

Abstract
The mitogen-activated protein (MAP) kinase extracellular signal-regulated kinase 5 (ERK5) plays a crucial role in cell proliferation, regulating gene transcription. ERK5 has a unique C-terminal tail which contains a transcriptional activation domain, and activates transcription by phosphorylating transcription factors and acting itself as a transcriptional coactivator. However, the molecular mechanisms that regulate its nucleocytoplasmatic traffic are unknown. We have used tandem affinity purification to identify proteins that interact with ERK5. We show that ERK5 interacts with the Hsp90-Cdc37 chaperone in resting cells, and that inhibition of Hsp90 or Cdc37 results in ERK5 ubiquitylation and proteasomal degradation. Interestingly, activation of cellular ERK5 induces Hsp90 dissociation from the ERK5-Cdc37 complex, leading to ERK5 nuclear translocation and activation of transcription, by a mechanism which requires the autophosphorylation at its C-terminal tail. Consequently, active ERK5 is no longer sensitive to Hsp90 or Cdc37 inhibitors. Cdc37 overexpression also induces Hsp90 dissociation and the nuclear translocation of a kinase-inactive form of ERK5 which retains transcriptional activity. This is the first example showing that ERK5 transcriptional activity does not require kinase activity. Since Cdc37 cooperates with ERK5 to promote cell proliferation, Cdc37 overexpression (as happens in some cancers) might represent a new, noncanonical mechanism by which ERK5 regulates tumor proliferation.
INTRODUCTION
Mitogen-activated protein kinases (MAPKs) are a family of protein serine/threonine (Ser/Thr) kinases that transduce a wide range of extracellular stimuli into intracellular responses, and are activated in response to growth factors and different forms of stress. Phosphorylating a broad range of substrates, MAPKs regulate many cellular functions, including cell proliferation, differentiation, metabolism, and apoptosis (1, 2). In mammals, four subfamilies of conventional MAPKs have been characterized: extracellular signal-regulated kinases 1 and 2 (ERK1/2), c-Jun N-terminal kinases (JNK), p38, and ERK5 (3).
ERK5 is activated in response to a wide range of growth factors and oxidative and hyperosmotic stresses (4, 5). ERK5 phosphorylates several transcription factors, including the members of the myocyte enhancer factor family, MEF2A, -C, and -D (4–6), and is required for epidermal growth factor (EGF)-induced cell proliferation and progression through the cell cycle (7).
ERK5 and its upstream activator, mitogen-activated protein kinase kinase 5 (MEK5), were independently cloned by different groups (8, 9). MEK5 activates ERK5 by dual phosphorylation of the Thr-Glu-Tyr (TEY) motif within the activation loop, and three findings indicate that MEK5 is the only kinase that activates ERK5: (i) MEK5 and ERK5 specifically interact with each other but not with other MAPKs (9); (ii) targeted deletion of the ERK5 and MEK5 genes renders identical phenotypes, with mice dying around embryonic day 10.5, due to angiogenic failure and cardiovascular defects (10–13); and (iii) in MEK5−/− MEF cells, EGF and stress fail to activate ERK5 but not other MAPKs (13).
ERK5 is twice the size of the other mammalian MAPKs (816 amino acids [aa] for human ERK5). ERK5 has a kinase domain located in the N-terminal half of the protein, homologous to the ERK2 kinase domain. In contrast with other MAPKs, ERK5 has a unique C-terminal tail that contains a transcriptional activation domain (residues 664 to 789 of the human protein [14]). Thus, ERK5 is able to activate transcription not only by direct phosphorylation of transcription factors but by acting itself as a transcriptional coactivator, as, for example, in the case for the activator protein 1 (AP-1) transcription factor (15). However, the molecular mechanisms that regulate nuclear-cytoplasmatic localization of ERK5 are unknown, and different cellular localizations, depending on the cell type studied, have been previously reported (16).
Members of the heat shock protein 90 (Hsp90) family are essential molecular chaperones expressed in the cytoplasm of mammalian cells, where they regulate the folding and the maturation of a wide array of proteins, comprising kinases, transcription factors, and steroid hormone receptors (17, 18). Hsp90 family members contain an ATP-binding site, required for the folding and release of the client proteins. Hsp90 inhibitors block the interaction with ATP, inducing the release of client proteins and its subsequent unfolding and degradation by the proteasome (19). Since many Hsp90 clients (such as Akt, Raf, ERBB2, and epidermal growth factor receptor [EGFR]) are proteins which are mutated or deregulated in cancer, Hsp90 inhibitors exhibit anticancer activity. Among them, geldanamycin derivatives such as 17-AGG are in clinical trials for the treatment of a wide variety of cancers (20).
The cochaperone cell division-cycle 37 (Cdc37) promotes the association of Hsp90 with a protein kinase subset of client proteins (21); therefore, some kinases require Hsp90 and Cdc37 chaperones for maintaining their stability. Via its N-terminal domain, Cdc37 first interacts with the catalytic domain of the kinase client, and then its C-terminal domain recruits Hsp90, generating a Cdc37-kinase-Hsp90 ternary complex (22). The superchaperone Hsp90-Cdc37 keeps the client protein kinases either active or in a conformation competent for activation (23). Interestingly, Cdc37 acts as an oncogene by stabilizing other oncogenes which are mutated or overexpressed in cancer cells, and inhibition of the Cdc37 function with compounds such as celastrol represents a new therapy for cancer treatment (24, 25). This compound, by inhibiting the Cdc37-Hsp90 interaction, destabilizes kinase clients and sensitizes tumors to Hsp90 inhibitors (26).
In this report, we provide for the first time biochemical and pharmacological data showing that ERK5 interacts with the superchaperone Hsp90-Cdc37. We also show that the Cdc37-ERK5-Hsp90 complex resides in the cytosol of resting cells and that ERK5 activation results in Hsp90 dissociation and nuclear translocation of the kinase. Unexpectedly, we found that overexpression of Cdc37, an event occurring in several types of cancers, induces nuclear translocation of a catalytically inactive but transcriptionally active form of ERK5.
MATERIALS AND METHODS
Materials.
MG-132, MDL28170, and Z-VAD inhibitors were from Calbiochem. Radicicol, celastrol, G418 antibiotic, N-ethylmaleimide, rabbit IgG-agarose, antihemagglutinin (anti-HA)-agarose, anti-FLAG-agarose, MBP, and EGF were from Sigma; geldanamycin was from Serva; polyethylenimine was from Polysciences; and protein G-Sepharose, glutathione-Sepharose, calmodulin-Sepharose 4B, p81 phosphocellulose paper, and [γ-32P]ATP were from GE Healthcare. Ni2+-nitrilotriacetic acid (NTA)-agarose was from Qiagen. The ERK5 competitive inhibitor XMD2-98 was kindly provided by N. Gray (Dana-Farber Cancer Institute, Boston, MA).
Antibodies.
The polyclonal antibody anti-ERK5 raised in sheep against human protein was from the Division of Signal Transduction Therapy (Dundee, Scotland), and anti-phospho-ERK5-pT218EY220 was from Biosource. Anti-ERK5 rabbit polyclonal antibody, anti-ERK1/2, anti-glutathione S-transferase (anti-GST), anti-FLAG, anti-HA, and antiactin monoclonal antibodies were from Sigma; anti-Cdc37, antiubiquitin, and anti-ERK5 antibody (catalog. no. Sc-5626) were from Santa Cruz Biotechnology; anti-Hsp90β was from Merck Biosciences; and mouse monoclonal antibromodeoxyuridine (anti-BrdU) was from BD Pharmingen. Antibodies to CREB, Akt, and phospho-S473-Akt were from Cell Signaling Technology, and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH) monoclonal antibody was from Ambion. Anti-MEK5 and secondary Alexa Fluor 594-conjugated antibodies were from Invitrogen.
DNA constructs.
Recombinant DNA procedures were performed using standard protocols. We used the pEBGFP-C2-TAP vector previously described (63) to generate the pEBGFP-C2-TAP-ERK5 wild type (pEBGFP-C2-TAP-ERK5wt) and the pEBGFP-C2-TAP-ERK5(1–490) used in this work. The vector encoding human C-terminal FLAG-tagged ERK5 was generated by inserting the human ERK5 full-length coding sequence into a pCMV-Tag4 vector, using the restriction sites BamHI and XhoI. Human ERK5 C terminus (aa 401 to 816) was cloned into the BamHI and NotI sites of pEBG-2T. pCDNA3 vector encoding FLAG-tagged Cdc37 was a gift of C. Scheidereit (Center for Molecular Medicine, Berlin, Germany) (64), pcDNA3.1 vector encoding HA-tagged Hsp90β was from A. Papapetropoulos (University of Athens, Greece) (65), pRBG4 encoding HA-ubiquitin was from S. D. Conzen (University of Chicago) (66), pCMV vector encoding 6×His-tagged ubiquitin was from A. C. Vertegaal (Leiden University, Netherlands) (67), and pEBG-2T vectors encoding GST-tagged human ERK5 (full length and N terminus) (aa 1 to 490) were a gift from P. Cohen (University of Dundee, United Kingdom) (28), whereas those encoding GST-tagged human ERK5 in which residues Ser567, Ser720, Ser731, Thr733, and Ser803 were mutated to Ala (ERK5-5A) or Glu (ERK5-5D) were from A. Cuenda (Centro Nacional Biotecnología, Spain) (36). pCMV plasmid encoding HA-tagged MEK5-DD was from E. Nishida (Kyoto University, Japan) (35). AP-1–luciferase vector was purchased from Stratagene and pRL-CMV-Renilla from Promega.
Cell culture, transfection, and lysis.
Cells were cultured at 37°C under conditions of humidified air (5% CO2). Human HEK293, HeLa, PC-3, and MEF MEK5−/− cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and antibiotics, and human neuroblastoma SH-SY5Y cells were cultured in DMEM F-12 Ham containing 15% FBS and antibiotics. HEK293, HeLa, and PC3 cells were transfected using polyethyleneimine as described before (68). MEF MEK5−/− cells (kindly provided by Cathy Tournier) were transfected using Lipofectamine 2000 and Opti-MEM medium supplemented with 2 mM l-glutamine. Three hours after transfection, the medium was changed to DMEM. Unless otherwise stated, cells were lysed in ice-cold lysis buffer (50 mM Tris-HCl [pH 7.5], 1 mM EGTA, 1 mM EDTA, 1% [wt/vol] NP-40, 1 mM sodium orthovanadate, 10 mM sodium-β-glycerophosphate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 0.27 M sucrose, 0.1% [vol/vol] 2-mercaptoethanol, and complete protease inhibitor cocktail). Lysates were centrifuged at 12,000 × g for 12 min at 4°C, and supernatants were stored at −20°C. Protein concentrations were determined by the Bradford method (69).
