

Abstract
The scaffolding adapter protein Gab2 (Grb2-associated binder) participates in the signaling response evoked by various growth factors and cytokines. Gab2 is overexpressed in several human malignancies, including breast cancer, and was shown to promote mammary epithelial cell migration. The role of Gab2 in the activation of different signaling pathways is well documented, but less is known regarding the feedback mechanisms responsible for its inactivation. We now demonstrate that activation of the Ras/mitogen-activated protein kinase (MAPK) pathway promotes Gab2 phosphorylation on basic consensus motifs. More specifically, we show that RSK (p90 ribosomal S6 kinase) phosphorylates Gab2 on three conserved residues, both in vivo and in vitro. Mutation of these phosphorylation sites does not alter Gab2 binding to Grb2, but instead, we show that Gab2 phosphorylation inhibits the recruitment of the tyrosine phosphatase Shp2 in response to growth factors. Expression of an unphosphorylatable Gab2 mutant in mammary epithelial cells promotes an invasion-like phenotype and increases cell motility. Taken together, these results suggest that RSK is part of a negative-feedback loop that restricts Gab2-dependent epithelial cell motility. On the basis of the widespread role of Gab2 in receptor signaling, these findings also suggest that RSK plays a regulatory function in diverse receptor systems.
INTRODUCTION
The binding of growth factors to receptor tyrosine kinases (RTKs) regulates a number of biological processes, such as cell cycle progression, migration, metabolism, survival, and differentiation (1). Upon activation, RTKs recruit several docking adapter proteins that play a key role in transducing extracellular signals to downstream signaling cascades, such as the Ras/mitogen-activated protein kinase (MAPK) pathway (2, 3). The adapter protein Grb2 (growth factor receptor-bound protein 2) links the receptors to the MAPK pathway by binding to the Ras-specific guanine nucleotide exchange factor (GEF) son of sevenless (SOS). Recruitment of SOS to the plasma membrane leads to the activation of Ras, which results in the sequential phosphorylation and activation of Raf, MEK1/2, and extracellular signal-regulated kinase 1/2 (ERK1/2) protein kinases (4). Once activated, ERK1/2 phosphorylate numerous cytoplasmic and nuclear substrates, including the p90 ribosomal S6 kinase (RSK) family of Ser/Thr kinases (5, 6), which often collaborate with ERK1/2 to regulate cell growth, survival, and proliferation.
In addition to SOS, Grb2 associates with a number of docking adapter proteins that modulate RTK signaling (7). Members of the Grb2-associated binder (Gab) family of proteins function downstream of a variety of RTKs and comprise three vertebrate members (Gab1 to Gab3) (8, 9). These proteins mainly function as RTK signal transducers that activate pathways involved in cell growth, proliferation, and motility. Gab proteins contain several highly conserved regions, including an N-terminal pleckstrin homology (PH) domain, a central proline-rich domain, and multiple phosphotyrosine residues (9). While the PH domain binds membrane-associated phospholipids, the proline-rich domain contains several PXXP motifs that serve as docking sites for Src homology 3 (SH3) domain-containing proteins, including Grb2 (10). The Gab proteins harbor several tyrosine residues that become phosphorylated upon RTK activation. Many of these phosphorylated tyrosines are capable of interacting with Src homology 2 (SH2) domain-containing proteins, including the protein tyrosine phosphatase Shp2 and the p85 subunit of phosphoinositide 3-kinase (PI3K). Recruitment of these proteins leads to the activation and potentiation of the Ras/MAPK and PI3K/Akt pathways, respectively (9).
The Gab proteins perform important functions in normal physiology, and certain Gab isoforms also contribute to human malignancies (11). The GAB2 gene is frequently amplified in human cancer and was identified to be a potential oncogene in breast and ovarian cancers, as well as leukemia and melanoma (12). While Gab2 appears to be insufficient to transform primary mammary epithelial cells, it was shown to cooperate with ErbB2 (Neu or HER2) to potentiate tumorigenic signaling (13–15). Gab2 seems to contribute to a metastatic phenotype in breast cancer, as its overexpression in human mammary epithelial cells results in increased proliferation, invasiveness, and motility (13–15). The mechanisms by which Gab2 contributes to breast cancer are not fully understood, but Shp2 recruitment and the subsequent activation of the Ras/MAPK pathway were shown to be required (14). Moreover, recent evidence indicates that Gab2 regulates cytoskeletal organization and mammary epithelial cell motility through the recruitment of Shp2 (16).
The main role of Gab2 is to activate downstream signaling cascades via tyrosine phosphorylation and SH2 domain interactions, such as with Shp2. Conversely, Gab2 phosphorylation on Ser/Thr residues was previously reported to play inhibitory roles. Akt was shown to regulate the phosphorylation of Ser159, resulting in reduced ErbB2-mediated tyrosine phosphorylation through unknown mechanisms (17). ERK1/2 also phosphorylates Gab2 on Ser613, which was found to modulate Shp2 recruitment in response to interleukin-2 (IL-2) (18). More recently, phosphorylation of Gab2 on Ser210 and Thr391 by an unknown protein kinase was shown to promote 14-3-3 binding, resulting in reduced Grb2 binding and tyrosine phosphorylation (19). In the current study, we describe the regulation of Gab2 phosphorylation on Ser/Thr residues in response to the Ras/MAPK pathway. Our results indicate that RSK directly phosphorylates Gab2 on three serine residues, both in vivo and in vitro. We show that RSK-mediated Gab2 phosphorylation inhibits Shp2 recruitment, suggesting that RSK mediates a negative-feedback loop that attenuates Gab2-dependent functions, including cell motility.
MATERIALS AND METHODS
DNA constructs and recombinant proteins.
The plasmids encoding hemagglutinin (HA)-tagged murine Gab1 and Gab2 were provided by Morag Park (McGill University, Canada) and Isabelle Royal (University of Montreal, Montreal, Quebec, Canada), respectively, and described previously (20, 21). The vectors encoding constitutively active forms of Ras (G12V) and MEK1 (MEK-DD) and the inactive form of Ras (S17N) were described previously (22, 23). All HA-tagged RSK1 constructs were described previously (24). To subclone murine Gab1 and Gab2 into pcDNA3.0-6myc, HA-tagged Gab1 and Gab2 were amplified by PCR using these primers: primers Gab1-sense (5′-GCTTAGAATTCTATGAGCGGCGGCGAAGTGG-3′) and Gab1-antisense (5′-GCATAGAATTCCTACTTCACATTCTTGGTGGGTG-3′) and primers Gab2-sense (5′-GCTTACTCGAGTATGAGCGGCGGCGGCGGCGACGACGT-3′) and Gab2-antisense (5′-GCATACTCGAGTCATTACAGCTTGGCACCCTTGGAAG-3′), respectively. All murine Gab2 mutants were generated using the QuikChange methodology (Stratagene, La Jolla, CA). To subclone murine Gab2 into pBabe-puro and produce retroviral particles used in the generation of stable cell lines, Myc-tagged wild-type (wt) and mutant Gab2 were amplified by PCR using these primers: Gab2-sense (5′-GCTTAGGATCCATTTAAAGCTATGGAGCAAAAGC-3′) and Gab2-antisense (5′-GCATAGGATCCTCATTACAGCTTGGCACCCTTGGAAG-3′).
Antibodies.
Antibodies targeted against Arg/Lys-X-X-pSer/Thr (RXXpS/T; X is any amino acid) and Arg/Lys-X-X-pSer/Thr-X-Pro (RXXpS/TXP) consensus sequences, Gab2, RSK1 to RSK3, phospho-Akt (S473), Akt, phospho-Gab2 (S159), p85, ERK1/2, phospho-ERK1/2 (T202/Y204), phospho-RSK (S380), and Shp2 were purchased from Cell Signaling Technologies (Beverly, MA). The Shp2 antibody used for immunoprecipitation was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). Anti-Myc and anti-HA monoclonal antibodies were purchased from Sigma-Aldrich (Oakville, Ontario, Canada). All secondary horseradish peroxidase (HRP)-conjugated antibodies used for immunoblotting were purchased from Chemicon (Temecula, CA).
Cell culture and transfection.
