

Abstract
The coordinated replication and transcription of pericentromeric repeats enable RNA interference (RNAi)-mediated transmission of pericentromeric heterochromatin in fission yeast, which is essential for the proper function of centromeres. Rad3/ATR kinase phosphorylates histone H2A on serine-128/-129 to create γH2A in pericentromeric heterochromatin during S phase, which recruits Brc1 through its breast cancer gene 1 protein (BRCA1) C-terminal (BRCT) domains. Brc1 prevents the collapse of stalled replication forks; however, it is unknown whether this activity influences centromere function. Here, we show that Brc1 localizes in pericentromeric heterochromatin during S phase, where it enhances Clr4/Suv39-mediated H3 lysine-9 dimethylation (H3K9me2) and gene silencing. Loss of Brc1 increases sensitivity to the microtubule-destabilizing drug thiabendazole (TBZ) and increases chromosome missegregation in the presence of TBZ. Brc1 retains significant function even when it cannot bind γH2A. However, elimination of the serine-121 site on histone H2A, a target of Bub1 spindle assembly checkpoint kinase, sensitizes γH2A-deficient and brc1Δ cells to replication stress and microtubule destabilization. Collective results suggest that Brc1-mediated stabilization of stalled replication forks is necessary for fully efficient transmission of pericentromeric heterochromatin, which is required for accurate chromosome segregation during mitosis.
INTRODUCTION
Maintenance of genome integrity during successive cycles of cell division is essential to prevent the accumulation of debilitating genetic alterations and chromosome rearrangements. The responsibility for protecting genome stability falls to high-fidelity DNA replication and repair systems acting in conjunction with an accurate chromosome distribution mechanism. DNA damage and mitotic spindle assembly checkpoints regulate these processes and couple their completion to cell cycle progression (1–3).
Accurate chromosome segregation depends on the assembly of kinetochores at a single centromere on each chromosome, which serve as the attachment site for microtubules that form the mitotic spindle. In the fission yeast Schizosaccharomyces pombe, the central core domain of each centromere is flanked by large inverted repeats that are themselves composed of shorter repeats. These repeats are the targets of RNA interference (RNAi)-mediated assembly of transcriptionally silent heterochromatin (4–6). A defining feature of this heterochromatin is the dimethylation of histone H3 at lysine 9 (H3k9me2) by the Clr4/Suv39 subunit of Rik1 complex. The heterochromatin protein Swi6/HP1 binds H3k9me2-modified heterochromatin. Failure to maintain pericentromeric heterochromatin impairs centromere function and sister chromatid cohesion near centromeres, leading to increased chromosome loss and sensitivity to the antifungal drug thiabendazole (TBZ), which destabilizes microtubules. Defects in pericentromeric heterochromatin also create a critical requirement for the spindle assembly checkpoint (7). This checkpoint is partly mediated through phosphorylation of serine-121 in the C terminus of histone H2A by Bub1 kinase, which recruits shugoshin (8).
Stabilizing and repairing replication forks are essential for maintaining genome integrity. These activities are regulated by ATR/Rad3/Mec1 checkpoint kinase (9, 10). A notable substrate is the SQ motif at the C terminus of histone H2A in yeast species and H2AX in mammals (11). Phospho-H2A(X), also known as γH2A(X), is formed in large chromosomal domains flanking DNA lesions, where it creates a recruitment platform for DNA repair and checkpoint mediator proteins. As first shown with mammalian MDC1, these proteins bind γH2A(X) through a phosphopeptide docking pocket formed by tandem breast cancer gene 1 protein (BRCA1) C-terminal (BRCT) domains (12). Fission yeast has two γH2A-binding proteins: Crb2, which mediates activation of Chk1 by Rad3 (13–15), and the 6-BRCT domain protein Brc1, which facilitates recovery from stalled and collapsed replication forks (16, 17). Brc1 is structurally related to budding yeast Rtt107/Esc4 and mammalian PTIP, both of which also bind γH2A(X) through BRCT domains (18, 19).