Cell proliferation assays.
For cell counting, PC-3 cells were plated in 12-well plates at a density of 105 per well. After 24 h, the cells were cotransfected with plasmids encoding GFP and the indicated proteins (0.33 μg of each plasmid, except for Cdc37, for which 1 μg was used), and cells were counted at 12, 24, 48, 72, and 96 h posttransfection. Briefly, cells were subjected to trypsinization, washed with FBS, and centrifuged at 1,000 × g for 5 min. The pellets were resuspended in phosphate-buffered saline (PBS), and GFP-positive cells were counted in a Beckman Coulter FC 500 flow cytometer. Results are presented as means ± standard deviations (SD) of the results of triplicates of a representative experiment. For BrdU labeling, PC-3 cells plated in 6-well plates were transfected with the indicated plasmids as described above. At 24 h after transfection, cells were incubated with 3 μg/ml BrdU for 16 h, harvested, and processed using an anti-BrdU primary antibody followed by an Alexa Fluor 488–donkey anti-mouse fluorescein isothiocyanate (FITC)-conjugated secondary antibody, according to the protocol of the manufacturer (Becton, Dickinson). Total and BrdU-positive cells were counted in a Beckman Coulter FC 500 flow cytometer.
Cdc37 knockdown (siRNA).
A heterogeneous mixture of small interfering RNAs (siRNAs) that target the same human Cdc37 mRNA sequence was purchased from Sigma (Mission siRNA; EHU104551). HeLa cells were transfected with 1 μg (per well of a six-well plate) of Cdc37 siRNAs or control (scrambled) siRNA.
Subcellular fractionation.
Three 10-cm-diameter dishes of cells were homogenized in 0.5 ml of buffer (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2) in a Potter-Elvehjem homogenizer, left 30 min at 4°C, and centrifuged at 15,000 × g for 15 min at 4°C. Supernatants (cytosolic fractions) were kept at −20°C, and pellets were resuspended with 100 μl of radioimmunoprecipitation assay (RIPA) buffer (25 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.1/% [wt/vol] SDS, 1% [wt/vol] NP-40, 0.5% [wt/vol] deoxycholic acid, 1 mM EGTA, 5 mM sodium pyrophosphate) and homogenized by passing them through a 1-ml syringe with a 25-gauge needle. After centrifugation for 10 min at 20,000 × g, supernatants (nuclear fractions) were saved and kept at −20°C until use.
Immunofluorescence microscopy.
HeLa and MEF cells grown on polylysine-coated coverslips were fixed with 4% paraformaldehyde for 20 min and mounted in medium with 4,6′-diamidino-2-phenylindole (DAPI) for DNA staining. To monitor endogenous ERK5, fixed cells were processed as described before (68) using anti-ERK5 antibody (C terminus; Santa Cruz) and the corresponding fluorescence-labeled secondary antibody. Cells were visualized by fluorescence microscopy in a Nikon Eclipse 90i epifluorescence microscope.
Generation of stable cell lines, tandem affinity purification (TAP), and nanoscale liquid chromatographic tandem mass spectrometry (nLC-MS/MS) analysis.
SH-SY5Y cells cultured in 10-cm-diameter dishes were transfected with 5 μg pEGFP-C2-TAP construct encoding no protein (control), full-length human ERK5, or human ERK5 aa 1 to 490 (N terminus). Transfected cells were selected by adding G418 antibiotic (300 μg/ml) 24 h posttransfection. At 20 days after transfection, individual colonies were visualized, selected, and expanded. Cell lines expressing low levels of GFP were analyzed by immunoblotting to ensure that expressed proteins migrated as a single polypeptide at the expected molecular weight.
For ERK5-TAP, we followed the method described by Al-Hakim et al. (63), which is a modification of the original method (27). For each TAP procedure, 140 dishes (150-mm diameter) were cultured for each stable cell line. Each dish was washed three times with phosphate-buffered saline (PBS) and lysed in 0.5 ml of ice-cold TAP buffer (10 mM Tris-HCl [pH 8], 150 mM NaCl, 0.1% NP-40, and complete protease inhibitor cocktail). Combined lysates were then centrifuged for 45 min at 40,000 × g, and the supernatant was incubated with 0.5 ml of rabbit IgG-agarose beads for 90 min at 4°C. IgG-agarose beads were then washed twice with buffer B (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 0.1% NP-40, and 1 mM dithiothreitol [DTT]), followed by two washes with buffer B containing 0.5 mM EDTA, prior incubation for 4 h at 4°C with 100 μg of tobacco etch virus (TEV) protease. TAP-ERK5 released protein was diluted with 3 vol of buffer C (50 mM Tris-HCl, [pH 8.0], 150 mM NaCl, 0.1% NP-40, 1 mM Mg2+-acetate, 1 mM imidazole, 2 mM CaCl2, and 1 mM DTT) and further incubated with 0.25 ml of calmodulin-Sepharose beads (pre-equilibrated in buffer C) for 1 h at 4°C. Beads were then washed three times with buffer C and twice with buffer C lacking NP-40 detergent. Then, 0.5 ml of 0.0125× NuPAGE-LDS sample buffer (6.8 mM Tris, 0.1% [wt/vol] LDS [lithium dodecyl sulfate], 0.5% [vol/vol] glycerol containing 1 mM DTT [final pH, 8.5]) was added to the washed beads, subjected to a vortex procedure, incubated 15 min at room temperature, and filtered through Spin-X filters (Costar) (0.45 μm pore size). Finally, filtrates containing the eluted proteins were concentrated to 5 μl by the use of a Speed Vacuum, resuspended in 25 μl of H2O Milli-Q containing 10 mM DTT, heated 5 min at 70°C, and subjected to electrophoresis on a precast 4%-to-12% Bis-Tris LDS-polyacrylamide gel (Invitrogen). Resolved proteins were stained with colloidal Coomassie blue (Pierce). All visible bands were excised, washed, and digested with trypsin as previously described (70). Protein digests were identified by LC-MS/MS on a 4000 Q-Trap system (Applied Biosystems) as described previously (71). Database searching was performed using Mascot 2.1 (Matrixscience) run on a local server, using both the Swiss-Prot and Celera human databases.
Immunoprecipitation and immunoblotting.
A 10-μl volume of protein G-Sepharose beads bound to 2 μg of the corresponding antibody was incubated with 0.5 mg of cell lysate for 2 h at 4°C. The immunoprecipitates were washed twice with lysis buffer containing 0.15 M NaCl and twice with buffer A (50 mM Tris-HCl [pH 7.5], 0.1 mM EGTA, 0.1% 2-mercaptoethanol), and the immune complexes were eluted in 2× sample buffer. GST-, HA-, and FLAG-tagged overexpressed proteins were immunoprecipitated as described above, using the appropriate resin. Immunoblotting was performed as described previously (72).
ERK5 ubiquitylation in vivo assay.
We followed the method described by Tatham et al. (73). Dishes (10-cm diameter) of HEK293 cells overexpressing His-tagged ubiquitin and the indicated form of ERK5 were resuspended in 5 ml of buffer 1 (6 M guanidinium-HCl, 10 mM Tris, 100 mM Na2HPO4-NaH2PO4 buffer, pH 8), sonicated for 1 min, and centrifuged at 5,000 × g for 5 min. The resulting supernatants were incubated with 20 μl of Ni2+-NTA-agarose beads for 2 h at room temperature with rotation. Beads were successively washed as follows: twice with 4 ml of buffer 1 plus 10 mM 2-mercaptoethanol; three times with 4 ml of buffer 2 (8 M urea, 10 mM Tris, 10 mM 2-mercaptoethanol, 100 mM Na2HPO4-NaH2PO4 buffer, pH 8); twice with 4 ml of buffer 3 (8 M urea, 10 mM Tris, 100 mM Na2HPO4-NaH2PO4 buffer, pH 6.3) containing 10 mM 2-mercaptoethanol; once with 1 ml of buffer 3 containing 0.2% Triton X-100; once with 1 ml of buffer 3 containing 0.1% Triton X-100 and 0.5 M NaCl; and three times with 1 ml of buffer 3. Finally, proteins were eluted by incubating the beads with 200 mM imidazole in 5% SDS, 0.15 M Tris-HCl, pH 6.7, 30% (vol/vol) glycerol, 0.72 M 2-mercaptoethanol for 1 h at 37°C with mixing.
ERK5 kinase activity assay.
HeLa or HEK293 cell extract (0.5 mg) overexpressing GST-ERK5 and the indicated proteins was incubated for 1 h at 4°C with 10 μl of glutathione-Sepharose beads. Beads were then washed twice with lysis buffer containing 0.15 M NaCl, followed by two washes with buffer A. A kinase activity assay was performed in an assay volume of 50 μl containing glutathione-Sepharose beads, buffer A, 10 mM magnesium acetate, and 0.1 mM [γ-32P]ATP (500 cpm/pmol), with 500 μM PIMtide (ARKKRRHPSGPPTA) or 10 μg MBP as the substrate. Assays were carried out for 45 min at 30°C and terminated by applying the reaction mixture onto p81 paper, and the incorporated radioactivity was measured as described previously (70). The amount of enzyme that catalyzes the phosphorylation of 1 pmol of PIMtide in 1 min was defined as 1 mU of activity.
Reporter luciferase assay.
Cells cultured in 12-well plates were transfected with 650 ng of DNA, which contained 100 ng of AP-1-driven luciferase reporter construct and 50 ng Renilla luciferase, and 100 ng of the indicated amounts of plasmids (except for Cdc37, for which 300 ng was used). After 24 h, a luciferase activity assay was performed using a dual-luciferase kit.
Gel filtration analysis.
