

Abstract
Alphaviruses such as Semliki Forest virus (SFV) are enveloped viruses whose surface is covered by an organized lattice composed of trimers of E2-E1 heterodimers. The E1 envelope protein, a class II fusion protein, contains the hydrophobic fusion loop and refolds to drive virus fusion with the endosome membrane. The E2 protein is synthesized as a precursor p62, whose processing by furin primes the heterodimer for dissociation during virus entry. Dissociation of the E2-E1 heterodimer is an essential step during low-pH-triggered fusion, while the dissociation of the immature p62-E1 dimer is relatively pH resistant. Previous structural studies described an “acid-sensitive region” in E2 that becomes disordered at low pH. Within this region, the conserved E2 H170 is in position to form a hydrogen bond with the underlying E1 S57. Here we experimentally tested the role of this interaction in regulating dimer dissociation in mature and immature virus. Alanine substitutions of E1 S57 and E2 H170 destabilized the heterodimer and produced a higher pH threshold for exposure of the E1 fusion loop and for fusion of the immature virus. E1 S57K or S57D mutations were lethal and caused transport and assembly defects that were partially abrogated by neutralization of the exocytic pathway. The lethal phenotype of E1 S57K was rescued by second-site mutations at E2 H170/M171. Together, our results define a key role for the E1 S57-E2 H170 interaction in dimer stability and the pH dependence of fusion and provide evidence for stepwise dissociation of the E2-E1 dimer at low pH.
INTRODUCTION
Alphaviruses are small enveloped plus-strand RNA viruses generally transmitted by mosquito vectors (1). Some alphaviruses cause severe illnesses in humans, including arthritis and fatal encephalitis, and are potential biodefense threats. For example, the alphavirus Chikungunya virus (CHIKV) has recently caused major human epidemics, infecting an estimated 7.5 million people in countries surrounding the Indian Ocean (2, 3). There are currently no vaccines or antiviral therapies available for the alphaviruses, and molecular information on their entry mechanisms may provide new approaches to inhibit this key step in the virus life cycle.
The alphavirus membrane is covered by a symmetrical lattice composed of the envelope glycoproteins E1 and E2, arranged in trimers of E2-E1 heterodimers (4, 5) (Fig. 1A). E1, the membrane fusion protein, is an elongated molecule containing 3 β-sheet-rich domains (DI to DIII), with the conserved hydrophobic fusion loop at the membrane distal tip (6, 7). E2 contains 3 β-barrel domains, consisting of the central A domain linked by a β-ribbon connector to the membrane-distal B domain and the membrane-proximal C domain (8, 9). On the virus particle, E2 covers most of E1, with the fusion loop clamped in the groove between E2 domains A and B. During entry, E2 binds to receptors on the cell surface to mediate virus internalization by clathrin-mediated endocytosis (10, 11). The low pH of the endosome then triggers conformational changes in which the E2-E1 dimers dissociate, the hydrophobic E1 fusion loop inserts into the endosome membrane, and E1 forms extended homotrimers connecting the viral and cellular membranes (Fig. 1A) (reviewed in reference 11). E1 then refolds into a postfusion hairpin-like structure, thus driving virus membrane fusion (12).
Fig 1.
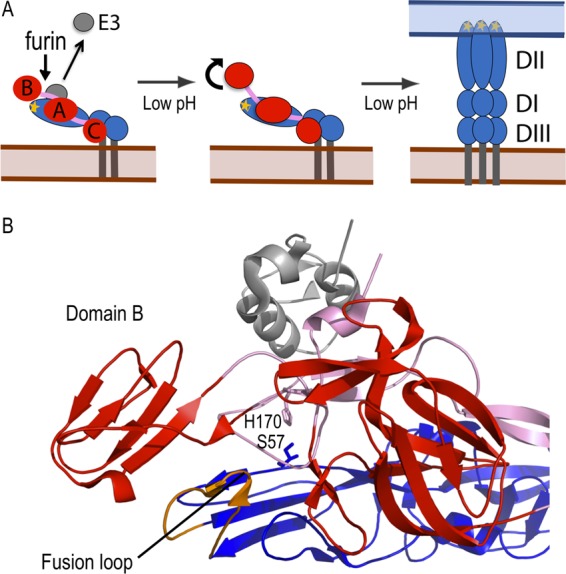
Model for low-pH-triggered conformational changes in the E2/E1 dimer. (A) Schematic model for conformational changes in the E2/E1 heterodimer. (Left panel) During virus biosynthesis, p62, the precursor to E2, is cleaved by furin in the late secretory pathway, allowing E3 release at neutral pH. (Middle panel) During virus entry, exposure to endosomal low pH causes E2 domain B to dissociate (shown as curved arrow), uncovering the E1 fusion loop. (Right panel) The fusion loop inserts into the endosomal membrane, and E1 forms an extended homotrimer. E2 is shown in red with domains A to C indicated and the beta-ribbon connector in pink, E3 is shown in gray, and E1 is shown in blue with domains I to III indicated and the fusion loop in orange. (B) A closeup view of the crystal structure of the CHIK p62/E1 dimer (Protein Data Bank [PDB] entry 3N41) (9) with the proteins colored as described for panel A. E1 S57 and E2 H170 are labeled and represented as sticks in blue and purple, respectively. This figure was prepared using Pymol (47).
E2 is initially synthesized as a precursor p62, which dimerizes with E1 in the endoplasmic reticulum (ER) and is required for proper folding of E1 (1). p62 is cleaved by furin late in the secretory pathway to produce the mature E2 protein and a smaller protein, E3 (13). The virus buds from the plasma membrane, and E3 is released at the neutral pH outside the cell (14). The pH threshold for fusion of the mature E2-containing Semliki Forest virus (SFV) is ∼6.2 (15). In contrast, immature p62-containing SFV requires a pH of ∼5.0 or lower to trigger fusion and infection (13, 16, 17). This differential pH sensitivity is due to the differential acid stabilities of the alphavirus p62-E1 and E2-E1 heterodimers, which must dissociate to free the E1 protein for fusion (16, 18).
The steps in low-pH-mediated heterodimer dissociation and the role of specific residues in regulating dimer stability are unclear. The Sindbis virus E2-E1 dimer was crystallized at pH 5.6 (8), while the CHIKV E2-E1 dimer (and associated E3) was crystallized at pH 8.0 (9). Comparison of these heterodimer structures reveals that at low pH, E2 domain B and about half of the β-ribbon connector are disordered. This suggests that an early intermediate in heterodimer dissociation is the movement of E2 domain B to “uncap” the E1 fusion loop (Fig. 1A). The missing “acid-sensitive region” of the flexible β-ribbon connector contains E2 H170, which is positioned to form a hydrogen bond with the underlying E1 S57 (9) (Fig. 1B). Both of these residues are well resolved in the dimer structure and are highly conserved among alphaviruses (9). In addition, the structure of the p62-E1 dimer suggests that the tethered E3 acts to clamp the acid-sensitive region of the connector in place, producing the increased acid stability of the immature dimer (9). Mutations in the E2 acid-sensitive region can rescue the infectivity of p62-containing virus (reviewed in reference 9) and can promote the production of Chikungunya virus-like particles (19). However, the role of interactions of the E2 acid-sensitive region with E1 during fusion loop uncapping, dimer dissociation, and subsequent conformational changes has not been examined.
