

Abstract
West Nile virus (WNV) is a neurotropic flavivirus that has emerged globally as a significant cause of viral encephalitis in humans. The WNV-induced innate immune response, including production of antiviral cytokines, is critical for controlling virus infection. The adaptor protein ASC mediates a critical step in innate immune signaling by bridging the interaction between the pathogen recognition receptors and caspase 1 in inflammasome complexes, but its role in WNV immunopathogenesis is not defined. Here, we demonstrate that ASC is essential for interleukin-1β (IL-1β) production and development of effective host immunity against WNV. ASC-deficient mice exhibited increased susceptibility to WNV infection, and reduced survival was associated with enhanced virus replication in the peripheral tissues and central nervous system (CNS). Infection of cultured bone marrow-derived dendritic cells showed that ASC was essential for the activation of caspase 1, a key component of inflammasome assembly. ASC−/− mice exhibited attenuated levels of proinflammatory cytokines in the serum. Intriguingly, infected ASC−/− mice also displayed reduced levels of alpha interferon (IFN-α) and IgM in the serum, indicating the overall protective role of ASC in restricting WNV infection. However, brains from ASC−/− mice displayed unrestrained inflammation, including elevated levels of proinflammatory cytokines and chemokines, such as IFN-γ, CCL2, and CCL5, which correlated with more pronounced activation of the astrocytes, enhanced infiltration of peripheral immune cells in the CNS, and increased neuronal cell death. Collectively, our data provide new insights into the role of ASC as an essential modulator of inflammasome-dependent and -independent immune responses to effectively control WNV infection.
INTRODUCTION
West Nile virus (WNV) is a neurotropic, enveloped, positive-strand RNA virus belonging to the family Flaviviridae and is related to other globally important viruses, such as dengue, Japanese encephalitis, and tick-borne encephalitis viruses (1). Though the number of cases of WNV infection reported to the Centers for Disease Control and Prevention declined between 2007 and 2011, there was an explosive increase in the cases of WNV infection in 2012 (5,387 cases, including 243 deaths, reported as of December 2012). WNV infection remains subclinical in most humans; however, ∼20 to 30% of patients develop symptoms of WNV disease ranging from fever and mild headaches to severe meningoencephalitis, including cognitive dysfunction, seizures, and flaccid paralysis (1, 2). Up to 70% of the WNV neuroinvasive disease survivors experience persistent neurological deficits for several months after infection (3). Viral neuropathogenesis is not completely understood, and there is no specific therapy approved for use in humans. In the central nervous system (CNS), neurons are the primary target of WNV replication, and virus-associated pathology is characterized by neuronal death, activation of glial cells, and massive infiltration of leukocytes in the perivascular space and parenchyma (4, 5). The global increase of WNV neuroinvasive disease warrants a greater understanding of the molecular mechanisms associated with virus detection, clearance, and neuroinflammation.
Studies using well-characterized WNV encephalitis mouse models show that WNV infection triggers the innate immune system, resulting in the rapid induction of type I interferons (IFNs) and inflammatory cytokines and chemokines, such as tumor necrosis factor alpha (TNF-α), interleukin-1 (IL-1), and CXCL10, some of which play a crucial role in protection against WNV (6, 7). IL-1β production is reported in human and mice following WNV infection (4, 8–10). WNV-induced migration of the skin Langerhans cells to the draining lymph nodes requires IL-1β, and treatment with an IL-1β-specific neutralizing antibody decreases the total number of immune cells being recruited to the lymph nodes of WNV-infected mice, indicating its role in virus control in the periphery (11).
The innate immune system relies on its capacity to rapidly detect pathogen- and damage-associated molecular patterns (PAMPs and DAMPs) and to eliminate them. Detection of these PAMPs and DAMPs by specific host pattern recognition receptors (PRRs) triggers downstream signaling pathways of innate immunity, which collectively work to restrict virus replication and modulate adaptive immune responses (12, 13). Several PRRs are characterized as recognizing RNA viruses, including Toll-like receptors (TLR), retinoic acid-inducible gene I (RIG-I), and NOD-like receptors containing pyrin domain (NLRPs) (12, 14). Recent reports clearly demonstrate that infection by pathogens can be detected by the PRRs, such as NLRP3 and absent in melanoma 2 (AIM2), resulting in the activation of caspase 1 via a multiprotein complex known as inflammasomes (15). In all these complexes, ASC (apoptosis-associated speck-like protein containing C-terminal caspase recruitment domain [CARD]) provides a link between these PRRs and the proform of caspase 1 (16, 17). Activation of pro-caspase 1 is required for the processing and subsequent secretion of the key proinflammatory cytokines pro-IL-1β and pro-IL-18 into their biologically active forms.
ASC is an adaptor molecule originally identified in the insoluble cytosolic fraction, called speck, of the cells undergoing apoptosis (18). It is composed of an N-terminal PYD domain and a CARD, which interacts directly with multiple PRRs, such as NLRPs, NLR caspase recruitment domain-containing protein (NLRC), and AIM2, to form caspase 1-activating platforms termed inflammasomes (15, 19). Several studies have recently revealed the in vivo role of ASC in different physiological and pathological events associated with inflammation (20). DNA viruses, including vaccinia virus, adenovirus, myxoma virus, and mouse cytomegalovirus (21–23), and RNA viruses, such as influenza A virus, Sendai virus, human respiratory syncytial virus, and encephalomyocarditis virus (EMCV), utilize the inflammasome assembly to induce IL-1β production (12, 24, 25). In another seminal study, Ichinohe et al. reported that ASC inflammasomes play a central role in innate and adaptive immunity to influenza virus (26). It was initially believed that ASC exerts its immune effects by mediating the interaction between NLRs and caspase 1 in inflammasome complexes. However, emerging evidence has indicated several inflammasome-independent roles of ASC in controlling immune responses. For example, host defense in chronic infection with Mycobacterium tuberculosis depends on ASC, but not on NLRP3 or caspase 1 (27). In addition, induction of antigen-specific IgG antibodies to influenza vaccines is impaired in mice deficient for ASC, but not NLRP3 or caspase 1 (28).
Although WNV infection results in IL-1β secretion (8, 10, 11), the specific signaling pathways associated with IL-1β production have not been characterized so far. Since ASC is a common adaptor protein in multiple inflammasome complexes, it is likely to play an important role in regulating IL-1β production and other immune functions in WNV-infected mice. Therefore, the objective of this study was to understand the in vivo role of ASC in host defense mechanisms against WNV infection. Our results describe the requirement for ASC in the production of IL-1β in mice, the absence of which resulted in impaired WNV replication and increased mortality of ASC−/− mice inoculated via the subcutaneous route. Mice that lacked ASC had a higher CNS virus burden and neuronal death following WNV infection, which was associated with altered levels of inflammatory cytokines, IFN-α, and virus-specific IgM. Collectively, these data provide the first evidence of the requirement for ASC as an essential modulator of immunity in vivo and demonstrate that ASC signaling orchestrates inflammasome-dependent and -independent immune responses to effectively control WNV infection.
MATERIALS AND METHODS
Virus and mouse experiments.