Rat brain or human HeLa cells were lysed in lysis buffer and centrifuged at 25,000 × g for 15 min to remove cellular debris and insoluble material. One milligram of each sample was loaded onto a Superdex-200HR-10/30 column on an ÄKTA system and equilibrated in buffer A containing 0.15 M NaCl at a flow rate of 0.3 ml/min, and 0.33-ml fractions were collected. Aliquots of each fraction were analyzed for ERK5 and MEK5 by immunoblotting. The column was calibrated using blue dextran (void volume) and gel filtration standards (Bio-Rad).
Statistical analysis.
Figures, tables, and statistical analyses were generated using Microsoft Excel, Prism 4.0, or Adobe Photoshop software. Statistical significance was determined using one-way analysis of variance (ANOVA) followed by Dunnett's test.
RESULTS
Isolation of ERK5-associated proteins by TAP.
We subjected lysates of rat brain and human SH-SY5Y cells to size exclusion chromatography and immunoblotted the eluted fractions for ERK5 and its upstream activator MEK5. As expected, endogenous rat and human MEK5 protein eluted with a molecular mass of ∼50 kDa (Fig. 1A). However, ERK5 eluted in two ranges: one corresponding to the expected molecular mass of ∼100 to 120 kDa, and another corresponding to a high molecular mass range, above 679 kDa (Fig. 1A). Quantification of the immunoblots revealed that ∼70% (rat brain) and 40% (human SH-SY5Y) of endogenous ERK5 eluted as a high-molecular-mass complex. This chromatographic analysis demonstrates that a pool of endogenous ERK5 exists as a high-molecular-mass protein complex.
Fig 1.

Gel filtration chromatography and tandem affinity purification for ERK5. (A) A 1-mg volume of rat brain or human SH-SYSY cell lysates was loaded onto a Superdex 200HR 10/30 column. A 15-μl volume of each fraction was immunoblotted for EKR5 or MEK5. The elution positions of protein standards are indicated as follows: thyroglobulin (670 kDa), gamma globulin (158 kDa), ovoalbumin (44 kDa), and myoglobin (17 kDa). Vo, void volume. Similar results were obtained in three separate experiments. (B) Tandem affinity purification of ERK5. SH-SY5Y cells stably expressing empty TAP-GFP vector (control), TAP-GFP-ERK5 (full length), or TAP-GFP-ERK5(1–490) were subjected to TAP as described in Materials and Methods. Purified proteins were resolved in 4% to 12% LDS-PAGE gels and visualized by colloidal-Coomassie blue staining. Bands were cut, subjected to in-gel digestion with trypsin, and analyzed by nLC-MS/MS. The designations of the bait proteins used are underlined. (C) Details of mass spectrometry analysis. GANAB, neutral alpha-glycosidase antibody; GRP-94, glucose-regulated protein 94; GRP-78, glucose-regulated protein 78; AtpA, ATP synthase alpha; PP2B, protein phosphatase 2B catalytic subunit; FSCN1, fascin homolog 1; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LDH, l-lactate dehydrogenase; MLCK2, skeletal myosin light-chain kinase 2. Scores refer to Mascot protein scores, and a value >70 was considered significant.
To identify the putative ERK5-interacting proteins, we used TAP (27). We generated human SH-SY5Y cells stably expressing green fluorescent protein (GFP)-TAP empty vector (control) or a GFP-TAP-tagged fusion full-length human ERK5 and an N-terminal form (aa 1 to 490) that contains a functional kinase domain, which can be phosphorylated and activated by MEK5 (28). Clones overexpressing low levels of TAP-ERK5 were selected, expanded, and subjected to tandem affinity purification under mild conditions to preserve the integrity of ERK5 interactors. Purified proteins were resolved by LDS-PAGE (Fig. 1B), and bands were excised, subjected to tryptic digestion, and identified using tandem mass spectroscopy. Twelve interacting proteins were identified for full-length and N-terminal ERK5s which were not detectable in the gel in the control purification, revealing the ERK5 N-terminal half as the region involved in the binding of those putative interactors. Six proteins also detected in the control sample were considered nonspecific contaminants.
Figure 1C summarizes the results obtained, which met the requirement of having three or more peptides matched to our data by the database-searching program. Importantly, four of those proteins correspond to different isoforms of the 14-3-3 protein, a known ERK5-interacting protein (29), thus validating the TAP approach for identification of physiological binding proteins. Of note, heat shock protein 90β rendered the highest peptide score and sequenced peptides covered 33% (full-length ERK5) and 55% [ERK5(1–490)] of the Hsp90β sequence. Therefore, we focused on Hsp90β in the ensuing experiments.
Hsp90β and Cdc37 interact with the N-terminal half of ERK5.
The cochaperone Cdc37 promotes the association of Hsp90 with a protein kinase subset of client proteins; therefore, many kinases require Hsp90 and Cdc37 chaperones for maintaining their stability. To check that Hsp90β and Cdc37 interact with ERK5 in vivo, we performed immunoprecipitation assays of endogenous proteins from SH-SY5Y cells and immunoblotted for each protein. The three proteins were detected in the corresponding ERK5, Hsp90β, and Cdc37 immunoprecipitates but not in control experiments lacking the antibody (Fig. 2A). We obtained identical results for human HeLa cells, mouse embryonic fibroblasts (MEF), and rat brain lysates (Fig. 2B), demonstrating that endogenous ERK5 is associated with Hsp90 and Cdc37 in vivo. We also observed the ERK5-Cdc37-Hsp90 trimeric complex in MEK5−/− MEF-deficient cells (Fig. 2B), indicating that chaperones Hsp90 and Cdc37 do not require MEK5 to interact with ERK5.
Fig 2.

ERK5 associates with Hsp90 and cochaperone Cdc37 through its N-terminal half. (A) One milligram of SH-SY5Y lysates was immunoprecipitated (IP) with anti-Cdc37, anti-ERK5, or anti-Hsp90β antibodies followed by immunoblotting of the immune complexes. (B) One-half milligram of the indicated cells and tissue lysates was immunoprecipitated with anti-ERK5 antibody and immunoblotted as described for panel A. (C to E) HEK293 cells were transfected with 1 μg of each of the indicated expression plasmids, and overexpressed ERK5 and Cdc37 were purified using glutathione-Sepharose and anti-FLAG-agarose resins, respectively. Immune complexes were immunoblotted for ERK5, Hsp90, and Cdc37. Similar results were obtained in three independent experiments.
To identify the region of ERK5 involved in the binding to Hsp90 and Cdc37, we transiently expressed these proteins in human HEK293 cells and performed immunoprecipitation assays. Overexpressed ERK5, Hsp90, and Cdc37 were detected in each of the pellets of the affinity-purified proteins (Fig. 2C). Thus, overexpressed EKR5, Hsp90, and Cdc37 proteins form a trimeric complex, as happens for the endogenous proteins. Next, we performed similar experiments in cells overexpressing ERK5 forms encoding either the N-terminal half (aa 1 to 490) or the C-terminal half (aa 401 to 816), in different combinations with Hsp90β and Cdc37. ERK5 and Cdc37 were subjected to affinity purification and immunoblotting for the three proteins studied. Panels D and E of Fig. 2 show that Hsp90 and Cdc37 interact with the N-terminal half of ERK5 but not with the C-terminal half.
Hsp90 inhibition or Cdc37 silencing induces loss of ERK5 expression.
The importance of Hsp90 for ERK5 stability was addressed by treating cells with two structurally unrelated Hsp90 inhibitors, geldanamycin and radicicol, which inhibit the chaperone function of Hsp90 by binding its ATP-binding pocket. Treatment of SH-SY5Y cells for 24 h with increasing amounts of either geldanamycin or radicicol resulted in a dose-dependent decrease in the expression of ERK5, which was completely abolished at 5 μM concentrations (Fig. 3A), without affecting the expression of the non-Hsp90 client protein kinases ERK1 and ERK2. Identical results were obtained for HeLa cells (results not shown).
Fig 3.

Hsp90 inhibition or Cdc37 silencing induces loss of ERK5 protein by promoting its ubiquitylation and proteasome-mediated degradation. (A) Hsp90 inhibition induces loss of ERK5 expression. SH-SY5Y cells were treated with the indicated concentrations of geldanamycin or radicicol for 24 h (upper panels) or with a 5 μM concentration for the indicated times (lower panels). After the cells were subjected to lysis in buffer containing 1% SDS, protein levels were determined by immunoblotting. (B) Cdc37 silencing induces loss of ERK5 expression. HeLa cells transfected with 1 μg of Cdc37 siRNA or nonspecific siRNA (siINS) were lysed at the times indicated in buffer containing 1% SDS. Lysates were immunoblotted with the indicated antibodies. NT, no transfection; MK, mock transfection. (C) Treatment with the proteasome inhibitor and geldanamycin (GA) results in ERK5 accumulation in insoluble cellular fractions. SH-SY5Y cells were pretreated for 3 h with 20 μM proteasome inhibitor MG-132, 10 μM calpain inhibitor MDL-28170, 10 μM caspase inhibitor Z-VAD, or 20 mM ammonium chloride (inhibitor of lysosomal degradation), followed by 24 h of treatment with 5 μM geldanamycin. Cells were lysed in Nonidet P-40 buffer and centrifuged, and the NP-40 insoluble fraction (pellet) was solubilized in 1% SDS buffer. ERK5 and actin levels were analyzed by immunoblotting. (D and E) Treatment with geldanamycin induces ERK5 ubiquitylation. HEK293 cells overexpressing the indicated proteins were treated with MG-132 alone or with geldanamycin, and His-tagged ubiquitins were purified using Ni2+-NTA-agarose beads and immunoblotted (IB) for ERK5. ERK5 and total ubiquitin expression levels are shown in the middle and bottom panels, respectively. Levels of overexpressed free His-tagged ubiquitin are also shown in panel D. Similar results were obtained in three separate experiments.
To confirm the role of Cdc37 in ERK5 stability, we undertook experiments to silence endogenous Cdc37. Transfection of a siRNA specific for Cdc37 resulted in complete knockdown of Cdc37 at day 5 posttransfection. Cdc37 depletion abolished the expression of ERK5 and Akt without affecting the levels of ERK1/2 (Fig. 3B). Control siRNA affected neither Cdc37 expression levels nor the levels of Akt and ERK5 proteins.
Hsp90 inhibition induces ERK5 ubiquitylation and proteasome-dependent degradation.