Here we evaluated the function of the E1 S57-E2 H170 interaction using single and double alanine substitutions of these residues in an SFV infectious clone. These mutations destabilized the envelope protein heterodimer, producing a higher pH threshold for uncapping of the E1 fusion loop and increasing virus growth in furin-deficient cells. Replacement of E1 S57 with bulky charged lysine or aspartic acid residues blocked virus assembly. Revertants of the E1 S57K and S57D mutants revealed that virus viability could be rescued by mutations at E2 H170/M171, thus defining the key interaction of these regions of E1 and E2 in the function of the alphavirus heterodimer.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells were maintained in complete BHK medium (Dulbecco's modified Eagle medium containing 5% fetal bovine serum, 10% tryptose phosphate broth, 100 U penicillin/ml, and 100 μg streptomycin/ml) at 37°C. The furin-deficient FD-11 CHO cell line and its parental cell line, CHO-K1, were kindly provided by Stephen H. Leppla at the National Institutes of Health and were cultured in α-minimal essential medium containing 10% fetal bovine serum, 100 U penicillin/ml, and 100 μg streptomycin/ml at 37°C (20).
Site-directed mutagenesis using the DG-1 and XYZ-1 subgenomic plasmids was performed as previously described (13, 21). In brief, the E1 S57A, S57K, and S57D mutations were introduced into DG-1 by circular mutagenesis using PrimeSTAR HS DNA polymerase (TaKaRa Bio Inc., Madison, WI). The mutated NsiI-SpeI fragments were subcloned into the pSP6-SFV4 infectious clone (22). The E2 H170A mutation was introduced into the XYZ-1 subgenomic plasmid (13). The E1 S57A-E2 H170A double mutant was constructed using the XYZ-1-containing E2 H170A as the template. Sequence analysis was used to confirm the sequence of the mutated region and the absence of other mutations in the structural proteins (Genewiz Inc., North Brunswick, NJ). Infectious RNAs were generated by in vitro transcription and electroporated into BHK-21 cells to produce virus infection or generate virus stocks (22). All plaque assays were performed on BHK cells. The resultant wild-type (WT) or mutant virus stocks showed similar titers, indicating similar efficiencies of particle production. No differences in the stability of the mutant virus stocks at 37°C were observed, although this point was not extensively tested.
[35S]methionine-cysteine-labeled virus was prepared by infecting BHK-21 cells with 50 PFU/cell of WT or E1 S57A-E2 H170A virus. Radiolabeled virus was purified on sucrose gradients as previously described (21).
Fusion-infection assay.
Appropriate dilutions of mature or immature viruses were adsorbed to BHK-21 cells on ice. Fusion with the plasma membrane was triggered by treatment of the prebound virus for 3 min at 37°C with media at the indicated pH. Cells were then cultured at 28°C overnight in media containing 20 mM NH4Cl to prevent secondary infection, and virus-infected cells were quantitated by immunofluorescence as described previously (23). The ratios of the virus titers at the maximal pH for fusion versus the titers derived by plaque assays were essentially equivalent (0.95 to 1.01), suggesting that the overall efficiencies of fusion of the WT and the three alanine mutants were similar.
pH dependence of dimer dissociation.
35S-labeled WT or E1 S57A-E2 H170A mature viruses were diluted in morpholineethanesulfonic acid (MES)-saline buffer (pH 8.0) containing 0.1% bovine serum albumin (BSA). For coimmunoprecipitation analysis, samples were treated at the indicated pH for 10 min on ice, solubilized with 1% Nonidet P-40 (NP-40) for 10 min on ice, and adjusted to pH 8.0 (18). Samples were immunoprecipitated with monoclonal antibodies (MAb) to E1 (E1-1) or E2 (E2-3) (24, 25). For tests of fusion loop exposure, samples were preincubated at 37°C for 3 min, adjusted to the indicated pH, and incubated at 37°C for an additional 5 min. Samples were adjusted to neutral pH and immunoprecipitated with MAb to the E1 fusion loop (MAb E1f) as described previously (26). All samples were analyzed by SDS-PAGE and quantitated by PhosphorImager analysis using ImageQuant version 1.2 software (Molecular Dynamics, Sunnyvale, CA).
Virus assembly assays.
Pulse-chase assays were used to evaluate virus assembly (21). BHK-21 cells were electroporated with either WT RNA or mutant RNA or infected with WT or mutant virus at 1 PFU/cell. After a 6-h incubation at 37°C, the infected cells were pulse-labeled with [35S]methionine-cysteine (PerkinElmer) for 30 min and chased in media without label. At the indicated times, the cell lysates and chase media were collected and analyzed by immunoprecipitation with a polyclonal antibody to the envelope glycoproteins. In order to recover intact virus particles, medium samples were precipitated in the absence of detergent. Samples were analyzed by SDS-PAGE and phosphorimaging. To determine if E1 was being trafficked to the Golgi compartment, endoglycosidase H (Endo H; New England BioLabs) digestions were performed. Lysate samples from the 0- and 1-h time points were digested with Endo H for 90 min at 37°C in 50 mM citrate buffer at pH 5.50 (25).
Immunofluorescence and pulse-chase assays of NH4Cl-treated cells.
BHK-21 cells were electroporated with WT or mutant RNA, incubated at 37°C for 2 h, and cultured in media ± 20 mM NH4Cl for ∼14 h at 37°C. Cells were fixed in 3% paraformaldehyde and stained with MAb E1-1. Alternatively, 4 h after the transfer to medium ± 20 mM NH4Cl, cells were pulse-labeled with [35S]methionine-cysteine for 30 min and then incubated in chase medium for 4 h at 37°C, all ± 20 mM NH4Cl, as indicated. Viral proteins released during the chase were analyzed as described above.
Isolation of E1 S57K and E1 S57D revertants.
E1 S57K and E1 S57D viral RNAs were electroporated into BHK-21 cells. Electroporated cells were mixed with nonelectroporated cells at various ratios and the cells cultured at 37°C for 4 days, when cytopathic effects were observed. Virus was plaque purified from the culture media, amplified on BHK cells for 24 h at 37°C, and pelleted by ultracentrifugation. Viral RNA was extracted and reverse transcribed, and the cDNA was sequenced as previously reported (27).
RESULTS
Generation and characterization of SFV E1 S57 and E2 H170 mutants.
Mutagenesis of the SFV infectious clone was used to characterize the role of the alphavirus E1 S57-E2 H170 interaction. The serine and histidine residues were each changed to alanine to produce the single mutants E1 S57A and E2 H170A and the double mutant E1 S57A-E2 H170A. Viral RNAs were transcribed in vitro and electroporated into BHK cells to characterize mutant phenotypes.
