

Abstract
Angiopoietin-like protein 4 (ANGPTL4/FIAF) has been proposed as a circulating mediator between the gut microbiota and fat storage. Here, we show that transcription and secretion of ANGPTL4 in human T84 and HT29 colon adenocarcinoma cells is highly induced by physiological concentrations of short-chain fatty acids (SCFA). SCFA induce ANGPTL4 by activating the nuclear receptor peroxisome proliferator activated receptor γ (PPARγ), as demonstrated using PPARγ antagonist, PPARγ knockdown, and transactivation assays, which show activation of PPARγ but not PPARα and PPARδ by SCFA. At concentrations required for PPARγ activation and ANGPTL4 induction in colon adenocarcinoma cells, SCFA do not stimulate PPARγ in mouse 3T3-L1 and human SGBS adipocytes, suggesting that SCFA act as selective PPARγ modulators (SPPARM), which is supported by coactivator peptide recruitment assay and structural modeling. Consistent with the notion that fermentation leads to PPAR activation in vivo, feeding mice a diet rich in inulin induced PPAR target genes and pathways in the colon. We conclude that (i) SCFA potently stimulate ANGPTL4 synthesis in human colon adenocarcinoma cells and (ii) SCFA transactivate and bind to PPARγ. Our data point to activation of PPARs as a novel mechanism of gene regulation by SCFA in the colon, in addition to other mechanisms of action of SCFA.
INTRODUCTION
In the last decade, interest in the role of the intestinal microbiota has grown exponentially. The functional role of the gut microbiota has been studied mainly in relation to intestinal health and immune function. However, there are growing speculations that the gut microbiota also influence other diseases, including type 1 diabetes, autism, and obesity, and impact more distant organs (1). Those notions have spurred the search for circulating factors that communicate between the intestinal microbiota and other parts of the body. One factor that was found to be strongly downregulated in intestine upon colonization of the gut of germ-free mice with microbiota and that appears to be important for microbiota-induced deposition of triglycerides in adipocytes is angiopoietin-like 4 (ANGPTL4), also referred to as FIAF (fasting-induced adipose factor) (2, 3). ANGPTL4 is secreted by a variety of tissues, including adipose tissue, liver, skeletal muscle, and intestine (4). It is released as a 50-kDa prohormone that is subsequently cleaved into N- and C-terminal fragments. The N-terminal fragment of ANGPTL4 blocks activity of the enzyme lipoprotein lipase (LPL), which catalyzes uptake of circulating lipids into tissues (5). Accordingly, ANGPTL4 overexpression raises circulating triglyceride levels (6–10). In cardiomyocytes and macrophages, induction of ANGPTL4 by fatty acids and subsequent inhibition of LPL is part of a feedback mechanism aimed at preventing cellular lipid overload and thus reducing lipotoxicity and inflammation (11, 12).
Expression of ANGPTL4 is under transcriptional control of peroxisome proliferator-activated receptors (PPARs). Indeed, ANGPTL4 was originally cloned as a target gene of PPARα and PPARγ (13, 14). Depending on the specific tissue, ANGPTL4 mRNA levels are governed primarily by PPARα (liver and small intestine) (13, 15), PPARδ (skeletal muscle, heart, and macrophages) (11, 12, 16), or PPARγ (adipocytes) (13, 14). In addition to the tissues mentioned above, ANGPTL4 is also expressed in human colon (17) (http://biogps.org/), but little is known about the factors regulating ANGPTL4 mRNA expression and protein secretion in this tissue. PPARδ is highly expressed in human colonocytes, followed by PPARγ and, to a lesser extent, PPARα, suggesting that PPARs are important regulators of ANGPTL4 expression (18). A similar expression profile is found in mouse colonocytes (19). Considering the status of ANGPTL4 as a putative mediator between the gut microbiota and adipose tissue, we were interested in identifying the microbiota-related factors that influence ANGPTL4 production in the colon. Previously, ANGPTL4 expression in the human colon cell line HT29 was found to be stimulated by heat-stable factors secreted by the probiotic bacteria Lactobacillus sp. strain F19 (20). We hypothesized that these factors corresponded to short-chain fatty acids (SCFA), which are compounds formed by intestinal microbial activity. Accordingly, we studied the regulation of ANGPTL4 by SCFA.
MATERIALS AND METHODS
Animal experiments.
Male C57BL/6J mice were purchased from Charles River Laboratories. They were housed in pairs on a 12-h light/dark cycle at 21°C with free access to feed and water throughout the experimental period. For 2 weeks before the start of the interventions, mice were fed a standard semisynthetic low-fat diet based on D12450B (Research Diets Inc., New Brunswick, NJ), with modified content of sucrose and corn starch (16.4 and 40.5%, wt/wt, respectively). In the first experiment, 12-week-old mice were mildly sedated with isoflurane at 9:00 a.m. Mice were kept under sedation and received either an 80-μl rectal infusion of saline (n = 4) or saline containing 100 mM sodium propionate (n = 4). Both solutions had a pH of 6.5. The solutions were administered by inserting a gel-loading tip 3 cm into the rectum and slowly pushing the solution out of the tip. The infusions were administered on 6 consecutive days. Four hours after the rectal infusion on day 6, mice were anesthetized with isoflurane and subsequently sacrificed by cervical dislocation. The colon was excised, and adhering fat was carefully removed. The epithelial lining of the colon was scraped and immediately frozen in liquid nitrogen to be stored at −80°C for subsequent RNA isolation. In the second experiment, mice were stratified according to their body weight and allocated to the control diet group (n = 5; low-fat diet as described above) or inulin diet group (n = 6; low-fat diet with 10% [wt/wt] corn starch replaced by inulin). After 10 days of dietary intervention, mice were fasted overnight and provided with 1 g of experimental diet. After 4 h, mice were anesthetized using isoflurane and sacrificed by cervical dislocation. Colon was removed, and luminal content and mucosal scrapings were collected, snap-frozen in liquid nitrogen, and stored at −80°C (scrapings) or −20°C (content). Luminal content was used for SCFA measurement by gas chromatography. Mucosal scrapings were used for RNA isolation. The experiments were authorized by the Local Committee for Care and Use of Laboratory Animals at Wageningen University.
SCFA measurement.
SCFA concentrations were measured in the luminal content of different parts of the gastrointestinal (GI) tract of C57BL/6 mice fed a low-fat diet. After sacrifice, luminal content was collected in tubes containing phosphoric (H3PO4) and isocaproic acid, after which the tubes were thoroughly mixed and stored at −20°C. For analysis, samples were thawed, mixed on a vortex, and centrifuged at 14,000 rpm for 5 min. The supernatant was collected and SCFA concentrations were determined by gas chromatography (Fisons HRGC Mega 2; CE Instruments, Milan, Italy) at 190°C using a glass column fitted with Chromosorb 101. The carrier gas was N2 saturated with methanoic acid, and isocaproic acid was used as an internal standard.
Cell culture.
T84 and HT29 human colonic carcinoma epithelial cells were cultured in Dulbecco's modified Eagle medium (DMEM) or DMEM containing Ham's F-12 medium (DMEM F-12) (Lonza, the Netherlands) containing 5 or 10% fetal calf serum (FCS), respectively, and 1% penicillin-streptomycin at 37°C in a humidified incubator under 95% air and 5% CO2. Cells were grown until 80 to 90% confluence and treated for the indicated time periods with sodium acetate, sodium propionate, and sodium butyrate from a stock in phosphate-buffered saline (PBS) to a final concentration of 1 or 8 mM. Alternatively, cells were treated with synthetic agonists for PPARα (Wy14643; 5 μM), PPARδ (GW501516; 1 μM), PPARγ (rosiglitazone; 1 μM), synthetic PPARγ antagonist GW9662 (5 μM), or trichostatin A (100 nM) from a stock in dimethylsulfoxide (DMSO). In one experiment, cells were preincubated with α-amanitin (10 μg/ml) from a stock in water for 1 h, followed by 24 h of coincubation with butyrate at 1 mM. After the indicated time points, medium was collected and used for analysis of ANGPTL4 by enzyme-linked immunosorbent assay (ELISA), and/or cells were harvested for subsequent RNA isolation.
siRNA transfection.
Silencing of the PPARγ gene in T84 cells was done using ON-TARGETplus SMARTpool for human PPARγ (Thermo Scientific Dharmacon, Etten-Leur, the Netherlands), representing a mixture of four short interfering RNAs (siRNAs): CAAAUCACCAUUCGUUAUC, GACAUGAAUUCCUUAAUGA, GAUAUCAAGCCCUUCACUA, and GACAGCGACUUGGCAAUAU. As a control, we used an ON-TARGETplus nontargeting pool (Thermo Scientific Dharmacon). Transfection was done according to the manufacturer's protocol using DharmaFECT 1 transfection reagent. The cells were transfected with 100 nM siRNA and incubated for 48 h, followed by treatment with butyrate (1 mM) for a period of 24 h. Thereafter, medium was collected for ANGPTL4 protein analysis by ELISA and the cells were used for RNA isolation and subsequent quantitative PCR (qPCR).