Next we studied the effect of inhibitors of proteasome (MG-132), calpain (MDL-128), caspase (Z-VAD), and lysosomal function (ammonium chloride) in preventing the geldanamycin-induced degradation of ERK5. We analyzed the soluble and insoluble fractions of SH-SY5Y cells lysed in NP-40 buffer. In cells treated with geldanamycin, MG-132 prevented the loss of ERK5 expression, resulting in the accumulation in the NP-40 insoluble fraction (Fig. 3C). In contrast, caspase, calpain, and lysosome inhibitors had no effect on geldanamycin-induced loss of ERK5 expression. These findings are in agreement with those obtained for other Hsp90 client proteins, in which Hsp90 inhibition results in the ubiquitylation and proteasomal degradation of the client protein (17, 30–33).
To determine whether ERK5 is ubiquitylated prior to proteasomal degradation, we overexpressed ERK5 and His-tagged ubiquitin in cells that were treated with or without geldanamycin and MG-132. Levels of ubiquitylation were determined by pulling down His ubiquitin with Ni2+-NTA-agarose beads and immunoblotting for ERK5. ERK5 slow-migrating species (corresponding to ubiquitylated forms) were detected in the His-ubiquitin pulldowns from cells treated with MG-132 that were more apparent in those from cells treated with geldanamycin and MG-132 (Fig. 3D). To identify the region of ERK5 that had been ubiquitylated, we set out to perform similar experiments overexpressing the N-terminal or the C-terminal half of ERK5. The N-terminal half, but not the C-terminal tail, became ubiquitylated after treatment of cells with MG-132 at the same level as that used for cells treated with geldanamycin and MG-132 (Fig. 3E). Interestingly, untreated cells also rendered faint bands of slow-migrating (ubiquitylated) species for the N-terminal-half form. These results show for the first time that Hsp90 inhibition results in the ubiquitylation of ERK5 at its N-terminal half, prior to degradation at the proteasome.
Activation of ERK5 causes Hsp90 dissociation from the Cdc37-ERK5 complex, by a mechanism that requires autophosphorylation of the ERK5 C-terminal tail.
Since inactive ERK5 associates with Hsp90 and Cdc37 in resting cells, we next asked if this was also the case for active ERK5. Treatment of HeLa cells with EGF activated ERK5, as measured by both electrophoretic mobility shift and phosphorylation of the T-loop (Fig. 4A, left panels). Unexpectedly, immunoprecipitation assays showed that activation of endogenous ERK5 by EGF resulted in Hsp90β dissociation from the ERK5-Cdc37 complex. To our knowledge, this is the first report showing that activation of a MAPK induces Hsp90 dissociation. As a control for a canonical Hsp90 protein kinase client, the right panels of Fig. 4A show that activation of Akt in response to EGF (measured by monitoring Ser473 phosphorylation [34]) did not induce Hsp90 dissociation.
Fig 4.

ERK5 activation induces Hsp90β dissociation from the ERK5-Cdc37 complex. (A) HeLa cells were starved for 16 h prior to stimulation with EGF. One milligram of lysate was immunoprecipitated with anti-ERK5 (left panels) or anti-Akt (right panels) antibodies, and the immunoprecipitates were immunoblotted for the indicated proteins. (B to D) HeLa cells were transfected with 1 μg of each of the indicated expression plasmids. (B and C) After 24 h, cells were lysed and GST-ERK5 complexes affinity purified and immunoblotted for the indicated protein. Panel B shows the results for ERK5 wt or the kinase-dead mutant (D200A), panel C shows the results obtained for the N-terminal half of ERK5, and panel D shows the results for a form of ERK5 in which the autophosphorylated residues at the C terminus are mutated to Ala (5A) or Glu (5E). Similar results were obtained in four separate experiments.
To check if kinase activity was necessary for Hsp90 dissociation, we overexpressed in cells Hsp90, Cdc37, and ERK5 or a ERK5 mutant which lacks kinase activity (D200A) (28), alone or together with a constitutive active human MEK5 mutant (MEK5DD, in which residues S311 and S315 are replaced by Asp). ERK5 was affinity purified, and associated proteins were visualized by immunoblotting. MEK5DD induced T-loop phosphorylation of the overexpressed-ERK5 wild type and the kinase-inactive mutant, but only the wild-type form showed an electrophoretic mobility shift (Fig. 4B). More importantly, activation of ERK5 resulted in Hsp90 dissociation, whereas the phosphorylated dead kinase still interacted with Hsp90. This suggests the requirement of the ERK5 kinase activity for Hsp90 dissociation. To further study this, we set out to perform similar experiments using the ERK5 N-terminal half, shown to bind Hsp90 and Cdc37 in Fig. 2D. Although MEK5DD induced T-loop phosphorylation of the N-terminal half, it did not induce Hsp90 dissociation (Fig. 4C), indicating that Hsp90 release from the complex requires the C-terminal half of the kinase. Overall, these results suggest the requirement of ERK5 kinase activity for Hsp90 dissociation, perhaps due to the autophosphorylation of the C-terminal tail. Of note, we checked that ERK5 does not phosphorylate Hsp90 or Cdc37 (data not shown), discarding the possibility that phosphorylation of these proteins mediates the dissociation of Hsp90 from the ERK5-Cdc37 complex.
After phosphorylation of the TEY motif by MEK5, ERK5 becomes active and autophosphorylates several residues at the C-terminal tail, inducing a conformational change that results in an open conformation (35). To study the role of ERK5 autophosphorylation of its C-terminal tail, we overexpressed Hsp90, Cdc37, and/or MEK5DD proteins together with ERK5 mutants in which 5 autophosphorylated residues at the C-terminal were mutated to Glu (ERK5-5E, to mimic phosphorylation) or to Ala (ERK5-5A, to prevent phosphorylation). We previously showed in HeLa cells that the wild-type and ERK5-5A forms are cytosolic, whereas ERK5-5E resides in the nucleus, mimicking ERK5 nuclear translocation after activation (36). The ERK5-5A mutant interacted with Cdc37 and Hsp90, even when active MEK5 was coexpressed, whereas in the ERK5-5E mutant, interaction with Hsp90 was lost (Fig. 4D). Taken together, these results show the requirement of the C-terminal autophosphorylation for Hsp90 dissociation from the ERK5-Cdc37 complex.
Active ERK5 is no longer sensitive to Hsp90 or Cdc37 inhibition.
To confirm that active ERK5 does not interact with Hsp90, we used the Hsp90 inhibitor radicicol in cells overexpressing MEK5DD and ERK5. Expression of active endogenous and overexpressed ERK5 (as judged by the electrophoretic mobility shift) is not affected by treating cells with radicicol, at a concentration that abolished the expression of Akt or inactive ERK5 (Fig. 5A). These results further demonstrate that active ERK5 does not associate with Hsp90 and, therefore, that it is no longer sensitive to Hsp90 inhibition.
Fig 5.

Active ERK5 is no longer sensitive to Hsp90 or Cdc37 inhibition. HeLa cells overexpressing GST-ERK5 and MEK5DD (A) or MEK5DD (B) were treated with 5 μM radicicol (A) or celastrol (B) for the indicated times. After the cells were subjected to lysis with buffer containing 1% SDS, the levels of the indicated proteins were monitored by immunoblotting. Similar results were obtained in three independent experiments. endog, endogenous.
We next addressed the role of Cdc37 in ERK5 stability. We used celastrol, a compound that binds Cdc37 and disrupts the Cdc37-Hsp90 complex, leading to the proteasomal degradation of the client protein (37). Celastrol induced a decrease in ERK5 and Akt protein levels in a time-dependent manner, and completely abolished expression of both kinases at 24 h without affecting Hsp90 or nonclient kinases ERK1 and -2 (Fig. 5B). Interestingly, and as seen with Hsp90 inhibitors, celastrol did not induce degradation of active ERK5, at a concentration and time that induced Akt degradation.
Overexpression of high levels of Cdc37 induces Hsp90β dissociation from the ERK5-Cdc37 complex and nuclear translocation of ERK5.
Since Cdc37 is overexpressed in a number of cancers (38), we asked if Cdc37 overexpression had any effect on ERK5 activity or function. To do so, we overexpressed in cells ERK5, Hsp90, and increasing quantities of Cdc37. ERK5 was affinity purified, and associated proteins were visualized by immunoblotting. Figure 6A shows the presence of Hsp90 in pellets from cells cotransfected with 1 μg of plasmid encoding Cdc37 (as shown in Fig. 2C) but not in those from cells cotransfected with 2 or 4 μg of the plasmid encoding Cdc37. Thus, expression of high levels of Cdc37 induces Hsp90 dissociation from the ERK5-Cdc37 complex, similar to what happened when ERK5 became activated in response to EGF stimulation (Fig. 4A). Since EGF also induces ERK5 nuclear translocation (16), we next studied whether this is also the case for Cdc37 overexpression.
Fig 6.

Cdc37 overexpression induces Hsp90 dissociation from the ERK5-Cdc37 complex and nuclear translocation of ERK5. (A) Cells were transfected with 1 μg of plasmids encoding GST-ERK5 and/or Hsp90β and the indicated amounts of plasmid encoding Cdc37. After 36 h, cells were lysed and GST-ERK5 was affinity purified, and precipitated proteins were immunoblotted using the indicated antibodies. The lower panels show the levels of overexpressed proteins (whole lysate). (B to E) HeLa cells were transfected with 1 μg of plasmids encoding the indicated proteins, except for Cdc37, for which 3 μg was used. (B) Cells overexpressing the indicated proteins were fractionated to obtain cytosolic (Cyt) and nuclear (Nuc) fractions, and each fraction was analyzed by immunoblotting. GAPDH and CREB proteins are used as markers of cytosolic and nuclear fractions, respectively. (C) Cells overexpressing GFP-ERK5 and MEK5DD or Cdc37 were fixed, and GFP-ERK5 (green) localization was visualized directly by GFP fluorescence. (D) HeLa cells transfected with the indicated plasmids were fixed and subjected to immunofluorescence staining for endogenous ERK5 (red). (E) HeLa cells overexpressing ERK5 C-terminal autophosphorylation-defective mutant GST-ERK5(5A) and MEK5DD or Cdc37 were fixed and subjected to immunofluorescence staining for GST (green). Nuclei are shown in blue (DAPI). Similar results were obtained in three independent experiments. Bars, 20 μm.