HEK293 and mouse embryonic fibroblasts (MEFs) were maintained at 37°C in Dulbecco's modified Eagle's medium (DMEM) with 4.5 g/liter glucose supplemented with 10% fetal bovine serum (FBS) and antibiotics. Wild-type and Gab2-deficient MEFs were described elsewhere (25). MCF-10A cells were cultured in DMEM–F-12 medium with growth medium (supplemented with 5% [vol/vol] horse serum [Invitrogen], 20 ng/ml human recombinant epidermal growth factor [EGF; R&D Systems, Minneapolis, MN], 0.5 μg/ml hydrocortisone [Sigma], 100 ng/ml cholera toxin [Sigma], 10 μg/ml bovine insulin [Sigma], 50 U/ml penicillin G [Invitrogen], and 50 μg/ml streptomycin sulfate [Invitrogen]). Gab2-deficient MEFs and MCF-10A stable cell lines were generated using pBabe-puro-derived retroviral particles, and expressing cells were selected using puromycin (2 μg/ml). HEK293 cells were transfected by calcium phosphate precipitation as previously described (26). Cells were grown for 24 h after transfection and serum starved using serum-free DMEM where indicated for 16 to 18 h. Starved cells were pretreated with PD184352 (10 μM), U0126 (20 μM), or BI-D1870 (10 μM) (Biomol, Plymouth Meeting, PA), where indicated, and stimulated with FBS (10%), phorbol myristate acetate (PMA; 25 to 100 ng/ml), or EGF (25 ng/ml) before being harvested. Unless indicated otherwise, all drugs and growth factors were purchased from Invitrogen (Burlington, Ontario, Canada). The proliferation rate was measured by the colorimetric 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide assay.
RNA interferences.
For small interfering RNA (siRNA)-mediated knockdown of RSK1 and RSK2, validated 21-nucleotide cRNAs with symmetrical 2-nucleotide overhangs were obtained from Qiagen. HEK293 cells were transfected using calcium phosphate and 50 nM siRNA per dish. At 24 h following transfection, cells were serum starved overnight before being harvested.
Immunoprecipitations and immunoblotting.
Cell lysates were prepared as previously described. Briefly, cells were washed three times with ice-cold phosphate-buffered saline (PBS) and lysed in CLB (10 mM K3PO4, 1 mM EDTA, 5 mM EGTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 0.5% Nonidet P-40, 0.1% Brij 35, 0.1% deoxycholic acid, 1 mM sodium orthovanadate [Na3VO4], 1 mM phenylmethylsulfonyl fluoride, and a cOmplete protease inhibitor cocktail tablet [Roche]). For immunoprecipitations, cell lysates were incubated with the indicated antibodies for 2 h, followed by a 1-h incubation with protein A–Sepharose CL-4B beads (GE Healthcare). Unless they were used for kinase assays, immunoprecipitates were washed three times in lysis buffer and beads were eluted and boiled in 2× reducing sample buffer (5× buffer is 60 mM Tris-HCl, pH 6.8, 25% glycerol, 2% SDS, 14.4 mM 2-mercaptoethanol, 0.1% bromophenol blue). Eluates and total cell lysates were subjected to 8 to 10% SDS-PAGE, and resolved proteins were transferred onto polyvinylidene fluoride (PVDF) membranes for immunoblotting. Densitometry analysis was conducted using identical areas for each lane of a given blot and inverted histograms in the Adobe Photoshop CS6 package. After subtracting the background, Shp2 levels were normalized to the levels of immunoprecipitated Gab2.
Protein phosphotransferase assays.
For RSK1 assays, transfected HA-tagged wt or kinase-inactive (kinase-deficient [kd]) RSK1 (K112/464R) was immunoprecipitated from cells lysed in BLB buffer, as previously described (26). Immunoprecipitates were washed thrice in BLB and twice in kinase buffer (25 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 5 mM β-glycerophosphate). Kinase assays were performed with immunopurified full-length Myc-tagged wt Gab2 or the S160/211/620A mutant (from here on termed the S3A mutant) as the substrates under linear assay conditions. Assays were performed for 10 min at 30°C in kinase buffer supplemented with 5 μCi [γ-32P]ATP per reaction. All samples were subjected to SDS-PAGE, and incorporation of radioactive phosphate (32P) was determined by autoradiography using a Fuji PhosphorImager with Multi-Gauge (version 3.0) software. The data presented are representative of at least three independent experiments.
Digestion, TMT labeling, and mass spectrometry analysis.
Following SDS-PAGE separation with Coomassie staining, bands corresponding to Gab2 were excised and digested in gel with sequencing-grade trypsin (Promega, Madison, WI) as described previously (27). Labeling with sixplex iobaric tandem mass tag (TMT6) reagents (Thermo Scientific) was accomplished as published previously with minimal modifications (28, 29). Labeled peptides were then mixed and underwent C18 solid-phase extraction as described previously (30). Finally, combined samples were resuspended in 5% acetonitrile–5% formic acid for mass spectrometry analysis. All liquid chromatography (LC)-mass spectrometry (MS) experiments were performed on an LTQ-Velos-Orbitrap column (Thermo Fisher Scientific) equipped with a Famos autosampler (LC Packings) and an Agilent 1100 binary high-pressure liquid chromatography pump (Agilent Technologies) essentially as described previously (29). The LTQ-Velos-Orbitrap was operated in the data-dependent mode and collected high-resolution Orbitrap MS/MS spectra after higher-energy C-trap dissociation for the top 10 most intense ions following each survey scan collected in the Orbitrap.
Peptide, protein, and phosphorylation site identification.
Following acquisition, individual MS/MS spectra were assigned to peptides using the Sequest program (31). To maximize phosphorylation site identifications, a two-stage database searching strategy was employed. In the first stage, all MS/MS spectra were searched against a database containing all protein sequences from the human International Protein Index database (version 3.6) in the forward and reverse orientations as well as sequences of common contaminants. Initial searches were performed with the following parameters: 25-ppm precursor ion tolerance; 0.02-Da product ion tolerance; fully tryptic digestion with up to two missed cleavages; static modifications Cys alkylation (+57.021464) and TMT labeling of Lys and peptide N termini (+229.162932); and dynamic modifications Met oxidation (+15.994915) and phosphorylation of Ser, Thr, and Tyr (+79.966330). The target-decoy approach (32) was then used to distinguish correct and incorrect peptide identifications using linear discriminant analysis based on several parameters, including Xcorr, dCn′, peptide length, precursor ion mass error, numbers of missed cleavages, peptide length, and charge state (33). After filtering to an initial 1% peptide-level false detection rate (FDR), peptides were then assembled into proteins, and proteins were scored and filtered to a final protein FDR of 1% (33).
In the second stage, relaxed parameters were used to match additional MS/MS spectra against a filtered database. Sequest was again employed, but this time the database was filtered to include only forward and reverse sequences for proteins that were identified in stage 1. Stage 2 search parameters included a 3.1-Da precursor ion tolerance; a 0.02-Da product ion tolerance; no enzyme specificity; static modifications Cys alkylation (+57.021464) and TMT labeling of Lys and peptide N termini (+229.162932); and dynamic modifications Met oxidation (+15.994915) and phosphorylation of Ser, Thr, and Tyr (+79.966330). The resulting peptides were again filtered via linear discriminant analysis, but this time, the number of tryptic ends was included as an additional feature and peptide mass errors were corrected to account for occasional incorrect monoisotopic mass assignments.
To evaluate phosphorylation site localization, all phosphopeptides matching Gab2 were scored using the Ascore algorithm (34) and peptides were grouped according to the phosphorylation sites that they contained. A minimum Ascore of 13 was required for phosphorylation site localization (P < 0.05), and phosphorylation site assignments were manually validated to ensure reliability.
Phosphorylation site quantification.
Relative quantification of each peptide was accomplished on the basis of the intensities observed for all six reporter ions from high-resolution Orbitrap MS/MS spectra, after correcting for batch-specific isotopic enrichments of each TMT reagent. Each peptide was required to have a minimum isolation specificity of 0.75 (29) and a summed reporter ion intensity of at least 500 with no more than four missing reporter ions. Individual sites were quantified on the basis of the summed reporter ion intensities for all matching peptides. Nonphosphorylated peptides matching Gab2 were combined to estimate unmodified protein abundance. Quantitative profiles for all phosphorylation sites were normalized to account for slight changes in Gab2 abundance. Finally, analysis of variance (ANOVA) was used to identify statistically significant, site-specific changes in protein phosphorylation. Within each experiment, all P values were adjusted to account for multiple-hypothesis testing via the method of Hochberg and Benjamini (35).
Epifluorescence microscopy.
For immunofluorescence analyses, 5 × 104 MCF-10A cells were seeded in 12-well plates containing coverslips. Twenty-four hours later, cells were washed twice in PBS and fixed in 3.7% formaldehyde for 10 min at room temperature. Cells were washed twice in PBS, permeabilized for 5 min in PBS containing 0.2% Triton X-100, and blocked with PBS containing 0.1% bovine serum albumin for 30 min. Cells were incubated for 2 h with anti-Myc antibodies, washed twice with PBS, and incubated for 1 h with a secondary Alexa Fluor 488-conjugated goat anti-mouse antibody (Invitrogen), Texas Red-phalloidin, and DAPI (4′,6-diamidino-2-phenylindole) diluted in PBS. Images were acquired on a Zeiss Axio Imager Z1 wide-field fluorescence microscope using a ×40 oil-immersion objective.