Wild-type (WT) C57BL/6 mice were bought from the Jackson Laboratory (Bar Harbor, ME), and ASC−/− mice were obtained from Vishwa Dixit (Genentech, CA) and did not contain a Dock2 mutation (data not shown), as reported in some ASC-deficient mouse lines (29). All mice were bred and genotyped in the animal facility at the John A. Burns School of Medicine, and the experiments were approved and conducted in accordance with the University of Hawaii at Manoa animal studies guidelines. All infections were conducted on 8- to 10-week-old mice by inoculation of 100 PFU of WNV (NY99, isolated from crow brain and passaged once in Vero cells) into either the footpad or cranium, as described previously (30). On specific days after infection, blood was collected by cardiac puncture, and the serum was separated by centrifugation and stored at −80°C. The mice were then extensively perfused with 20 ml cold phosphate-buffered saline (PBS) under anesthesia, and the harvested organs (spleen, kidney, and brain), after flash freezing, were stored at −80°C until further processing. Alternatively, groups of mice were monitored for survival for 21 days after infection.
WNV infection of mouse bone marrow-derived dendritic cells (BMDCs).
Eight-week-old WT and ASC−/− mice were sacrificed, and bone marrow cells were isolated from the hind limbs as described previously (31). For BMDCs, bone marrow cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 20 ng/ml of recombinant granulocyte colony-stimulating factor (GM-CSF) (R&D Systems), and IL-4 (R&D Systems). The cells were fed with fresh medium on days 2 and 4. At day 7, the cells were checked for purity by staining with anti-CD11c (eBioscience) using flow cytometry and infected with WNV at a multiplicity of infection (MOI) of 0.1. Cell lysates were collected at various time points after infection for the Western blot analysis of caspase 1 and IL-1β.
Quantitation of the virus load in blood and tissues.
The virus titers were analyzed in the serum and homogenates of spleen and kidney by plaque assays and by quantitative real-time PCR (qRT-PCR) in the brain, as described previously (30). qRT-PCR was conducted using primers and 6-carboxyfluorescein (FAM)- and 6-carboxytetramethylrhodamine (TAMRA)-labeled probes specific for the WNV envelope region, and the standard curve was generated using RNA extracted from previously titrated WNV dilutions (10,000 to 0.1 PFU), as described by Lanciotti et al. (32). The data are expressed as WNV PFU equivalents/μg of RNA.
Quantitation by qRT-PCR and Western blots.
The mRNA levels of multiple host genes were determined using qRT-PCR, and the fold change in infected brains compared to controls was calculated after normalizing to the GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene (33). The primer sequences and annealing temperatures used for qRT-PCR are listed in Table 1. The expression profiles of multiple cytokines, chemokines, and their receptors in the brain were analyzed using a commercial RT2 Profiler inflammatory cytokine and receptor PCR array (SABiosciences) following the manufacturer's instructions. Briefly, 500 ng of total RNA was reverse transcribed into cDNA using an RT2 mRNA First Strand Kit (Qiagen) according to the manufacturer's instructions. One microgram of total cDNA pooled from two mouse brains from each group was mixed with RT2 SYBR green Fluor qPCR Master Mix (Qiagen). Twenty-five microliters of this mix was loaded in each well of the 96-well plate, and qPCR was carried out on a Bio-Rad iCycler real-time PCR machine. The qPCR data obtained were analyzed using RT2 Profiler PCR Array Data Analysis version 3.5 (SABiosciences). Cycle threshold (Ct) values of >35 were considered to be nonspecific, and the fold change in the brains of infected mice compared to mock-infected mice of the same genotype was calculated after normalizing to the housekeeping genes. The data are expressed as the mean of a total of four animals from two independent arrays for each group. For Western blotting, total cellular protein was extracted from the brain and BMDCs, and 20 to 30 μg protein was separated on SDS-PAGE, transferred onto nitrocellulose membranes, and incubated overnight with polyclonal antibodies against IL-1β, ASC, caspase 1 (Cell Signaling), and IgG (AbCam) as described previously (33). Following incubation with secondary antibodies conjugated with IRDye 800 and IRDye 680 (Li-Cor Biosciences), the membranes were scanned using the Odyssey infrared imager (Li-Cor Biosciences) (30).
Table 1.
Primer sequences used for qRT-PCR
| Gene (ID) | Primer |
|
|---|---|---|
| Forward | Reverse | |
| IL-1β (NM_008361) | CCTCACAAGCAGAGCACAAG | AAACAGTCCAGCCCATACTTTAG |
| Caspase 1 (NM_009807) | GGAAGCAATTTATCAACTCAGTG | GCCTTGTCCATAGCAGTAATG |
| ASC (NM_023258) | CAGCAACACTCCGGTCAG | AGCTGGCTTTTCGTATATTGTG |
| GFAP (NM_010277) | GTGGATTTGGAGAGAAAG | GTATTGAGTGCGAATCTC |
| CD45 (L36091) | GCCCAAACAAATTACACAT | TTAGGCGTTTCTGGAATC |
| CD4 (NM_013488) | GGAAGACTCTCAGACTTAT | GAAGGTCACTTTGAACAC |
| CD8 (NM_009857) | GTAATGAGCAGACTGTAAC | CTATATGATGGGCAGACA |
Measurement of WNV-specific antibodies.
The levels of WNV-specific IgM antibodies were measured in the serum using microsphere immunoassay (MIA) for WNV envelope (E) protein (L2 Diagnostics), as described previously (34, 35). Briefly, serum samples were incubated with magnetic microspheres (MagPlexTM-C) coated with a recombinant WNV E antigen for 30 min, followed by secondary goat anti-mouse IgG or IgM conjugated to red phycoerythrin (Jackson ImmunoResearch) for 45 min. The fluorescence intensity of the microspheres was analyzed with a Luminex 100 instrument (Millipore Corp.).
Measurement of IFN-α, cytokines, and chemokines.
The levels of alpha interferon (IFN-α) were measured in the sera using the VeriKine Mouse Interferon-Alpha ELISA (enzyme-linked immunosorbent assay) Kit (PBL Interferon Source), as described previously (34). The levels of cytokines and chemokines were measured in the sera and brain homogenates by Luminex assay using a Milliplex Map Mouse Cytokine/Chemokine kit (Millipore).
Infiltration of leukocytes in the CNS.
Brains and spleens from three mice per experimental group were isolated, pooled, and homogenized using a Miltenyi gentleMACS cell dissociator. Infiltrated leukocytes from the brains of WT and ASC−/− mice at day 8 after infection were isolated by discontinuous Percoll gradient centrifugation, as described previously (30), washed twice, and counted. Cells were resuspended in fluorescence-activated cell sorter (FACS) buffer and stained for CD45, CD11b, CD3, CD4, and CD8 using directly conjugated antibodies (eBioscience) for 30 min at 4°C and then fixed with 4% paraformaldehyde (PFA) at 4°C for 15 min. Samples were analyzed by multicolor flow cytometry on a FACS Aria, and the data were analyzed with FlowJo software (version 9.4.11), as described previously (30).
Immunohistochemistry.