γH2A has been studied mostly in the context of double-strand breaks (DSBs), but genome-wide mapping revealed that γH2A is also formed at specific chromosomal locations during an unperturbed DNA synthesis (S) phase (20, 21). These regions include pericentromeric heterochromatin in fission yeast, suggesting that replication forks stall in these chromosomal domains. Indeed, a recent study detected X-shaped DNA structures in pericentromeric heterochromatin (22). These DNA structures are absent in clr4Δ cells, suggesting that the alternating pattern of replication origins and noncoding RNA in the pericentromeric heterochromatin of fission yeast results in collisions between the replication and transcription machineries. RNAi-mediated release of RNA polymerase II was proposed to favor fork restart by replisomes associated with Rik1 complex, thereby ensuring efficient transmission of heterochromatin-specific histone modifications in each S phase (22, 23). This model predicts that replication stress responses may contribute to maintenance of pericentromeric heterochromatin and accurate chromosome segregation. Here, we probe the involvement of Brc1 in centromere function.
MATERIALS AND METHODS
Strains, media, genetic analysis, and general procedures.
Strains are listed in Table 1. Growth media for S. pombe were prepared and genetic procedures performed as described previously (24). Growth assays were performed by spotting 5- or 10-fold serial dilutions of exponentially growing cells onto yeast extract with glucose and supplements (YES) plates in the absence or presence of the indicated agents. Plates were incubated at 30°C and photographed after 2 to 7 days of growth. Chromatin immunoprecipitation (ChIP) assays were performed as described previously (21) using anti-dimethyl-histone H3-K9 antibody (UpState Biotechnology) or anti-green fluorescent protein (anti-GFP) antibody (Roche). The percentage of immunoprecipitated DNA (%IP) in the ChIP samples was calculated relative to the amount of DNA in the input samples. ChIP fold enrichment was calculated relative to act1. Sequences of qPCR primers are available upon request. Gene-silencing assays at the centromere were performed as described previously (25–28). RNA preparation and quantitative real-time PCR (qRT-PCR) experiments were carried out as previously described (27, 29, 30). All error bars in figures represent the standard errors from a minimum of 3 experiments.
Table 1.
Genotypes of strains used in this study
| Strain | Genotype | Source |
|---|---|---|
| JW107 | h− ura4-D18 leu1-32 pRep41-N-GFP-brc1+ | Lab stock |
| JW397 | h+ leu1-32 brc1::hphMx | Lab stock |
| PR109 | h− leu1-32 ura4-D18 | Lab stock |
| PR110 | h+ leu1-32 ura4-D18 | Lab stock |
| SR118 | h− leu1-32 ura4-D18 his3-D1 cdc25-22 | Lab stock |
| SR163 | h+ leu1-32 ade6-210 ura4-DS/E otr1R(SphI)::ura4+ | Lab stock |
| SR164 | h+ leu1-32 ade-ura4-DS/E imr1R(dg-glu) NcoI::ura4+ | Lab stock |
| SR273 | h+ leu1-32 ade6-210 ura4-DS/E otr1R(SphI)::ura4+ clr4::natMX | This study |
| SR357 | h+ leu1-32 ura4-D18 ade6-M216 swi6::ura4+ | Lab stock |
| SR442 | h+ leu1-32 ura4-D18 clr4::natMX6 pRep41-N-GFP-brc1+ | Lab stock |
| SY01 | h+ leu1-32 ade6-210 ura4-DS/E otr1R(SphI)::ura4+ brc1-T672A::natMX | This study |
| SY02 | h− leu1-32 ade-ura4-DS/E imr1R(dg-glu) NcoI::ura4+ brc1-T672A::natMX | This study |
| SY03 | h− leu1-32 ade-ura4-DS/E imr1R(dg-glu) NcoI::ura4+ clr4::natMX | This study |
| SY04 | h+ leu1-32 ade6-210 ura4-DS/E otr1R(SphI)::ura4+ brc1::hphMx | This study |
| SY05 | h− leu1-32 ade-ura4-DS/E imr1R(dg-glu) NcoI::ura4+ brc1::hphMx | This study |
| SY06 | h− leu1-32 ura4-D18 his3-D1 cdc25-22 brc1::hphMx | This study |
| SY07 | h+ leu1-32 ura4-D18 ade6-M216 swi6::ura4+ brc1::kanMx | This study |
| SY08 | h− ura4-D18 leu1-32 hta1- S121A-S129A::natMX hta2-S121A-S128A::kanMx | This study |
| SY09 | h− ura4-D18 leu1-32 hta1-S121A::natMX hta2-S121A::kanMx | This study |
| SY10 | h− ura4-D18 leu1-32 hta1-S129A::natMX hta2-S128A::kanMx | This study |
| SY11 | h− ura4-D18 leu1-32 brc1::hphMx hta1- S121A::natMX hta2-S121::kanMx | This study |
| SY12 | h− ura4-D18 leu1-32 brc1::hphMx hta1- S129A::natMX hta2-S128::kanMx | This study |
| SY13 | h+ leu1-32 ura4-D18 pRep41-N-GFP-brc1 hta1- S129A::natMX hta2-S128::kanMx | This study |
| SY14 | h+ leu1-32 ura4-D18 pRep41-N-GFP-brc1 hta1- S121A::natMX hta2-S121::kanMx | This study |
| SY15 | h+ leu1-32 ura4-D18 pRep41-N-GFP-brc1 hta1- S121A-S129A::natMX hta2-S121A-S128A::kanMx | This study |
Microscopy.