PPARγ ChIP.
T84 cells were grown in 10-cm plates to a density of approximately 5 million cells/dish and treated with butyrate (8 mM) or rosiglitazone (1 μM). After 1 h, medium was removed and cells were fixed in 1% formaldehyde in PBS. After 10 min, cross-linking was stopped by the addition of glycine to a final concentration of 0.125 M. The cells were washed three times with ice-cold PBS and resuspended in 0.3 ml of lysis buffer (0.1% SDS, 1% Triton X-100, 0.15 M NaCl, 1 mM EDTA, 20 mM Tris, pH 8.0). The lysate was frozen at −80°C until further analysis. Chromatin immunoprecipitation (ChIP) was carried out as described by Siersbæk et al. (21), with the exceptions that disuccinimidyl glutarate was not used for cross-linking and sonication was performed in 2 rounds of 10 on/off cycles of 30 s each. The PPARγ antibody used was PPARγ H100 (sc-7196; Santa Cruz Biotechnologies).
PPARγ stable reporter assay.
The PPARγ CALUX cell line was obtained from BioDetection Systems B.V. (Amsterdam, the Netherlands). PPARγ CALUX cells are based on human osteoblastic (U2-OS) cells (American Type Culture Collection [ATCC], Manassas, VA) stably transfected with a human PPARγ expression plasmid and a luciferase reporter construct (22). The cells were cultured as described previously in a 1:1 mixture of DMEM and Ham's F-12 medium (Invitrogen, Breda, the Netherlands) supplemented with 7.5% fetal bovine serum (FBS) (Invitrogen), 1% nonessential amino acids (Invitrogen), and penicillin-streptomycin (Invitrogen) to final concentrations of 10 U/ml and 10 μg/ml, respectively (22, 23). Once per month 200 μg/ml G418-disulfate was added to the culture medium.
The PPARγ CALUX assay was performed as described previously (22, 24). In short, for the exposure experiments, PPARγ-CALUX cells were plated in the 96-well plates with phenolred-free 1:1 mixture of DMEM and Ham's F-12 medium supplemented with 5% dextran-coated charcoal-stripped FBS (Invitrogen) at a volume of 200 μl per well. The next day, medium was refreshed and cells were incubated in triplicates with test compounds added to the culture medium. After 24 h, the cells were checked visually for cytotoxicity, the medium was removed, and the cells were lysed in luciferase cell culture lysis reagent (Promega, Leiden, the Netherlands). Luciferase activity was measured in cellular extracts using a Synergy HT multidetection microplate reader (BioTek Instruments Inc.). For each test compound at last two independent experiments were performed.
GAL4-PPAR stable reporter assay.
The GAL4-PPAR stable reporter assay was carried out in HeLa cells stably expressing a chimeric protein containing the ligand binding domain (LBD) of human PPARα, PPARδ, or PPARγ fused to the yeast transactivator GAL4 DNA binding domain (DBD) and a luciferase reporter gene driven by a pentamer of the GAL4 recognition sequence in front of a β-globin promoter. The cells were seeded in 96-well plates and incubated the following day with tested compounds for 24 h. At the end of the incubation, the luciferase activity was measured using a BMG LUMIstar Galaxy luminometer. Results expressed as relative light units (RLU) were obtained from experiments performed in triplicate. Rosiglitazone, GW7647, and L165041 (all at 1 μM) were used as positive controls for activation of PPARα, PPARδ, and PPARγ, respectively. The measurements were performed as a commercial service by Tebu-bio Laboratories (Le Perray-en-Yvelines, France).
ANGPTL4 ELISA.
ANGPTL4 levels in the medium were measured by ELISA as detailed previously (17). Briefly, 96-well plates were coated with anti-human ANGPTL4 polyclonal goat IgG antibody (AF3485; R&D Systems) and incubated overnight at 4°C. Plates were washed extensively between each step. After blocking, 100 μl of medium of cells was applied, followed by 2 h of incubation at room temperature. A standard curve was prepared using recombinant human ANGPTL4 (3485-AN; R&D Systems) at 0.3 to 2.1 ng/well. One hundred microliters of diluted biotinylated anti-human ANGPTL4 polyclonal goat IgG antibody (BAF3485; R&D Systems) then was added for 2 h, followed by addition of streptavidin-conjugated horseradish peroxidase for 20 min and tetramethyl benzidine substrate reagent for 6 min. The reaction was stopped by the addition of 50 μl of 10% H2SO4, and the absorbance was measured at 450 nm.
Cofactor recruitment assay.
Nuclear receptor PamChip arrays (PamGene, s'Hertogenbosch, the Netherlands) were used as described previously (25). Upon binding a ligand, PPARs undergo a conformational change that promotes the formation of a cofactor binding pocket, subsequently allowing interaction with the so-called LXXLL motif within some coregulators. The PamChip arrays consist of 53 peptides encompassing the LXXLL motifs of 21 different coregulator proteins. The sequences are provided as supplementary data in Koppen et al. (25). Briefly, the arrays were incubated with glutathione S-transferase-tagged PPARγ LBD (Invitrogen, Breda, the Netherlands) in the presence and absence of the ligands, i.e., sodium acetate, sodium propionate, sodium butyrate (each at 40 mM), and rosiglitazone (1 μM). Quantification of the interaction between PPARγ and coregulators was made using Alexa 488-conjugated anti-glutathione S-transferase rabbit polyclonal antibody (Invitrogen).
Affymetrix GeneChip microarray analysis.
Microarray analysis was performed on T84 cells treated with 0.1 μM rosiglitazone or 1 mM butyrate for 24 h. Total RNA was prepared using TRIzol reagent (Invitrogen) and purified on columns using the RNeasy Minikit (Qiagen, Venlo, the Netherlands). RNA quality and integrity were verified with the RNA 6000 Nano assay on an Agilent 2100 Bioanalyzer (Agilent Technologies, Amsterdam, the Netherlands). Hybridization, washing, and scanning of the Affymetrix Human Gene 1.1 ST array plate was performed on Affymetrix GeneTitan. Scans of the Affymetrix arrays were processed using packages from the Bioconductor project (26). Raw signal intensities were obtained by robust multiarray (RMA) normalization (27). Probe sets were defined according to Dai et al. using remapped chip definition file (CDF), version 15, based on data from the Entrez gene database (28).
Microarray analysis of colon of mice fed inulin was carried out as described above using the Affymetrix Mouse Gene 1.1 ST array plate.
3T3-L1 adipogenesis.
3T3-L1 fibroblasts were amplified in DMEM-10% FCS and subsequently seeded into six-well plates. Two days after the cells reached confluence, the medium was changed to DMEM-10% fetal calf serum containing 0.5 mM 3-isobutyl-1-methylxanthine, 2 μg/ml insulin (Actrapid), 0.5 μM dexamethasone, and either the short-chain fatty acids or rosiglitazone (1 μM). After 2 days, the medium was changed to DMEM-10% fetal calf serum containing 2 μg/ml insulin and the short-chain fatty acids or rosiglitazone. Cells were harvested after two more days for RNA isolation (day 4). This mild adipogenesis protocol permits assessment of PPARγ agonist activity of added compounds (29). Oil red O staining was performed at day 10 using a standard protocol.
In a second protocol, 2 days after the cell confluence (i.e., day 0), the medium was changed to DMEM-10% fetal calf serum containing 0.5 mM 3-isobutyl-1-methylxanthine, 5 μg/ml insulin, and 1 μM dexamethasone. After 3 days, the medium was changed to DMEM-10% fetal calf serum containing 5 μg/ml insulin. The medium was subsequently changed every 3 days, and no further insulin was added after 6 days. At day 10, 3T3-L1 adipocytes were treated with rosiglitazone (1 μM) and the short-chain fatty acids (8 mM) for 24 h.
Adipogenesis in SGBS.
Human Simpson-Golabi-Behmel syndrome (SGBS) adipocytes were cultured and grown to confluence in 0F medium (DMEM–F-12–biotin–panthotenate–penicillin-streptomycin) plus 10% FCS. One day postconfluence, the medium was changed to Quick-diff medium, which is 3FC medium (0F medium plus 0.01 mg/ml human apotransferrin, 2 × 10−8 M insulin, 0.1 μM cortisol, 0.2 nM T3) with 50 nM dexamethasone, 500 μM 3-isobutyl-1-methylxanthine, and 2 μM rosiglitazone or 8 mM SCFA. On day 4, medium was changed to 3FC medium and changed every 4 days until day 15. Expression of differentiation markers and PPARγ targets was determined by qPCR at day 15. DMSO was used as a control.
RNA isolation and qPCR.
RNA was isolated from T84 cells using RNeasy columns or TRIzol. One μg of total RNA was reverse transcribed with iScript (Bio-Rad, Veenendaal, the Netherlands) or a Fermentas cDNA synthesis kit according to the instructions from the manufacturer. cDNA was amplified on a Bio-Rad CFX384 real-time system using Sensimix (Bioline, GC Biotech, Alphen aan de Rijn, the Netherlands). Cyclophilin or 36B4 was used as a housekeeping gene. PCR primer sequences were taken from the PrimerBank and ordered from Eurogentec (Seraing, Belgium). Primer sequences are presented in Table 1.