Proliferation assays.
For proliferation assays, MCF-10A cells were grown in medium supplemented with 10% FBS. The relative number of viable cells was measured every 24 h during four consecutive days using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS), cell proliferation assay from Promega, as shown elsewhere (36). The absorbance was measured at 490 nm using a Tecan GENios Plus microplate reader, and the results displayed represent the mean of triplicates ± standard error (SE).
Cell migration assays.
For the Transwell assays, MCF-10A cells stably expressing wt Gab2 or the S3A mutant were starved overnight, and 4 × 104 cells suspended in 100 μl of EGF-free medium were seeded in the top chamber of polyethylene terephthalate Transwells (24-well insert; pore size, 8 μm; Becton, Dickinson). Cells were allowed to migrate toward 20 ng/ml EGF for 24 h at 37°C in the presence of dimethyl sulfoxide or PD184352 (1 μM), fixed with 10% Formalin, and stained with hematoxylin-eosin (H&E). Cells that did not migrate (top side of the filter) were wiped with a cotton swab. Filters were scanned, and migrated cells were automatically counted using the VisiomorphDP tool (Visiopharm).
For the live-cell-imaging wound-healing assays, an Oris cell migration assay kit (Platipus Technologies) was used. Cells were seeded at 100% confluence (1 × 104 cells) into 96 wells containing a round disk in the center, for creation of the detection zone, and allowed to adhere overnight. Disks were then removed, allowing cells to migrate to the center of the well. Cells were filmed at 37°C with a DeltaVision microscope (Olympus U Plan S-Apo ×20/0.75 numerical aperture) using softWoRx software equipped with a camera (CoolSNAP HQ2). Images were acquired every 20 min for 24 h. Tracking was performed on phase-contrast image series using ImageJ software (http://rsb.info.nih.gov/ij/).
RESULTS
Identification of Gab2 as a substrate of the Ras/MAPK pathway.
To determine if the Ras/MAPK pathway promotes Gab2 phosphorylation on basic consensus motifs, we used two phosphorylation site-specific antibodies directed against similar consensus sequences: Arg/Lys-X-X-pSer/Thr (RXXpS/T) and Arg/Lys-X-X-pSer/Thr-X-Pro (RXXpS/TXP). These consensus sequences are often found in substrates of AGC family kinases, such as Akt, RSK, and S6K1 (37). HEK293 cells transfected with Myc-tagged mouse Gab2 were serum starved overnight and stimulated with different agonists. Immunoprecipitated Gab2 was then analyzed for phosphorylation by immunoblotting using the anti-RXXpS/T and anti-RXXpS/TXP antibodies. With this approach, we found that treatment of cells with agonists of the Ras/MAPK pathway, including the phorbol ester PMA, epidermal growth factor (EGF), and serum (10%), led to the phosphorylation of Gab2 on basic consensus motifs (Fig. 1A). Gab2 phosphorylation correlated with the phosphorylation of both ERK1/2 (T202/Y204) and RSK (S380), suggesting that this pathway converges on Gab2 to promote its phosphorylation.
Fig 1.
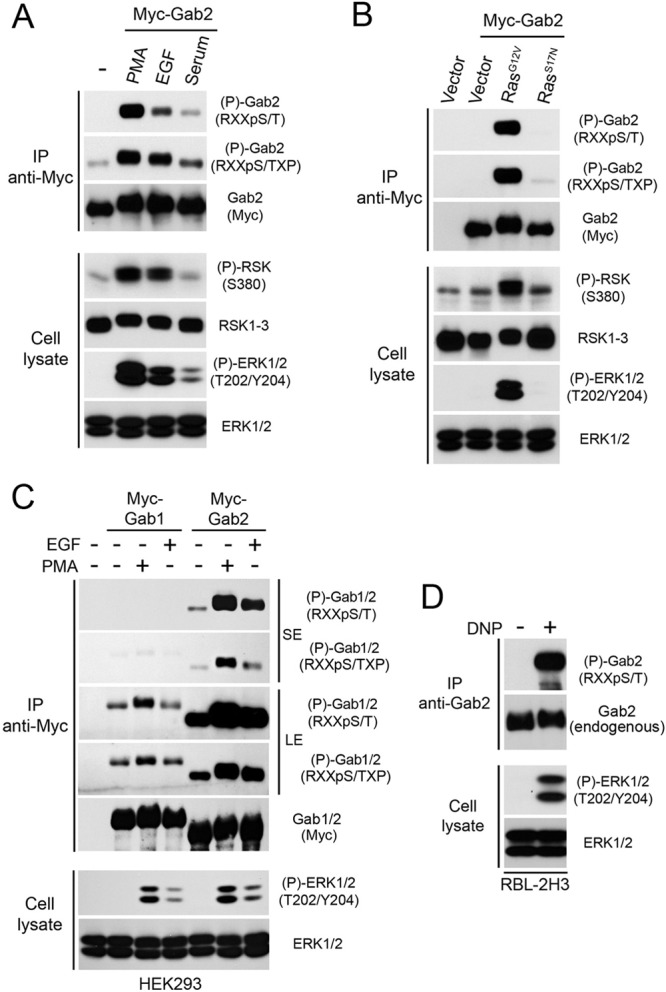
Identification of Gab2 as a target of Ras/MAPK signaling. (A and C) HEK293 cells were transfected with an empty vector or Myc-tagged Gab1 or Gab2, serum starved overnight, and stimulated for 30 min with PMA (100 ng/ml) or 10 min with EGF (25 ng/ml) or fetal bovine serum (10%). Immunoprecipitated (IP) Myc-Gab1 or Myc-Gab2 was then assayed for phosphorylation with phosphomotif antibodies that recognize the RXXpS/T and RXXpS/TXP consensus motifs. Phosphorylated and total levels of RSK and ERK1/2 were assayed by immunoblotting on total cell lysates. SE, short exposure; LE, long exposure. (B) As for panel A, except that HEK293 cells were cotransfected with Myc-Gab2 and a constitutively active (G12V) or inactive (S17N) form of Ras. (D) RBL-2H3 cells were incubated overnight with anti-2,4-dinitrophenol IgE and stimulated with 2,4-dinitrophenol for 10 min prior to endogenous Gab2 immunoprecipitation. Phosphorylation of endogenous Gab2 was assayed using a phosphomotif antibody that recognizes RXXpS/T sequences.
The involvement of the Ras/MAPK pathway was further confirmed using constitutively activated (G12V) and dominant-negative (S17N) forms of H-Ras. We found that expression of RasG12V strongly stimulated Gab2 phosphorylation in the absence of serum or growth factors (Fig. 1B), indicating that Ras-dependent signaling is sufficient to promote Gab2 phosphorylation. To determine if the Ras/MAPK pathway specifically targets the Gab2 isoform, we performed a similar analysis of Gab1 and Gab3, which display ∼38% amino acid identity with Gab2. While we found that Gab3 is not sufficiently soluble to allow efficient immunoprecipitation, our results indicated that Gab1 is not significantly phosphorylated on basic consensus sites compared to the level of phosphorylation of Gab2 (Fig. 1C). To determine if endogenous Gab2 is also regulated by the Ras/MAPK pathway, we used the RBL-2H3 leukemic cell line, which was previously shown to express high levels of Gab2 (38). Importantly, we found that stimulation of the Ras/MAPK pathway in these cells using dinitrophenyl produced a robust increase in endogenous Gab2 phosphorylation on basic consensus sites (Fig. 1D). Together, these findings demonstrate that growth factors and mitogens promote the specific phosphorylation of Gab2 through a pathway that likely involves ERK1/2 signaling.
RSK phosphorylates Gab2 at basic consensus motifs in vivo and in vitro.
RSK is the most likely basophilic protein kinase operating downstream of the MAPK pathway (5). To test its potential involvement in the regulation of Gab2 phosphorylation, we used a MEK1/2 inhibitor (PD184352), which prevents ERK1/2 from activating RSK, as well as an RSK inhibitor (BI-D1870) to directly block its activity (Fig. 2A). We found that treatment of cells with PD184352 or BI-D1870 strongly prevented Gab2 phosphorylation in response to PMA stimulation (Fig. 2B), suggesting that Gab2 is an RSK substrate in cells. The involvement of ERK/RSK signaling was further confirmed by expressing a constitutively activated form of MEK1 (MEK-DD; S212/218D), which was found to be sufficient to promote Gab2 phosphorylation in serum-starved cells (Fig. 2C). We found that treatment of these cells with PD184532 or BI-D1870 completely prevented Gab2 phosphorylation induced by MEK-DD expression, suggesting that RSK is the predominant basophilic kinase regulating Gab2 phosphorylation downstream of the MAPK pathway. Consistent with this, we found that knockdown of RSK1 and RSK2 significantly reduced Gab2 phosphorylation induced by expression of MEK-DD (Fig. 2D) or PMA stimulation (Fig. 2E), indicating that RSK1 and RSK2 are required for the phosphorylation of Gab2 in cells.