At day 8 after infection, mice were transcardially perfused with 20 ml of PBS, followed by 20 ml of 4% PFA; brains were harvested, cryoprotected in 30% sucrose (Sigma), and embedded, and frozen sections were cut. Tissue preparation and staining with various primary antibodies were performed as previously described (30, 31). Terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining was performed using an in situ cell death detection kit (Roche) according to the manufacturer's instructions (8). Images were acquired using a fluorescence microscope (Zeiss Axiovert 200). Tissue sections were also stained with anti-GFAP (DakoCytomation), a marker for astrocyte activation; fluorescein isothiocyanate (FITC)-conjugated anti-CD8 (eBiosciences); and biotinylated anti-NeuN (Millipore) (30).
Statistical analysis.
For survival analyses, GraphPad Prism 5.0 was used to perform a Kaplan-Meier log-rank test to compare curves. Comparison of means was carried out with the Mann-Whitney test and unpaired Student t test using Prism 5.0. Differences with P values of <0.05 were considered significant.
RESULTS
Inflammasome components are induced in the brain during WNV infection.
Increased production of IL-1β has been previously established in the sera and brains of WNV-infected mice (4, 9); however, the involvement of the inflammasome complex in IL-1β induction is unknown. Consistent with the previous results, we observed that the processing of pro-IL-1β to its active form increased in the brains of WT mice at day 8 after infection (Fig. 1A). To begin to dissect the in vivo importance of ASC in WNV infection, we first examined IL-1β processing and its relationship with the activation profile of caspase 1 in BMDCs from WT and ASC−/− mice. WNV infection (MOI = 0.1) of BMDCs induced the mRNA expression of IL-1β and caspase 1 at 48 h after infection, and this induction was comparable between WT and ASC−/− mice (Fig. 1B). As seen in Fig. 1C, Western blot analysis showed an increase in the cleaved form of IL-1β in mouse BMDCs upon WNV infection; however, this increase was significantly reduced in the ASC−/− BMDCs. Since the activation of caspase 1 requires inflammasome assembly, we next tested caspase 1 activation in the BMDCs and whether lack of ASC would result in its inhibition. BMDCs recovered from WT mice exhibited a dramatic increase in the P20 subunit of caspase 1 at 48 and 72 h after infection, but this activation was markedly reduced in infected ASC−/− BMDCs, which correlated well with the reduced levels of cleaved IL-1β (Fig. 1D). The fold increase of caspase 1 mRNA did not differ between WT and ASC−/− BMDCs, suggesting that the deficiency of ASC affects the activation of caspase 1 and not transcription of the gene. These results collectively suggest that WNV induced inflammasome assembly and that ASC was required for caspase 1 activation.
Fig 1.

ASC is essential for WNV-induced IL-1β maturation and caspase 1 activation. (A) WT mice were mock infected (M) or infected with 100 PFU of WNV via the footpad, total cell lysates prepared from brains at day 8 (D8) after infection were resolved by SDS-PAGE, and IL-1β maturation was measured by Western blotting. (B) Total RNA extracted from WNV-infected BMDCs (MOI, 0.1) from WT and ASC−/− mice was used for the analysis of mRNAs of IL-1β, ASC, and caspase 1 (Casp-1) at 48 h after infection. The data are normalized to the values for GAPDH and are expressed as the relative fold increase compared to uninfected controls. ND, not detected. (C) Cell lysates (15 to 30 μg) prepared from WNV-infected BMDCs were immunoblotted with antibodies against IL-1β, ASC, and caspase 1. (D) Quantitative analysis of Western blot results. All values are relative to β-actin and are representative of samples from at least two independent experiments. *, P < 0.05 compared to mock-infected controls. The data are expressed as means ± standard deviations.
ASC is required for survival after sublethal WNV challenge.
To characterize the in vivo physiological relevance of the findings depicted in Fig. 1 and the importance of ASC in immune-mediated protection, we assessed the survival of mice deficient in ASC against two different infectious doses of WNV. ASC−/− mice were highly susceptible to WNV infection following subcutaneous challenge with 10 and 100 PFU of virus (Fig. 2). While the infectious dose of 10 PFU (Fig. 2A) resulted in 10 to 15% mortality in WT mice, mortality in ASC−/− mice was 40 to 45% (P < 0.05). Similarly ASC−/− mice inoculated with 100 PFU exhibited significantly higher mortality (85%) than WT mice (55% mortality; P < 0.05) (Fig. 2B). There was no significant difference between the median survival times of infected WT and ASC−/− mice (11 days for a 100-PFU dose).
Fig 2.

Survival analysis of WT and ASC−/− mice following WNV infection. Eight- to 10-week-old WT and ASC−/− mice were inoculated subcutaneously with 10 PFU (A) or 100 PFU (B) of WNV and monitored twice daily for mortality. The data shown are pooled from two independent experiments (n = 19 per group for 10 PFU and n = 36 per group for 100 PFU). The survival difference between WT and ASC−/− mice was statistically significant. *, P < 0.05.
ASC-dependent signaling modulates WNV replication, tissue tropism, and CNS invasion.
To better understand how a deficiency of ASC caused higher mortality following WNV infection, we challenged WT and ASC−/− mice with 100 PFU of WNV via the footpad route, and the viral loads in serum, spleen, and kidney were measured at early (day 2), middle (day 4), and late (day 6) stages of infection in the periphery. As shown in Fig. 3, no statistically significant difference in the WNV titer was observed in the sera from WT and ASC−/− mice at days 2 and 4 after infection, when the viremia is high in the mouse model. However, in the spleen, while 60% of the WT mice had detectable WNV, 90% of the ASC−/− mice showed splenic infection at day 4 after infection. In addition, ASC−/− mouse spleens had significantly higher virus titers than WT mice (Fig. 3). Virus titers assayed in the kidneys also followed a similar trend, and significantly higher virus replication was observed in ASC−/− mice than in WT mice at day 4 after infection (Fig. 3). To understand the consequences of higher virus replication in the peripheral tissues at the level of CNS invasion, virus titers were measured by qRT-PCR in the brains harvested at days 4, 6, and 8 after infection. WNV is typically detected in the CNS of mice between 4 and 6 days after WNV challenge via the footpad. Consistent with this trend, infected WT mice demonstrated a detectable viral load in the brain by day 6 after infection. Virus replication in the brains of ASC−/− mice was slightly higher at day 6 after infection, but the difference between WT and ASC−/− mice was not statistically significant. However, at day 8 after infection, we observed significantly higher levels of WNV in the ASC−/− mouse brains than in WT mice (P < 0.05) (Fig. 3). Thus, the absence of ASC caused increased virus dissemination and replication after subcutaneous inoculation.
Fig 3.

Viral-burden analysis in WT and ASC−/− mice. Eight- to 10-week-old WT and ASC−/− mice were inoculated subcutaneously with 100 PFU of WNV, and the viral loads in the serum, spleen, and kidney were measured by plaque assay using Vero cells. The WNV copy number in the brain was determined by qRT-PCR. Each data point represents an individual mouse from two independent experiments. The data for mock-inoculated mice were negative for WNV and are not shown on the graphs. The horizontal dotted lines denote the detection limit of the plaque assay. The solid horizontal lines signify the median of 7 to 10 mice per group. *, P < 0.05. D, day.
Serum cytokine responses are blunted in ASC−/− mice.