Table 1.
List of primers used
| Name | Primer sequence |
|
|---|---|---|
| Forward | Reverse | |
| m36B4 | ATGGGTACAAGCGCGTCCTG | GCCTTGACCTTTTCAGTAAG |
| Fabp4 | AAGAAGTGGGAGTGGGCTTT | AATCCCCATTTACGCTGATG |
| Gyk | ATCCGCTGGCTAAGAGACAACC | TGCACTGGGCTCCCAATAAGG |
| Slc2a4 | GGAAGGAAAAGGGCTATGCTG | TGAGGAACCGTCCAAGAATGA |
| Adipoq | GCAGAGATGGCACTCCTGGA | CCCTTCAGCTCCTGTCATTCC |
| Angptl4 | GTTTGCAGACTCAGCTCAAGG | CCAAGAGGTCTATCTGGCTCTG |
| ANGPTL4 | CACAGCCTGCAGACACAACTC | GGAGGCCAAACTGGCTTTGC |
| PPARG | GAGCCCAAGTTTGAGTTTGC | CAGGGCTTGTAGCAGGTTGT |
| PPARA | CAGAACAAGGAGGCGGAGGTC | TTCAGGTCCAAGTTTGCGAAGC |
| PPARD | TGGCTTTGTCACCCGTGAGT | ACAGAATGATGGCCGCAATGAA |
| PLIN2 | ATGGCATCCGTTGCAGTTGAT | GATGGTCTTCACACCGTTCTC |
| UCP2 | TGCCCTCCTGAAAGCCAAC | CTTGACCACGTCTACAGGGGA |
| AQP8 | GCGAGTGTCCTGGTACGAAC | CAGGCACCCGATGAAGATGAA |
| LGALS1 | TCGCCAGCAACCTGAATCTC | GCACGAAGCTCTTAGCGTCA |
| h36B4 | CGGGAAGGCTGTGGTGCTG | GTGAACACAAAGCCCACATTCC |
Modeling of butyrate binding to PPARγ.
For modeling the structure of the complex between PPARγ and butyrate, HADDOCK version 2.1 was used (30). HADDOCK is a highly successful modeling approach that makes use of structural knowledge when available to drive the docking procedure. In this case, the X-ray crystal structure of PPARγ with bound decanoic acid (DA; 3U9Q) was used to identify the likely binding site for short-chain fatty acids such as butyrate (31). The docking was performed using the web server version of HADDOCK (32), with the following modifications. The high-temperature rigid-body torsion angle dynamics and the first rigid-body cooling stage were not performed (i.e., the number of steps was set to 0), and the second cooling phase of torsion angle dynamics with flexible side chains at the interface was started with a reduced initial temperature of 500K instead of the default 1,000K.
All calculations were performed with CNS1.2 (33). Nonbonded interactions were calculated with the OPLS force field using a cutoff of 8.5 Å (34). The electrostatic potential (Eelec) was calculated by using a shift function, while a switching function (between 6.5 and 8.5Å) was used to define the Van der Waals potential (EvdW). The HADDOCK score (in arbitrary units [AU]) was used to rank the generated poses. It is a weighted sum of intermolecular electrostatic (Eelec), van der Waals (EvdW), desolvation (Edesolv), and ambiguous interaction restraint (AIR) energies with weight factors of 0.2, 1.0, 1.0, and 0.1, respectively.
The ambiguous interaction restraints used to drive the docking of butyrate to PPARγ were derived from the X-ray crystal structure of PPARγ with bound DA (3U9Q), where protein residues near the decanoic acid were identified as the butyrate binding site. All protein residues within 3.9 Å of the decanoic acid (residues 278, 281, 282, 285, 289, 323, 356, 360, 363, 449, 469, and 473) were defined as active restraints for the rigid-body docking phase and as passive restraints for the subsequent semiflexible refinement stage. The ligand was considered active for both docking phases. This strategy effectively pulls the butyrate ligand into the binding site during rigid-body docking while allowing a thorough exploration of the binding pocket during the refinement stage. With the protein active-site residues defined as passive during the semiflexible refinement stage, the ligand is not restrained to any particular orientation or location within the defined binding site. For comparison, a second docking trial was performed with active residues for the entire PPARγ binding site, defined as amino acids within 3.9Å of either the partial agonist decanoic acid (from 3U9Q, residues 278, 281, 282, 285, 289, 323, 356, 360, 363, 449, 469, and 473) or the full agonist-nitrosylated fatty acid (from 3CWD, residues 285, 286, 288, 289, 326, 327, 330, 340, 341, 364, 449, and 473). Docking with this wider binding pocket definition did not give additional solutions.
At the end of the docking protocol, clustering based on ligand pairwise root mean square deviation (RMSD) criteria was performed, and the best-scoring structure of the best-scoring cluster was taken as the best solution. The clustering distance cutoff was reduced from the default 7.5-Å cutoff for protein-protein complexes to 1.75 Å, which is more suitable for protein-ligand complexes.
The interactions between decanoic acid and PPARγ were analyzed by submitting the X-ray crystal structure 3U9Q to the HADDOCK refinement server (32), which only performs a gentle refinement in explicit solvent and returns statistics similar to those of a full docking run.
Microarray data accession numbers.
The microarray data were submitted to Gene Expression Omnibus under accession no. GSE40706 and GSE43065.
RESULTS
ANGPTL4 production is stimulated by short-chain fatty acids.
One set of molecules that may mediate the effect of microbiota on ANGPTL4 expression in the colon are the short-chain fatty acids (SCFA) acetate, propionate, and butyrate. To estimate the concentration of fatty acids to be used for in vitro experiments, we determined the total SCFA concentration in the lumen of different parts of the gastrointestinal (GI) tract of mice fed a low-fat diet (Fig. 1A). Total SCFA concentration varied from <10 mM in the proximal small intestine to >40 mM in the cecum. In the colon the highest concentrations were found for acetate, followed by propionate and butyrate (Fig. 1B). In the literature, even higher concentrations have been reported that exceed 100 mM (35). Based on these data, we used low- to medium-millimolar SCFA concentrations.
Fig 1.

Physiological concentrations of SCFA stimulate ANGPTL4 mRNA and protein secretion in colon adenocarcinoma cells. (A) Total SCFA concentration in different sections of the intestine of mice fed a low-fat diet. Errors bars represent SEM. (B) Concentration of individual SCFA in colon of mice fed a low-fat diet. Errors bars represent SEM. (C) ANGPTL4 concentration in medium of T84 and HT29 cells treated with SCFA for 24 h at the indicated concentrations. (D) ANGPTL4 mRNA in T84 and HT29 cells treated with SCFA for 24 h at the indicated concentrations. (E) Time course of induction of ANGPTL4 mRNA in T84 cells by butyrate (1 mM). (F) Inhibitory effect of α-amanitin (10 μg/ml) on induction of ANGPTL4 secretion by 1 mM butyrate in T84 cells. (G) Effect of rectal infusion of propionate on Angptl4 mRNA in epithelial scrapings of mouse colon. Errors bars represent SEM. (H) Time course of regulation of ANGPTL4 protein in medium of T84 and HT29 cells by trichostatin A (100 nM) and butyrate (8 mM). (I) Stimulatory effect of trichostatin A (100 nM) on LGALS1 mRNA in T84 cells. Error bars represent standard deviations (SD) except where indicated otherwise. An asterisk indicates a result significantly different from that of the control according to Student's t test (P < 0.05).
To study regulation of ANGPTL4 production in colonocytes, we used the human colonic adenocarcinoma cell lines T84 and HT29. Remarkably, all SCFA significantly increased ANGPTL4 secretion by T84 and HT29 cells (Fig. 1C). The strongest effects were observed for butyrate, followed by propionate and acetate. Increased ANGPTL4 secretion was mirrored by a pronounced increase in ANGPTL4 mRNA (Fig. 1D), suggesting SCFA stimulate ANGPTL4 transcription, which was further supported by the rapid time course of induction of ANGPTL4 mRNA by butyrate (Fig. 1E). Furthermore, induction of ANGPTL4 secretion by butyrate was almost completely abolished by α-amanitin, an inhibitor of RNA polymerase II (Fig. 1F). In support of in vivo regulation of Angptl4 gene expression by SCFA, rectal infusion of propionate significantly increased Angptl4 mRNA expression in mouse colon (Fig. 1G).
Butyrate and, to a lesser extent, propionate are able to inhibit histone deacetylase (HDAC) activity, while acetate is ineffective (36, 37). To study the impact of HDAC inhibition on ANGPTL4 secretion, we treated T84 and HT29 cells with the specific HDAC inhibitor trichostatin A, which, similar to butyrate, inhibits class I and class II HDACs. Unlike butyrate, trichostatin A had no effect on ANGPTL4 secretion (Fig. 1H), indicating that the stimulatory effect of butyrate on ANGPTL4 is likely independent of HDAC inhibition. The effectiveness of trichostatin A was demonstrated by the marked induction of the LGALS1 gene (Fig. 1I).