Immunofluorescent staining showed that all three mutants efficiently expressed the E1 and E2 proteins on the cell surface and caused secondary infection of cocultured nonelectroporated BHK cells (data not shown). We generated virus stocks in BHK cells and compared mutant and WT virus growth kinetics on control and furin-deficient CHO (FD11) cells. Cells were infected at a low multiplicity of infection (MOI) to allow evaluation of multiple cycles of infection. FD11 cells produce the immature p62 form of the virus, which can be efficiently quantitated by plaque assays on BHK cells due to cleavage by furin during entry (13). This system was previously used to select for and define mutations that compensate for the lack of p62 processing (13, 28, 29).
The mutants showed growth kinetics in control CHO cells similar to the kinetics seen with the WT virus (Fig. 2A). In contrast, all 3 mutant viruses had a growth advantage in FD11 cells (Fig. 2B). While the mutant viruses produced only slightly higher titers than the WT at early time points in infection, after 20 h, all mutants showed a strong growth advantage, with maximal titers 1 to 2 logs higher than that of WT. These results suggest that the mutations compensated for the more acid-stable p62-E1 dimer produced in FD11 cells, allowing more efficient secondary infection.
Fig 2.
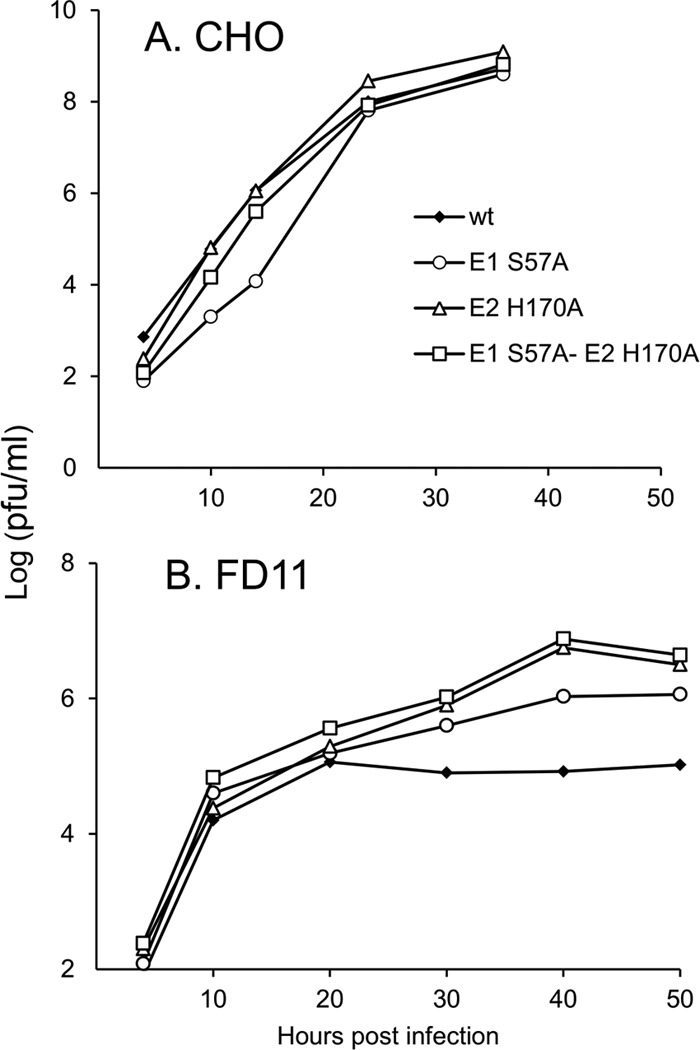
Growth kinetics of WT and mutant SFV on CHO and FD11 cells. Control (A) and FD11 furin-deficient (B) CHO cells were infected with WT or mutant viruses at a multiplicity of infection of 0.01 PFU/cell for 90 min at 37°C. After infection, cells were washed twice with serum-free media. At the indicated time postinfection, the virus secreted into the media was collected and the titer was determined by plaque assay on BHK cells. Data represent the results of 2 independent experiments.
pH dependence of WT and mutant membrane fusion.
A possible cause of the growth advantage of the mutants in the immature p62 form is a change in the pH threshold of fusion. This was directly tested using fusion-infection assays of WT and mutant viruses in the mature and immature forms. Viruses were prebound to BHK-21 cells in the cold to prevent endocytosis and then treated at low pH for 3 min at 37°C to trigger fusion at the plasma membrane. Infected cells were quantitated by immunofluorescence. The mature forms of WT and the 3 mutants showed similar fusion activities, with maximal fusion at pH 6.0 (Fig. 3A). As previously observed, immature WT SFV showed maximal fusion at pH 4.5 and a fusion pH threshold of ∼4.9 (Fig. 3B) (13, 29). While the mutants also showed maximal fusion at pH 4.5, they showed dramatic increases in fusion efficiency at higher pH values. The double mutant, E1 S57A-E2 H170A, had the strongest phenotype, showing 75% maximal fusion at pH 5.3.
Fig 3.
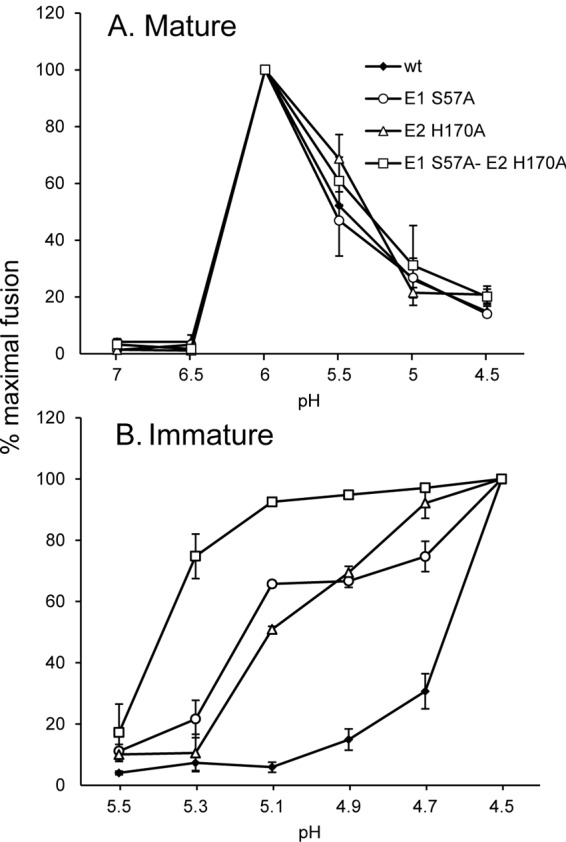
pH dependence of fusion of WT and mutant SFV. Mature, E2-containing virus produced by BHK cells (A) and immature, p62-containing virus produced by FD11 cells (B) were adsorbed to BHK cells on ice. Virus-plasma membrane fusion was triggered by treatment at the indicated pH at 37°C for 3 min. Virus-infected cells were then quantitated by immunofluorescence and the results shown as the percentage of maximal fusion for each virus. Entry via endocytosis is low under these conditions, as shown by pH 7.0 controls (see, e.g., panel A). Data represent averages and standard deviations of the results of 3 independent experiments. In Fig. 3B, the difference in the percentage of fusion between WT and E1 S57A-E2 H170A was statistically significant from pH 4.7 to 5.3, and between WT and the mutants E1 S57A and E2 H170A from pH 4.7 to 5.1 (P < 0.05 using a two-tailed t test).
pH dependence of dimer dissociation and E1 fusion loop exposure.