Fig 2.

Activation of the Ras/MAPK pathway induces RSK-dependent phosphorylation of Gab2. (A) Schematic representation of the agonists and pharmacological inhibitors used in this study. (B) HEK293 cells were transfected with an empty vector or Myc-tagged Gab2 and serum starved overnight. Cells were pretreated with PD184352 (10 μM) or BI-D1870 (10 μM) for 30 min prior to stimulation with PMA (100 ng/ml) for 30 min. Immunoprecipitated Myc-Gab2 was then assayed for phosphorylation using phosphomotif antibodies that recognize the RXXpS/T and RXXpS/TXP consensus sequences. (C) As for panel B, except that cells were also cotransfected with a constitutively activated form of MEK1 (MEK-DD). (D) HEK293 cells were transfected with Flag-tagged MEK-DD and either a scrambled siRNA or siRNAs targeting RSK1 and/or RSK2. Cells were serum starved overnight, and immunoprecipitated Myc-Gab2 was assayed for phosphorylation using a phosphomotif antibody that recognizes the RXXpS/T consensus sequence. (E) As for panel D, except that cells were stimulated with PMA instead of being transfected with MEK-DD. In addition, total cell lysates were immunoblotted with a phosphorylation site-specific antibody against Ser160. (F) Wild-type and kinase-deficient RSK1 (K112/464R) were immunoprecipitated from HEK293 cells stimulated with PMA (100 ng/ml), and the resulting immunoprecipitates were incubated in a kinase reaction with [γ-32P]ATP and immunopurified full-length Myc-Gab2. The resulting samples were run on SDS-polyacrylamide gels and analyzed by exposing dried film on an autoradiogram. Levels of both RSK1 and Gab2 are shown on the Coomassie-stained gel.
To determine if RSK directly phosphorylates Gab2, we performed in vitro kinase assays with purified proteins and [γ-32P]ATP. HEK293 cells were transiently transfected with wt or kinase-deficient (K112/464R) HA-tagged RSK1, and purified RSK1 from unstimulated or PMA-treated cells was incubated in a reaction buffer with full-length Myc-Gab2 immunopurified from serum-starved cells. Although low levels of 32P label incorporation were detected in purified Gab2 incubated with unstimulated RSK1, we found that activated RSK1 robustly increased 32P label incorporation (∼12-fold) in purified Gab2 (Fig. 2F). The phosphotransferase activity of RSK1 was found to be necessary for this effect, as the kinase-deficient form of RSK1, which retained some ability to autophosphorylate, did not have significantly increased 32P label incorporation in Gab2. Taken together, our results indicate that RSK directly promotes Gab2 phosphorylation in vivo and in vitro in response to Ras/MAPK pathway activation.
Identification of Ser160, Ser211, and Ser620 as RSK-dependent phosphorylation sites.
To identify the RSK-dependent phosphorylation sites in Gab2, we analyzed the sequence surrounding all Ser/Thr residues for similarities to phosphorylation sites in known substrates of RSK (5). We located six potential consensus phosphorylation sites (RXXpS/T), consisting of Ser160, Ser211, Thr256, Thr388, Ser434, and Ser620 (according to mouse Gab2 numbering). To determine whether RSK phosphorylates these sites in cells, each one was individually mutated to an unphosphorylatable alanine residue and Gab2 phosphorylation was assessed using phosphorylation site-specific antibodies against basic consensus sequences. While mutation of Thr256, Thr388, and Ser434 did not affect Gab2 phosphorylation, we found that mutation of Ser160, Ser211, and Ser620 partly prevented Gab2 phosphorylation induced by PMA stimulation, as detected by the anti-RXXpS/TXP or the anti-RXXpS/T antibodies (Fig. 3A). These results suggested that these three serine residues are directly phosphorylated by RSK in cells.
Fig 3.

Identification of Ser160, Ser211, and Ser620 as being regulated by Ras/MAPK signaling. (A) HEK293 cells were transfected with wt Gab2 or potential RSK phosphorylation site mutants (S160A, S211A, T256A, T388A, S434A, and S620A), serum starved overnight, and stimulated with PMA (100 ng/ml) for 30 min. Immunoprecipitated Myc-Gab2 was then assayed for phosphorylation with phosphomotif antibodies that recognize the RXXpS/T and RXXpS/TXP consensus motifs. (B) HEK293 cells were transfected with wt or a kinase-inactive form of RSK1, serum starved overnight, and stimulated with PMA (25 ng/ml) for 15 min. Endogenous Gab2 phosphorylation at Ser160 was determined with a phosphorylation site-specific antibody. (C) As for panel B, except that phosphorylation of endogenous Gab2 at Ser160 was monitored in HEK293 cells pretreated with UO126 (20 μM), PD184352 (10 μM), or BI-D1870 (10 μM) for 30 min prior to PMA stimulation. (D) Phosphorylation of murine Gab2 was confirmed via high-resolution MS/MS sequencing. Gab2 was immunoprecipitated in biological duplicate from cells following either mock treatment, treatment with PMA, or treatment with PMA and PD184352. In a separate experiment, cells were either transfected with an empty vector or different activated (G12V) and inactivated (S17N) alleles of Ras. After subsequent SDS-PAGE separation, bands corresponding to Gab2 were excised and proteins were digested in gel with trypsin followed by TMT labeling for relative quantitation. (E) The spectrum identifies a tryptic peptide bearing phosphorylation at Ser620, as well as TMT labels on both lysine side chains and the peptide N terminus; localization of the phosphorylation site is indicated by the presence of the y9 ion, which matches its theoretical mass to approximately 1 ppm. (Inset) Relative abundance of Ser620 across the six samples normalized with respect to overall Gab2 abundance. PD, PD184352. (F) The spectrum identifies a tryptic peptide bearing two TMT labels as well as phosphorylation of Ser211. Phosphorylation site localization is confirmed by the y5 and y6 ions and b1 and b2 ions, as well as the b3 and b4 ions with subsequent neutral loss of H3PO4. (Inset) Relative levels of Gab2 phosphorylation at Ser211, after normalization on the basis of overall Gab2 abundance. Red and blue lines within the peptide sequences indicate y and b ions, respectively.
To further validate the link between Gab2 phosphorylation and RSK, we made use of a phosphorylation site-specific antibody raised against Ser160 (Ser159 in human Gab2), a site previously shown to be regulated by Akt (17). HEK293 cells were transfected with wt or kinase-deficient RSK1 (K112/464R), and endogenous Gab2 phosphorylation at Ser160 was assessed by immunoblotting on total cell lysates. Compared to control cells, we found that expression of wt RSK1 specifically increased Gab2 phosphorylation at Ser160 after PMA stimulation (Fig. 3B), suggesting that Gab2 is a substrate of RSK in cells. Expression of wt RSK1 induced Gab2 phosphorylation even in the absence of serum and PMA stimulation, consistent with the idea that expression of the wt protein increases basal RSK activity (Fig. 3B). RSK1 phosphotransferase activity was found to be required for stimulating Gab2 phosphorylation, as an RSK1 mutant with inactivating mutations in both kinase domains (K112/464R) did not increase Gab2 phosphorylation to a level over the level already stimulated by endogenous RSK activity (Fig. 3B). In order to validate that ERK/RSK signaling is required for Gab2 phosphorylation at Ser160 in cells, we pretreated cells with MEK1/2 inhibitors (PD184352, UO126) or an RSK inhibitor (BI-D1870) prior to stimulation with PMA. We found that both MEK1/2 and RSK inhibitors almost completely abrogated PMA-induced Gab2 phosphorylation at Ser160 (Fig. 3C), suggesting that RSK activity is required for Gab2 phosphorylation in cells. Residual levels of Ser160 phosphorylation may be due to other basophilic kinases, such as Akt, which was previously shown to phosphorylate this site in Gab2 (17).