Given that inflammasome activation is a critical step in IL-1β maturation, we compared the serum IL-1β levels between WT and ASC−/− mice. The production of IL-1β was completely abolished in ASC−/− mice, suggesting that WNV induces IL-1β in an ASC-dependent manner (Fig. 4A). Since IL-1β is postulated to modulate the expression of multiple inflammatory molecules, we next assessed the levels of downstream cytokines and chemokines in the sera of WT and ASC−/− mice. We did not observe any differences in the basal cytokine and chemokine levels in the sera from uninfected WT and ASC−/− mice (Fig. 4). However, compared to WT mice, infected ASC−/− mice produced significantly lower levels of proinflammatory cytokines, such as IL-6 and IFN-γ, at days 2 and 4 after infection (Fig. 4B and C). On the other hand, the comparison of levels of serum chemokines, such as CCL2 (MCP-1), CCL5 (RANTES), and CXCL1 (MIP-2), between WT and ASC−/− mice showed no significant differences in their levels at any time point after infection (Fig. 4D to F). These results demonstrate that the absence of ASC results in reduced systemic cytokine response to WNV infection in the periphery.
Fig 4.

Impaired levels of cytokines in the sera from WNV-infected ASC−/− mice. Sera were collected from mock (M)- and WNV-infected WT and ASC−/− mice (100 PFU via the footpad) at days 2, 4, and 6 after infection. Levels of cytokines IL-1β, IL-6, and IFN-γ (A to C) and chemokines CCL2, CCL5, and CXCL1 (D to F) were measured using a multiplex Luminex assay and expressed as the mean concentrations (pg/ml) ± standard errors of the mean (SEM) and are representative of two independent experiments (n = 7 per group). *, P < 0.05; **, P < 0.001.
Antiviral responses are altered in ASC−/− mice.
The robust production of type I IFN is required to restrict WNV dissemination in the periphery and entry into the CNS (6). Since IL-1β is not directly involved in regulating type I IFN levels, we predicted that the deficiency of ASC would not lead to differences in IFN-α levels between WT and ASC−/− mice. However, as demonstrated in Fig. 5A, we observed a trend of slightly reduced levels of IFN-α in the sera from ASC−/− mice at day 2 after infection, which became statistically significant at day 4 after infection. Similarly, virus-specific IgM and IgG antibody responses are critical for the clearance of WNV, and previous studies have indicated an inflammasome-independent role of ASC in adaptive immunity (28). We measured the levels of WNV-specific antibodies in WT and ASC−/− mice. Serum IgM levels were elevated in the WT mice at days 6 and 8 after infection; however, this response was markedly blunted in ASC−/− mice (Fig. 5B) (P < 0.05). In contrast, the difference in the serum IgG responses between WT and ASC−/− mice was not statistically significant (data not shown), indicating that ASC produces an inflammasome-independent induction of isotype-specific antibody response to WNV.
Fig 5.
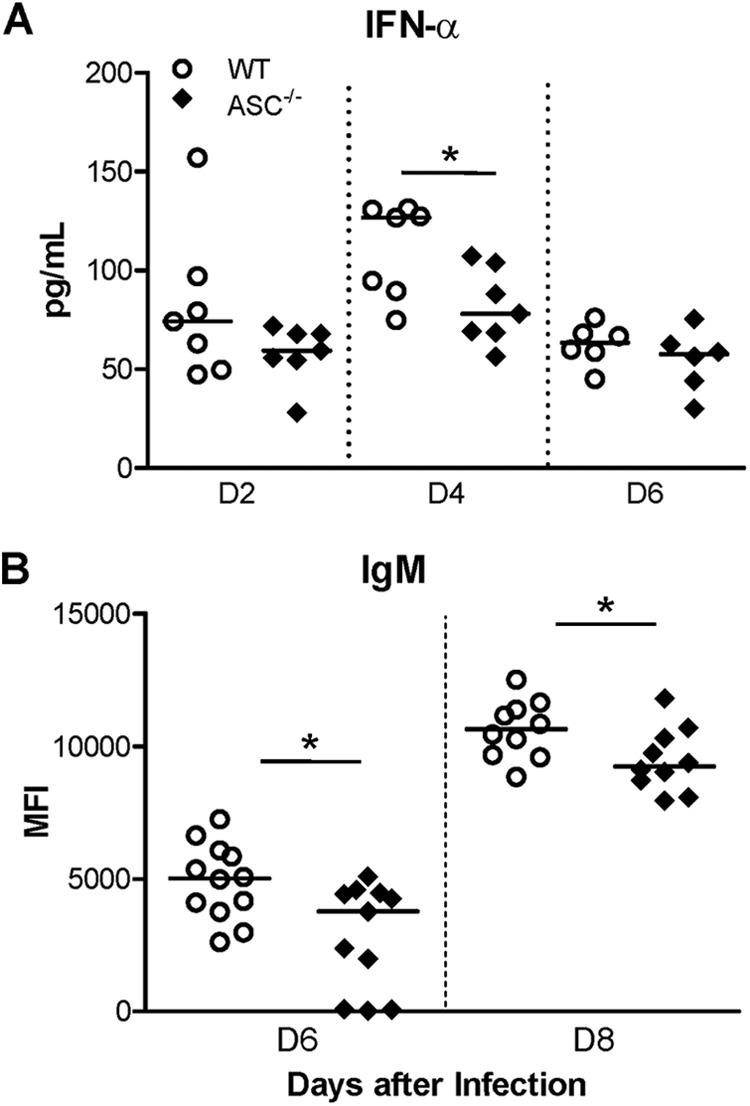
WNV-specific immune responses are impaired in the sera from ASC−/− mice. (A) IFN-α levels in the sera from WT and ASC−/− mice following WNV challenge were measured on the indicated days after infection using a mouse IFN-α ELISA kit. The data depicted are representative of two independent experiments. The solid horizontal lines signify the median of seven mice per group. *, P < 0.05. (B) Sera were collected from WT and ASC−/− mice at the indicated time points, and the WNV-specific IgM response was measured by MIA using WNV E antigen. The values represent the median fluorescence intensities (MFI) of individual infected mice minus the mean MFI ± 3 standard deviations of the respective mock group (n = 8). The data are depicted as scattered points representing individual mice, with the median represented by the horizontal lines, and are representative of two independent experiments (n = 10 to 12). *, P < 0.05.
ASC−/− mice exhibit increased inflammation in the CNS.
Given that the deficiency of ASC affected the proinflammatory cytokine response in the periphery, we assessed the levels of multiple inflammatory cytokines and chemokines in the brain homogenates from WT and ASC−/− mice following WNV inoculation via the footpad. As expected, IL-1β levels increased significantly in the homogenates from WT mouse brains at day 8 after infection, but this increase was absent in the brains of ASC−/− mice (Fig. 6A). The levels of TNF-α and IFN-γ were greatly elevated in the brains of WT mice at day 8 after infection; however, this increase was more pronounced and statistically significant in ASC−/− mice (P < 0.05) (Fig. 6B and C). Moreover, while the chemokine responses in the sera from WT and ASC−/− mice did not differ, the levels of important chemokines, such as CXCL1, CCL2, CCL5, and CCL3 (MIP-1), were significantly higher in the brains of ASC−/− mice at day 8 after infection (Fig. 6E to H). The increase of proinflammatory mediators in the brain at the mRNA level was also validated using inflammation pathway-specific arrays. As shown in Table 2, mRNA changes observed in the brains correlated well with the protein levels of cytokines and chemokines in the brain homogenates at day 8 after infection. Compared to WT mice, the mRNA fold increase of chemokines such as CXCL1, CCL2, CCL5, and CCL3 and their corresponding receptors, CCR1, -4, and -7, were markedly elevated in the brains of ASC−/− mice.