We hypothesized that the E1 S57 and E2 H170 mutations act by destabilizing the p62/E2-E1 heterodimer, thus facilitating virus fusion. We tested this model in the mature virus, using purified 35S-labeled WT and mutant viruses to evaluate the pH dependence of E2-E1 dimer dissociation. Radiolabeled virus was treated at various pH values on ice, solubilized in nonionic detergent, and adjusted to pH 8.0. Coimmunoprecipitation of the glycoproteins was used to monitor dimer dissociation. As previously observed, the WT heterodimer was efficiently immunoprecipitated after treatment at pH 8.0 to 7.0, and dissociation was observed after treatment at pH 6.5 to 6.0 (Fig. 4A). The E1 S57A-E2 H170A dimer was significantly destabilized, and only ∼15% of E2 was coprecipitated with an E1 MAb even after treatment at pH 8.0 (Fig. 4A). Similar results were observed when the coprecipitation was performed with an E2 MAb (data not shown).
Fig 4.
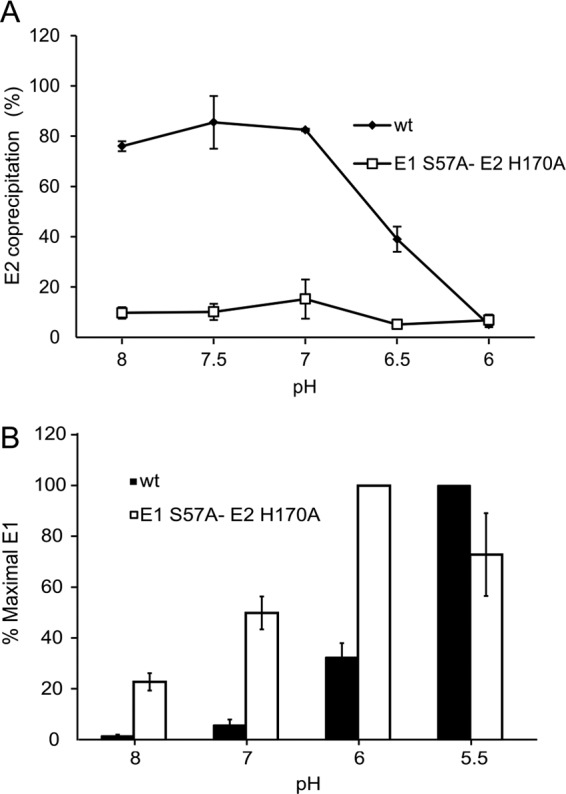
pH dependence of WT and S57A-H170A dimer dissociation. (A) Purified 35S-labeled WT or S57A-H170A virus was treated at the indicated pH values for 10 min on ice. Samples were solubilized in 1% NP-40, adjusted to pH 8.0, and immunoprecipitated using MAb to E1. Samples were analyzed by SDS-PAGE and quantitated by phosphorimaging. The ratio of E2 signal to E1 signal was determined for each pH point and represented as percentage of E2 coprecipitated. Data represent averages and ranges of the results of 2 independent experiments. (B) Purified 35S-labeled WT (black bars) or S57A-H170A (white bars) virus was treated at the indicated pH for 5 min at 37°C. Samples were adjusted to neutral pH and immunoprecipitated with MAb to the E1 fusion loop. Samples were analyzed by SDS-PAGE and quantitated by phosphorimaging and the results shown as the percentage of maximal immunoprecipitation (ranging from 50% to 80% of total E1). Data represent averages and standard deviations of the results of 3 independent experiments. The difference in immunoprecipitation between the WT and E1 S57A-E2 H170A was statistically significant at pH 8, 7, and 6 (P < 0.05 using a two-tailed t test).
Based on the crystal structure of the E2/E1 dimer, it is proposed that an early step in fusion is the dissociation of the acid-sensitive region, allowing the movement of E2 domain B to expose the E1 fusion loop (8, 9). To determine the role of the E1 S57-E2 H170 interaction in this process, we carried out immunoprecipitations of virus particles with MAb E1f against the E1 fusion loop (Fig. 4B) (26). In WT virus, exposure of the E1 fusion loop showed a pH threshold of ∼6.0 and was maximal at pH 5.5. In contrast, the E1 S57-E2 H170 double mutant showed significant exposure of the E1 fusion loop even at pH 8, and maximal exposure at pH 6. Our data thus indicate that the E1 S57-E2 H170 interaction is important in controlling the early stages of dimer dissociation during membrane fusion.
Characterization of E1 S57K and E1 S57D mutants.
We extended our study by generating two additional substitutions for E1 S57, S57K and S57D. We hypothesized that the positively charged lysine in the E1 S57K mutant might destabilize the dimer even more drastically whereas the negatively charged aspartic acid in the E1 S57D mutant might significantly stabilize the dimer by formation of a salt bridge with E2 H170. Initial characterization showed that neither virus mutant caused secondary infection, and that the mutations inhibited cell surface expression of the E1 but not the E2 envelope protein in infected cells (data not shown). While cell surface transport and assembly of some alphavirus mutants can be rescued by incubation at 28°C (30, 31), these conditions did not rescue E1 S57K or E1 S57D secondary infection or E1 transport (data not shown).
Pulse-chase assays of infected cells were used to define the E1 S57K and E1 S57D transport defects. Results for the mutants were identical, and data for E1 S57K are shown in Fig. 5. Production of mutant virus particles was blocked (Fig. 5A). After a 1-h or 4-h chase, the E2 protein in mutant-infected cells migrated more slowly than that in WT-infected cells. Digestion with N-glycosidase F demonstrated that this difference was due to aberrant E2 glycosylation (data not shown), as previously observed for SFV mutants with impaired E1 transport (32). WT E1 became resistant to Endo H digestion after a 1-h chase, reflecting the traffic of E1 to the medial Golgi compartment (Fig. 5B). In contrast, the mutant E1 proteins remained sensitive to Endo H digestion, suggesting that E1 transport was blocked prior to the medial Golgi compartment. In keeping with previous studies, the WT and mutant E2 proteins remained Endo H sensitive due to retention of a high-mannose carbohydrate chain (33).
Fig 5.