Cells were photographed using a Nikon Eclipse E800 microscope equipped with a Photometrics Quantix CCD camera. GFP-Brc1 was expressed from pRep42-GFP-brc1+ or pREP41-GFP-brc1+ plasmids (16). Cells were grown in Edinburgh minimal medium (EMM2) in the absence of thiamine for 18 to 20 h at 30°C. For analysis of mitotic abnormalities using the cdc25-22 arrest and release protocol, cells were grown to log phase at 25°C, shifted to 36°C for 4 h, and then released from the arrest by transfer to 25°C in the absence or presence of 5 μg/ml TBZ. Cells were harvested 60 min later. DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (500 μg/ml), and the spindle was stained with an antitubulin antibody (TAT1).
RESULTS
Heterochromatin-dependent enrichment of Brc1 at centromeres.
Brc1 localization at pericentromeric repeat sequences depends largely on γH2A, which appears in these sequences during S phase (21). γH2A enrichment in pericentromeric heterochromatin is largely abolished in clr4Δ cells (21). These findings predict that the enrichment of Brc1 in pericentromeric regions should also depend on Clr4. We used ChIP experiments to test this prediction. For these experiments, we expressed Brc1-GFP and performed ChIP assays with anti-GFP antibodies. Using unsynchronized cells from wild-type (clr4+) log-phase cultures, we detected ∼3-fold enrichments of Brc1 using cen-dh and cen-dg primers relative to the actin (act1) gene locus (Fig. 1A; see Fig. 2 for a centromere map). These Brc1 enrichments in the otr repeats were abolished in clr4Δ cells, indicating that they likely occur through replication fork stalling in the pericentromeric heterochromatin (Fig. 1A).
Fig 1.
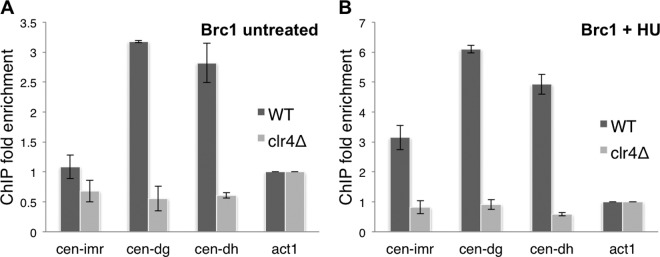
HU treatment increases Clr4-dependent Brc1 enrichment at centromeres. ChIP analysis of Brc1-GFP in log-phase cells (A) and cells treated with 12 mM HU for 4 h (B). Enrichment was calculated relative to the act1 (actin) locus (see Materials and Methods).
Fig 2.

Brc1 enhances gene silencing in pericentromeric heterochromatin. (A) Diagram of centromere I (cen1) with ura4+ insertion sites (top panel). Gene-silencing assays in nonselective (N/S), selective (−URA), and counterselective (5-FOA) media (bottom panels). (B) qRT-PCR analysis of ura4 (otr::ura4+) transcript levels relative to a control transcript, act1+, normalized to the wild type.