We first studied the cellular localization of exogenous and endogenous ERK5 in HeLa cells overexpressing Cdc37. Subcellular fractionation showed most of the ERK5 in the cytosolic fraction of resting cells, and cells coexpressing MEK5DD showed active ERK5 mainly located at the nuclear fraction (Fig. 6B). Overexpression of Cdc37 induced the nuclear localization of both exogenous and endogenous ERK5 proteins. We further confirmed these results by fluorescence microscopy. Both overexpressed ERK5 and endogenous ERK5 mainly localized throughout the cytoplasm of HeLa cells, while expression of MEK5DD induced ERK5 nuclear translocation (Fig. 6C and D). Consistent with results obtained by subcellular fractionation, overexpression of Cdc37 induced nuclear translocation of endogenous and exogenous ERK5. Finally, overexpression of Cdc37, but not of MEK5DD, induced nuclear translocation of the C-terminal autophosphorylation-defective mutant GST-ERK5(5A) (Fig. 6E).
Cdc37 overexpression enhances ERK5-mediated AP-1 transcriptional activity, without affecting ERK5 kinase activity.
Activation of ERK5 induces its translocation to the nucleus, where, among other effects, it enhances the activity of the AP-1 transcriptional complex (39, 40). We tested if overexpression of Cdc37 and/or Hsp90 had any effect on AP-1 activity using a luciferase-based reporter assay. In HeLa and HEK293 cells, coexpression of ERK5 and MEK5DD enhanced AP-1 activity by ∼25-fold over the levels seen with the control (ERK5 or MEK5DD alone) (Fig. 7A). Interestingly, coexpression of ERK5 with Cdc37, but not with Hsp90, increased AP-1 activity to the same extent as MEK5DD did (Fig. 7A). Overexpression of Hsp90 or Cdc37 had a negligible effect on AP-1 transcriptional activity. It is worth noting that coexpression of Hsp90 abolished the effect on Cdc37-induced AP-1 activity via ERK5, indicating that an excess of Cdc37 over Hsp90 is required to observe the effect. This is probably due to fact that the high levels of Hsp90 would sequester Cdc37 and therefore impair its action directed at the ERK5 protein. Importantly, Cdc37 overexpression also increases the AP-1 transcriptional activity of the ERK5 mutant defective for autophosphorylation of its C-terminal ERK5(5A) (Fig. 7B).
Fig 7.


Cdc37 overexpression induces ERK5-mediated AP-1 transcriptional activity without affecting ERK5 kinase activity. (A and B) Cdc37 overexpression increases ERK5-mediated AP-1 transcriptional activity of ERK5 (A) and ERK5 C-terminal autophosphorylation-defective mutant ERK5(5A) (B). pAP-1–luciferase reporter and pRL-CMV-Renilla plasmids were cotransfected with the indicated plasmids (see Materials and Methods), and 24 h later, lysates were subjected to a dual-luciferase assay. Each value is the mean ± SD of the results of four different determinations, each performed in triplicate, and normalized using the Renilla values. (C) Hsp90β or Cdc37 has no effect on ERK5 kinase activity. Cells overexpressing the indicated proteins were lysed, and GST-ERK5 proteins were affinity purified and assayed for ERK5 activity, using 100 μM [γ-32P]ATP and 500 μM PIMtide or 10 μg MBP as the substrate. The activity data are the means ± SD of the results of three separate experiments, with each determination performed in triplicate. Cell lysates were also immunoblotted for ERK5 and phospho-ERK5. (D) MEK5−/− MEF cells were transfected with plasmid encoding GFP-Cdc37 or the empty vector, and 36 h later, cells were fixed and stained for immunofluorescence for endogenous ERK5 (red). GFP-Cdc37 (green) was visualized directly by GFP fluorescence. Nuclei are shown in blue (DAPI). The white arrow indicates an untransfected cell (that does not overexpress Cdc37) showing cytosolic ERK5. Similar results were obtained in three independent experiments. (E) HeLa cells transfected with plasmids encoding GFP-ERK5 (1 μg) alone or in combination with MEK5DD (1 μg) or Cdc37 (3 μg) were fixed, and GFP-ERK5 was visualized directly by GFP fluorescence. Where indicated, 4 h after transfection, cells were treated with 10 μM XMD8-92 (the ERK5 inhibitor). Similar results were obtained in three independent experiments. (F) pAP-1–luciferase reporter and pRL-CMV-Renilla plasmids were cotransfected in HEK293 cells with the indicated plasmids, and 24 h later, lysates were subjected to a dual-luciferase assay. Where indicated, cells were treated with 10 μM XMD8-92 4 h after transfection. Each value is the mean ± SD of the results of three different determinations, each performed in triplicate, and normalized using the Renilla values. (G) The pAP-1–luciferase reporter and pRL-CMV-Renilla plasmids were cotransfected in HEK293 cells with the ERK5 kinase-dead mutant D200A and/or MEK5DD and/or Cdc37, and 24 h later, lysates were subjected to the dual-luciferase assay. Each value is the mean ± SD of the results of three different determinations, each performed in triplicate.
To check if ERK5 kinase activity mediates the increase of AP-1 transcriptional activity in cells oeverexpressing Cdc37, we expressed ERK5, Cdc37, and Hsp90 proteins in cells and performed kinase assays with affinity-purified ERK5 complexes using two different substrates (MBP and PIMtide). Overexpression of Cdc37 or/and Hsp90 had no effect on ERK5 activity, phosphorylation of the T-loop, or autophosphorylation (Fig. 7C). As a control, ERK5 phosphorylation and activation in response to MEK5DD overexpression are shown.
To further demonstrate that Ccd37 induces nuclear translocation of a catalytically inactive form of ERK5, we used MEK5−/− MEF cells. As MEK5 is the only upstream kinase for ERK5, ERK5 cannot be activated in these cells (13). Cdc37 overexpression induced endogenous ERK5 nuclear translocation in MEK5−/− MEF cells (Fig. 7D). We also used the ERK5 competitive inhibitor XMD8-92, which blocks ERK5 activity and autophosphorylation (41). XMD8-92 did not prevent Cdc37-induced ERK5 nuclear translocation, at a concentration that blocked the ERK5 nuclear entry induced by MEK5 (Fig. 7E). According to those results, XMD8-98 inhibited the AP-1 transcriptional activity induced by active MEK5 but had no significant effect on that induced by Cdc37 overexpression (Fig. 7F). Furthermore, Cdc37 also induced AP-1 transcriptional activity of the ERK5 kinase-dead mutant D200A (Fig. 7G).
All together, our results show that overexpression of Cdc37 induces ERK5 nuclear translocation and enhances ERK5-mediated AP-1 transcriptional activity by a mechanism which does not require ERK5 kinase activity or C-terminal phosphorylation.
Cdc37 cooperates with ERK5 to promote cell proliferation.
As the ERK5 pathway is critical for cell proliferation, we next tested if overexpressed Cdc37 collaborates with ERK5 to promote cell proliferation. We used human prostate cancer PC-3 cells, since prostatic cells that overexpress MEK5 or EKR5 show an enhanced proliferation rate and enhanced efficiency in forming tumors in vivo (42–44). PC-3 cells were cotransfected with plasmids encoding GFP or GFP-ERK5, alone or together with those encoding MEK5DD or Cdc37, and GFP-positive cells were counted at different days in a flow cytometer. No morphological changes between the transfected cells and control cells were apparent (data not shown). Overexpression of MEK5DD or Cdc37 resulted in a slight but significant (P < 0.01) increase in PC-3 cell proliferation after 4 days (Fig. 8A), as previously shown (42, 45). Interestingly, overexpression of ERK5 alone had no effect but its coexpression with MEK5 or Cdc37 resulted in a robust increase (P < 0.001) in cell proliferation (Fig. 8A).
Fig 8.
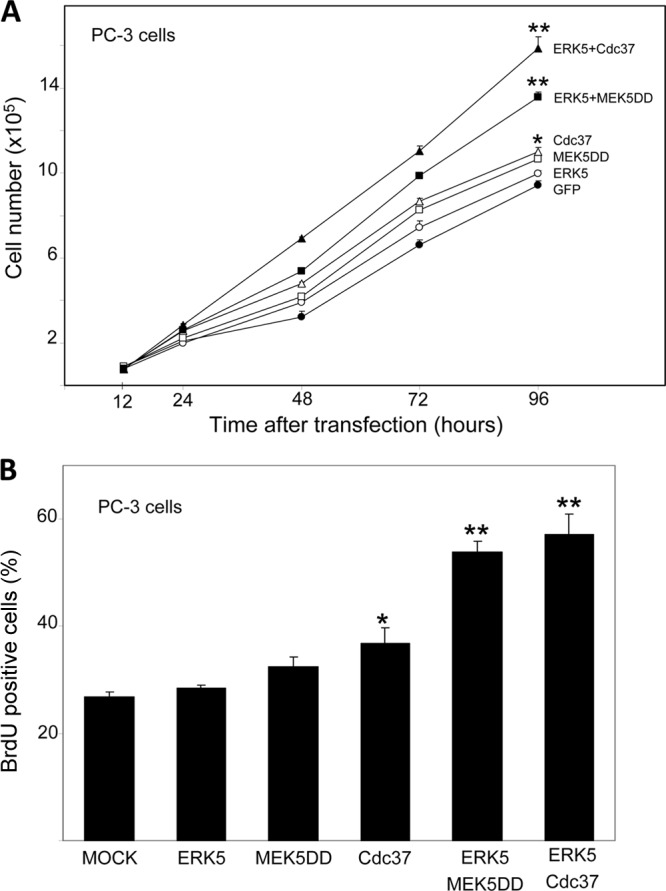
Cdc37 collaborates with ERK5 to promote cell proliferation. (A) PC3 prostate cancer cells were transfected with plasmids encoding proteins GFP, GFP-ERK5, MEK5-DD and GFP, Cdc37 and GFP, GFP-ERK5 and MEK5-DD, or GFP-ERK5 and Cdc37, as described in Materials and Methods. At the indicated times, green fluorescent cells were counted by flow cytometry. Each value is the mean ± SD of the results determined for three different transfected cell dishes. Similar results were obtained in three independent experiments. *, P < 0.01; **, P < 0.001 (for values at 96 h after transfection compared with the GFP results at 96 h). (B) PC-3 cells were transfected with empty plasmid (MOCK) or with plasmids encoding the indicated proteins as described for panel A. Cells were treated with BrdU, and incorporation of BrdU was detected by flow cytometry. Ratios of numbers of BrdU-positive cells to total numbers of live cells were determined in triplicate samples, and data are expressed as the mean ± SD of the results determined for two independent experiments. *, P < 0.01; **, P < 0.001 (in comparison to the mock transfection data).