Fig 6.

Enhanced production of cytokines and chemokines in the brains of ASC−/− mice after WNV infection. Brains were harvested from mock (M)- and WNV-infected WT and ASC−/− mice at days 4, 6, and 8 after infection, and homogenized brain lysates were used to measure the levels of multiple cytokines (A to D) and chemokines (E to H), expressed as the mean concentrations (pg/ml) ± SEM, using a multiplex Luminex assay. The data are representative of two independent experiments (n = 7 per group). *, P < 0.05.
Table 2.
Fold changes in the mRNA levels of inflammatory molecules in the brains of WNV-infected WT and ASC−/− mice
| Gene | mRNA fold changea |
|
|---|---|---|
| WT | ASC−/− | |
| Casp1 | 5.01 ± 0.35 | 2.64 ± 0.45 |
| Ccl2 (MCP-1) | 85.92 ± 10.20 | 588.13 ± 31.61 |
| Ccl3 (MIP-α) | 6.17 ± 0.67 | 55.72 ± 2.52 |
| Ccl4 (MIP-β) | 23.02 ± 1.21 | 84.45 ± 4.82 |
| Ccl5 (RANTES) | 52.89 ± 4.23 | 90.51 ± 10.50 |
| Ccr1 | 7.09 ± 0.42 | 9.85 ± 1.16 |
| Ccr4 | 1.17 ± 0.04 | 3.48 ± 0.30 |
| Ccr7 | 1.77 ± 0.16 | 6.06 ± 0.76 |
| Cxcl10 (IP-10) | 641.36 ± 13.34 | 831.75 ± 37.13 |
| Cxcl11 (IP-9) | 9.35 ± 0.85 | 45.25 ± 0.4 |
| IFN-γ | 11.51 ± 1.51 | 18.38 ± 1.98 |
| IL-10 | 18.70 ± 2.64 | 10.56 ± 1.05 |
| IL-4 | 3.80 ± 0.41 | 1.87 ± 0.05 |
| TNF-α | 13.22 ± 1.1 | 22.63 ± 2.13 |
The data are expressed as means ± standard deviations for a total of four animals from two independent arrays for each group.
Enhanced inflammation associates with increased neuropathology in ASC−/− mice.
Enhanced virus replication and neuroinflammation in the brain is directly linked to increased neuronal death and activation of glia. As ASC−/− mice exhibited increased virus replication and inflammatory responses in the brain, we hypothesized that the decreased-survival phenotype of ASC−/− mice could be due to increased neuronal death. To assess this, brain sections from WT and ASC−/− mice at day 8 after infection were evaluated for the presence of TUNEL-positive cells. WT and ASC−/− mice showed TUNEL-positive cells within the cortex, hippocampus, or cerebellum region. However, the numbers were significantly higher in the brains of ASC−/− mice (Fig. 7). Similarly, compared to the WT mice, increased immunoreactivity to GFAP, a marker of the activation of astrocytes, was more pronounced in the brains from the ASC−/− mice (Fig. 7D to G). Analysis of the mRNA level of GFAP by qRT-PCR further demonstrated an 8-fold increase in ASC−/− mice compared to the 2.2-fold increase in WT mice. In addition, the extravasation of peripheral IgGs in the CNS as a marker of blood-brain barrier (BBB) disruption was analyzed in the WT and ASC−/− mice at day 8 after inoculation. As shown in Fig. 7H, both WT and ASC−/− mouse brains had significant extravasation of systemic IgG compared to mock controls at day 8 after infection; however, the difference between WT and ASC−/− mice was not statistically significant (Fig. 7I).
Fig 7.

Increased neuropathology in the brains of ASC−/− mice. (A and B) A TUNEL assay was conducted on PFA-fixed brain sections from WNV-infected WT (A) and ASC−/− (B) mice at day 8 after infection to evaluate neuronal apoptosis. The photomicrographs show representative images obtained from two independent experiments (n = 4 per group). (C) Quantitative representation of TUNEL-positive cells from 15 different brain areas per section from two independent experiments (n = 4 per group). The number of TUNEL-positive cells was significantly higher in the ASC−/− mouse brain. *, P < 0.05. The data are expressed as means ± standard deviations. (D and E) PFA-fixed brain sections from WNV-infected WT (D) and ASC−/− (E) mice at day 8 after infection were stained for GFAP, a marker for activated astrocytes. The immunoreactivity of GFAP was higher in WNV-infected ASC−/− mice. White arrows indicate activated astrocytes. (F and G) The sections from mock-infected WT (F) and ASC−/− (G) mice did not show reactivity for GFAP. The photomicrographs show representative images obtained from two independent experiments (n = 4 per group). (H) Brain lysates were resolved by SDS-PAGE, and Western blotting was conducted to detect heavy and light chains of IgG as a marker of increased BBB permeability. (I) Quantitative analysis of Western blot results. All values are relative to β-actin and are representative of four mice per group from at least two independent experiments.
Effect of ASC on leukocyte infiltration in the CNS.
Another hallmark of WNV encephalitis is the accumulation of inflammatory immune cells in the brain (5). Since chemokines in the brain modulate trafficking of leukocytes, we next assessed the total number of leukocytes in the WT and ASC−/− mouse brains. While no difference was observed between the total leukocytes in the WT and ASC−/− spleens, they increased by >2-fold in the brains from ASC−/− mice (Fig. 8A and B). To better characterize this response, we analyzed the numbers and phenotypes of different cell populations in WT and ASC−/− mice by flow cytometry. ASC−/− mouse brains had significantly higher numbers of CD45+ and CD11b+ macrophages than WT mouse brains (Fig. 8C and D). Additionally, the total number of CD3+ CD4+ and CD3+ CD8+ T cells in the brains of ASC−/− mice was also >3-fold higher than in WT mouse brains (P < 0.01) (Fig. 8E to G). To verify these results, we also measured the levels of CD45+, CD4+, and CD8+ cells in the brain by qRT-PCR analysis. Consistent with the flow cytometry results, compared to the WT mice, the fold increase of these genes was higher in ASC−/− mouse brains (Fig. 8H). To further confirm the ability of CD8+ T cells to traffic across the perivascular space, brain sections from ASC−/− mice were also immunostained for CD8+ T cells. Figure 8I demonstrates that infiltrated CD8+ T cells were able to cross the BBB and perivascular space and traffic to the neurons. These results suggest that the absence of ASC exacerbates leukocyte trafficking and accumulation in the brain, thereby contributing to increased neuroinflammation and neuronal death.
Fig 8.