While there are no phosphorylation site-specific antibodies against Ser211 and Ser620, we sought to confirm that these RSK-dependent phosphorylation sites are modulated in cells using a quantitative mass spectrometry (MS)-based approach. HEK293 cells were transfected with Myc-tagged Gab2, serum starved overnight, and pretreated with vehicle or the MEK1/2 inhibitor PD184352, prior to being stimulated with PMA (Fig. 3D). Alternatively, cells were cotransfected with an empty vector or constitutively activated (G12V) or dominant-negative (S17N) Ras for 48 h prior to serum starvation and cell lysis (Fig. 3D). Immunoprecipitated Gab2 was then isolated via SDS-PAGE, digested in-gel with trypsin, and labeled with 6-plex isobaric tandem mass tags (TMT6), as done previously (28). Samples were then combined and analyzed by liquid chromatography-assisted tandem MS, and the relative abundances of all identified phosphopeptides were measured across experimental conditions (Fig. 3D). Using this approach, we found that two phosphopeptides containing basic consensus sequences showed significant changes (ANOVA; FDR < 5%) between the activated (PMA or RasG12V) and inactive (PD184352 or RasS17N) conditions (Fig. 3E and F, insets). These phosphopeptides were found to contain Ser211 and Ser620, and identification of these residues as being phosphorylated was obtained by MS/MS sequencing, as depicted by the annotated high-resolution MS/MS spectra (Fig. 3E and F). While previous studies have reported Ser160 as being phosphorylated in cells, we were not able to identify this phosphopeptide in the current study, likely due to the large size of the tryptic peptide containing Ser160 (44 amino acids). Together, our data indicate that RSK regulates the phosphorylation of Ser160, Ser211, and Ser620.
Next, we combined the three RSK-dependent phosphorylation sites identified as described above into a single Gab2 molecule (S3A) and determined its phosphorylation levels in cells treated with agonists of the Ras/MAPK pathway. Importantly, we found that mutation of the three sites completely abrogated Gab2 phosphorylation at basic consensus motifs in response to PMA (Fig. 4A) and EGF (Fig. 4B) stimulation, as well as MEK-DD expression (Fig. 4C), indicating that Ser160/211/620 are the predominant RSK-dependent sites in Gab2. We performed in vitro kinase assays to confirm that RSK predominantly phosphorylates Ser160/211/620 in vitro. Importantly, we found that RSK-mediated 32P label incorporation was strongly reduced when mutant Gab2 (S3A) was used as the substrate (Fig. 4D), indicating that RSK primarily promotes Gab2 phosphorylation on Ser160/211/620. Some level of phosphorylation was also detected with the Gab2 S3A mutant, suggesting that RSK may target additional residues in vitro. While Ser160 and Ser211 are located between the PH domain and the first Grb2-SH3 domain-binding site, it is interesting to note that Ser620 lies between the two Shp2-SH2 domain-binding sites (Tyr603, and Tyr632) (Fig. 4E), suggesting that RSK may modulate Shp2 recruitment in response to growth factors. All three phosphorylation sites are evolutionarily conserved along with the basic residues forming the consensus motif for RSK phosphorylation (Fig. 4F), suggesting that they play important functions.
Fig 4.

RSK predominantly phosphorylates Ser160, Ser211, and Ser620 in cells and in vitro. (A) HEK293 cells were transfected with Myc-tagged wt Gab2 or a triple mutant with all three serine residues converted to alanines (S160/211/620A [S3A]), serum starved overnight, and stimulated with PMA (100 ng/ml) for 30 min. Immunoprecipitated Myc-Gab2 was then assayed for phosphorylation with phosphomotif antibodies that recognize the RXXpS/T and RXXpS/TXP consensus motifs. (B) As for panel A, except that cells were treated with EGF (25 ng/ml) for 10 min. (C) As for panel A, except that cells were transfected with MEK-DD prior to serum starvation and cell lysis. (D) Wild-type RSK1 was immunoprecipitated from HEK293 cells stimulated with PMA (100 ng/ml), and the resulting immunoprecipitates were incubated in a kinase reaction with [γ-32P]ATP and immunopurified wt or S3A Myc-Gab2. The resulting samples were run on SDS-polyacrylamide gels and analyzed by exposing dried film on an autoradiogram. Levels of both RSK1 and Gab2 are shown on the Coomassie-stained gel. (E) Schematic representation of Gab2 with known phosphorylation binding sites. The three phosphorylation sites identified in this study are shown with circles labeled P. (F) Alignment of Gab2 amino acid sequences from different vertebrate species which demonstrate evolutionary conservation of all three phosphorylation sites.
The Ras/MAPK pathway modulates Shp2 recruitment in an RSK-dependent manner.
To address whether RSK-mediated Gab2 phosphorylation modulates its interaction with protein partners, we analyzed Gab2 immunoprecipitates from HEK293 cells stimulated with EGF for different times. As expected, we identified Grb2 within Gab2 immunoprecipitates but did not find that their interaction was modulated by either EGF or inhibitor treatments (Fig. 5A), consistent with the fact that this interaction is mediated via SH3 domains (10). Interestingly, we found that the EGF-dependent recruitment of Shp2 was significantly enhanced in cells exposed to PD184352, suggesting that the Ras/MAPK pathway negatively regulates Shp2 recruitment (Fig. 5A). To further address the role of Gab2 phosphorylation in the recruitment of Shp2, we pretreated HEK293 cells with PMA to promote Gab2 phosphorylation at basic consensus motifs (as for Fig. 1A) and then exposed cells to EGF for 5 or 10 min prior to Gab2 immunoprecipitation. Whereas PMA treatment by itself did not promote Shp2 recruitment (Fig. 5B, first two lanes), we found that pretreatment of cells with PMA severely decreased the ability of Gab2 to associate with Shp2 in response to EGF stimulation (Fig. 5B). Consistent with a role for the MAPK pathway, we found that inhibition of MEK1/2 using PD184352 rescued the inhibitory effect of PMA on Shp2 recruitment (Fig. 5B, last two lanes). The role of MEK1/2-dependent signaling was further addressed by expressing a constitutively activated form of MEK1 (MEK-DD) prior to EGF stimulation. Using this approach, we found that the constitutive activation of the MAPK pathway decreased Gab2-mediated recruitment of Shp2 in response to EGF stimulation (Fig. 5C). To determine if Shp2 recruitment to Gab2 was regulated in an RSK-dependent manner, we pretreated HEK293 cells with the RSK inhibitor BI-D1870 prior to a time course of EGF stimulation. Importantly, we found that inhibition of endogenous RSK activity increased Shp2 recruitment induced by EGF treatment (Fig. 5D), indicating that the Ras/MAPK pathway negatively regulates Gab2-mediated Shp2 recruitment in an RSK-dependent manner. Notably, inhibition of RSK activity using BI-D1870 was found to increase ERK1/2 phosphorylation in response to EGF stimulation, consistent with a previous report showing that RSK negatively regulates ERK1/2 signaling by directly phosphorylating SOS1 (39).
Fig 5.

The Ras/MAPK pathway modulates Shp2 recruitment in an RSK-dependent manner. (A) HEK293 cells were transfected with Myc-Gab2, serum starved overnight, pretreated with PD184352 (10 μM), and stimulated with EGF (25 ng/ml) over a time course. Associated endogenous Grb2 and Shp2 within Myc-Gab2 immunoprecipitates were assayed by immunoblotting. (B) HEK293 cells were transfected with Myc-tagged Gab2, serum starved overnight, and pretreated with PD184352 (10 μM) and/or PMA (100 ng/ml) for 30 min, prior to treatment with EGF (25 ng/ml) for 5 or 10 min. Associated Shp2 was assayed as described for panel A. (C) As for panel A, except that cells were cotransfected with MEK-DD, serum starved overnight, and stimulated with EGF over a time course. (D) As for panel A, except that cells were pretreated with the RSK inhibitor BI-D1870 (10 μM) for 30 min prior to EGF stimulation.