We did not detect Brc1 enrichment using cen-imr probes (Fig. 1A). This result was consistent with the ChIP analysis of γH2A using asynchronous cells (21). To investigate whether synchronizing cells in S phase reveals Brc1 enrichment with the imr or otr probes, we repeated the ChIP assays using cells exposed to 12 mM hydroxyurea (HU) for 4 h, which arrests cell cycle progression in early S phase by inhibiting ribonucleotide reductase, required for deoxynucleoside triphosphate (dNTP) biosynthesis. This approach was used previously to characterize enrichment of RNAi and DNA repair factors during S phase (22, 31). S-phase arrest increased Brc1 enrichment at cen-dh and cen-dg 5- to 6-fold relative to act1 (Fig. 1B). Brc1 increased 3-fold at cen-imr in HU-treated cells (Fig. 1B). As seen for the asynchronous cells, Brc1 enrichment at pericentromeric loci was abolished in HU-treated clr4Δ cells that are unable to form heterochromatin (Fig. 1B). The requirement for Clr4 indicates that Brc1 enrichment at pericentromeric loci is not solely a consequence of HU-induced fork arrest but instead depends on replication fork stalling in pericentromeric heterochromatin.
Brc1 promotes gene silencing in the centromeric repeats.
Replication and transcription of pericentromeric repeats are coupled to enable RNAi-mediated transmission of heterochromatin and consequent gene silencing (22, 23). The localization of Brc1 at pericentromeric repeats and its role in stabilizing replication forks suggested that it might have a role in gene silencing. To investigate this possibility, we assessed whether centromeric gene silencing is diminished in brc1Δ cells. With a ura4+ reporter gene inserted in the outer (otr::ura4+) or innermost (imr::ura4+) repeat regions (6, 32), we monitored ura4+ expression by plating serial dilutions of cells on nonselective (N/S), selective (−URA), or counterselective (5-fluoro-orotic acid [5-FOA]) plates. 5-FOA is a toxigenic substrate for the Ura4 protein. Deletion of brc1+ decreased gene silencing at both the otr::ura4+ and imr::ura4+ insertion sites, as indicated by brc1Δ colony formation on −URA plates and their retarded growth on FOA plates (Fig. 2A). Consistent with these findings, quantitative real-time PCR revealed that ura4+ transcripts from otr::ura4+ were increased ∼4-fold in brc1Δ cells relative to the wild type (Fig. 2B). As measured by both types of assays, the gene silencing defects in brc1Δ cells were partial compared to those in clr4Δ cells, which are completely deficient in pericentromeric heterochromatin (Fig. 2A and B).
We also analyzed gene silencing in a brc1-T672A mutant that has a missense mutation in the BRCT5,6 region that impairs binding to γH2A (16). In this mutant we detected a silencing defect with imr::ura4+, although the 5-FOA effect was weaker than that in the brc1Δ cells (Fig. 2A). The brc1-T672A effect on otr::ura4+ silencing was even weaker (Fig. 2A). As discussed below, these data indicate that Brc1 retains significant activity even when binding to γH2A is impaired, as previously observed (16).
Brc1 is required for efficient histone H3 lysine-9 methylation in pericentromeric heterochromatin.
Methylation of histone H3 on Lys9 (H3K9me2) by Clr4 methyltransferase is a key step in heterochromatin formation. H3K9me subsequently recruits Swi6/HP-1 to facilitate heterochromatin spreading (33, 34). H3K9 dimethylation is also a prominent mark of transcriptional repression. As our data indicated that Brc1 is involved in gene silencing at pericentromeric regions, we used ChIP to measure H3K9me2. Consistent with the silencing defect, reduced H3K9me2 levels at cen-dg were observed in brc1Δ cells (Fig. 3). As seen for the gene-silencing assays, the defect in brc1Δ cells was partial compared to that in clr4Δ cells. We repeated the assay with cells arrested in early S phase by HU treatment and found that the H3K9me2 signal was further diminished in brc1Δ cells (Fig. 3). These results are consistent with the gene-silencing assays and suggest that Brc1 is required for efficient maintenance of heterochromatin-specific modifications of histone H3 in pericentromeric regions.
Fig 3.
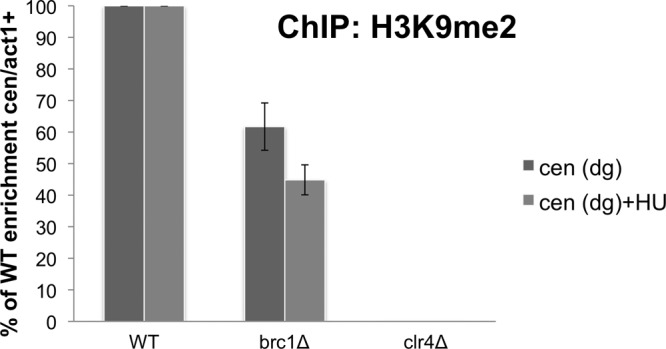
Brc1 assists heterochromatin-specific modification of the histone H3 tail at the centromere. ChIP analysis using anti-dimethyl-histone H3K9 (H3K9me2) antibodies at cen(dg) relative to act1+, normalized to the wild type. Cells were either untreated or exposed to 12 mM HU for 4 h.