PPARγ mediates induction of ANGPTL4 by SCFA.
Because the medium-chain fatty acid (MCFA) decanoic acid was recently shown to be a direct ligand of PPARγ and ANGPTL4 is a sensitive target of PPARγ in colonocytes (31, 38), we hypothesized that butyrate upregulates ANGPTL4 expression in colonocytes by activating PPARγ. To test the responsiveness of ANGPTL4 to PPAR activation in T84 and HT29 cells, the cells were treated with synthetic agonists for PPARα, PPARδ, and PPARγ. The PPARγ agonist rosiglitazone was the most potent inducer of ANGPTL4 protein secretion (Fig. 2A) and mRNA (Fig. 2B) in both cell types, followed by the PPARδ agonist GW501516 and the PPARα agonist Wy14643. These results are consistent with the high expression of PPARG mRNA in T84 cells (Fig. 2C, top) and, to a lesser extent, in HT29 cells (Fig. 2C, lower) and the corresponding protein levels of the three PPARs in the two cell lines (39–43). Upon incubation with rosiglitazone, a time- and dose-dependent increase in ANGPTL4 protein was observed in T84 cells (Fig. 2D and E), indicating that ANGPTL4 production is highly sensitive to PPARγ activation. Accordingly, we considered PPARγ as a possible candidate mediating induction of ANGPTL4 mRNA by butyrate.
Fig 2.

PPARγ potently stimulates ANGPTL4 in colon adenocarcinoma cells. Synthetic agonists for PPARα (Wy14643 [WY]; 5 μM), PPARδ (GW501516 [GW]; 1 μM), and PPARγ (rosiglitazone [Rosi]; 1 μM) stimulate ANGPTL4 secretion (A) and mRNA expression (B) in T84 and HT29 cells. Cntl, control. (C) Amplification curve of PPARα, PPARδ, and PPARγ mRNA as determined by qPCR of T84 and HT29 cells. Sizes of amplicons varied less than 10%. (D) Time course of induction of ANGPTL4 protein in medium by rosiglitazone (1 μM). (E) Dose-dependent induction of ANGPTL4 protein in medium by rosiglitazone. Unless indicated otherwise, cells were treated for 24 h. Error bars represent SD. An asterisk indicates a result significantly different from that of the control according to Student's t test (P < 0.05).
To investigate whether the stimulatory effect of butyrate on ANGPTL4 secretion occurs via PPARγ, we used the PPARγ antagonist GW9662. Induction of ANGPTL4 secretion by butyrate in T84 cells was strongly diminished by GW9662 (Fig. 3A), suggesting that butyrate increases ANGPTL4 secretion mainly by activating PPARγ. Similarly, siRNA-mediated knockdown of PPARγ mRNA by about 60% led to a very significant 40% decrease in the butyrate-induced ANGPTL4 mRNA level (Fig. 3B) and butyrate-induced ANGPTL4 protein secretion (Fig. 3C), again suggesting that the effect of butyrate on ANGPTL4 is largely mediated by PPARγ. Stimulation of PPARγ mRNA by butyrate (Fig. 3B) likely reflects PPARγ autoregulation. To further pursue the role of PPARγ in ANGPTL4 regulation by butyrate, we performed chromatin immunoprecipitation to study binding of PPARγ to the ANGPTL4 gene. The results indicate that PPARγ occupies the PPAR binding site within intron 3 of the ANGPTL4 gene (Fig. 3D), which is known to mediate PPAR responsiveness (44, 45). Occupancy was modestly induced by both butyrate and rosiglitazone. Taken together, these data strongly suggest that induction of ANGPTL4 by butyrate in T84 cells is mediated by PPARγ.
Fig 3.

Induction of ANGPTL4 by butyrate is mediated by PPARγ. (A) Inhibitory effect of PPARγ antagonist GW9662 (5 μM) on induction of ANGPTL4 secretion in medium by rosiglitazone (10 nM) and butyrate (1 mM) in T84 cells. Effect of siRNA-mediated PPARγ knockdown (B, left) in T84 cells on butyrate-induced upregulation of ANGPTL4 mRNA (B, right) and ANGPTL4 protein in medium (C). (D) PPARγ ChIP-qPCR on selected loci in T84 cells treated with butyrate (8 mM) or rosiglitazone (1 μM) for 24 h. Bars represent the mean recovery plus ranges from two independent experiments. Error bars represent SD except when indicated otherwise. An asterisk indicates a significantly different result from that of the control according to Student's t test (P < 0.05).
To investigate whether butyrate acts as a general inducer of PPARγ-dependent gene regulation, we performed microarray analysis on T84 cells treated either with butyrate (1 and 8 mM) or rosiglitazone. Remarkably, effects of butyrate on global gene expression were much more pronounced than those of rosiglitazone, which is likely linked to its property as a potent HDAC inhibitor (Fig. 4A; also see Fig. S1 in the supplemental material). Interestingly, more overlap was observed between 1 mM butyrate and rosiglitazone than between 8 mM butyrate and rosiglitazone (see Fig. S1). Cluster analysis identified clusters of genes induced by rosiglitazone and butyrate (Fig. 4A). Genes in one cluster, which, besides ANGPTL4, included PPAR targets HMOX1, PDK4, and UCP2 (46–48), were induced by 1 and 8 mM butyrate. Genes in another cluster, which included other PPAR targets, including AQP8 and PLIN2 (49, 50), were induced by 1 mM butyrate and surprisingly were not induced or even suppressed by 8 mM butyrate. The role of PPARγ in mediating the stimulatory effect of 1 mM butyrate on expression of PLIN2 and UCP2, representing two distinct clusters, was verified by siRNA (Fig. 4B).
Fig 4.

Global effects of butyrate in T84 cells. T84 cells were treated with rosiglitazone (1 μM) or butyrate (1 and 8 mM) for 24 h and subjected to Affymetrix microarray analysis. (A) Hierarchical clustering based on Pearson's correlation with average linkage. (B) Effect of siRNA-mediated PPARγ knockdown in T84 cells on induction of PLIN2 and UCP2 mRNA by butyrate. AQP8 mRNA (C) and PLIN2 mRNA (D) levels were determined in T84 cells treated with SCFA for 24 h at the indicated concentrations. Error bars represent SD. An asterisk indicates a result significantly different from that of the control according to Student's t test (P < 0.05).
The biphasic regulation of AQP8 and PLIN2 by butyrate but not by propionate and acetate was confirmed by qPCR (Fig. 4C). Such a biphasic regulation is possible only if a gene is regulated by at least two distinct mechanisms: a stimulatory effect, presumably via PPARγ, which is already active at lower butyrate concentrations, and a suppressive effect impacting a subset of PPAR targets at higher butyrate concentrations. Overall, these data indicate that SCFA acts as a general inducer of PPARγ-dependent gene regulation in T84 cells. However, especially at higher concentrations, the main effects of SCFA in T84 cells are independent of PPARγ.
SCFA function as PPARγ agonists.
To further study activation of PPARγ by SCFA, we employed a stable PPARγ reporter assay. HeLa cells stably transfected with a fusion construct between the DNA binding domain of Gal4 and the ligand binding domain of PPARγ, PPARα, or PPARδ were incubated with increasing concentrations of SCFA. Remarkably, PPARγ was strongly activated by butyrate and, to a lesser extent, by propionate (Fig. 5A). In contrast, PPARα and PPARδ were not activated by any of the SCFA, even at higher concentrations.
Fig 5.

SCFA are agonists of PPARγ. (A) Stable GAL4-PPAR chimera reporter assay showing activation of PPARγ but not PPARα and PPARδ by SCFA at concentrations of ≥1 mM. The dotted lines represent the levels of luciferase activity reached upon incubation with synthetic PPAR agonists. Cells were treated for 24 h. Error bars represent SD. (B) Stable PPARγ reporter assay showing activation of PPARγ by SCFA at concentrations of ≥500 μM. Cells were treated for 24 h. Note the different x axis for rosiglitazone and the SCFA. Error bars represent SD. (C) A nuclear receptor PamChip assay was used to measure the interaction between PPARγ and immobilized peptides corresponding to specific coregulator-nuclear receptor binding regions in the presence and absence of rosiglitazone (1 μM), butyrate (40 mM), or acetate (40 mM). Representative images are shown. (D) Quantitation of the PamChip assay results for rosiglitazone and butyrate compared to the control. Arrows point to the same peptides as those described for panel C.
To corroborate activation of PPARγ by SCFA, we performed an alternative reporter assay in U2OS cells stably transfected with a human PPARγ expression construct and a PPRE-luciferase reporter construct. Similar to the results described above, propionate, but especially butyrate, markedly activated PPARγ reporter activity, while acetate had minimal effect (Fig. 5B). The threshold for stimulating reporter activity was 0.5 mM for butyrate and 10 nM for rosiglitazone, indicating that butyrate has relatively weak PPARγ agonist activity.