Next, we wanted to determine whether Gab2 phosphorylation on Ser160/211/620 was directly responsible for the reduction in Shp2 recruitment. To assess this, HEK293 cells were transfected with wt Gab2 or the Gab2 S3A mutant (S160/211/620A), treated with EGF over a time course, and Gab2 immunoprecipitated. As shown in Fig. 6A, we found that the Gab2 S3A mutant more efficiently recruited Shp2 in response to EGF treatment, suggesting that RSK-dependent phosphorylation of Ser160/211/620 regulates Shp2 binding. This effect appeared to be specific to Shp2, as no modulations were observed in the recruitment of p85 in response to EGF stimulation (Fig. 6B). The increase induced by the Gab2 S3A mutant was quantified by densitometry and found to be significantly different from that induced by wt Gab2 at the 5-, 10-, and 30-min time points (Fig. 6C). Interestingly, we also found that expression of the Gab2 S3A mutant potentiated ERK1/2 phosphorylation in response to EGF stimulation (Fig. 6A), consistent with the established role of Shp2 as a positive regulator of Ras/MAPK signaling. We also determined whether constitutive activation of the MAPK pathway using MEK-DD could similarly negatively regulate Shp2 recruitment to Gab2. Whereas MEK-DD expression strongly suppressed Shp2 recruitment to wt Gab2, we found that the Gab2 S3A mutant was mostly insensitive to the activation of the Ras/MAPK pathway (Fig. 6D). To further validate these findings, we made use of Gab2-deficient MEFs (25) to stably express Myc-tagged wt Gab2 or the Gab2 S3A mutant in a background without endogenous Gab2 expression (Fig. 6E). To determine the ability of exogenous Gab2 to recruit Shp2, we analyzed the levels of Myc-tagged Gab2 within endogenous Shp2 immunoprecipitates. Compared to wt Gab2, we found that the Gab2 S3A mutant recruited more Shp2 in response to EGF stimulation (Fig. 6F). We also determined which of the three RSK-dependent phosphorylation sites participated in the regulation of Shp2 binding and found that mutation of Ser211 or Ser620 was sufficient to increase Shp2 recruitment in response to EGF stimulation (Fig. 6G). Together, these results provide robust evidence that RSK-mediated phosphorylation of Gab2 on Ser211 and Ser620 impedes Shp2 recruitment and function in response to growth factors.
Fig 6.

A Gab2 mutant that cannot be phosphorylated on Ser160/211/620 promotes Shp2 recruitment. (A) HEK293 cells were transfected with wt Myc-Gab2 or the S3A mutant, serum starved overnight, and stimulated with EGF (25 ng/ml) over a time course. Associated Shp2 within Gab2 immunoprecipitates was evaluated by immunoblotting. (B) HEK293 cells were transfected with wt Myc-Gab2 or the S3A mutant, serum starved overnight, and stimulated with EGF (25 ng/ml) over a time course. Associated p85 was evaluated within Gab2 immunoprecipitates by immunoblotting, as well as Akt phosphorylation at Ser473. (C) The bar graph shows the relative Shp2 recruitment quantified by densitometry (as described in Materials and Methods) from three independent experiments similar to the one whose results are shown in panel A. The data are expressed as the mean fold increase in Shp2 recruitment comparing immunoprecipitations with wt Gab2 and the S3A mutant. Results are means ± SEMs. One-tailed unequal variance Student's t test P values are indicated for a comparison of the means of the wt and mutant (S3A) Gab2 levels. *, P < 0.01. (D) HEK293 cells were cotransfected with wt Myc-Gab2 or the S3A mutant, with or without MEK-DD, serum starved overnight, and stimulated with EGF (25 ng/ml) over a time course. Shp2 recruitment was evaluated as described for panel A. (E) Wild-type and Gab2-deficient MEFs were evaluated for the presence of endogenous Gab2 by immunoblotting on total cell lysates. (F) Gab2-deficient MEFs stably expressing wt Gab2 or the S3A mutant were seeded at a similar density and serum starved overnight, prior to EGF (25 ng/ml) stimulation over a time course. Associated Myc-Gab2 was assayed within endogenous Shp2 immunoprecipitates by immunoblotting. (G) HEK293 cells were transfected with wt Myc-Gab2 or each of the Gab2 mutants (S160A, S211A, or S620A), serum starved overnight, and stimulated with EGF (25 ng/ml) over a time course. Associated Shp2 was evaluated within Gab2 immunoprecipitates by immunoblotting.
RSK-mediated Gab2 phosphorylation impairs mammary epithelial cell migration.
Gab2-mediated Shp2 recruitment has previously been shown to promote the migration of mammary epithelial cells (14, 16). We thus used this biological system to address the function of RSK-mediated Gab2 phosphorylation. We generated stable MCF-10A cells, which express very low endogenous levels of Gab2, to express an empty vector, wt Gab2, or the Gab2 S3A mutant. Upon generation of pooled populations of stably expressing cells, we readily observed a change in morphology between parental and Gab2-expressing cells (Fig. 7A). This change in morphology induced by the expression of wt Gab2 was previously shown to correlate with enhanced cell motility (16). Interestingly, we also observed a change in morphology between cells expressing wt Gab2 and the S3A mutant (Fig. 7A), with the latter having a more elongated cell shape reminiscent of mesenchymal morphology. Immunofluorescence experiments indicated that wt Gab2 and the S3A mutant were expressed at similar levels, which was confirmed by immunoblotting the total cell lysates of all three cell lines (Fig. 7B). The differences in morphology between the three stable cell lines were quantified, and the S3A Gab2-expressing cells were found to be 2 and 7 times more likely to have an elongated shape than wt Gab2 and empty vector-expressing cells, respectively (Fig. 7C). We also performed an assay to verify whether Shp2 recruitment was modulated in MCF-10A cells and found that the Gab2 S3A mutant recruited more Shp2 than wt Gab2 in response to EGF stimulation (Fig. 7D).
Fig 7.

Stable expression of the Gab2 S3A mutant alters MCF-10A cell morphology. (A) Confocal images of growing MCF-10A cells stably expressing an empty vector, wt Gab2, or the S3A mutant. Cells were stained with phalloidin to visualize the actin cytoskeleton, anti-Myc to monitor Gab2 expression, and DAPI to visualize nuclei. (B) Stable MCF-10A cells were lysed and immunoblotted for Gab2 (anti-Myc) to show equal expression levels between wt Gab2 and the S3A mutant, as well as phosphorylated ERK1/2 (T202/Y204) to show increased activation in Gab2 S3A-expressing cells. The ratio of ERK1/2 phosphorylation over total ERK1/2 levels was calculated on the basis of densitometric values. EV, empty vector. (C) Quantification of elongated cells from immunostaining experiments similar to the one shown in panel A. (D) MCF-10A cells stably expressing an empty vector, wt Myc-Gab2, or the S3A mutant were serum starved overnight and stimulated with EGF (25 ng/ml) over a time course. Associated Myc-Gab2 within endogenous Shp2 immunoprecipitates was evaluated by immunoblotting. The ratio of ERK1/2 phosphorylation over total ERK1/2 levels was calculated on the basis of densitometric values.
Based on the known role of Gab2 in cell motility, we determined whether Gab2 phosphorylation affected MCF-10A cell migration using a Transwell assay. While we confirmed previous data by showing that expression of wt Gab2 increases MCF-10A migration (16), we found that cells expressing the Gab2 S3A mutant exhibited a significant increase in cell migration (∼1.5-fold compared to wt Gab2-expressing cells) (Fig. 8A and B). As shown in Fig. 8C, we confirmed that this effect was not a result of increased cell proliferation. To further characterize this apparent gain of function of mutant Gab2, we performed live-cell-imaging wound-healing assays by tracking migrating cells. Quantifications of cell paths revealed that MCF-10A cells expressing the Gab2 S3A mutant exhibited a robust increase in displacement from the point of origin (Fig. 8D). Indeed, the speed of movement of Gab2 S3A-expressing cells was found to be ∼1.6-fold higher than that of wt Gab2-expressing cells, reaching 0.45 μm/min (Fig. 8E). As shown in Fig. 7B and D, we found that cells expressing the Gab2 S3A mutant displayed increased ERK1/2 phosphorylation, consistent with the role of Shp2 in the regulation of the Ras/MAPK pathway. To determine whether this pathway was partly responsible for the observed increased motility of cells expressing the Gab2 S3A mutant, we treated cells with the MEK1/2 inhibitor PD184352 during image acquisitions. As shown in Fig. 8E, we found that PD184352 treatment abrogated the increased motility of Gab2 S3A-expressing cells, consistent with a role of the Ras/MAPK pathway. Taken together, our results indicate that RSK-mediated phosphorylation of Gab2 inhibits cell elongation and Gab2-dependent migration of mammary epithelial cells. These findings suggest that RSK-mediated regulation of Gab2 function represents a novel negative-feedback pathway that restricts cell migration occurring in response to Ras/MAPK signaling (Fig. 9).
Fig 8.

Stable expression of the Gab2 S3A mutant promotes MCF-10A cell migration. (A) MCF-10A cells stably expressing an empty vector, wt Gab2, or the S3A mutant were seeded on top of Boyden chambers in EGF-free medium. Cells were allowed to migrate toward 20 ng/ml EGF for 24 h, fixed, and stained using H&E. (B) Filters processed as described for panel A were scanned, and migrated cells were automatically counted, as described in Materials and Methods. (C) Analysis of the proliferation of stable MCF-10A cells using the MTS assay over a time course of 72 h. The data are expressed as the mean fold increase in cell proliferation. A.U., absorbance units. (D) Live-cell-imaging wound-healing assays were performed using MCF-10A cells stably expressing wt Gab2 or the S3A mutant. Tree graphs show the length (μm), direction, and displacement (distance from the origin) of more than 70 cell paths. (E) Quantification of cell speed (μm/min) based on the total displacement calculated in wound-healing assays. An unpaired Student's t test was performed to determine that the differences between wt and S3A Gab2 conditions and the effect of PD184352 treatment were statistically significant (*, P < 0.00001).