Enhanced neuroinflammation is associated with increased leukocyte infiltration in the brains of ASC−/− mice during WNV infection. WT and ASC−/− mice were infected subcutaneously with 100 PFU of WNV, and brains and spleens were harvested after extensive perfusion at day 8 after infection. (A and B) Leukocytes were recovered from brains (A) and spleens (B), and absolute cell counts were determined. (C to F) Total numbers of specific inflammatory cells, microglia/infiltrating macrophages (CD45+) (C) and CD11b+ (D), CD3+ CD4+ (E), and CD3+ CD8+ (F) T cells, were determined by flow cytometry. The data represent the means and SEM for six mice per group from two experiments (*, P < 0.05). (G) Representative flow cytometry profile of CD3 and CD8 staining of brain leukocytes from WT and ASC−/− mice at day 8 after infection. PE, phycoerythrin; APC, allophycocyanin. (H) Total RNA extracted from the brains of WT and ASC−/− mice at day 8 after infection via the footpad was used for qRT-PCR analysis of CD45, CD4, and CD8 mRNAs. The data represent the means and SEM for at least four mice per group. (I) To determine the ability of CD8+ T cells to traffic to the neurons, the brain sections of WNV-infected ASC−/− mice were costained for CD8+ T cells using anti-CD8–FITC (a), for neurons using biotinylated anti-NeuN antibody (b), and for nuclei using DAPI (4′,6-diamidino-2-phenylindole) (blue) (c). Representative photomicrographs from the same field show that CD8+ T cells (green; white arrows) are able to cross the BBB and the perivascular space to traffic to the neurons (red). The arrowhead indicates a blood capillary.
ASC does not control virus replication after intracranial infection of WNV.
To further understand whether ASC played a direct role in limiting virus replication in the CNS, we inoculated WT and ASC−/− mice with 100 PFU of WNV via the intracranial route and monitored the virus burden on days 2, 4, and 6 after infection. Intracranial infection with WNV resulted in rapid replication in the brain, and the titers reached 108 PFU/μg of RNA by day 6 after infection. However, there was no difference between the virus titers in WT and ASC−/− mouse brains (Fig. 9), suggesting that ASC does not directly control virus replication in the CNS tissues.
Fig 9.

Virus replication kinetics in the brains of WT and ASC−/− mice following intracranial inoculation. Eight- to 10-week-old WT and ASC−/− mice were inoculated intracranially with 100 PFU of WNV, and the brains were harvested at days 4, 6, and 8 after infection. WNV copy numbers in the brains were determined by qRT-PCR. Each data point represents an individual mouse, and data from two independent experiments are depicted. The solid horizontal lines signify the median of four mice per group. *, P < 0.05.
DISCUSSION
ASC is a key adaptor protein of multiple inflammasome complexes and has emerged as an important mediator of the inflammatory response during host defense in several infection scenarios. Upon detection of the viral PAMPs or DAMPs, PRRs such as NLR proteins bind to the PYD domain of ASC, which then recruits pro-caspase 1 through its CARD to the inflammasome complex, ultimately processing pro-IL-1β to mature IL-1β. WNV infection induces IL-1β production (9); however, the precise molecular mechanisms associated are not clearly understood. We initiated this study because mouse BMDC experiments demonstrated a significant increase in the cleavage of pro-IL-1β into its active form. Caspase 1 is a central component of the inflammasome; therefore, its activation in WT BMDCs is the first indication of formation of the inflammasome complex during WNV infection. Our finding that ASC is required for caspase 1 activation and IL-1β maturation (Fig. 1C) further implies that ASC is a critical component of the WNV-induced inflammasome and that the production of IL-1β is dependent on ASC.
Here, we report the molecular and cellular immune responses in ASC−/− mice following WNV infection and demonstrate that the expression of ASC is crucial for virus clearance and survival in the mouse model of WNV disease. The route of virus inoculation is known to influence the host immune response, thereby affecting WNV replication kinetics. Infection via the subcutaneous route stimulates early host responses in the periphery, resulting in an effective and balanced innate immune response that restricts WNV dissemination (36). On the other hand, intracranial inoculation induces rapid replication of virus and inflammation in the CNS. The lack of a significant difference in WNV levels in the brains of WT and ASC−/− mice after intracranial inoculation (Fig. 9) suggests that in the absence of ASC, WNV is not efficiently cleared from the periphery. This notion is further supported by several experimental observations. First, increased virus burden in the peripheral tissues, such as the spleen and kidney, indicates impaired clearance of WNV in the periphery in ASC−/− mice. Second, reduced cytokine (IL-1β, IL-6, and IFN-γ) and IFN-α levels in the sera from ASC−/− mice suggest an impaired innate intracellular immune response in the periphery, and finally, decreased levels of IgM further identify the role of ASC in modulating adaptive immunity during WNV infection. Collectively, our data suggest that ASC-dependent signaling functions primarily to establish a balanced and protective innate immune response during WNV infection.
Restricted inflammation is essential to clear pathogens and trigger an effective adaptive immune response. IL-1β is a critical cytokine that initiates cascades of events, such as amplification of the inflammatory response, modulation of T-helper 1-associated responses, and activation of endothelial cells (37). IL-1β production involves two discrete signals: signal 1 induces gene expression of pro-IL-1β, and signal 2 triggers its processing via inflammasome assembly (38). Our data demonstrate reduced caspase 1 cleavage in ASC−/− BMDCs (Fig. 1) despite comparable levels of IL-1β and caspase 1 mRNAs in WT and ASC−/− BMDCs following WNV infection, suggesting that the deficiency of ASC affects signal 2 of IL-1β production. IL-1β promotes inflammation by inducing important secondary cytokines, such as IL-6, IFN-γ, and COX-2 (39). While IFN-γ possesses antiviral activity, IL-6 governs antibody production and activation of T cells. Therefore, a possible explanation for the attenuated inflammatory response in the periphery (Fig. 4) could be the reduced IL-1β production in the ASC−/− mice following WNV infection. The requirement for inflammasome activation for an adequate inflammatory response has also been shown for influenza A virus, where deficiency of caspase 1 resulted in decreased levels of IL-1β, IL-6, and TNF-α (40). Another interesting observation of our study is the significantly reduced production of IFN-α in the ASC−/− mice (Fig. 6A). The robust induction of type I IFN via the TLR3 and RIG-I signaling pathway is a major hallmark of host antiviral response to WNV (6). While recent studies by Guarda et al. demonstrate inhibition of IL-1β maturation and inflammasome activation by type I IFN (41), the role of ASC in governing IFN-α/β levels has not been reported previously. The mechanisms underlying the reduced IFN-α levels in the infected ASC−/− mice is unclear; however, it is likely that ASC may indirectly affect IFN-α/β production by interacting with other PRRs involved in the IFN signaling cascade.
Accumulating evidence of the role of ASC in innate immunity has led to studies describing its function outside the realm of the inflammasome. ASC has been identified as the regulator of NF-κB and MAPK/ERK-2 activity in murine and human monocytes/macrophages (42, 43). The disease progression in the mouse model of autoimmune arthritis is attenuated in mice lacking ASC, but not in caspase 1−/− mice (44, 45). Further, Ichinohe et al. reported that nasal IgA and systemic IgG responses against influenza virus required ASC, but not NLRP3 (26). In another study, the absence of ASC significantly reduced H5N1-specific IgG antibody responses after vaccination with MF59-adjuvanted influenza vaccines, while they were intact in NLRP3−/− and caspase 1−/− mice (28). These findings allow important new insights into the role of ASC in adaptive immunity and suggest that ASC-dependent signals are required to elicit adequate IgM response in mice following WNV challenge.