To explore the possibility that SCFA serve as direct PPARγ agonists, we used nuclear receptor PamChip arrays. In this system, the interaction between soluble nuclear receptors and 53 immobilized peptides corresponding to specific coregulator-nuclear receptor binding regions is studied. Using this system for PPARγ, we have previously generated ligand-specific coregulator interaction profiles (25). Both rosiglitazone and butyrate promoted the interaction between PPARγ and numerous coactivator peptides (e.g., CBP) (Fig. 5C). However, in contrast to rosiglitazone, butyrate did not relieve the interaction between PPARγ and the NCoR1 and NCoR2 corepressor peptides. Consistent with the PPARγ reporter data, acetate had little effect. Quantitative analysis of the PamChip arrays is presented in Fig. 5D. These data suggest that butyrate functions as a selective PPARγ modulator (SPPARM) (51).
Modeling of butyrate bound to PPARγ.
To find support for SCFA behaving as selective PPARγ modulators, we performed structural modeling of the binding of butyrate to PPARγ using HADDOCK (30, 32) (Fig. 6A). Docking of butyrate into PPARγ resulted in three different clusters of solutions, each showing butyrate bound in the ligand-binding pocket of the protein. The best cluster in terms of HADDOCK score was also the largest one (179/200 calculated structures) (Table 2) and contained the best scoring models. It reveals a binding mode very similar to that of decanoic acid within the crystal structure of PPARγ (3U9Q) (31). Specifically, the average RMSD between the butyrate and decanoic acid common atoms upon aligning the backbone atoms of the PPARγ (ligand RMSD) was 1.55 ± 0.29 Å for the best four cluster members. The remaining clusters have less favorable interaction energy.
Fig 6.

Modeling of butyrate into the PPARγ binding pocket. (A) The model reveals the complex between butyrate and PPARγ with the best HADDOCK score (butyrate is shown as cyan sticks), overlaid with the crystal structure of the decanoic acid complex with PPARγ (3U9Q, a decanoic acid, is shown as green sticks) by aligning the protein backbone atoms of the two structures (ribbon displayed for the HADDOCK model). The displayed protein side chains are shown as thin cyan or green sticks and the side chains making contacts with the docked butyrate or decanoic acid, respectively. Hydrogen bond contacts between the butyrate and the protein are shown as yellow dashed lines. (B) Comparison of the binding location for coactivator peptide PGC-1α (blue ribbon) bound to PPARγ (HADDOCK model of the PPARγ-butyrate complex, left) with the binding site for the SMRT corepressor peptide (purple ribbon) binding to PPARα (1KKQ; right) (69). In both structures, the C-terminal portion of the PPAR molecule that forms helix AF-2 is colored pink for comparison. The structures were aligned using the backbone atoms of the receptors.
Table 2.
Characteristics of PPARγ complexes with fatty acidsa
| Complex | BSA (Å2) | HADDOCK scoreb (AU) | Ligand RMSDc (Å) | Size of clusterd |
|---|---|---|---|---|
| 3U9Q | 478 ± 6 | −22.1 ± 2.9 | 0.0 | NA |
| Cluster 1-butyrate | 286 ± 14 | −7 ± 3 | 1.55 ± 0.29 | 179 |
| Cluster 2-butyrate | 280 ± 6 | 0.1 ± 4 | 4.70 ± 0.77 | 13 |
| Cluster 3-butyrate | 275 ± 6 | 4.6 ± 4 | 1.99 0.13 | 6 |
Averages and standard deviations calculated from the best four members (in terms of HADDOCK score) of a set of structures are reported. 3U9Q is the HADDOCK-refined crystal structures of the PPARγ-decanoic acid complex, while the other three clusters were obtained by HADDOCK modeling of the butyrate-PPARγ complex. NA, not applicable.
HADDOCK score as defined in Materials and Methods.
Positional RMSD of the ligand atoms calculated after superimposition of the protein backbone atoms.
Number of calculated structures (out of 200 total) that were clustered together.
Interactions between butyrate and PPARγ are dominated by electrostatic interactions and hydrogen bonds between the carboxylate group and several hydrogen bond-donating protein side chains, which are very similar to those found in the structure with decanoic acid (Fig. 6A). In the latter can be found hydrogen bonds to four different side chains near the carboxylate group, namely, His323 HE2, Ser289 OG, His449 HE2, and Tyr473 OH. The top four structures of the best cluster (in terms of HADDOCK score) reveal specific hydrogen bonds from the butyrate oxygens to His323, Tyr473, and His449. The hydrogen bond to Ser289 is never observed, while in two of the structures a fourth hydrogen bond is made to Tyr327 OH. The average buried surface area (BSA) for the best four docking solutions is 286 ± 14 Å2, and the average HADDOCK score for the best four structures is −7 ± 3. In comparison, the buried surface area and HADDOCK score calculated for the decanoic acid-PPARγ structure are 478 Å2 and −22.1, respectively.
We next explored the possible influence of butyrate on coactivator and corepressor binding (Fig. 6B). Fortunately, the structure of PPARγ with decanoic acid also contained a bound PGC1α coactivator peptide, allowing us to model butyrate docked within the crystal structure of PPARγ complexed with the PGC-1α peptide. However, no structures were available for corepressor (peptides) binding to PPARγ, which limited us to the crystal structure of PPARα complexed with an antagonist and the SMRT corepressor peptide (1KKQ). As shown in Fig. 6B, the binding sites for the coactivator (left, shown in blue) and corepressor (right, shown in purple) peptides occupy structurally analogous positions on PPARγ and PPARα and are likely to be mutually exclusive binding events. The C-terminal AF2 helix of PPARγ (shown in pink) forms part of the binding site for the coactivator peptide PGC-1α, and stabilization of this helix is known to promote binding of coactivator peptides (31). As can be seen from the structural comparison, the AF2 helix of PPARγ also occludes part of the structurally analogous binding site for the corepressor peptide binding to PPARα. The butyrate molecule in the HADDOCK model makes several contacts with the AF2 helix (such as the hydrogen bond to Tyr473 OH) that are very similar to the contacts that were observed between decanoic acid and the AF2 helix in the X-ray crystal structure of the decanoic acid-PPARγ complex (3U9Q), which is consistent with our finding using a coactivator peptide binding assay that butyrate can promote at least partial agonism of the PPARγ receptor but does not promote dissociation of corepressor peptides.
SCFA do not induce PPARγ-mediated adipogenesis.
Given that SCFA can activate PPARγ-mediated transcription in reporter assays but only partially overlap a full PPARγ agonist in their coregulator binding profile, SCFA may not display the same capacity as full agonists in cell-based assays. Since PPARγ is a critical regulator of adipogenesis and mediates stimulation of adipogenesis by rosiglitazone, we subsequently used stimulation of 3T3-L1 adipogenesis as an in vitro readout for PPARγ activation by SCFA. It has been shown that NCoR1 represses 3T3-L1 adipogenesis and that dismissal of NCoR1 is essential for induction of adipogenesis by PPARγ agonists (52, 53). Since SCFA/butyrate did not release the association between NCoR1 and PPARγ as observed using the PamChip array, it may be expected that SCFA are unable to mimic the adipogenic effects of PPARγ agonists (29). Consistent with this notion and in contrast to rosiglitazone, SCFA did not induce 3T3-L1 adipogenesis, as revealed by oil red O staining (Fig. 7A) and gene expression analysis of adipogenesis markers, which also represent PPARγ targets (Fig. 7B). In fact, at higher concentrations butyrate and propionate suppressed adipogenesis (Fig. 7C). Furthermore, in contrast to rosiglitazone, butyrate did not upregulate PPARγ targets in differentiated 3T3-L1 adipocytes (Fig. 7D). Similarly, in contrast to rosiglitazone, SCFA were unable to induce adipogenesis in the human SGBS adipocyte model, as revealed by lack of induction of adipogenesis markers (Fig. 7E). Together with the data from colon cell lines and PamChip arrays, these data support the classification of SCFA as selective PPARγ modulators.
Fig 7.

Butyrate and propionate inhibit 3T3-L1 adipogenesis. (A) Oil red O staining of 3T3-L1 adipocytes at day 10 treated with SCFA (8 mM) from day 0. The mix contained 2.67 mM of each of the SCFA. (B) Expression of differentiation markers and PPARγ targets was determined by qPCR at day 4. (C) Concentration-dependent effect of butyrate on 3T3-L1 differentiation when added at day 0, as determined by expression of differentiation markers at day 4. (D) Effect of SCFA (8 mM) and rosiglitazone (1 μM) on expression of PPARγ targets in fully differentiated 3T3-L1 adipocytes. Cells were treated for 24 h. (E) Effect of SCFA (8 mM) and rosiglitazone (1 μM) added on day 1 on human SGBS adipocyte differentiation and expression of adipogenesis marker genes at day 15. Error bars represent SD. An asterisk indicates a result significantly different from that of the control according to Student's t test (P < 0.05).
Consumption of inulin leads to PPAR activation in mouse colon.