To quantify the percentage of proliferating cells, PC-3 cells overexpressing different combinations of proteins were labeled with the nucleotide analogue BrdU, a method used to quantify cells undergoing DNA synthesis. Similar to data shown in Fig. 8A, ERK5, MEK5, or Cdc37 overexpression had little or no effect on the number of BrdU-positive cells, whereas cells overexpressing both ERK5 and MEK5 or Cdc37 showed a consistent marked increase of cell proliferation (Fig. 8B). All in all, our results show that Cdc37 cooperates with ERK5 to promote cell proliferation.
DISCUSSION
We used tandem affinity purification to identify ERK5-interacting proteins. This methodology identified the chaperone protein Hsp90β as the major protein associated with ERK5 (Fig. 1). Immunoprecipitation assays revealed that endogenous Hsp90 and ERK5 proteins interact. Cdc37, the canonical Hsp90 cochaperone for many kinase clients, also interacted with ERK5, resulting in an Hsp90-Cdc37-ERK5 trimeric complex in cells (Fig. 2). Our analysis also identified other ERK5-interacting proteins, whose physiological relevance is being currently investigated.
The importance of the interaction of Hsp90 with ERK5 was established by the fact that two structurally unrelated Hsp90 inhibitors (geldanamycin and radicicol) promoted ERK5 degradation. Although Truman et al. observed that yeast Hsp90 binds human ERK5 (46), other authors excluded ERK5 as a client of mammalian Hsp90 since the expression of this kinase was not affected by treatment of COS7 cells with the Hsp90 inhibitor 17-AAG (47). This was probably due to the use of a relatively low dose (1 μM) of inhibitor, since we noticed that ERK5 expression is not affected by low concentrations of Hsp90 inhibitors that alter other Hsp90 clients. For instance, 1 μM geldanamycin induces degradation of LKB1 (48), JAK1/JAK2 (49), CHK1 (50), or IRAK-1 (51) kinases after 4 h of treatment, a concentration that has almost no effect on ERK5 after 18 h. The use of 5 μM geldanamycin was necessary to induce complete degradation of ERK5 (Fig. 3A). This is not surprising, since closely related kinases show differential levels of sensitivity to geldanamycin, as is the case for the receptor tyrosine kinases ErbB1 and ErbB2. Both proteins require Hsp90 for proper folding, but only ErbB2 binds Hsp90 in the mature state, making ErbB2 more sensitive to geldanamycin (52). We also show the requirement of Cdc37 for ERK5 stability. Treatment of cells with the cdc37 inhibitor celastrol or silencing the Cdc37 protein results in loss of ERK5 expression (Fig. 3B and 5B). Celastrol inhibits Cdc37-Hsp90 interaction, preventing the recruitment of Hsp90 to the kinase domain of the client protein (22), therefore resulting in the proteasomal degradation of this protein (26).
ERK5 comprises two well-differentiated regions; the N-terminal region contains the kinase domain, and the C-terminal region contains a nuclear localization signal and a transcriptional transactivation domain. We show that Hsp90 and Cdc37 bind the N-terminal region but not the C-terminal tail (Fig. 2D and E). The fact that the N-terminal half is ubiquitylated in the absence of proteasomal inhibitor (Fig. 3E) suggests that this half becomes unstable in the absence of the C-terminal region.
Our most striking finding is the observation that activation of ERK5 (in response to either EGF stimulation or active MEK5 overexpression) results in Hsp90 dissociation from the ERK5-Cdc37 complex (Fig. 4A and B). According to this, active ERK5 is not sensitive to Hsp90 inhibitors (Fig. 5A). These results suggest that ERK5 is an unstable kinase in its inactive form and that binding to Hsp90 stabilizes it, whereas its activation results in a stable protein that does not require Hsp90 stabilization. Although unusual, a transient interaction of Hsp90 with certain kinase clients, such as the cyclin-dependent kinase CDK4, has been reported. Inactive CDK4 binds Hsp90 and Cdc37, and binding of cyclin D induces the release of the Hsp90-Cdc37 superchaperone and CDK4 activation (53). In this case, Hsp90-Cdc37 promotes stabilization until cyclin D takes over this activity by interacting with (and displacing Hsp90-Cdc37 from) the N-terminal lobe of the kinase domain. For other kinases, only the mutated forms show Hsp90 dependency. For instance, wild-type B-Raf does not bind Hsp90 but the mutant active form most commonly found in cancer B-RafV600E shows a strong Hsp90 dependence for its stability (30). The substitution of Val600 for Glu within the T-loop provokes a conformational change of the N-terminal lobe, exposing the chaperone binding site.
Here we show that a kinase-dead mutant of ERK5 (D200A) remains associated with Hsp90 even after being phosphorylated at the TEY motif by MEK5 (Fig. 4B). Thus, ERK5 kinase activity is necessary for the dissociation of Hsp90 but not the phosphorylation of its TEY motif. On the other hand, a functional form of ERK5(1–490) that lacks the C-terminal region remains associated with Hsp90 after MEK5 phosphorylation (Fig. 4C). Finally, an ERK5 mutant in which 5 autophosphorylatable residues are mutated to Glu does not bind Hsp90 under basal conditions (Fig. 4D). Taken together, these results indicate that autophosphorylation of the C-terminal region induces the release of Hsp90 from the ERK5-Cdc37 complex. The ERK5 C-terminal tail might work in a manner analogous to that of cyclin D for CDK4: upon ERK5 activation, autophosphorylation of its C-terminal region results in a conformational change of this tail that induces the release of Hsp90 and takes over its chaperone function in the stabilization of the N-terminal lobe of the kinase domain. However, and unlike what happens for CDK4, Cdc37 remains bound to active ERK5.
Unexpectedly, expression of high levels of Cdc37 in cells induced the release of Hsp90 from the ERK5-Cdc37 complex (Fig. 6A), by a mechanism that does not involve enzyme activation and C-terminal autophosphorylation (Fig. 7). Thus, Cdc37 mimics ERK5 C-terminal autophosphorylation, probably inducing a conformational change that results in the release of Hsp90. This was an unexpected result, since it is accepted that Cdc37 promotes the association of Hsp90 with client proteins (21, 22). One possibility might be that high levels of expression of Cdc37 result in the binding of a second molecule to the ERK5 protein, displacing the Hsp90 by an unknown, noncanonical mechanism. Interestingly, Fig. 6A shows that larger amounts of Cdc37 bound to ERK5 in those cells that overexpress higher levels of Cdc37. We are currently investigating the mechanism by which Cdc37 displaces Hsp90 from the trimeric complex.
We show that MEK5 activation or overexpression of Cdc37 results in three common events: (i) Hsp90 dissociation from the ERK5-Cdc37 complex; (ii) ERK5 nuclear translocation; and (iii) an increase of ERK5-mediated AP-1 transcriptional activity. Several studies have addressed the issue of subcellular location of ERK5. ERK5 resides in the cytosol of HeLa cells and migrates to the nuclei after activation in response to EGF (35, 54) or active MEK5 overexpression (55). However, other studies stated that the cellular location of ERK5 depends on the cell type and can even be nuclear in unstimulated cells (16, 56). Kondoh et al. proposed an accepted mechanism for the nucleocytoplasmic transport of ERK5 (35). The C-terminal region contains a nuclear localization sequence (NLS), and ERK5 itself has nuclear export (NES) activity, so subcellular localization of ERK5 depends on the balance between nuclear export and import: the N-terminal and C-terminal halves bind each other, and this binding is necessary for cytoplasmatic localization. Thus, in resting cells, the two halves would interact, forming either a putative NES or a domain that binds a cytoplasmatic anchor protein. After activation, ERK5 autophosphorylates its C-terminal half, an event that would disrupt the intramolecular interaction between the C-terminal and N-terminal regions, resulting in the loss of the NES activity, exposition of the NLS, and nuclear translocation. Supporting the idea of a role of C-terminal phosphorylation in ERK5 nuclear localization, an ERK5 that is catalytically inactive (but phosphorylated at its C terminus) that resides in the nucleus during mitosis has been previously reported (36, 57).
Our results suggest that Hsp90 might be the proposed cytosolic anchor, since dissociation of Hsp90 is the common event implicated in two different mechanisms that induce ERK5 nuclear translocation, i.e., MEK5 activation and Cdc37 overexpression (Fig. 9). Thus, Hsp90 might act as it does for steroid receptors. In the absence of the hormone, steroid receptors reside in the cytosol complexed with Hsp90 and this interaction maintains the receptor in a state capable of binding the hormone. Binding the steroid leads to dissociation of Hsp90 and receptor nuclear translocation (58). In an analogous way, the Hsp90-ERK5 complex resides in the cytosol and ERK5 activation induces Hsp90 dissociation and nuclear translocation of the kinase, configuring a kinase activity-dependent shuttling (Fig. 9). In this paradigm, it is likely that Hsp90 would keep cytosolic/basal ERK5 properly folded for MEK5 recognition and activation.
Fig 9.

A model for the role of chaperones in the mechanism of nucleocytoplasmatic transport of ERK5. In the resting state, the N-terminal half of ERK5 interacts intramolecularly with the C-terminal half, generating a region responsible for the interaction with the cytoplasmic anchor protein Hsp90. This interaction would keep ERK5 in the cytosol, even though the nuclear localization signal (NLS) is present at the ERK5 C-terminal tail. Upon MEK5-mediated phosphorylation of the TEY motif and activation, ERK5 autophosphorylation of its C-terminal half disrupts the intramolecular interaction, inducing a conformational change that results in dissociation of Hsp90, exposition of the NLS, and ERK5 nuclear translocation. In contrast, overexpression of Cdc37 (as happens in some cancers) induces the nuclear translocation of a catalytically inactive form of ERK5 that is transcriptionally active. TAD, transcriptional-activation domain.