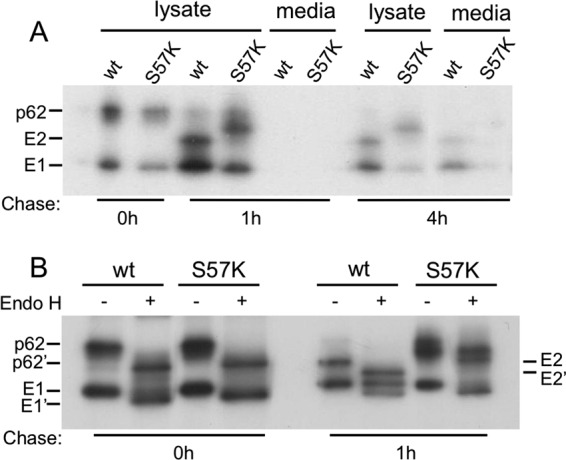
Assembly properties of S57K virus. BHK cells were electroporated with WT or mutant viral RNA and incubated at 37°C for 6 h. The cells were then pulse-labeled for 30 min with [35S]methionine-cysteine and chased for the indicated times at 37°C. (A) The cell lysates and media were immunoprecipitated with a polyclonal antibody to the envelope glycoproteins and analyzed by SDS-PAGE. (B) After immunoprecipitation, an aliquot of the lysates was digested with endoglycosidase H as indicated. p62′, E2′, and E1′ indicate the positions of the protein after Endo H digestion. Data represent the results of 2 independent experiments.
Ammonium chloride treatment of E1 S57K- and E1 S57D-infected cells.
We hypothesized that the lack of cell surface expression of the mutant E1 proteins might reflect the loss of E1's pH protection due to disruption of heterodimer interactions. We therefore tested the effect of neutralization of the secretory pathway by adding NH4Cl to the culture medium 2 h after electroporation of BHK-21 cells with viral RNA. E1 surface expression was significantly rescued in E1 S57K-infected cells, but not detectably increased in E1 S57D-infected cells (Fig. 6A). Pulse-chase analysis showed that NH4Cl treatment decreased production of WT virus particles (Fig. 6B), in keeping with the known effects of NH4Cl on protein secretion (34, 35). Mutant virus particle production was not rescued by NH4Cl treatment of either E1 S57K- or E1 S57D-infected cells (Fig. 6B). However, a soluble truncated form of E1 (E1s) was released from NH4Cl-treated mutant-infected cells. E1s is produced in cells in which budding is inhibited and/or the E2-E1 dimer is destabilized (31, 36, 37). Production of E1s can occur after E1 delivery to the cell surface and requires E1's exit from the ER (36). Thus, taken together, our data suggest that neutralization of exocytic pH promotes transport of the mutant E1 proteins but that heterodimer interactions remain insufficient to support efficient virus budding.
Fig 6.
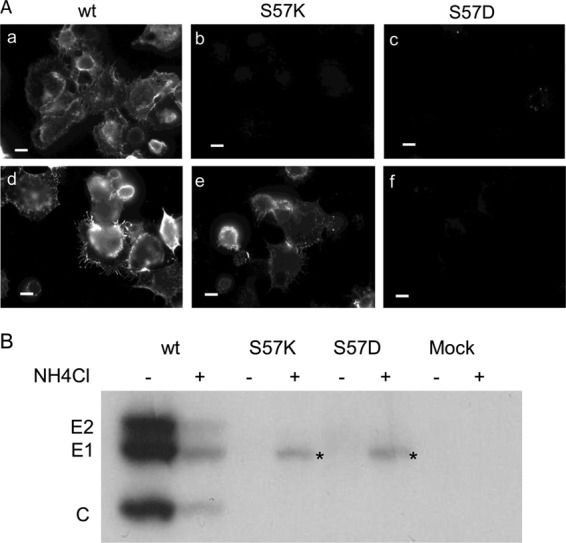
Effect of neutralization of the exocytic pH on S57K- and S57D-infected cells. (A) BHK cells were electroporated with WT or mutant viral RNA and incubated at 37°C for 2 h. Medium without (a to c) or with (d to f) 20 mM ammonium chloride was then added to the cells and the incubation continued for an additional 12 h. Cells were then fixed with paraformaldehyde without permeabilization, and indirect immunofluorescence staining was performed with MAb to E1. Fluorescence microscopy images were acquired with the same exposure time. Data represent the results of 3 independent experiments. Bar = 10 μM. (B) Two hours postelectroporation, medium with or without 20 mM ammonium chloride was added to WT or mutant-infected cells. After 4 h at 37°C, the cells were pulse-labeled for 30 min and incubated in chase medium ± 20 mM NH4Cl for an additional 4 h at 37°C. Chase media were immunoprecipitated with a polyclonal antibody to the envelope glycoproteins in the absence of detergent, and analyzed by SDS-PAGE. Bands labeled with an asterisk represent E1s, a truncated form of E1. Data represent the results of 3 independent experiments.
Isolation of E1 S57K and E1 S57D revertants.
To obtain information about the permissible interactions in the E1 S57 region, we selected for revertants that rescued the lethal phenotype of the E1 S57K and E1 S57D mutants (Table 1). Note that these revertants include pseudorevertants at E1 position 57 and second-site revertants; we refer to them generally here as revertants.
Table 1.
S57D and S57K revertants
| Revertant group | S57 residue | Second site(s) | No. of independent isolates |
|---|---|---|---|
| S57D revertants | A | None | 2 |
| G | None | 2 | |
| G | E1 Q196H | 1 | |
| G | E1 D109Y | 1 | |
| S57K revertants | Q | None | 6 |
| A | None | 1 | |
| S | None | 1 | |
| Sa | None | 1 | |
| N | None | 2 | |
| N | E2 S1I | 1 | |
| N | E2 H170Y | 1 | |
| K | E2 H170Y, M171T | 1 |
Two independent serine revertants were isolated, each with distinct codon changes that differed from the WT virus sequence.
Four distinct revertants were isolated from E1 S57D-infected cells. In each case, E1 D57 mutated to alanine or glycine via a single nucleotide change, with some revertants containing additional second-site mutations. As discussed above, the E1 S57A mutant efficiently assembled and caused secondary infection. Similarly, all of the glycine revertants showed efficient growth kinetics with maximal titers similar to that seen with the WT, and growth was not affected by the second-site mutations E1 Q196H and E1 D109Y (data not shown).
Eight distinct revertants were isolated from E1 S57K-infected cells. These included E1 S57A, as observed above, and a true revertant containing the original E1 S57 residue. K57 also changed to glutamine or asparagine, either of which can hydrogen bond with histidine. Two of the asparagine revertants also contained single second-site mutations in E2, S1I, or H170Y. Importantly, a single revertant maintained the E1 K57 mutation but acquired 2 second-site mutations in E2, H170Y plus M171T. Growth and fusion studies of the glutamine revertant showed that it had properties similar to those of the WT virus (data not shown). In contrast, growth curves showed that the maximal titers of revertants E1 S57N-E2 S1I and E1 S57N-E2 H170Y were ∼2 logs lower than that of the WT, while those of revertants E1 S57N and E2 H170Y plus M171T were ∼3.5 logs lower than that of the WT (data not shown). We therefore focused on characterizing the phenotypes of the asparagine and E2 H170Y-plus-M171T revertants.
Maturation and fusion properties of revertants.