To find support for activation of PPARγ by SCFA in vivo, we studied the effect of dietary fiber on PPAR activation in mouse colon. To that end, C57BL/6 mice were fed either a control diet containing no soluble fiber or the same diet containing 10% inulin for 10 days. Inulin feeding markedly increased concentrations of acetate and propionate in the colon, which reached significance for propionate (Fig. 8A). To study the effect of inulin on PPAR target gene expression, colons were subjected to microarray analysis followed by gene set enrichment analysis. Remarkably, the most significantly induced gene set was PPAR targets (false discovery rate q-value, 0; normalized enrichment score, 2.44) (54). It should be noted that PPAR target genes cannot be separated according to PPAR isotype. Representation of the individual expression changes of the most highly ranked genes within PPAR targets in a heatmap reveals the pronounced and consistent induction of numerous PPAR targets by inulin (Fig. 8B). These data suggest that gut microbial activity and resultant formation of SCFA leads to activation of PPARs.
Fig 8.
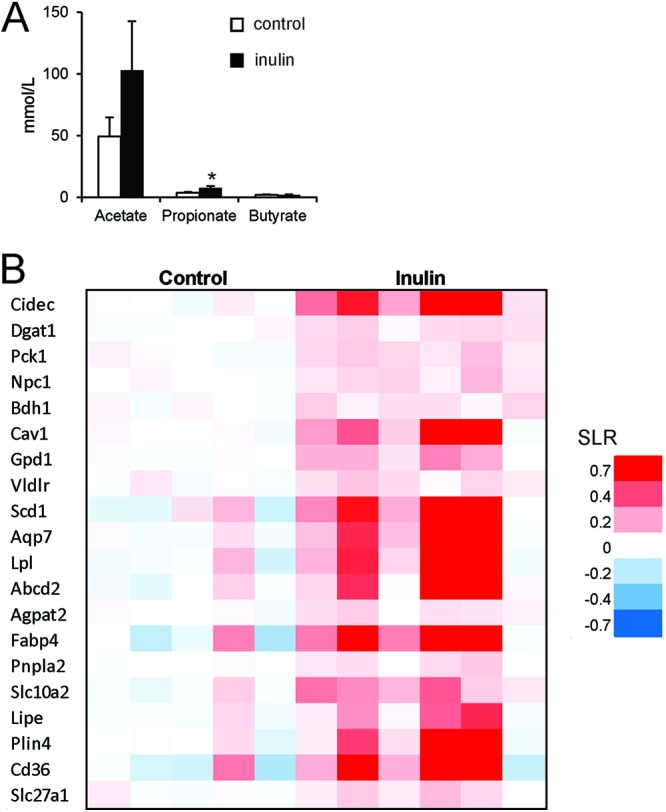
Inulin feeding activates PPAR in colon. Mice were fed a diet enriched with inulin for 10 days. (A) Lumenal concentration of SCFA in the colon as determined by gas chromatography. Error bars represent SEM. (B) Gene expression changes in colon illustrated by heat map of genes belonging to the most significantly induced gene set, termed PPAR targets. SLR, signal log ratio.
DISCUSSION
Here, we explored the mechanisms involved in regulation of ANGPTL4 synthesis in human colon. The two major findings are (i) ANGPTL4 synthesis is highly stimulated by SCFA and (ii) SCFA transactivate and bind to PPARγ, likely by serving as selective PPAR modulators. Overall, the data indicate that SCFA induce ANGPTL4 mRNA expression and protein secretion in colon cells by activating PPARγ. Butyrate was the strongest activator followed by propionate, whereas acetate only weakly stimulated PPARγ and ANGPTL4.
Butyrate is a potent histone deacetylase inhibitor, which likely accounts for most of the observed effects of butyrate in colon adenocarcinoma cells as revealed by microarray. Indeed, a far larger number of genes was regulated by butyrate than by rosiglitazone, suggesting that PPARγ activation is quantitatively a relatively minor pathway in gene regulation by SCFA, at least in T84 cells.
Two previous studies have hinted at potential activation of PPARγ by butyrate (55, 56), yet the concept has largely eluded recognition in the field. In contrast, long-chain (unsaturated) fatty acids (LCFA) are well-known activators of PPARγ (57). They activate PPARγ at concentrations in the low- to medium-micromolar range and thus serve as high-affinity agonists of PPARγ. In contrast, concentrations of SCFA needed to activate PPARγ are in the high-micromolar to low-millimolar range. Due to the low affinity, the in vivo relevance of PPARγ activation by SCFA is likely insignificant in most human tissues, including adipose tissue. However, the situation is different in the GI tract and in liver (58). Indeed, SCFA concentrations approaching or even exceeding 100 mM have been reported in human colon and cecum (59). In mouse intestine, we measured total SCFA concentrations of around 40 mM, which would result in substantial activation of PPARγ. Accordingly, we believe that activation of PPARγ by SCFA is physiologically meaningful only in human cecum and colon and perhaps in liver.
Interestingly, medium-chain fatty acids (MCFA; C8 to C10) were recently shown to act as modulators of PPARγ (31). Similar to the data reported here for SCFA, the MCFA decanoic acid bound and (trans)activated PPARγ, and contrary to synthetic PPARγ agonists and LCFA, decanoic acid inhibited adipogenesis. Furthermore, it was shown that the hydrocarbon tail of decanoic acid occupies a completely different pocket than the tail of LCFA or rosiglitazone. Remarkably, even though no specific orientational or positional restraints were used to guide the binding of butyrate in the large PPARγ binding site, the most favorable solution to the docking was very similar to that of decanoic acid. In general, the best solutions of the docking protocol displayed high-quality interactions with the PPARγ receptor, with a slightly different orientation of the butyrate carboxylic acid group in the binding site compared to decanoic acid. The model shows that butyrate is stabilized in the binding site by interactions with protein side chains. Since the buried surface area and interaction energy with the receptor are less than those for decanoic acid, the affinity of the complex with butyrate is predicted to be weaker than that with decanoic acid.
PPARγ has an antineoplastic effect in many different tumor types, yet its role in colorectal tumors remains controversial (60). In contrast, the anti-inflammatory effect of PPARγ in the colon is well recognized (61). PPARγ ligands were shown to suppress inflammatory gene expression in colonic cell lines by suppressing NF-κB and reduce inflammation in a mouse model of inflammatory bowel disease (62, 63). In addition, PPARγ in colonic epithelial cells was shown to protect against experimental inflammatory bowel disease (49). Similarly, SCFA, especially butyrate, seem to have broad anti-inflammatory properties by altering immune cell migration, adhesion, and cytokine expression and by affecting cell proliferation and apoptosis (64). Accordingly, it can be hypothesized that the anti-inflammatory properties of SCFA in the colon are at least partially conveyed by PPARγ (65).
Previously, ANGPTL4 expression in the human colon cell line HT29 was found to be stimulated by heat-stable factors secreted by the probiotic bacteria Lactobacillus strain F19 in a PPARα- and PPARγ-dependent manner (20). Based on data presented here, it seems highly plausible that the secreted factors represent SCFA. Thus, probiotic and resident microbiota may be able to influence ANGPTL4 production via production of SCFA and subsequent activation of PPARγ. Backhed reported that colonization of the gut of germ-free mice with microbiota reduces Angptl4 expression in mouse intestine (2). Inasmuch as SCFA stimulate ANGPTL4 expression, the suppressive effect of colonization on ANGPTL4 must be mediated by a mechanism other than SCFA.
It has been suggested that alterations in intestinal Angptl4 expression influence adipose LPL activity and thereby impact adipose mass (2). SCFA may thus inhibit fat storage by stimulating release of ANGPTL4. Whether ANGPTL4 also has a functional role in the intestine is unclear. Since the intestine does not express LPL, the local role of ANGPTL4 in intestine must extend beyond LPL inhibition. LPL inhibition is conferred exclusively by the N-terminal domain of ANGPTL4, whereas the C-terminal fragment of ANGPTL4 acts as a ligand for integrins to alter cellular signaling (66–68).
In conclusion, we show that SCFA potently stimulate ANGPTL4 production in human colon cell lines via PPARγ. Our data point to activation of PPARs as a novel mechanism of gene regulation by SCFA in the colon, in addition to other mechanisms of action of SCFA.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the Netherlands Nutrigenomics Centre, the Netherlands Organization for Scientific Research (NWO) (VICI grant 700.56.442 to A.B., visitor travel grant 040.11.299 to J.G., and TOP grant 40-00812-98-08030 to S.K.), and the Netherlands Consortium for Systems Biology.
S.A., H.R., S.M., E.K., M.M., G.J.H., and S.K. designed research; S.A., K.L., T.A., J.S.G., E.S., A.K., A.K.H., K.M., D.H., and S.A.-L. performed research; R.H. and B.V.D.B. contributed analytic tools; S.A., K.L., T.A., J.S.G., E.S., A.K., A.K.H., H.R., A.M.J.J.B., E.K., G.J.H., and S.K. analyzed data; S.K. wrote the initial draft of the paper. All authors read and edited the paper.