One of the most intriguing findings of our study was the difference in the inflammatory responses observed in the serum and brain. While the deficiency of ASC did not affect chemokine responses in the serum, they were significantly higher in the ASC−/− brains (Fig. 4 and 6). Likewise, IFN-γ levels, which were reduced in the serum, increased significantly in the brains of ASC−/− mice. One explanation for this observation is that CNS cells may respond differently to the deficiency of ASC. Several studies have demonstrated, astrocytosis, gliosis, and activation of astrocytes and microglia as a hallmark of WNV-CNS infection in mouse and human studies (30, 46–48). Activated astrocytes, along with microglia and infiltrating macrophages, have been associated with producing inflammatory mediators in several neuroinflammatory scenarios (49). We and others have previously shown that WNV-infected and activated astrocytes can produce high levels of cytokines and chemokines (10, 50). Thus, it is possible that more pronounced activation of astrocytes observed in infected ASC−/− brains (Fig. 7) may result in an increased chemokine and cytokine response in the brain; however, the possibility of infiltrating leukocytes and microglia contributing to increased inflammation cannot be ruled out. Although infiltration of leukocytes, specifically CD4+ and CD8+ T cells, in the CNS is critical for clearance of WNV, our results (Fig. 8) suggest that increased infiltration of immune cells is not enough to efficiently clear the virus in the brains of ASC−/− mice. It has been shown previously that increased CD8+ T cells contribute to immunopathology with high doses of WNV challenge (51). Further, recent studies by Ramos et al. have also demonstrated a positive correlation between increased WNV replication in the CNS and increased total and antigen-specific CD8+ T cells within the CNS of IL-1r−/− mice compared to WT mice at day 10 after WNV infection (9). Therefore, it is likely that increased chemokine responses and leukocyte recruitment in the brains of ASC−/− mice may contribute to severe immunopathology, such as increased TUNEL-positive apoptotic cells (Fig. 7). This study did not address the specific PRR involved in the activation of the ASC inflammasome; however, the increased mortality in WNV-infected NLRP3−/− mice observed by Ramos et al. indicates the role of NLRP3 in mediating protective immunity against WNV (9). Moreover, based on prior studies reporting detection of RNA viruses by NLRP3 (24, 26), it is likely that WNV detection by NLRP3 recruits ASC and mediates IL-1β production via caspase 1 activation.
Collectively, the results from this as well as other studies suggest that the role of inflammasomes in the host defense against RNA viruses is complex. The inflammasome activity was protective against influenza virus; however, despite the ability of the inflammasomes to detect EMCV or vesicular stomatitis virus, WT and caspase 1-deficient mice were equally susceptible to infection with both viruses (24). To our knowledge, our data for the first time demonstrate a critical role of ASC during WNV infection. We propose that ASC coordinates a well-regulated innate and adaptive immune response to WNV, ultimately restricting immunopathogenesis during infection. Considering the contribution of ASC to both inflammatory and antiviral IgM responses in mice, it may play an equally profound role in host protection upon WNV infection in humans. Future studies are warranted to understand the mechanisms underlying the role of ASC in modulating adaptive immune responses, and they will have important implications for the design of an ideal formulation for future WNV vaccines.
ACKNOWLEDGMENTS
This study was partially supported by grants from the Hawaii Community Foundation (20050405), the Centers of Biomedical Research Excellence (P20GM103516), the National Institute of General Medical Sciences, the National Institutes of Health (NIH), and institutional funds.
We thank Miyoko Bellinger, Histopathology Core staff of the Research Centers in Minority Institutions Program (G12MD007601), National Institute on Minority Health and Health Disparities, NIH, for assistance with histology. We thank Genentech, CA, for providing ASC−/− mice and Michael S. Diamond for providing the positive control and protocol for Dock2 genotyping. We also thank Melissa Valdez for helping with the manuscript preparation and Alexandra Gurary and Madhuri Namekar for technical assistance with the flow cytometry and antibody assays, respectively.
Footnotes
Published ahead of print 9 January 2013
REFERENCES
- 1. Hayes EB, Gubler DJ. 2006. West Nile virus: epidemiology and clinical features of an emerging epidemic in the United States. Annu. Rev. Med. 57:181–194 [DOI] [PubMed] [Google Scholar]
- 2. Diamond MS. 2009. Progress on the development of therapeutics against West Nile virus. Antiviral Res. 83:214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA, Fleischauer A, Leis AA, Stokic DS, Petersen LR. 2003. Neurologic manifestations and outcome of West Nile virus infection. JAMA 290:511–515 [DOI] [PubMed] [Google Scholar]
- 4. Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM. 2005. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 202:1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. 2005. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J. Virol. 79:11457–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Diamond MS, Gale M., Jr 2012. Cell-intrinsic innate immune control of West Nile virus infection. Trends Immunol. 33:522–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shrestha B, Zhang B, Purtha WE, Klein RS, Diamond MS. 2008. Tumor necrosis factor alpha protects against lethal West Nile virus infection by promoting trafficking of mononuclear leukocytes into the central nervous system. J. Virol. 82:8956–8964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kumar M, Verma S, Nerurkar VR. 2010. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J. Neuroinflammation 7:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale M., Jr 2012. IL-1beta Signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog. 8:e1003039 doi:10.1371/journal.ppat.1003039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Verma S, Kumar M, Nerurkar VR. 2011. Cyclooxygenase-2 inhibitor blocks the production of West Nile virus-induced neuroinflammatory markers in astrocytes. J. Gen. Virol. 92:507–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Byrne SN, Halliday GM, Johnston LJ, King NJ. 2001. Interleukin-1beta but not tumor necrosis factor is involved in West Nile virus-induced Langerhans cell migration from the skin in C57BL/6 mice. J. Investig. Dermatol. 117:702–709 [DOI] [PubMed] [Google Scholar]
- 12. Kanneganti TD. 2010. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 10:688–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamkanfi M, Dixit VM. 2009. Inflammasomes: guardians of cytosolic sanctity. Immunol. Rev. 227:95–105 [DOI] [PubMed] [Google Scholar]
- 14. Kawai T, Akira S. 2006. Innate immune recognition of viral infection. Nat. Immunol. 7:131–137 [DOI] [PubMed] [Google Scholar]
- 15. Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10:417–426 [DOI] [PubMed] [Google Scholar]
- 16. Bryan NB, Dorfleutner A, Rojanasakul Y, Stehlik C. 2009. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 182:3173–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martinon F, Mayor A, Tschopp J. 2009. The inflammasomes: guardians of the body. Annu. Rev. Immunol. 27:229–265 [DOI] [PubMed] [Google Scholar]
- 18. Taniguchi S, Sagara J. 2007. Regulatory molecules involved in inflammasome formation with special reference to a key mediator protein, ASC. Semin. Immunopathol. 29:231–238 [DOI] [PubMed] [Google Scholar]