The revertant E1 S57N-E2 S1I contains a second-site mutation at position +1 of the furin cleavage site. A serine at this position was shown to promote furin processing in Sindbis virus, while an isoleucine inhibited processing (38). Pulse-chase analysis was used to test the p62-processing phenotype of all three E1 S57N revertants plus the E2 H170Y-plus-M171T revertant (Fig. 7). Processing of p62 was undetectable for the E1 S57N-E2 S1I revertant, whereas WT SFV and the other three revertants showed efficient processing. We then determined the pH dependence of virus fusion for these revertants (Fig. 8). WT SFV had a pH threshold of ∼6.0 and showed decreased fusion below pH 6.0 due to acid inactivation. The pH thresholds of the E1 S57N and E1 S57N-E2 H170Y revertants were similar to that of the WT, suggesting that dimer stability was restored in these revertants. In contrast, the E1 S57N-E2 S1I revertant had a strongly acid-shifted phenotype, with maximal fusion at pH 5.5 and more than 50% fusion seen at a pH as low as 4.5. This phenotype is in keeping with the p62 processing defect in this revertant. The E2 H170Y-plus-M171T revertant showed a pH threshold of 5.5 and maximal fusion at pH 5.0. Thus, even in the presence of the E1 S57K substitution, the second-site mutations in the E2 protein of this revertant stabilize the E2-E1 dimer to produce a fusion threshold lower than that of WT virus.
Fig 7.
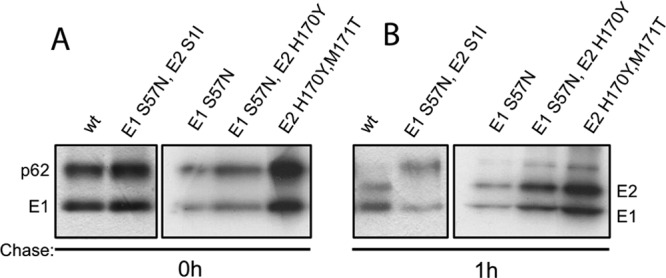
p62 processing of S57K revertants. BHK cells were infected with WT or mutant virus at a multiplicity of infection of 1 PFU/cell for 90 min at 37°C. After a 5-h incubation, cells were pulse-labeled for 30 min with [35S]methionine-cysteine and chased for 0 (A) or 1 (B) h. Cell lysates were immunoprecipitated with a polyclonal antibody to the envelope glycoproteins and analyzed by SDS-PAGE. Revertants are referred to by the amino acid in the wt SFV sequence, its position, and the revertant amino acid replacement. Data represent the results of 2 independent experiments.
Fig 8.
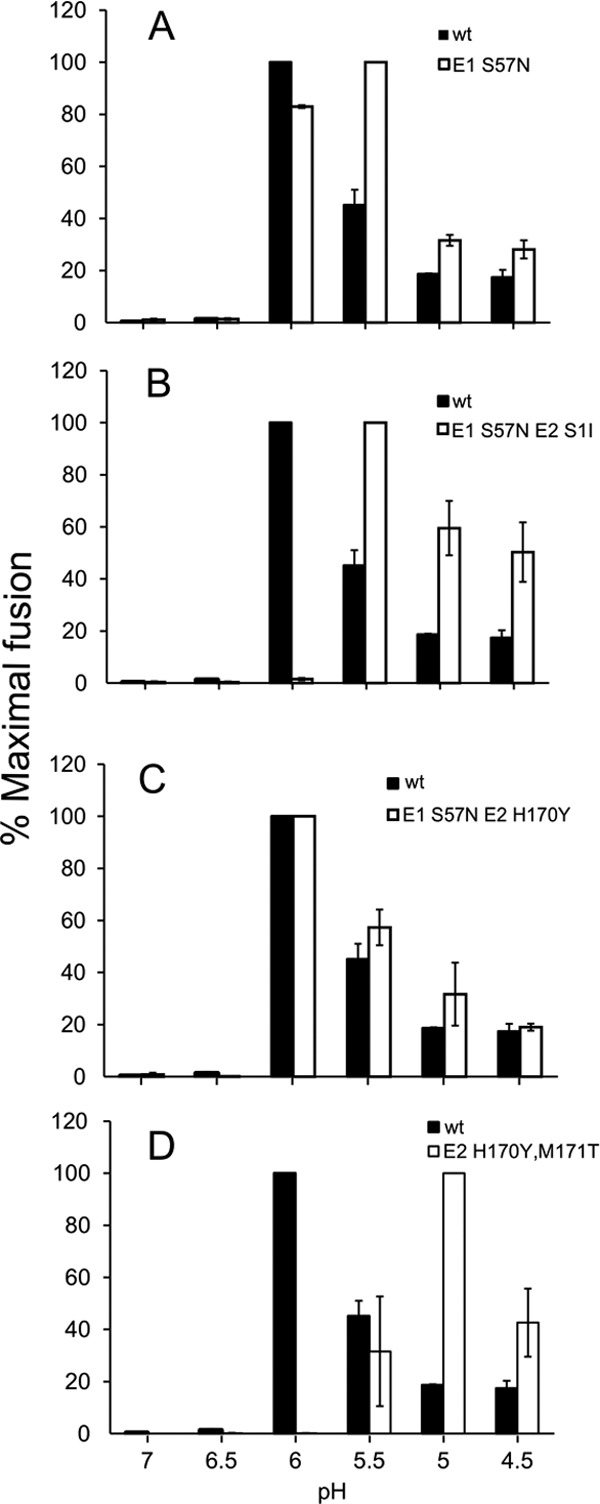
pH dependence of fusion of S57K revertant viruses. Virus stocks were produced in BHK cells. Serial dilutions of virus were adsorbed to BHK cells for 90 min on ice. Virus-plasma membrane fusion was triggered by treatment at the indicated pH at 37°C for 3 min. Virus-infected cells were then quantitated by immunofluorescence and the results shown as the percentage of maximal fusion for each virus. Data represent averages and standard deviations of the results of 3 independent experiments.
DISCUSSION
Together, our studies delineate the important role of the E1 S57-E2 H170 interaction in the low-pH-mediated regulation of alphavirus fusion. Alanine substitutions of these residues destabilized the E2-E1 heterodimer in coimmunoprecipitation assays, and produced a more basic pH threshold for exposure of the E1 fusion loop on mature virus particles. These mutations also appear to destabilize the immature p62-E1 heterodimer, as evinced by an increased efficiency of infection and a more basic pH threshold for fusion. The more radical E1 substitutions S57K and S57D blocked E1 transport to the cell surface and virus assembly. The E1 transport block could be partially overcome by NH4Cl treatment, although virus particle production was still inhibited. The lethal E1 S57K mutation could be rescued by second-site mutations at E2 H170/M171, confirming the key role of the E1 S57-E2 H170 interaction.
Role of the E2-E1 dimer during alphavirus biogenesis.
The structure of the p62-E1 heterodimer reveals extensive interactions of E2 along the length of E1 DII, with the E1 fusion loop clamped in the groove between E2 domains A and B (9). There are also extensive interactions at the membrane-proximal side of the heterodimer. In particular, the stem region of E1 interacts with E2 through insertion of a strand into E2 domain C (9), and cryo-electron microscopy studies showed that the transmembrane anchors of the envelope proteins associate closely within the virus membrane (39, 40). These various dimeric interactions help to promote the folding and transport of E1, prevent its premature triggering, and mediate the assembly of the dimer into the virus particle (reviewed in references 1, 11, and 14). Processing of p62 severs the furin linker that tethers E3 to E2 and thus allows E3 release at neutral pH (14), but p62 maturation does not cause other significant changes in the overall heterodimer structure or interactions (9).