Chromosome missegregation in brc1Δ cells.
Maintenance of pericentromeric heterochromatin is essential for robust cohesion of chromosome arms in pericentromeric regions and for proper centromere function (35, 36). Defects in centromeric heterochromatin cause sensitivity to TBZ. Interestingly, we found that brc1Δ cells are sensitive to TBZ (Fig. 4A). This TBZ-sensitive phenotype of brc1Δ cells was moderate, but it was strongly synergistic with elimination of Swi6/HP1 (Fig. 4A).
Fig 4.

TBZ sensitivity in brc1Δ cells. (A) TBZ sensitivity caused by brc1Δ is additive to that caused by swi6Δ. Tenfold serial dilutions were incubated at 30°C for 2 days. (B) Mitotic abnormalities in brc1Δ cells released from a cdc25-22 arrest in the absence or presence of 5 μg/ml TBZ. Micrographs were taken at 60 min after release from the arrest. DNA was stained with DAPI and antitubulin antibody (TAT1). The arrow points to a lagging chromosome (or chromosome fragment) in brc1Δ cells treated with TBZ.
Genetic assays established that Brc1 is required for mitotic chromosome stability, which is indicative of a role in chromosome segregation (37). To further investigate the effect of TBZ in brc1Δ cells, we monitored chromosome segregation in cultures synchronized by the cdc25-22 block and release protocol, which arrests cells in late G2 phase and synchronously releases them into mitosis. Cells were released from the G2 arrest in either the presence or the absence of a low concentration (5 μg/ml) of TBZ. DNA and tubulin staining revealed a large increase of lagging chromosomes in TBZ-treated brc1Δ cells compared to wild-type cells (16.4% in brc1Δ cells versus 3.0% in wild-type cells) (Fig. 4B and C). These data suggest that defects in centromere function sensitize brc1Δ cells to TBZ.
γH2A defect enhances TBZ sensitivity of spindle assembly checkpoint mutant.
In contrast to brc1Δ cells, TBZ sensitivity was not observed in htaAQ (hta1-S129A hta2-S128A) cells lacking the C-tail SQ phosphorylation site in both H2A genes (Fig. 5A). This relationship was also observed in replication stress survival assays (Fig. 5A) (16), implying that Brc1 retains substantial activity in the absence of γH2A. This relationship is shared with other γH2A-binding proteins (13, 18, 38) and is likely explained by their γH2A-independent scaffolding functions (39) and the compensatory activities of other genome protection mechanisms (21). To investigate the latter possibility, we tested whether elimination of γH2A enhances the TBZ-sensitive phenotype in cells lacking the histone H2A serine-121 site phosphorylated by Bub1. Replacement of serine-121 by alanine (hta1-S121A hta2-S121A) to generate the htaSA genotype causes substantial sensitivity to TBZ (8) (Fig. 5A). Combining the htaSA and htaAQ mutations to generate an htaSA.AQ strain (hta1-S121,129A hta2-S121,128A) further enhanced sensitivity to TBZ (Fig. 5A). Interestingly, the htaAQ and brc1Δ mutations had similar effects in the htaSA background, and htaSA increased sensitivity to replication stress agents in the htaAQ and brc1Δ backgrounds (Fig. 5A). Importantly, the htaSA genotype did not reduce Brc1 localization at cen-imr or cen-dh (Fig. 5B). These data indicate independent compensatory activities of the γH2A-Brc1 replication stress response and the Bub1-mediated spindle assembly checkpoint.
Fig 5.

Loss of γH2A or Brc1 increases TBZ sensitivity in cells lacking the serine-121 site in histone H2A that is phosphorylated by Bub1. (A) Histone H2A mutants lacking the γH2A site (htaAQ [hta1-S129A hta2-S128A]) or the Bub1 phosphorylation site (htaSA [hta1-S121A hta2-S121A]) alone, or in combination (htaSA.AQ), or with brc1Δ were tested with TBZ or the indicated genotoxins. Cells were incubated at 30°C for 2 days. (B) Phosphorylation of histone H2A at serine-121 is not required for localization of Brc1 at centromeres. ChIP analysis of Brc1-GFP in cells treated with 12 mM HU for 4 h. Enrichment was calculated relative to the act1 locus (see Materials and Methods).