Footnotes
Published ahead of print 22 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00858-12.
REFERENCES
- 1. Sekirov I, Russell SL, Antunes LC, Finlay BB. 2010. Gut microbiota in health and disease. Physiol. Rev. 90: 859–904 [DOI] [PubMed] [Google Scholar]
- 2. Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. 2004. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U. S. A. 101: 15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Backhed F, Manchester JK, Semenkovich CF, Gordon JI. 2007. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U. S. A. 104: 979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lichtenstein L, Kersten S. 2010. Modulation of plasma TG lipolysis by Angiopoietin-like proteins and GPIHBP1. Biochim. Biophys. Acta 1801: 415–420 [DOI] [PubMed] [Google Scholar]
- 5. Yoshida K, Shimizugawa T, Ono M, Furukawa H. 2002. Angiopoietin-like protein 4 is a potent hyperlipidemia-inducing factor in mice and inhibitor of lipoprotein lipase. J. Lipid Res. 43: 1770–1772 [DOI] [PubMed] [Google Scholar]
- 6. Ge H, Yang G, Yu X, Pourbahrami T, Li C. 2004. Oligomerization state-dependent hyperlipidemic effect of angiopoietin-like protein 4. J. Lipid Res. 45: 2071–2079 [DOI] [PubMed] [Google Scholar]
- 7. Koster A, Chao YB, Mosior M, Ford A, Gonzalez-DeWhitt PA, Hale JE, Li D, Qiu Y, Fraser CC, Yang DD, Heuer JG, Jaskunas SR, Eacho P. 2005. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology 146: 4943–4950 [DOI] [PubMed] [Google Scholar]
- 8. Mandard S, Zandbergen F, van Straten E, Wahli W, Kuipers F, Muller M, Kersten S. 2006. The fasting-induced adipose factor/angiopoietin-like protein 4 is physically associated with lipoproteins and governs plasma lipid levels and adiposity. J. Biol. Chem. 281: 934–944 [DOI] [PubMed] [Google Scholar]
- 9. Xu A, Lam MC, Chan KW, Wang Y, Zhang J, Hoo RL, Xu JY, Chen B, Chow WS, Tso AW, Lam KS. 2005. Angiopoietin-like protein 4 decreases blood glucose and improves glucose tolerance but induces hyperlipidemia and hepatic steatosis in mice. Proc. Natl. Acad. Sci. U. S. A. 102: 6086–6091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yu X, Burgess SC, Ge H, Wong KK, Nassem RH, Garry DJ, Sherry AD, Malloy CR, Berger JP, Li C. 2005. Inhibition of cardiac lipoprotein utilization by transgenic overexpression of Angptl4 in the heart. Proc. Natl. Acad. Sci. U. S. A. 102: 1767–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Georgiadi A, Lichtenstein L, Degenhardt T, Boekschoten MV, van Bilsen M, Desvergne B, Muller M, Kersten S. 2010. Induction of cardiac Angptl4 by dietary fatty acids is mediated by peroxisome proliferator-activated receptor beta/delta and protects against fatty acid-induced oxidative stress. Circ. Res. 106: 1712–1721 [DOI] [PubMed] [Google Scholar]
- 12. Lichtenstein L, Mattijssen F, de Wit NJ, Georgiadi A, Hooiveld GJ, van der Meer R, He Y, Qi L, Koster A, Tamsma JT, Tan NS, Muller M, Kersten S. 2010. Angptl4 protects against severe proinflammatory effects of saturated fat by inhibiting fatty acid uptake into mesenteric lymph node macrophages. Cell Metab. 12: 580–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kersten S, Mandard S, Tan NS, Escher P, Metzger D, Chambon P, Gonzalez FJ, Desvergne B, Wahli W. 2000. Characterization of the fasting-induced adipose factor FIAF, a novel peroxisome proliferator-activated receptor target gene. J. Biol. Chem. 275: 28488–28493 [DOI] [PubMed] [Google Scholar]
- 14. Yoon JC, Chickering TW, Rosen ED, Dussault B, Qin Y, Soukas A, Friedman JM, Holmes WE, Spiegelman BM. 2000. Peroxisome proliferator-activated receptor gamma target gene encoding a novel angiopoietin-related protein associated with adipose differentiation. Mol. Cell. Biol. 20: 5343–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bunger M, van den Bosch HM, van der Meijde J, Kersten S, Hooiveld GJ, Muller M. 2007. Genome-wide analysis of PPARalpha activation in murine small intestine. Physiol. Genomics 30: 192–204 [DOI] [PubMed] [Google Scholar]
- 16. Staiger H, Haas C, Machann J, Werner R, Weisser M, Schick F, Machicao F, Stefan N, Fritsche A, Haring HU. 2009. Muscle-derived angiopoietin-like protein 4 is induced by fatty acids via peroxisome proliferator-activated receptor (PPAR)-delta and is of metabolic relevance in humans. Diabetes 58: 579–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kersten S, Lichtenstein L, Steenbergen E, Mudde K, Hendriks HF, Hesselink MK, Schrauwen P, Muller M. 2009. Caloric restriction and exercise increase plasma ANGPTL4 levels in humans via elevated free fatty acids. Arterioscler. Thromb. Vasc. Biol. 29: 969–974 [DOI] [PubMed] [Google Scholar]
- 18. Modica S, Gofflot F, Murzilli S, D'Orazio A, Salvatore L, Pellegrini F, Nicolucci A, Tognoni G, Copetti M, Valanzano R, Veschi S, Mariani-Costantini R, Palasciano G, Schoonjans K, Auwerx J, Moschetta A. 2010. The intestinal nuclear receptor signature with epithelial localization patterns and expression modulation in tumors. Gastroenterology 138: 636–648 [DOI] [PubMed] [Google Scholar]
- 19. Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. 2006. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 126: 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Aronsson L, Huang Y, Parini P, Korach-Andre M, Hakansson J, Gustafsson JA, Pettersson S, Arulampalam V, Rafter J. 2010. Decreased fat storage by Lactobacillus paracasei is associated with increased levels of angiopoietin-like 4 protein (ANGPTL4). PLoS One 5: e13087 doi:10.1371/journal.pone.0013087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Siersbaek MS, Loft A, Aagaard MM, Nielsen R, Schmidt SF, Petrovic N, Nedergaard J, Mandrup S. 2012. Genome-wide profiling of peroxisome proliferator-activated receptor gamma in primary epididymal, inguinal, and brown adipocytes reveals depot-selective binding correlated with gene expression. Mol. Cell. Biol. 32: 3452–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gijsbers L, Man HY, Kloet SK, de Haan LH, Keijer J, Rietjens IM, van der Burg B, Aarts JM. 2011. Stable reporter cell lines for peroxisome proliferator-activated receptor gamma (PPARgamma)-mediated modulation of gene expression. Anal. Biochem. 414: 77–83 [DOI] [PubMed] [Google Scholar]
- 23. Sonneveld E, Riteco JA, Jansen HJ, Pieterse B, Brouwer A, Schoonen WG, van der Burg B. 2006. Comparison of in vitro and in vivo screening models for androgenic and estrogenic activities. Toxicol. Sci. 89: 173–187 [DOI] [PubMed] [Google Scholar]
- 24. Sonneveld E, Jansen HJ, Riteco JA, Brouwer A, van der Burg B. 2005. Development of androgen- and estrogen-responsive bioassays, members of a panel of human cell line-based highly selective steroid-responsive bioassays. Toxicol. Sci. 83: 136–148 [DOI] [PubMed] [Google Scholar]
- 25. Koppen A, Houtman R, Pijnenburg D, Jeninga EH, Ruijtenbeek R, Kalkhoven E. 2009. Nuclear receptor-coregulator interaction profiling identifies TRIP3 as a novel peroxisome proliferator-activated receptor gamma cofactor. Mol. Cell. Proteomics 8: 2212–2226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5: R80 doi:10.1186/gb-2004-5-10-r80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264 [DOI] [PubMed] [Google Scholar]
- 28. Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. 2005. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 33: e175 doi:10.1093/nar/gni179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scarsi M, Podvinec M, Roth A, Hug H, Kersten S, Albrecht H, Schwede T, Meyer UA, Rucker C. 2007. Sulfonylureas and glinides exhibit peroxisome proliferator-activated receptor gamma activity: a combined virtual screening and biological assay approach. Mol. Pharmacol. 71: 398–406 [DOI] [PubMed] [Google Scholar]
- 30. Dominguez C, Boelens R, Bonvin AM. 2003. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125: 1731–1737 [DOI] [PubMed] [Google Scholar]
- 31. Malapaka RR, Khoo S, Zhang J, Choi JH, Zhou XE, Xu Y, Gong Y, Li J, Yong EL, Chalmers MJ, Chang L, Resau JH, Griffin PR, Chen YE, Xu HE. 2012. Identification and mechanism of 10-carbon fatty acid as modulating ligand of peroxisome proliferator-activated receptors. J. Biol. Chem. 287: 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Vries SJ, van Dijk M, Bonvin AM. 2010. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5: 883–897 [DOI] [PubMed] [Google Scholar]
- 33. Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. 1998. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905–921 [DOI] [PubMed] [Google Scholar]
