cell line from David Fitzgerald, the 33L1-7 antibody from Martin Sapp, the Rab7a and 9a constructs from Richard Pagano, and the Rab7b constructs from Cecilia Bucci.
Footnotes
Published ahead of print 23 January 2013
REFERENCES
- 1. Liu WJ, Gissmann L, Sun XY, Kanjanahaluethai A, Muller M, Doorbar J, Zhou J. 1997. Sequence close to the N-terminus of L2 protein is displayed on the surface of bovine papillomavirus type 1 virions. Virology 227:474–483 [DOI] [PubMed] [Google Scholar]
- 2. Buck CB, Thompson CD, Pang YY, Lowy DR, Schiller JT. 2005. Maturation of papillomavirus capsids. J. Virol. 79:2839–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richards RM, Lowy DR, Schiller JT, Day PM. 2006. Cleavage of the papillomavirus minor capsid protein, L2, at a furin consensus site is necessary for infection. Proc. Natl. Acad. Sci. U. S. A. 103:1522–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kämper N, Day PM, Nowak T, Selinka HC, Florin L, Bolscher J, Hilbig L, Schiller JT, Sapp M. 2006. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J. Virol. 80:759–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bienkowska-Haba M, Williams C, Kim SM, Garcea RL, Sapp M. 2012. Cyclophilins facilitate dissociation of the human papillomavirus type 16 capsid protein L1 from the L2/DNA complex following virus entry. J. Virol. 86:9875–9887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Florin L, Becker KA, Lambert C, Nowak T, Sapp C, Strand D, Streeck RE, Sapp M. 2006. Identification of a dynein interacting domain in the papillomavirus minor capsid protein L2. J. Virol. 80:6691–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schneider MA, Florin L, Lambert C. 2011. Identification of the dynein light chains required for human papillomavirus infection. Cell. Microbiol. 13:32–46 [DOI] [PubMed] [Google Scholar]
- 8. Pyeon D, Pearce SM, Lank SM, Ahlquist P, Lambert PF. 2009. Establishment of human papillomavirus infection requires cell cycle progression. PLoS Pathog. 5:e1000318 doi:10.1371/journal.ppat.1000318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Day PM, Baker CC, Lowy DR, Schiller JT. 2004. Establishment of papillomavirus infection is enhanced by promyelocytic leukemia protein (PML) expression. Proc. Natl. Acad. Sci. U. S. A. 101:14252–14257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buck CB, Pastrana DV, Lowy DR, Schiller JT. 2005. Generation of HPV pseudovirions using transfection and their use in neutralization assays. Methods Mol. Med. 119:445–462 [DOI] [PubMed] [Google Scholar]
- 11. Chiron MF, Fryling CM, FitzGerald D. 1997. Furin-mediated cleavage of Pseudomonas exotoxin-derived chimeric toxins. J. Biol. Chem. 272:31707–31711 [DOI] [PubMed] [Google Scholar]
- 12. Day PM, Pang YY, Kines RC, Thompson CD, Lowy DR, Schiller JT. 2012. A human papillomavirus (HPV) in vitro neutralization assay that recapitulates the in vitro process of infection provides a sensitive measure of HPV L2 infection-inhibiting antibodies. Clin. Vaccine Immunol. 19:1075–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Roden RB, Greenstone HL, Kirnbauer R, Booy FP, Jessie J, Lowy DR, Schiller JT. 1996. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 70:5875–5883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sapp M, Kraus U, Volpers C, Snijders PJ, Walboomers JM, Streeck RE. 1994. Analysis of type-restricted and cross-reactive epitopes on virus-like particles of human papillomavirus type 33 and in infected tissues using monoclonal antibodies to the major capsid protein. J. Gen. Virol. 75(Part 12):3375–3383 [DOI] [PubMed] [Google Scholar]
- 15. Choudhury A, Dominguez M, Puri V, Sharma DK, Narita K, Wheatley CL, Marks DL, Pagano RE. 2002. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 109:1541–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Progida C, Cogli L, Piro F, De Luca A, Bakke O, Bucci C. 2010. Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci. 123:1480–1491 [DOI] [PubMed] [Google Scholar]