Our data underscore the multiple roles of p62/E2 during the regulation and biogenesis of the E1 protein. The E1 S57-E2 H170 interaction was important in both the immature and mature heterodimer, affecting the low-pH dependence of dimer dissociation and of fusion. The relatively drastic E1 S57K and S57D substitutions blocked transport of E1 to the plasma membrane. We hypothesized that these more disruptive amino acid changes led to the loss of the pH protection p62 normally provides to E1. This model was supported by the restoration of surface expression of E1 protein and the secretion of E1s observed in NH4Cl-treated cells. Nonetheless, virus production was not rescued by NH4Cl treatment, suggesting that while the mutant E1 was able to escape low-pH inactivation, its dimer interactions were still insufficient to support virus assembly.
Revertant analyses.
We then asked if same-site or second-site mutations could rescue the pH protection and assembly defects of the E1 S57D and S57K mutants (Table 1). All of the S57D revertants contained single nucleotide changes that replaced the aspartate with either alanine or glycine. Although these small apolar residues cannot form hydrogen bonds, our results with both E1 S57A and E1 S57G indicate that they are well tolerated at this position. The S57K revertants were more varied, probably due to the requirement for at least two nucleotide changes for reversion of K to S, G, or A. We isolated two true revertants containing the parental S57 residue. Ten pseudorevertants mutated the lysine to glutamine or asparagine, each of which can act as a donor or acceptor to form a hydrogen bond with histidine. These results indicate that steric matching at this position is not required, since both Q and N are considerably larger than the parental S. Influences of the side-chain rotamer also probably play an important role in determining the efficacy of substitutions at this E1-E2 interface. The Q revertant had growth and fusion properties similar to those of the WT and was independently isolated 6 times. The N revertant grew significantly less efficiently than the WT, although it had only a slightly lower pH dependence for fusion. These results suggest that the E1 N57-E2 H170 interaction may stabilize the dimer more strongly than the E1 S57-E2 H170 interaction, but it may also have other effects during virus fusion or biogenesis. The E1 N57 revertant was also isolated along with the E2 second-site mutation S1I or H170Y. The E2 S1I mutation inhibited p62 cleavage and caused a further acid shift in the pH threshold for fusion. The E1 S57N-E2 H170Y revertant showed a fusion pH dependence similar to that of the WT, although previous results had indicated that the E1 S57-E2 Y170 pair could destabilize the immature heterodimer (29). In combination with E1 S57N, either the E2 S1I or E2 H170Y mutation promoted virus growth that was more efficient than that seen with E1 N57 alone. Thus, the E1 N57 mutation may have arisen first and caused partial rescue, followed by acquisition of the E2 S1I or E2 H170Y mutation.
We isolated one true second-site revertant containing both E1 K57 and the E2 mutations H170Y and M171T. This revertant grew less efficiently than the WT, and its pH threshold for fusion was shifted to ∼5.5. This increased acid stability may have been due to the formation of a hydrogen bond between E1 K57 and the tyrosine at E2 position 170, or to alternative interactions, such as cation-π or hydrophobic interactions between these two residues. Structural studies indicate that the methionine at position 171 in E2 has van der Waals contacts with residues in E3 and points away from E1, suggesting that a threonine at this position is not likely to interact directly with E1 (9).
Stages of alphavirus low-pH-dependent conformational changes.
Even when detailed structural information is available, the identification of specific residues involved in the low-pH dependence of protein conformational changes is often complex (reviewed in reference 41). In the case of alphaviruses, a series of low-pH-triggered conformational changes occurs, starting with dissociation of the E2-E1 heterodimer. Our results support a model in which the dimer is optimally stabilized by a hydrogen bond between E1 S57 and E2 H170, while the absence of this interaction affects the movement of the β-ribbon connector and E2 domain B, and thus changes dimer stability and the pH dependence of E1 fusion loop exposure (8, 9). Note, however, that the control of heterodimer dissociation (and of fusion) is clearly more complicated than this single interaction. For example, our engineered E1 S57A-E2 H170A mutations abrogated the hydrogen bond but produced a change in pH dependence only when p62 cleavage was blocked. The E1 S57N-E2 H170Y revertant had pH dependence for fusion comparable to that of WT SFV, although its reduced growth showed that these substitutions decreased viral fitness. Which other residues might affect the pH-sensitive interactions between E2 and E1? Although their amino acid positions are not conserved among alphaviruses, histidine residues are enriched in the interfaces between E2 and E1, particularly around the fusion loop and membrane-proximal contact sites (8, 9). In addition, the interactions of other residues can also be pH sensitive in this range (reviewed in references 41 and 42). Thus, while our results support the idea of a role for E1 S57 and E2 H170 in the initial uncapping of the E1 fusion loop, they also suggest additional steps in the overall heterodimer conformational change.
The E1 fusion protein independently requires low pH to induce the conformational changes involving its membrane interaction and trimerization (43, 44). Despite their locations in regions of E1 known to rearrange during trimerization (7), alanine substitutions of E1 H125, H133, and H333 did not produce significant effects on the pH dependence of fusion (45). In contrast, a conserved H3 histidine on E1 DI plays an important role in the pH dependence of the E1 refolding reaction. E1 H3 is part of a network of interactions between residues in DI and the DI-DIII linker region, helping to drive the formation of the postfusion trimer (46). Thus, even when the dimer is destabilized, E1's pH dependence probably compensates for this misregulation and helps to prevent fusion at neutral pH.
Here, our results indicate that several potential hydrogen bond pairs can substitute for the important interaction between E1 S57 and E2 H170. In the overall life cycle of the virus, however, the highly conserved serine-histidine pair has apparently been evolutionarily selected to provide the right balance of dimer stability. The heterodimer needs to be optimized to permit the complex to fold and assemble correctly, and to promote the conformation of the dimer needed for budding. Conversely, the dimer needs to be able to readily dissociate once it reaches the low pH of the endosome during entry. This dissociation process is complex and involves both the E1 S57-E2 H170 bond and additional interactions that may regulate the rearrangements of both proteins on the highly organized virus particle during fusion.
ACKNOWLEDGMENTS
We thank all the members of our laboratory for helpful discussions, experimental suggestions, and comments on the manuscript. We thank Jonathan Lai for helpful comments on the manuscript and Sonu Nanda and Youqing Xiang for excellent technical assistance.
The data in this paper are from a thesis to be submitted by W.F. in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.
This work was supported by a grant to M.K. from the National Institute of Allergy And Infectious Diseases (R01-AI075647) and by Cancer Center Core Support grant NIH/NCI P30-CA13330. W.F. was supported in part by the Training Program in Cellular and Molecular Biology and Genetics, T32 GM007491.