- 34. Jorgensen WL, Tirado-Rives J. 1988. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110: 1657–1666 [DOI] [PubMed] [Google Scholar]
- 35. Macfarlane GT, Gibson GR, Cummings JH. 1992. Comparison of fermentation reactions in different regions of the human colon. J. Appl. Bacteriol. 72: 57–64 [DOI] [PubMed] [Google Scholar]
- 36. Boffa LC, Vidali G, Mann RS, Allfrey VG. 1978. Suppression of histone deacetylation in vivo and in vitro by sodium butyrate. J. Biol. Chem. 253: 3364–3366 [PubMed] [Google Scholar]
- 37. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. 2008. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J. Nutr. Biochem. 19: 587–593 [DOI] [PubMed] [Google Scholar]
- 38. Harmon GS, Dumlao DS, Ng DT, Barrett KE, Dennis EA, Dong H, Glass CK. 2010. Pharmacological correction of a defect in PPAR-gamma signaling ameliorates disease severity in Cftr-deficient mice. Nat. Med. 16: 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han X, Benight N, Osuntokun B, Loesch K, Frank SJ, Denson LA. 2007. Tumour necrosis factor alpha blockade induces an anti-inflammatory growth hormone signalling pathway in experimental colitis. Gut 56: 73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee SK, Kim YW, Chi SG, Joo YS, Kim HJ. 2009. The effect of Saccharomyces boulardii on human colon cells and inflammation in rats with trinitrobenzene sulfonic acid-induced colitis. Digest. Dis. Sci. 54: 255–263 [DOI] [PubMed] [Google Scholar]
- 41. Narravula S, Colgan SP. 2001. Hypoxia-inducible factor 1-mediated inhibition of peroxisome proliferator-activated receptor alpha expression during hypoxia. J. Immunol. 166: 7543–7548 [DOI] [PubMed] [Google Scholar]
- 42. Shimada T, Kojima K, Yoshiura K, Hiraishi H, Terano A. 2002. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut 50: 658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang JB, Qi LL, Zheng SD, Wang HZ, Wu TX. 2009. Curcumin suppresses PPARdelta expression and related genes in HT-29 cells. World J. Gastroenterol. 15: 1346–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kaddatz K, Adhikary T, Finkernagel F, Meissner W, Muller-Brusselbach S, Muller R. 2010. Transcriptional profiling identifies functional interactions of TGF beta and PPAR beta/delta signaling: synergistic induction of ANGPTL4 transcription. J. Biol. Chem. 285: 29469–29479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mandard S, Zandbergen F, Tan NS, Escher P, Patsouris D, Koenig W, Kleemann R, Bakker A, Veenman F, Wahli W, Muller M, Kersten S. 2004. The direct peroxisome proliferator-activated receptor target fasting-induced adipose factor (FIAF/PGAR/ANGPTL4) is present in blood plasma as a truncated protein that is increased by fenofibrate treatment. J. Biol. Chem. 279: 34411–34420 [DOI] [PubMed] [Google Scholar]
- 46. Camirand A, Marie V, Rabelo R, Silva JE. 1998. Thiazolidinediones stimulate uncoupling protein-2 expression in cell lines representing white and brown adipose tissues and skeletal muscle. Endocrinology 139: 428–431 [DOI] [PubMed] [Google Scholar]
- 47. Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, Bochkov VN, Exner M, Binder BR, Leitinger N. 2007. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler. Thromb. Vasc. Biol. 27: 1276–1282 [DOI] [PubMed] [Google Scholar]
- 48. Wu P, Inskeep K, Bowker-Kinley MM, Popov KM, Harris RA. 1999. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 48: 1593–1599 [DOI] [PubMed] [Google Scholar]
- 49. Adachi M, Kurotani R, Morimura K, Shah Y, Sanford M, Madison BB, Gumucio DL, Marin HE, Peters JM, Young HA, Gonzalez FJ. 2006. Peroxisome proliferator activated receptor gamma in colonic epithelial cells protects against experimental inflammatory bowel disease. Gut 55: 1104–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lytle C, Tod TJ, Vo KT, Lee JW, Atkinson RD, Straus DS. 2005. The peroxisome proliferator-activated receptor gamma ligand rosiglitazone delays the onset of inflammatory bowel disease in mice with interleukin 10 deficiency. Inflamm. Bowel Dis. 11: 231–243 [DOI] [PubMed] [Google Scholar]
- 51. Higgins LS, Depaoli AM. 2010. Selective peroxisome proliferator-activated receptor gamma (PPARgamma) modulation as a strategy for safer therapeutic PPARgamma activation. Am. J. Clin. Nutr. 91: 267S–272S [DOI] [PubMed] [Google Scholar]
- 52. Guan HP, Ishizuka T, Chui PC, Lehrke M, Lazar MA. 2005. Corepressors selectively control the transcriptional activity of PPARgamma in adipocytes. Genes Dev. 19: 453–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yu C, Markan K, Temple KA, Deplewski D, Brady MJ, Cohen RN. 2005. The nuclear receptor corepressors NCoR and SMRT decrease peroxisome proliferator-activated receptor gamma transcriptional activity and repress 3T3-L1 adipogenesis. J. Biol. Chem. 280: 13600–13605 [DOI] [PubMed] [Google Scholar]
- 54. Rakhshandehroo M, Knoch B, Muller M, Kersten S. 2010. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010: 612089 doi:10.1155/2010/612089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kinoshita M, Suzuki Y, Saito Y. 2002. Butyrate reduces colonic paracellular permeability by enhancing PPARgamma activation. Biochem. Biophys. Res. Commun. 293: 827–831 [DOI] [PubMed] [Google Scholar]
- 56. Schwab M, Reynders V, Loitsch S, Steinhilber D, Stein J, Schroder O. 2007. Involvement of different nuclear hormone receptors in butyrate-mediated inhibition of inducible NF kappa B signalling. Mol. Immunol. 44: 3625–3632 [DOI] [PubMed] [Google Scholar]
- 57. Georgiadi A, Kersten S. 2012. Mechanisms of gene regulation by fatty acids. Adv. Nutr. 3: 127–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bugaut M, Bentejac M. 1993. Biological effects of short-chain fatty acids in nonruminant mammals. Annu. Rev. Nutr. 13: 217–241 [DOI] [PubMed] [Google Scholar]
- 59. Cummings JH, Pomare EW, Branch WJ, Naylor CP, Macfarlane GT. 1987. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 28: 1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dai Y, Wang WH. 2010. Peroxisome proliferator-activated receptor gamma and colorectal cancer. World J. Gastrointest. Oncol. 2: 159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dubuquoy L, Rousseaux C, Thuru X, Peyrin-Biroulet L, Romano O, Chavatte P, Chamaillard M, Desreumaux P. 2006. PPARgamma as a new therapeutic target in inflammatory bowel diseases. Gut 55: 1341–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kelly D, Campbell JI, King TP, Grant G, Jansson EA, Coutts AG, Pettersson S, Conway S. 2004. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-gamma and RelA. Nat. Immunol. 5: 104–112 [DOI] [PubMed] [Google Scholar]
- 63. Su CG, Wen X, Bailey ST, Jiang W, Rangwala SM, Keilbaugh SA, Flanigan A, Murthy S, Lazar MA, Wu GD. 1999. A novel therapy for colitis utilizing PPAR-gamma ligands to inhibit the epithelial inflammatory response. J. Clin. Investig. 104: 383–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Meijer K, de Vos P, Priebe MG. 2010. Butyrate and other short-chain fatty acids as modulators of immunity: what relevance for health? Curr. Opin. Clin. Nutr. Metab. Care 13: 715–721 [DOI] [PubMed] [Google Scholar]
- 65. Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. 2008. Review article: the role of butyrate on colonic function. Aliment. Pharmacol. Ther. 27: 104–119 [DOI] [PubMed] [Google Scholar]
- 66. Goh YY, Pal M, Chong HC, Zhu P, Tan MJ, Punugu L, Lam CR, Yau YH, Tan CK, Huang RL, Tan SM, Tang MB, Ding JL, Kersten S, Tan NS. 2010. Angiopoietin-like 4 interacts with integrins beta1 and beta5 to modulate keratinocyte migration. Am. J. Pathol. 177: 2791–2803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goh YY, Pal M, Chong HC, Zhu P, Tan MJ, Punugu L, Tan CK, Huang RL, Sze SK, Tang MB, Ding JL, Kersten S, Tan NS. 2010. Angiopoietin-like 4 interacts with matrix proteins to modulate wound healing. J. Biol. Chem. 285: 32999–33009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sukonina V, Lookene A, Olivecrona T, Olivecrona G. 2006. Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. U. S. A. 103: 17450–17455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, McKee DD, Galardi CM, Plunket KD, Nolte RT, Parks DJ, Moore JT, Kliewer SA, Willson TM, Stimmel JB. 2002. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature 415: 813–817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.