- 17. Spoden G, Freitag K, Husmann M, Boller K, Sapp M, Lambert C, Florin L. 2008. Clathrin- and caveolin-independent entry of human papillomavirus type 16—involvement of tetraspanin-enriched microdomains (TEMs). PLoS One 3:e3313 doi:10.1371/journal.pone.0003313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doms RW, Russ G, Yewdell JW. 1989. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J. Cell Biol. 109:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56:801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y. 1986. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 261:11398–11403 [PubMed] [Google Scholar]
- 21. Damke H, Klumperman J, von Figura K, Braulke T. 1991. Effects of brefeldin A on the endocytic route. Redistribution of mannose 6-phosphate/insulin-like growth factor II receptors to the cell surface. J. Biol. Chem. 266:24829–24833 [PubMed] [Google Scholar]
- 22. Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD. 1991. Brefeldin A's effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 67:601–616 [DOI] [PubMed] [Google Scholar]
- 23. Sáenz JB, Sun WJ, Chang JW, Li J, Bursulaya B, Gray NS, Haslam DB. 2009. Golgicide A reveals essential roles for GBF1 in Golgi assembly and function. Nat. Chem. Biol. 5:157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Day PM, Lowy DR, Schiller JT. 2003. Papillomaviruses infect cells via a clathrin-dependent pathway. Virology 307:1–11 [DOI] [PubMed] [Google Scholar]
- 25. Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT. 1992. Papillomavirus L1 major capsid protein self-assembles into virus-like particles that are highly immunogenic. Proc. Natl. Acad. Sci. U. S. A. 89:12180–12184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Campos SK, Chapman JA, Deymier MJ, Bronnimann MP, Ozbun MA. 2012. Opposing effects of bacitracin on human papillomavirus type 16 infection: enhancement of binding and entry and inhibition of endosomal penetration. J. Virol. 86:4169–4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonifacino JS, Rojas R. 2006. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 7:568–579 [DOI] [PubMed] [Google Scholar]
- 28. Lombardi D, Soldati T, Riederer MA, Goda Y, Zerial M, Pfeffer SR. 1993. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 12:677–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Riederer MA, Soldati T, Shapiro AD, Lin J, Pfeffer SR. 1994. Lysosome biogenesis requires Rab9 function and receptor recycling from endosomes to the trans-Golgi network. J. Cell Biol. 125:573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schelhaas M, Shah B, Holzer M, Blattmann P, Kuhling L, Day PM, Schiller JT, Helenius A. 2012. Entry of human papillomavirus type 16 by actin-dependent, clathrin- and lipid raft-independent endocytosis. PLoS Pathog. 8:e1002657 doi:10.1371/journal.ppat.1002657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chia PZ, Gasnereau I, Lieu ZZ, Gleeson PA. 2011. Rab9-dependent retrograde transport and endosomal sorting of the endopeptidase furin. J. Cell Sci. 124:2401–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ishii Y, Tanaka K, Kondo K, Takeuchi T, Mori S, Kanda T. 2010. Inhibition of nuclear entry of HPV16 pseudovirus-packaged DNA by an anti-HPV16 L2 neutralizing antibody. Virology 406:181–188 [DOI] [PubMed] [Google Scholar]
- 33. Donaldson JG, Lippincott-Schwartz J, Bloom GS, Kreis TE, Klausner RD. 1990. Dissociation of a 110-kD peripheral membrane protein from the Golgi apparatus is an early event in brefeldin A action. J. Cell Biol. 111:2295–2306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Helms JB, Rothman JE. 1992. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nature 360:352–354 [DOI] [PubMed] [Google Scholar]
- 35. Robinson MS, Kreis TE. 1992. Recruitment of coat proteins onto Golgi membranes in intact and permeabilized cells: effects of brefeldin A and G protein activators. Cell 69:129–138 [DOI] [PubMed] [Google Scholar]