The content of this paper is solely our responsibility and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Published ahead of print 16 January 2013
REFERENCES
- 1. Kuhn RJ. 2007. Togaviridae: the viruses and their replication, p 1001–1022 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 1 Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 2. Enserink M. 2007. Infectious diseases. Chikungunya: no longer a third world disease. Science 318:1860–1861 [DOI] [PubMed] [Google Scholar]
- 3. Schwartz O, Albert ML. 2010. Biology and pathogenesis of Chikungunya virus. Nat. Rev. Microbiol. 8:491–500 [DOI] [PubMed] [Google Scholar]
- 4. Mancini EJ, Clarke M, Gowen BE, Rutten T, Fuller SD. 2000. Cryo-electron microscopy reveals the functional organization of an enveloped virus, Semliki forest virus. Mol. Cell 5:255–266 [DOI] [PubMed] [Google Scholar]
- 5. Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. 2006. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure 14:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lescar J, Roussel A, Wien MW, Navaza J, Fuller SD, Wengler G, Rey FA. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137–148 [DOI] [PubMed] [Google Scholar]
- 7. Roussel A, Lescar J, Vaney M-C, Wengler G, Wengler G, Rey FA. 2006. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 14:75–86 [DOI] [PubMed] [Google Scholar]
- 8. Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712 [DOI] [PubMed] [Google Scholar]
- 10. Helenius A, Kartenbeck J, Simons K, Fries E. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84:404–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibbons DL, Vaney M-C, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325 [DOI] [PubMed] [Google Scholar]
- 13. Zhang X, Fugere M, Day R, Kielian M. 2003. Furin processing and proteolytic activation of Semliki Forest virus. J. Virol. 77:2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sjöberg M, Lindqvist B, Garoff H. 2011. Activation of the alphavirus spike protein is suppressed by bound E3. J. Virol. 85:5644–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. White J, Helenius A. 1980. pH-dependent fusion between the Semliki Forest virus membrane and liposomes. Proc. Natl. Acad. Sci. U. S. A. 77:3273–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Salminen A, Wahlberg JM, Lobigs M, Liljeström P, Garoff H. 1992. Membrane fusion process of Semliki Forest virus II: Cleavage-dependent reorganization of the spike protein complex controls virus entry. J. Cell Biol. 116:349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smit JM, Klimstra WB, Ryman KD, Bittman R, Johnston RE, Wilschut J. 2001. PE2 cleavage mutants of Sindbis virus: correlation between viral infectivity and pH-dependent membrane fusion activation of the spike heterodimer. J. Virol. 75:11196–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wahlberg JM, Boere WAM, Garoff H. 1989. The heterodimeric association between the membrane proteins of Semliki Forest virus changes its sensitivity to low pH during virus maturation. J. Virol. 63:4991–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Akahata W, Nabel GJ. 2012. A specific domain of the Chikungunya virus E2 protein regulates particle formation in human cells: implications for alphavirus vaccine design. J. Virol. 86:8879–8883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. 1995. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 63:82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chanel-Vos C, Kielian M. 2004. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J. Virol. 78:13543–13552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liljeström P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 65:4107–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liao M, Kielian M. 2005. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J. Cell Biol. 171:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Justman J, Klimjack MR, Kielian M. 1993. Role of spike protein conformational changes in fusion of Semliki Forest virus. J. Virol. 67:7597–7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kielian M, Jungerwirth S, Sayad KU, DeCandido S. 1990. Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J. Virol. 64:4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gibbons DL, Ahn A, Liao M, Hammar L, Cheng RH, Kielian M. 2004. Multistep regulation of membrane insertion of the fusion peptide of Semliki Forest virus. J. Virol. 78:3312–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chanel-Vos C, Kielian M. 2006. Second-site revertants of a Semliki Forest virus fusion-block mutation reveal the dynamics of a class II membrane fusion protein. J. Virol. 80:6115–6122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang X, Kielian M. 2005. An interaction site of the envelope proteins of Semliki Forest virus that is preserved after proteolytic activation. Virology 337:344–352 [DOI] [PubMed] [Google Scholar]
- 29. Zhang X, Kielian M. 2004. Mutations that promote furin-independent growth of Semliki Forest virus affect p62-E1 interactions and membrane fusion. Virology 327:287–296 [DOI] [PubMed] [Google Scholar]
- 30. Chatterjee PK, Eng CH, Kielian M. 2002. Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J. Virol. 76:12712–12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Duffus WA, Levy-Mintz P, Klimjack MR, Kielian M. 1995. Mutations in the putative fusion peptide of Semliki Forest virus affect spike protein oligomerization and virus assembly. J. Virol. 69:2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Levy-Mintz P, Kielian M. 1991. Mutagenesis of the putative fusion domain of the Semliki Forest virus spike protein. J. Virol. 65:4292–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hubbard SC. 1988. Regulation of glycosylation. The influence of protein structure on N-linked oligosaccharide processing. J. Biol. Chem. 263:19303–19317 [PubMed] [Google Scholar]
- 34. Neblock DS, Berg RA. 1982. Lysosomotropic agents ammonium chloride and chloroquine inhibit both the synthesis and secretion of procollagen by freshly isolated embryonic chick tendon cells. Biochem. Biophys. Res. Commun. 105:902–908 [DOI] [PubMed] [Google Scholar]
- 35. Paroutis P, Touret N, Grinstein S. 2004. The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda) 19:207–215 [DOI] [PubMed] [Google Scholar]
- 36. Lu YE, Eng CH, Shome SG, Kielian M. 2001. In vivo generation and characterization of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 75:8329–8339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao H, Garoff H. 1992. Role of cell surface spikes in alphavirus budding. J. Virol. 66:7089–7095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heidner HW, Johnston RE. 1994. The amino-terminal residue of Sindbis virus glycoprotein E2 influences virus maturation, specific infectivity for BHK cells, and virulence in mice. J. Virol. 68:8064–8070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang R, Hryc CF, Cong Y, Liu X, Jakana J, Gorchakov R, Baker ML, Weaver SC, Chiu W. 2011. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 30:3854–3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang W, Mukhopadhyay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG. 2002. Placement of the structural proteins in Sindbis virus. J. Virol. 76:11645–11658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Harrison SC. 2008. The pH sensor for flavivirus membrane fusion. J. Cell Biol. 183:177–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sánchez-San Martín C, Liu CY, Kielian M. 2009. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 17:514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Glomb-Reinmund S, Kielian M. 1998. fus-1, a pH-shift mutant of Semliki Forest virus, acts by altering spike subunit interactions via a mutation in the E2 subunit. J. Virol. 72:4281–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klimjack MR, Jeffrey S, Kielian M. 1994. Membrane and protein interactions of a soluble form of the Semliki Forest virus fusion protein. J. Virol. 68:6940–6946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qin ZL, Zheng Y, Kielian M. 2009. Role of conserved histidine residues in the low-pH dependence of the Semliki Forest virus fusion protein. J. Virol. 83:4670–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zheng Y, Sanchez-San Martin C, Qin ZL, Kielian M. 2011. The domain I-domain III linker plays an important role in the fusogenic conformational change of the alphavirus membrane fusion protein. J. Virol. 85:6334–6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeLano WL. 2002. The PyMOL user's manual. DeLano Scientific, San Carlos, CA [Google Scholar]