We propose that Cdc37 overexpression represents an alternative, noncanonical mechanism of ERK5 nuclear translocation. Unlike the results seen with MEK5, overexpression of Cdc37 does not result in activation of ERK5 (Fig. 7C) or in a shift of its electrophoretic mobility, and it induces the nuclear translocation of a catalytically inactive form of ERK5 that retains its transcriptional activity (Fig. 9). This finding is strongly supported by the genetic and pharmacological data (Fig. 7): (i) Cdc37 overexpression induces ERK5 nuclear translocation in MEF MEK5−/− cells, (ii) ERK5 inhibitor XMD2-98 blocks ERK5 nuclear translocation and transcriptional activity induced by active MEK5 but not by Cdc37 overexpression, and (iii) overexpression of Cdc37 induces transcriptional activity of two ERK5 mutants unable to autophosphorylate, D200A and ERK5(5A). Interestingly, Cdc37 overexpression also increases nuclear translocation of other proteins, such as the intracellular domain of the tyrosine kinase receptor Ryk, after being cleaved by γ-secretase (59). How Cdc37 overexpression induces the release of Hsp90 remains to be studied, but it could be the consequence of Cdc37 either directly displacing Hsp90 or inducing a conformational change in ERK5 that results in Hsp90 dissociation. It is important to study if the different ERK5 cytosolic/nuclear localizations observed in several cell lines are, in fact, the consequence of a different Cdc37 expression level.
Cdc37 acts as an oncogene, stabilizing other oncogenes that are mutated or overexpressed in cancer cells (25); it is overexpressed in a number of cancers (60), and silencing Cdc37 reduces cell growth and invasive capability (24, 61). On the other hand, MEK5 and ERK5 represent a key pathway associated with some cancers, such as breast and prostate cancer. Tumors showing a strong nuclear ERK5 localization have a poor specific disease factor (42, 43, 54, 62), and silencing ERK5 or Cdc37 expression induces growth arrest and inhibits the invasive capability of cancer cells (44). In accordance with the results described above, we show that, in prostate cancer cells, Cdc37 cooperates with ERK5 to promote cell proliferation, as it does proliferation of active MEK5 (Fig. 8). It is important to establish if ERK5 and Cdc37 also cooperate to regulate the progression and invasiveness of cancer. If so, our observation that active ERK5 is no longer sensitive to Hsp90 or Cdc37 inhibitors supports the idea that the ERK5-Cdc37 interaction might represent a new target for therapeutic intervention in some cancers.
ACKNOWLEDGMENTS
We thank Philip Cohen and Maria Deak (MRC PPU, Dundee, United Kingdom) for ERK5 antibody and the generation of the TAP-tagged ERK5 vectors, respectively. We also thank Nathanael Gray (Dana-Farber Cancer Institute, Boston, MA) for providing us with the ERK5 competitive inhibitor XMD2-98 and Cathy Tournier (The University of Manchester, United Kingdom) for the MEF MEK5−/− cells. We are grateful to Ana Cuenda for helpful discussions, Cristina Gutierrez for tissue culture assistance, Anna Vilalta for technical support, and Isabelle Gelot for editorial assistance. We are grateful to the Servei de Genòmica from the UAB.
This work was supported by grants from the Spanish Ministerio Educación (BFU2004-00757 and BFU2007-60268).
Footnotes
Published ahead of print 19 February 2013
REFERENCES
- 1. Kyriakis JM, Avruch J. 2001. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol. Rev. 81:807–869 [DOI] [PubMed] [Google Scholar]
- 2. Johnson GL, Lapadat R. 2002. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 298:1911–1912 [DOI] [PubMed] [Google Scholar]
- 3. Raman M, Chen W, Cobb MH. 2007. Differential regulation and properties of MAPKs. Oncogene 26:3100–3112 [DOI] [PubMed] [Google Scholar]
- 4. Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. 1998. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature 395:713–716 [DOI] [PubMed] [Google Scholar]
- 5. English JM, Pearson G, Baer R, Cobb MH. 1998. Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J. Biol. Chem. 273:3854–3860 [DOI] [PubMed] [Google Scholar]
- 6. Yang CC, Ornatsky OI, McDermott JC, Cruz TF, Prody CA. 1998. Interaction of myocyte enhancer factor 2 (MEF2) with a mitogen-activated protein kinase, ERK5/BMK1. Nucleic Acids Res. 26:4771–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kamakura S, Moriguchi T, Nishida E. 1999. Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signaling pathway to the nucleus. J. Biol. Chem. 274:26563–26571 [DOI] [PubMed] [Google Scholar]
- 8. Lee JD, Ulevitch RJ, Han J. 1995. Primary structure of BMK1: a new mammalian map kinase. Biochem. Biophys. Res. Commun. 213:715–724 [DOI] [PubMed] [Google Scholar]
- 9. Zhou G, Bao ZQ, Dixon JE. 1995. Components of a new human protein kinase signal transduction pathway. J. Biol. Chem. 270:12665–12669 [DOI] [PubMed] [Google Scholar]
- 10. Sohn SJ, Sarvis BK, Cado D, Winoto A. 2002. ERK5 MAPK regulates embryonic angiogenesis and acts as a hypoxia-sensitive repressor of vascular endothelial growth factor expression. J. Biol. Chem. 277:43344–43351 [DOI] [PubMed] [Google Scholar]
- 11. Regan CP, Li W, Boucher DM, Spatz S, Su MS, Kuida K. 2002. Erk5 null mice display multiple extraembryonic vascular and embryonic cardiovascular defects. Proc. Natl. Acad. Sci. U. S. A. 99:9248–9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yan L, Carr J, Ashby PR, Murry-Tait V, Thompson C, Arthur JS. 2003. Knockout of ERK5 causes multiple defects in placental and embryonic development. BMC Dev. Biol. 3:11 doi:10.1186/1471-213X-3-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang X, Merritt AJ, Seyfried J, Guo C, Papadakis ES, Finegan KG, Kayahara M, Dixon J, Boot-Handford RP, Cartwright EJ, Mayer U, Tournier C. 2005. Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol. Cell. Biol. 25:336–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kasler HG, Victoria J, Duramad O, Winoto A. 2000. ERK5 is a novel type of mitogen-activated protein kinase containing a transcriptional activation domain. Mol. Cell. Biol. 20:8382–8389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chiariello M, Marinissen MJ, Gutkind JS. 2000. Multiple mitogen-activated protein kinase signaling pathways connect the cot oncoprotein to the c-jun promoter and to cellular transformation. Mol. Cell. Biol. 20:1747–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Borges J, Pandiella A, Esparis-Ogando A. 2007. Erk5 nuclear location is independent on dual phosphorylation, and favours resistance to TRAIL-induced apoptosis. Cell Signal. 19:1473–1487 [DOI] [PubMed] [Google Scholar]
- 17. Pearl LH, Prodromou C. 2006. Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu. Rev. Biochem. 75:271–294 [DOI] [PubMed] [Google Scholar]
- 18. McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. 2007. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell 131:121–135 [DOI] [PubMed] [Google Scholar]
- 19. Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. 1997. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90:65–75 [DOI] [PubMed] [Google Scholar]
- 20. Neckers L, Workman P. 2012. Hsp90 molecular chaperone inhibitors: are we there yet? Clin. Cancer Res. 18:64–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kimura Y, Rutherford SL, Miyata Y, Yahara I, Freeman BC, Yue L, Morimoto RI, Lindquist S. 1997. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 11:1775–1785 [DOI] [PubMed] [Google Scholar]
- 22. Roe SM, Ali MM, Meyer P, Vaughan CK, Panaretou B, Piper PW, Prodromou C, Pearl LH. 2004. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116:87–98 [DOI] [PubMed] [Google Scholar]
- 23. Karnitz LM, Felts SJ. 2007. Cdc37 regulation of the kinome: when to hold 'em and when to fold 'em. Sci. STKE 2007:pe22 doi:10.1126/stke.3852007pe22 [DOI] [PubMed] [Google Scholar]
- 24. Gray PJ, Jr, Stevenson MA, Calderwood SK. 2007. Targeting Cdc37 inhibits multiple signaling pathways and induces growth arrest in prostate cancer cells. Cancer Res. 67:11942–11950 [DOI] [PubMed] [Google Scholar]
- 25. Gray PJ, Jr, Prince T, Cheng J, Stevenson MA, Calderwood SK. 2008. Targeting the oncogene and kinome chaperone CDC37. Nat. Rev. Cancer 8:491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith JR, Clarke PA, de Billy E, Workman P. 2009. Silencing the cochaperone CDC37 destabilizes kinase clients and sensitizes cancer cells to HSP90 inhibitors. Oncogene 28:157–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B. 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 17:1030–1032 [DOI] [PubMed] [Google Scholar]
- 28. Mody N, Campbell DG, Morrice N, Peggie M, Cohen P. 2003. An analysis of the phosphorylation and activation of extracellular-signal-regulated protein kinase 5 (ERK5) by mitogen-activated protein kinase kinase 5 (MKK5) in vitro. Biochem. J. 372:567–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zheng Q, Yin G, Yan C, Cavet M, Berk BC. 2004. 14-3-3beta binds to big mitogen-activated protein kinase 1 (BMK1/ERK5) and regulates BMK1 function. J. Biol. Chem. 279:8787–8791 [DOI] [PubMed] [Google Scholar]
- 30. Grbovic OM, Basso AD, Sawai A, Ye Q, Friedlander P, Solit D, Rosen N. 2006. V600E B-Raf requires the Hsp90 chaperone for stability and is degraded in response to Hsp90 inhibitors. Proc. Natl. Acad. Sci. U. S. A. 103:57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. 2002. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 277:39858–39866 [DOI] [PubMed] [Google Scholar]