- 36. Laniosz V, Dabydeen SA, Havens MA, Meneses PI. 2009. Human papillomavirus type 16 infection of human keratinocytes requires clathrin and caveolin-1 and is brefeldin A sensitive. J. Virol. 83:8221–8232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bergant Marušič M, Ozbun MA, Campos SK, Myers MP, Banks L. 2012. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 13:455–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bousarghin L, Touze A, Sizaret PY, Coursaget P. 2003. Human papillomavirus types 16, 31, and 58 use different endocytosis pathways to enter cells. J. Virol. 77:3846–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Selinka HC, Giroglou T, Sapp M. 2002. Analysis of the infectious entry pathway of human papillomavirus type 33 pseudovirions. Virology 299:279–287 [DOI] [PubMed] [Google Scholar]
- 40. Mizuno-Yamasaki E, Rivera-Molina F, Novick P. 2012. GTPase networks in membrane traffic. Annu. Rev. Biochem. 81:637–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chavrier P, Parton RG, Hauri HP, Simons K, Zerial M. 1990. Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 62:317–329 [DOI] [PubMed] [Google Scholar]
- 42. Gorvel JP, Chavrier P, Zerial M, Gruenberg J. 1991. rab5 controls early endosome fusion in vitro. Cell 64:915–925 [DOI] [PubMed] [Google Scholar]
- 43. Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. 2000. Rab7: a key to lysosome biogenesis. Mol. Biol. Cell 11:467–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ng EL, Gan BQ, Ng F, Tang BL. 2012. Rab GTPases regulating receptor trafficking at the late endosome-lysosome membranes. Cell Biochem. Funct. 30:515–523 [DOI] [PubMed] [Google Scholar]
- 45. Smith JL, Campos SK, Wandinger-Ness A, Ozbun MA. 2008. Caveolin-1-dependent infectious entry of human papillomavirus type 31 in human keratinocytes proceeds to the endosomal pathway for pH-dependent uncoating. J. Virol. 82:9505–9512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Progida C, Nielsen MS, Koster G, Bucci C, Bakke O. 2012. Dynamics of Rab7b-dependent transport of sorting receptors. Traffic 13:1273–1285 [DOI] [PubMed] [Google Scholar]
- 47. Carlton J, Bujny M, Rutherford A, Cullen P. 2005. Sorting nexins—unifying trends and new perspectives. Traffic 6:75–82 [DOI] [PubMed] [Google Scholar]
- 48. Böttcher RT, Stremmel C, Meves A, Meyer H, Widmaier M, Tseng HY, Fassler R. 2012. Sorting nexin 17 prevents lysosomal degradation of beta1 integrins by binding to the beta1-integrin tail. Nat. Cell Biol. 14:584–592 [DOI] [PubMed] [Google Scholar]
- 49. Steinberg F, Heesom KJ, Bass MD, Cullen PJ. 2012. SNX17 protects integrins from degradation by sorting between lysosomal and recycling pathways. J. Cell Biol. 197:219–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yin W, Liu LDN, Xu L, Li S, Lin S, Shu X, Pei D. 2012. SNX17 regulates Notch pathway and pancreas development through the retromer-dependent recycling of Jag1. Cell Regeneration 1:1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tai AW, Chuang JZ, Sung CH. 1998. Localization of Tctex-1, a cytoplasmic dynein light chain, to the Golgi apparatus and evidence for dynein complex heterogeneity. J. Biol. Chem. 273:19639–19649 [DOI] [PubMed] [Google Scholar]
- 52. Palmer KJ, Hughes H, Stephens DJ. 2009. Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol. Biol. Cell 20:2885–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yeh TY, Chuang JZ, Sung CH. 2005. Dynein light chain rp3 acts as a nuclear matrix-associated transcriptional modulator in a dynein-independent pathway. J. Cell Sci. 118:3431–3443 [DOI] [PubMed] [Google Scholar]
- 54. Herzig RP, Andersson U, Scarpulla RC. 2000. Dynein light chain interacts with NRF-1 and EWG, structurally and functionally related transcription factors from humans and drosophila. J. Cell Sci. 113(Part 23):4263–4273 [DOI] [PubMed] [Google Scholar]