- 19. Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, Sagara J. 1999. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 274:33835–33838 [DOI] [PubMed] [Google Scholar]
- 20. Yamamoto M, Yaginuma K, Tsutsui H, Sagara J, Guan X, Seki E, Yasuda K, Akira S, Nakanishi K, Noda T, Taniguchi S. 2004. ASC is essential for LPS-induced activation of procaspase-1 independently of TLR-associated signal adaptor molecules. Genes Cells 9:1055–1067 [DOI] [PubMed] [Google Scholar]
- 21. Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J. 2008. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452:103–107 [DOI] [PubMed] [Google Scholar]
- 22. Rahman MM, McFadden G. 2011. Myxoma virus lacking the pyrin-like protein M013 is sensed in human myeloid cells by both NLRP3 and multiple Toll-like receptors, which independently activate the inflammasome and NF-kappaB innate response pathways. J. Virol. 85:12505–12517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rathinam VA, Jiang Z, Waggoner SN, Sharma S, Cole LE, Waggoner L, Vanaja SK, Monks BG, Ganesan S, Latz E, Hornung V, Vogel SN, Szomolanyi-Tsuda E, Fitzgerald KA. 2010. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 11:395–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajan JV, Rodriguez D, Miao EA, Aderem A. 2011. The NLRP3 inflammasome detects encephalomyocarditis virus and vesicular stomatitis virus infection. J. Virol. 85:4167–4172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Segovia J, Sabbah A, Mgbemena V, Tsai SY, Chang TH, Berton MT, Morris IR, Allen IC, Ting JP, Bose S. 2012. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One 7:e29695 doi:10.1371/journal.pone.0029695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. 2009. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 206:79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McElvania Tekippe E, Allen IC, Hulseberg PD, Sullivan JT, McCann JR, Sandor M, Braunstein M, Ting JP. 2010. Granuloma formation and host defense in chronic Mycobacterium tuberculosis infection requires PYCARD/ASC but not NLRP3 or caspase-1. PLoS One 5:e12320 doi:10.1371/journal.pone.0012320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ellebedy AH, Lupfer C, Ghoneim HE, DeBeauchamp J, Kanneganti TD, Webby RJ. 2011. Inflammasome-independent role of the apoptosis-associated speck-like protein containing CARD (ASC) in the adjuvant effect of MF59. Proc. Natl. Acad. Sci. U. S. A. 108:2927–2932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ippagunta SK, Malireddi RK, Shaw PJ, Neale GA, Walle LV, Fukui Y, Green DR, Lamkanfi M, Kanneganti TD. 2012. Addendum: defective Dock2 expression in a subset of ASC-deficient mouse lines. Nat. Immunol. 13:701–702 [DOI] [PubMed] [Google Scholar]
- 30. Roe K, Kumar M, Lum S, Orillo B, Nerurkar VR, Verma S. 2012. West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 93:1193–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Verma S, Hoffmann FW, Kumar M, Huang Z, Roe K, Nguyen-Wu E, Hashimoto AS, Hoffmann PR. 2011. Selenoprotein K knockout mice exhibit deficient calcium flux in immune cells and impaired immune responses. J. Immunol. 186:2127–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lanciotti RS, Kerst AJ, Nasci RS, Godsey MS, Mitchell CJ, Savage HM, Komar N, Panella NA, Allen BC, Volpe KE, Davis BS, Roehrig JT. 2000. Rapid detection of West Nile virus from human clinical specimens, field-collected mosquitoes, and avian samples by a TaqMan reverse transcriptase-PCR assay. J. Clin. Microbiol. 38:4066–4071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Verma S, Kumar M, Gurjav U, Lum S, Nerurkar VR. 2010. Reversal of West Nile virus-induced blood-brain barrier disruption and tight junction proteins degradation by matrix metalloproteinases inhibitor. Virology 397:130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar M, Roe K, Nerurkar PV, Namekar M, Orillo B, Verma S, Nerurkar VR. 2012. Impaired virus clearance, compromised immune response and increased mortality in type 2 diabetic mice infected with West Nile virus. PLoS One 7:e44682 doi:10.1371/journal.pone.0044682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Namekar M, Kumar M, O'Connell M, Nerurkar VR. 2012. Effect of serum heat-inactivation and dilution on detection of anti-WNV antibodies in mice by West Nile virus E-protein microsphere immunoassay. PLoS One 7:e45851 doi:10.1371/journal.pone.0045851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Samuel MA, Diamond MS. 2006. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J. Virol. 80:9349–9360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dinarello CA. 2005. Interleukin-1beta. Crit. Care Med. 33:S460–S462 [DOI] [PubMed] [Google Scholar]
- 38. Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH, van de Veerdonk FL, Ferwerda G, Heinhuis B, Devesa I, Funk CJ, Mason RJ, Kullberg BJ, Rubartelli A, van der Meer JW, Dinarello CA. 2009. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113:2324–2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dinarello CA. 2005. Blocking IL-1 in systemic inflammation. J. Exp. Med. 201:1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, Doherty PC, Kanneganti TD. 2009. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 30:566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, Farlik M, Decker T, Du Pasquier RA, Romero P, Tschopp J. 2011. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 34:213–223 [DOI] [PubMed] [Google Scholar]
- 42. Khweek AA, Amer A. 2010. Replication of Legionella pneumophila in human cells: why are we susceptible? Front. Microbiol. 1:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taxman DJ, Holley-Guthrie EA, Huang MT, Moore CB, Bergstralh DT, Allen IC, Lei Y, Gris D, Ting JP. 2011. The NLR adaptor ASC/PYCARD regulates DUSP10, mitogen-activated protein kinase (MAPK), and chemokine induction independent of the inflammasome. J. Biol. Chem. 286:19605–19616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kolly L, Busso N, Palmer G, Talabot-Ayer D, Chobaz V, So A. 2010. Expression and function of the NALP3 inflammasome in rheumatoid synovium. Immunology 129:178–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD. 2010. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J. Immunol. 184:4610–4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kelley TW, Prayson RA, Ruiz AI, Isada CM, Gordon SM. 2003. The neuropathology of West Nile virus meningoencephalitis. A report of two cases and review of the literature. Am. J. Clin. Pathol. 119:749–753 [DOI] [PubMed] [Google Scholar]
- 47. Petzold A, Groves M, Leis AA, Scaravilli F, Stokic DS. 2010. Neuronal and glial cerebrospinal fluid protein biomarkers are elevated after West Nile virus infection. Muscle Nerve 41:42–49 [DOI] [PubMed] [Google Scholar]
- 48. Szretter KJ, Samuel MA, Gilfillan S, Fuchs A, Colonna M, Diamond MS. 2009. The immune adaptor molecule SARM modulates tumor necrosis factor alpha production and microglia activation in the brainstem and restricts West Nile Virus pathogenesis. J. Virol. 83:9329–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ovanesov MV, Ayhan Y, Wolbert C, Moldovan K, Sauder C, Pletnikov MV. 2008. Astrocytes play a key role in activation of microglia by persistent Borna disease virus infection. J. Neuroinflammation 5:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. van Marle G, Antony J, Ostermann H, Dunham C, Hunt T, Halliday W, Maingat F, Urbanowski MD, Hobman T, Peeling J, Power C. 2007. West Nile virus-induced neuroinflammation: glial infection and capsid protein-mediated neurovirulence. J. Virol. 81:10933–10949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang Y, Lobigs M, Lee E, Mullbacher A. 2003. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J. Virol. 77:13323–13334 [DOI] [PMC free article] [PubMed] [Google Scholar]