Fig 9.
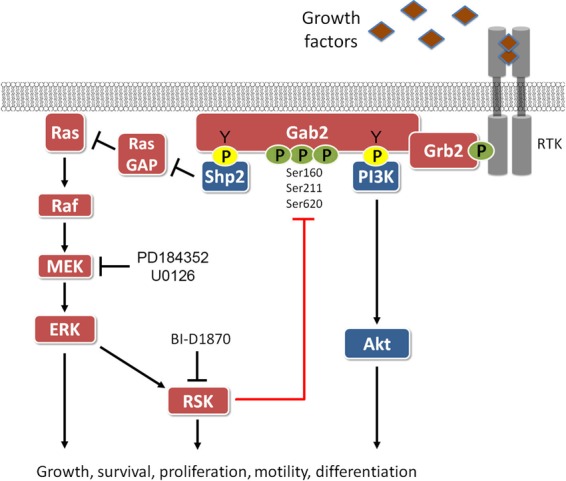
Schematic representation of Gab2 signaling and its negative regulation by RSK. While Gab2 phosphorylation at tyrosine residues promotes Shp2 recruitment and activation of the Ras/MAPK pathway, RSK-mediated phosphorylation of Gab2 on Ser/Thr residues inhibits Shp2 binding and Shp2-dependent signaling. RSK-mediated phosphorylation of Gab2 likely plays a role in tightly regulating Gab2-dependent biological functions, such as the regulation of cell motility.
DISCUSSION
The role of the Gab2 adapter protein has been widely investigated in hematopoietic cells and in cancer progression, where it functions to regulate the activation of several signaling pathways, including Ras/MAPK and PI3K/Akt. In this study, we demonstrate in different cell types that Gab2 phosphorylation is regulated by the Ras/MAPK pathway in an RSK-dependent manner (Fig. 1 to 2). Using phosphomotif antibodies, mass spectrometry, and site-directed mutagenesis, we found that RSK phosphorylates Gab2 on three serine residues which fall into the RXXpS/T consensus sequence (Ser160, Ser211, Ser620), both in vivo and in vitro (Fig. 3 to 4). Of note, we found that the closely related Gab1 isoform is not significantly phosphorylated by RSK in response to growth factors (Fig. 1C), which is indicative of the specificity of the regulation of Gab2. Using both gain- and loss-of-function approaches, we showed that RSK-mediated Gab2 phosphorylation decreases Shp2 binding in response to EGF stimulation (Fig. 5 to 7), suggesting that RSK negatively regulates the Gab2-Shp2 axis. Using mammary epithelial cells that stably express exogenous Gab2 alleles, we found that Gab2 phosphorylation on Ser160/211/620 inhibits cell motility (Fig. 8), suggesting a model whereby RSK limits Gab2-dependent cell migration upon activation of the Ras/MAPK pathway. Based on the widespread role of Gab2 in receptor signaling, our results suggest that RSK may regulate Gab2 function in response to many types of growth factors and cytokines.
Whereas Gab2 tyrosine phosphorylation and activation of downstream signaling pathways are well characterized, relatively little is known about the mechanisms involved in Gab2 inactivation by Ser/Thr phosphorylation. Our data demonstrate that the Ras/MAPK pathway and, more specifically, RSK phosphorylate Gab2 on three serine residues to inhibit Shp2 recruitment. A previous report has shown that Akt phosphorylates Gab2 on Ser159 (Ser160 is the homologous site in mouse Gab2) and thereby reduces Gab2 tyrosine phosphorylation via unknown mechanisms (17). While Ser159 is one of the three phosphorylation sites that we found to be regulated by RSK, we did not observe that Ser159 phosphorylation modulates Gab2 tyrosine phosphorylation in response to EGF stimulation. More recently, Gab2 was shown to be phosphorylated on two additional residues (Ser210 and Thr391, according to the human numbering) in response to growth factors, but the actual protein kinase involved in these phosphorylation events remains elusive (19). Interestingly, phosphorylation of these sites was shown to mediate 14-3-3 binding and to inhibit Grb2 binding, but again, the mechanisms involved have not been determined. While we found that Ser210 (Ser211 is the homologous site in mouse Gab2) is regulated by RSK (Fig. 3), we did not observe that RSK-mediated phosphorylation of Gab2 altered Grb2 or p85 binding (Fig. 5A and 6B). Our results also indicate that Thr391 is not regulated by RSK, consistent with the idea that RSK is mostly a serine kinase and that very few substrates of RSK have been shown to be phosphorylated on threonine residues (5).
We found that RSK phosphorylates a novel site in Gab2 (Ser620), which is located between the two SH2 domain-binding sites for Shp2 (Tyr603 and Tyr632) (Fig. 3 and 4). Our results indicate that RSK-mediated phosphorylation of Gab2 inhibits Shp2 recruitment, suggesting that phosphorylated Ser620 hinders Shp2 interaction to the SH2 domain-binding sites. Another possibility is that phosphorylation of Ser160/211/620 causes a conformational change in Gab2 that reduces its affinity for Shp2, but more experimentation will be required to fully understand the molecular events taking place upon RSK-mediated phosphorylation. Interestingly, ERK1/2 was also shown to modulate Shp2 recruitment in response to IL-2 stimulation (18). ERK1/2 was found to phosphorylate Ser613, which is also located in close proximity to the SH2 domain-binding motif of Gab2. Although we have not been able to identify phosphorylated Ser613 in our MS experiments, these results suggest that RSK may collaborate with ERK1/2 to inhibit Shp2 recruitment in response to agonists of the Ras/MAPK pathway. Based on the fact that RSK-mediated phosphorylation of Gab2 does not perturb recruitment of the p85 subunit of PI3K or Grb2 binding, our results suggest that Gab2 phosphorylation on Ser160/211/620 specifically regulates Shp2 recruitment. Our findings indicate that RSK is part of a negative-feedback loop that restricts Gab2-Shp2 signaling, similar to the recently described loop involving RSK and SOS1 (39). Based on the established role of Shp2 in the regulation of Ras/MAPK signaling (40), our results highlight a second RSK-dependent inhibitory loop that negatively regulates ERK1/2 signaling.
The mechanisms by which Gab2 contributes to breast cancer are not fully understood, but several studies implicated Shp2 in Gab2-dependent cell proliferation and motility (14, 16). Similar to our findings, expression of Gab2 in MCF-10A cells has been shown to promote a change in morphology and cell motility (16), providing an excellent system to analyze the impact of Gab2 phosphorylation on its function. We found that mutation of Ser160/211/620 potentiated the effect of wt Gab2 on cell motility, which is consistent with the important role that Shp2 plays in cytoskeletal organization and cell migration (16). In addition to regulating the Ras/MAPK pathway, Shp2 has recently been shown to modulate RhoA activation (16, 41). Indeed, the recruitment of Shp2 to the plasma membrane was found to promote the dephosphorylation of Rho kinase II (ROCKII), resulting in modulation of RhoA-induced cell rounding (41). Based on the known roles of the Ras/MAPK pathway in cell motility in diverse systems (42), these findings indicate that RSK likely regulates the involvement of Shp2 in both of these pathways to promote a change in cell shape and cell motility.
In summary, we have identified a novel mechanism by which growth factors regulate Gab2 function. Based on the involvement of Gab2 in the pathogenesis of breast cancer, our results provide important information about the complexity of its regulation. Our findings also have potential clinical implication, as RSK1 and RSK2 have been shown to be upregulated in both breast and prostate cancers (43). While these two RSK isoforms likely play important proproliferative functions (5, 6), our results indicate that they are also involved in the negative regulation of RTK signaling.
ACKNOWLEDGMENTS
We deeply appreciate all the members of our laboratories for their insightful discussions and comments on the manuscript and the data. We also specifically acknowledge Chris Jahns (HMS Taplin Biological Mass Spectrometry Facility) for assistance with preparation of in-gel digests.
This work was supported by grants from the Canadian Institutes of Health Research (CIHR; MOP123408), the Canadian Cancer Society Research Institute (700878), and the Cancer Research Society (F121153) (to P.P.R), as well as the United States National Institutes of Health (to S.P.G.; HG3456) and the CIHR (to S.C.; MOP89877). P. P. Roux holds the Canada Research Chair in Signal Transduction and Proteomics, and S. Carreno holds a Chercheur Boursier award from the Fonds de Recherche du Québec en Santé (FRQS). X. Zhang holds a China Scholarship Council studentship. J. A. Galan holds a postdoctoral fellowship from the Canadian Institutes of Health Research (CIHR). IRIC core facilities are supported in part by the FRQS.