- 32. Zsebik B, Citri A, Isola J, Yarden Y, Szollosi J, Vereb G. 2006. Hsp90 inhibitor 17-AAG reduces ErbB2 levels and inhibits proliferation of the trastuzumab resistant breast tumor cell line JIMT-1. Immunol. Lett. 104:146–155 [DOI] [PubMed] [Google Scholar]
- 33. Prince T, Sun L, Matts RL. 2005. Cdk2: a genuine protein kinase client of Hsp90 and Cdc37. Biochemistry 44:15287–15295 [DOI] [PubMed] [Google Scholar]
- 34. Lizcano JM, Alessi DR. 2002. The insulin signalling pathway. Curr. Biol. 12:R236–R238 [DOI] [PubMed] [Google Scholar]
- 35. Kondoh K, Terasawa K, Morimoto H, Nishida E. 2006. Regulation of nuclear translocation of extracellular signal-regulated kinase 5 by active nuclear import and export mechanisms. Mol. Cell. Biol. 26:1679–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Iñesta-Vaquera FA, Campbell DG, Tournier C, Gomez N, Lizcano JM, Cuenda A. 2010. Alternative ERK5 regulation by phosphorylation during the cell cycle. Cell Signal. 22:1829–1837 [DOI] [PubMed] [Google Scholar]
- 37. Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. 2008. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 7:162–170 [DOI] [PubMed] [Google Scholar]
- 38. Stepanova L, Finegold M, DeMayo F, Schmidt EV, Harper JW. 2000. The oncoprotein kinase chaperone CDC37 functions as an oncogene in mice and collaborates with both c-myc and cyclin D1 in transformation of multiple tissues. Mol. Cell. Biol. 20:4462–4473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Terasawa K, Okazaki K, Nishida E. 2003. Regulation of c-Fos and Fra-1 by the MEK5-ERK5 pathway. Genes Cells 8:263–273 [DOI] [PubMed] [Google Scholar]
- 40. Morimoto H, Kondoh K, Nishimoto S, Terasawa K, Nishida E. 2007. Activation of a C-terminal transcriptional activation domain of ERK5 by autophosphorylation. J. Biol. Chem. 282:35449–35456 [DOI] [PubMed] [Google Scholar]
- 41. Yang Q, Deng X, Lu B, Cameron M, Fearns C, Patricelli MP, Yates JR, III, Gray NS, Lee JD. 2010. Pharmacological inhibition of BMK1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 18:258–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mehta PB, Jenkins BL, McCarthy L, Thilak L, Robson CN, Neal DE, Leung HY. 2003. MEK5 overexpression is associated with metastatic prostate cancer, and stimulates proliferation, MMP-9 expression and invasion. Oncogene 22:1381–1389 [DOI] [PubMed] [Google Scholar]
- 43. McCracken SR, Ramsay A, Heer R, Mathers ME, Jenkins BL, Edwards J, Robson CN, Marquez R, Cohen P, Leung HY. 2008. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 27:2978–2988 [DOI] [PubMed] [Google Scholar]
- 44. Ramsay AK, McCracken SR, Soofi M, Fleming J, Yu AX, Ahmad I, Morland R, Machesky L, Nixon C, Edwards DR, Nuttall RK, Seywright M, Marquez R, Keller E, Leung HY. 2011. ERK5 signalling in prostate cancer promotes an invasive phenotype. Br. J. Cancer 104:664–672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schwarze SR, Fu VX, Jarrard DF. 2003. Cdc37 enhances proliferation and is necessary for normal human prostate epithelial cell survival. Cancer Res. 63:4614–4619 [PubMed] [Google Scholar]
- 46. Truman AW, Millson SH, Nuttall JM, King V, Mollapour M, Prodromou C, Pearl LH, Piper PW. 2006. Expressed in the yeast Saccharomyces cerevisiae, human ERK5 is a client of the Hsp90 chaperone that complements loss of the Slt2p (Mpk1p) cell integrity stress-activated protein kinase. Eukaryot. Cell 5:1914–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Citri A, Harari D, Shohat G, Ramakrishnan P, Gan J, Lavi S, Eisenstein M, Kimchi A, Wallach D, Pietrokovski S, Yarden Y. 2006. Hsp90 recognizes a common surface on client kinases. J. Biol. Chem. 281:14361–14369 [DOI] [PubMed] [Google Scholar]
- 48. Boudeau J, Deak M, Lawlor MA, Morrice NA, Alessi DR. 2003. Heat-shock protein 90 and Cdc37 interact with LKB1 and regulate its stability. Biochem. J. 370:849–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shang L, Tomasi TB. 2006. The heat shock protein 90-CDC37 chaperone complex is required for signaling by types I and II interferons. J. Biol. Chem. 281:1876–1884 [DOI] [PubMed] [Google Scholar]
- 50. Nomura M, Nomura N, Yamashita J. 2005. Geldanamycin-induced degradation of Chk1 is mediated by proteasome. Biochem. Biophys. Res. Commun. 335:900–905 [DOI] [PubMed] [Google Scholar]
- 51. De Nardo D, Masendycz P, Ho S, Cross M, Fleetwood AJ, Reynolds EC, Hamilton JA, Scholz GM. 2005. A central role for the Hsp90. Cdc37 molecular chaperone module in interleukin-1 receptor-associated-kinase-dependent signaling by toll-like receptors. J. Biol. Chem. 280:9813–9822 [DOI] [PubMed] [Google Scholar]
- 52. Xu W, Yuan X, Xiang Z, Mimnaugh E, Marcu M, Neckers L. 2005. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 12:120–126 [DOI] [PubMed] [Google Scholar]
- 53. Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massague J, Pavletich NP. 1995. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 376:313–320 [DOI] [PubMed] [Google Scholar]
- 54. Esparís-Ogando A, Díaz-Rodriguez E, Montero JC, Yuste L, Crespo P, Pandiella A. 2002. Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol. Cell. Biol. 22:270–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD. 1997. BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 16:7054–7066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Raviv Z, Kalie E, Seger R. 2004. MEK5 and ERK5 are localized in the nuclei of resting as well as stimulated cells, while MEKK2 translocates from the cytosol to the nucleus upon stimulation. J. Cell Sci. 117:1773–1784 [DOI] [PubMed] [Google Scholar]
- 57. Díaz-Rodríguez E, Pandiella A. 2010. Multisite phosphorylation of Erk5 in mitosis. J. Cell Sci. 123:3146–3156 [DOI] [PubMed] [Google Scholar]
- 58. Picard D. 2006. Chaperoning steroid hormone action. Trends Endocrinol. Metab. 17:229–235 [DOI] [PubMed] [Google Scholar]
- 59. Lyu J, Wesselschmidt RL, Lu W. 2009. Cdc37 regulates Ryk signaling by stabilizing the cleaved Ryk intracellular domain. J. Biol. Chem. 284:12940–12948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Stepanova L, Yang G, DeMayo F, Wheeler TM, Finegold M, Thompson TC, Harper JW. 2000. Induction of human Cdc37 in prostate cancer correlates with the ability of targeted Cdc37 expression to promote prostatic hyperplasia. Oncogene 19:2186–2193 [DOI] [PubMed] [Google Scholar]
- 61. Pang X, Yi Z, Zhang J, Lu B, Sung B, Qu W, Aggarwal BB, Liu M. 2010. Celastrol suppresses angiogenesis-mediated tumor growth through inhibition of AKT/mammalian target of rapamycin pathway. Cancer Res. 70:1951–1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Montero JC, Ocana A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparis-Ogando A. 2009. Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One 4:e5565 doi:10.1371/journal.pone.0005565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Al-Hakim AK, Goransson O, Deak M, Toth R, Campbell DG, Morrice NA, Prescott AR, Alessi DR. 2005. 14-3-3 cooperates with LKB1 to regulate the activity and localization of QSK and SIK. J. Cell Sci. 118:5661–5673 [DOI] [PubMed] [Google Scholar]
- 64. Hinz M, Broemer M, Arslan SC, Otto A, Mueller EC, Dettmer R, Scheidereit C. 2007. Signal responsiveness of IkappaB kinases is determined by Cdc37-assisted transient interaction with Hsp90. J. Biol. Chem. 282:32311–32319 [DOI] [PubMed] [Google Scholar]
- 65. Papapetropoulos A, Zhou Z, Gerassimou C, Yetik G, Venema RC, Roussos C, Sessa WC, Catravas JD. 2005. Interaction between the 90-kDa heat shock protein and soluble guanylyl cyclase: physiological significance and mapping of the domains mediating binding. Mol. Pharmacol. 68:1133–1141 [DOI] [PubMed] [Google Scholar]
- 66. Belova L, Brickley DR, Ky B, Sharma SK, Conzen SD. 2008. Hsp90 regulates the phosphorylation and activity of serum- and glucocorticoid-regulated kinase-1. J. Biol. Chem. 283:18821–18831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schimmel J, Larsen KM, Matic I, van Hagen M, Cox J, Mann M, Andersen JS, Vertegaal AC. 2008. The ubiquitin-proteasome system is a key component of the SUMO-2/3 cycle. Mol. Cell. Proteomics 7:2107–2122 [DOI] [PubMed] [Google Scholar]
- 68. Rodríguez-Asiain A, Ruiz-Babot G, Romero W, Cubi R, Erazo T, Biondi RM, Bayascas JR, Aguilera J, Gomez N, Gil C, Claro E, Lizcano JM. 2011. Brain specific kinase-1 BRSK1/SAD-B associates with lipid rafts: modulation of kinase activity by lipid environment. Biochim. Biophys. Acta 1811:1124–1135 [DOI] [PubMed] [Google Scholar]
- 69. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 70. Lizcano JM, Deak M, Morrice N, Kieloch A, Hastie CJ, Dong L, Schutkowski M, Reimer U, Alessi DR. 2002. Molecular basis for the substrate specificity of NIMA-related kinase-6 (NEK6). Evidence that NEK6 does not phosphorylate the hydrophobic motif of ribosomal S6 protein kinase and serum- and glucocorticoid-induced protein kinase in vivo. J. Biol. Chem. 277:27839–27849 [DOI] [PubMed] [Google Scholar]
- 71. Williamson BL, Marchese J, Morrice NA. 2006. Automated identification and quantification of protein phosphorylation sites by LC/MS on a hybrid triple quadrupole linear ion trap mass spectrometer. Mol. Cell. Proteomics 5:337–346 [DOI] [PubMed] [Google Scholar]
- 72. Lizcano JM, Alrubaie S, Kieloch A, Deak M, Leevers SJ, Alessi DR. 2003. Insulin-induced Drosophila S6 kinase activation requires phosphoinositide 3-kinase and protein kinase B. Biochem. J. 374:297–306 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73. Tatham MH, Rodriguez MS, Xirodimas DP, Hay RT. 2009. Detection of protein SUMOylation in vivo. Nat. Protoc. 4:1363–1371 [DOI] [PubMed] [Google Scholar]