We have no conflict of interest to declare.
Footnotes
Published ahead of print 11 February 2013
REFERENCES
- 1. Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell 141:1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy LO, Blenis J. 2006. MAPK signal specificity: the right place at the right time. Trends Biochem. Sci. 31:268–275 [DOI] [PubMed] [Google Scholar]
- 3. Roux PP, Blenis J. 2004. ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 68:320–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cargnello M, Roux PP. 2011. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 75:50–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Romeo Y, Zhang X, Roux PP. 2012. Regulation and function of the RSK family of protein kinases. Biochem. J. 441:553–569 [DOI] [PubMed] [Google Scholar]
- 6. Carriere A, Ray H, Blenis J, Roux PP. 2008. The RSK factors of activating the Ras/MAPK signaling cascade. Front. Biosci. 13:4258–4275 [DOI] [PubMed] [Google Scholar]
- 7. Brummer T, Schmitz-Peiffer C, Daly RJ. 2010. Docking proteins. FEBS J. 277:4356–4369 [DOI] [PubMed] [Google Scholar]
- 8. Gu H, Neel BG. 2003. The “Gab” in signal transduction. Trends Cell Biol. 13:122–130 [DOI] [PubMed] [Google Scholar]
- 9. Wohrle FU, Daly RJ, Brummer T. 2009. Function, regulation and pathological roles of the Gab/DOS docking proteins. Cell Commun. Signal 7:22 doi:10.1186/1478-811X-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wohrle FU, Daly RJ, Brummer T. 2009. How to Grb2 a Gab. Structure 17:779–781 [DOI] [PubMed] [Google Scholar]
- 11. Nishida K, Hirano T. 2003. The role of Gab family scaffolding adapter proteins in the signal transduction of cytokine and growth factor receptors. Cancer Sci. 94:1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Celebi JT, Adams SJ, Aydin IT. 2012. GAB2—a scaffolding protein in cancer. Mol. Cancer Res. 10:1265–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brummer T, Schramek D, Hayes VM, Bennett HL, Caldon CE, Musgrove EA, Daly RJ. 2006. Increased proliferation and altered growth factor dependence of human mammary epithelial cells overexpressing the Gab2 docking protein. J. Biol. Chem. 281:626–637 [DOI] [PubMed] [Google Scholar]
- 14. Bentires-Alj M, Gil SG, Chan R, Wang ZC, Wang Y, Imanaka N, Harris LN, Richardson A, Neel BG, Gu H. 2006. A role for the scaffolding adapter GAB2 in breast cancer. Nat. Med. 12:114–121 [DOI] [PubMed] [Google Scholar]
- 15. Bocanegra M, Bergamaschi A, Kim YH, Miller MA, Rajput AB, Kao J, Langerod A, Han W, Noh DY, Jeffrey SS, Huntsman DG, Borresen-Dale AL, Pollack JR. 2010. Focal amplification and oncogene dependency of GAB2 in breast cancer. Oncogene 29:774–779 [DOI] [PubMed] [Google Scholar]
- 16. Herrera Abreu MT, Hughes WE, Mele K, Lyons RJ, Rickwood D, Browne BC, Bennett HL, Vallotton P, Brummer T, Daly RJ. 2011. Gab2 regulates cytoskeletal organization and migration of mammary epithelial cells by modulating RhoA activation. Mol. Biol. Cell 22:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lynch DK, Daly RJ. 2002. PKB-mediated negative feedback tightly regulates mitogenic signalling via Gab2. EMBO J. 21:72–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Arnaud M, Crouin C, Deon C, Loyaux D, Bertoglio J. 2004. Phosphorylation of Grb2-associated binder 2 on serine 623 by ERK MAPK regulates its association with the phosphatase SHP-2 and decreases STAT5 activation. J. Immunol. 173:3962–3971 [DOI] [PubMed] [Google Scholar]
- 19. Brummer T, Larance M, Herrera Abreu MT, Lyons RJ, Timpson P, Emmerich CH, Fleuren ED, Lehrbach GM, Schramek D, Guilhaus M, James DE, Daly RJ. 2008. Phosphorylation-dependent binding of 14-3-3 terminates signalling by the Gab2 docking protein. EMBO J. 27:2305–2316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caron C, Spring K, Laramee M, Chabot C, Cloutier M, Gu H, Royal I. 2009. Non-redundant roles of the Gab1 and Gab2 scaffolding adapters in VEGF-mediated signalling, migration, and survival of endothelial cells. Cell Signal. 21:943–953 [DOI] [PubMed] [Google Scholar]
- 21. Lock LS, Maroun CR, Naujokas MA, Park M. 2002. Distinct recruitment and function of Gab1 and Gab2 in Met receptor-mediated epithelial morphogenesis. Mol. Biol. Cell 13:2132–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Romeo Y, Moreau J, Zindy PJ, Saba-El-Leil M, Lavoie G, Dandachi F, Baptissart M, Borden KL, Meloche S, Roux PP. 16 July 2012. RSK regulates activated BRAF signalling to mTORC1 and promotes melanoma growth. Oncogene [Epub ahead of print.] doi:10.1038/onc.2012.312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Carriere A, Romeo Y, Acosta-Jaquez HA, Moreau J, Bonneil E, Thibault P, Fingar DC, Roux PP. 2011. ERK1/2 phosphorylate Raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). J. Biol. Chem. 286:567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carriere A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, Roux PP. 2008. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 18:1269–1277 [DOI] [PubMed] [Google Scholar]
- 25. Gu H, Saito K, Klaman LD, Shen J, Fleming T, Wang Y, Pratt JC, Lin G, Lim B, Kinet JP, Neel BG. 2001. Essential role for Gab2 in the allergic response. Nature 412:186–190 [DOI] [PubMed] [Google Scholar]
- 26. Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. 2004. Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc. Natl. Acad. Sci. U. S. A. 101:13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shevchenko A, Wilm M, Vorm O, Mann M. 1996. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal. Chem. 68:850–858 [DOI] [PubMed] [Google Scholar]
- 28. Cargnello M, Tcherkezian J, Dorn JF, Huttlin EL, Maddox PS, Gygi SP, Roux PP. 2012. 4E-T phosphorylation by JNK promotes stress-dependent P-body assembly. Mol. Cell. Biol. 32:4572–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ting L, Rad R, Gygi SP, Haas W. 2011. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods 8:937–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rappsilber J, Ishihama Y, Mann M. 2003. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75:663–670 [DOI] [PubMed] [Google Scholar]
- 31. Yates JR, III, Eng JK, McCormack AL, Schieltz D. 1995. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67:1426–1436 [DOI] [PubMed] [Google Scholar]
- 32. Elias JE, Gygi SP. 2007. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 4:207–214 [DOI] [PubMed] [Google Scholar]
- 33. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. 2010. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 143:1174–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. 2006. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 24:1285–1292 [DOI] [PubMed] [Google Scholar]
- 35. Hochberg Y, Benjamini Y. 1990. More powerful procedures for multiple significance testing. Stat. Med. 9:811–818 [DOI] [PubMed] [Google Scholar]
- 36. Julien LA, Carriere A, Moreau J, Roux PP. 2010. mTORC1-activated S6K1 phosphorylates Rictor on threonine 1135 and regulates mTORC2 signaling. Mol. Cell. Biol. 30:908–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. 1996. Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett. 399:333–338 [DOI] [PubMed] [Google Scholar]
- 38. Huang F, Tong X, Deng H, Fu L, Zhang R. 2008. Inhibition of the antigen-induced activation of RBL-2H3 cells by Gab2 siRNA. Cell. Mol. Immunol. 5:433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Saha M, Carriere A, Cheerathodi M, Zhang X, Lavoie G, Rush J, Roux PP, Ballif BA. 2012. RSK phosphorylates SOS1 creating 14-3-3-docking sites and negatively regulating MAPK activation. Biochem. J. 447:159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Neel BG, Gu H, Pao L. 2003. The ‘Shp'ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28:284–293 [DOI] [PubMed] [Google Scholar]
- 41. Lee HH, Chang ZF. 2008. Regulation of RhoA-dependent ROCKII activation by Shp2. J. Cell Biol. 181:999–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mendoza MC, Er EE, Zhang W, Ballif BA, Elliott HL, Danuser G, Blenis J. 2011. ERK-MAPK drives lamellipodia protrusion by activating the WAVE2 regulatory complex. Mol. Cell 41:661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romeo Y, Roux PP. 2011. Paving the way for targeting RSK in cancer. Expert Opin. Ther. Targets 15:5–9 [DOI] [PubMed] [Google Scholar]