DISCUSSION
The key conceptual advance that emerges from this study is that Brc1 contributes to centromere function in fission yeast. Our experimental evidence suggests that Brc1 enhances the maintenance of pericentromeric heterochromatin and thereby increases the probability of proper chromosome segregation, especially when spindle function is impaired. The discovery of this relationship is consistent with evidence that γH2A and Brc1 are enriched in pericentromeric heterochromatin during S phase in a Clr4-dependent manner (21). The data are furthermore consistent with studies indicating that RNAi-mediated release of RNA Pol II is necessary to complete replication of pericentromeric heterochromatin with replisomes that remain associated with the Rik1 holocomplex (22). Taken together, the data indicate that the γH2A-Brc1 module and the RNAi pathway work together to ensure the efficient transmission of pericentromeric heterochromatin during each S phase.
These studies indicate a physiologically significant role for Brc1 in the maintenance of pericentromeric heterochromatin, but pericentromeric gene silencing is not fully ablated in brc1Δ cells. In this respect, Brc1 is similar to 12 other genes recently identified in a targeted-deletion screen for loss of pericentromeric silencing (40). Several of these candidates are involved in replication stress responses, including the nonessential Nse5 subunit of Smc5-Smc6 structural maintenance of the chromosome holocomplex (31). As seen with γH2A and Brc1, the Smc5/6 holocomplex is transiently enriched at pericentromeric heterochromatin during S phase in a Clr4-dependent manner (41). Brc1 is a high-copy-number suppressor of smc6-74 and is required for viability in strains having hypomorphic mutations of essential subunits of Smc5/6 holocomplex (17, 37). These data suggest that multiple proteins involved in the response to replication stress contribute to the maintenance of pericentromeric heterochromatin.
Eliminating γH2A does not sensitize cells to TBZ, implying that Brc1 retains significant activity without binding γH2A. This result is consistent with our previous study showing that Brc1 enrichment at cen-dh is reduced in htaAQ cells but it remains above the Brc1 signal at the act1 locus (21). As mentioned above, γH2A(X)-independent activity occurs for many γH2A(X)-binding proteins, and for Crb2 this activity is at least partly explained by its multiple independent protein interactions (39). However, genetic interaction tests can reveal subphenotypic effects of mutations, as we have observed when the htaSA and htaAQ genotypes are combined. This particular genetic interaction also highlights how distinct genome protection pathways are propagated through C-tail phosphorylations of histone H2A.
Brc1's involvement in heterochromatic gene silencing may extend to its Saccharomyces cerevisiae homolog RTT107. This gene was initially identified through its regulation (i.e., repression) of ty1 retrotransposon transposition (42). Chromatin structure strongly impacts retrotransposon integration frequencies and site selection (43). Indeed, defects in chromatin assembly factor 1 (CAF-1) and HIR chromatin assembly complexes synergistically increase Ty1 transposition rates (44). Interestingly, centromeres from many plant species contain sequences related to retrotransposon long terminal repeats (45). RTT107 was later uncovered as ESC4 in a screen for genes that can establish silent chromatin at the HMR silent mating type cassette (46). The C-terminal BRCT5,6 domains of Esc4 bound Sir3, a subunit of the silent information regulator holocomplex that is required for chromatin silencing at transcriptionally repressed mating type donor cassettes and telomeric heterochromatin (47). These observations suggest that Rtt107/Esc4 may have retained chromatin assembly and modification functions even as the mechanism of chromatin-mediated transcriptional repression has diverged in S. cerevisiae.
The involvement of H2AX-binding DNA damage response proteins in gene silencing is not limited to yeast species. Mammalian Mdc1, which binds γH2AX through its C-terminal BRCT domains and functions in checkpoint signaling (12, 48), is critical for inactivation of sex chromosomes in male mouse meiosis (49).
Genetic and biochemical evidence supports a model in which collisions between the replisome and the RNA polymerase II transcription complex stall replication forks in pericentromeric heterochromatin of fission yeast (22, 23) (Fig. 6). According to this model, RNAi releases Pol II by processing pre-small interfering RNA (pre-siRNA) transcripts, allowing the completion of DNA replication. The leading-strand DNA polymerase associates with the Rik1 histone modification complex, which includes Clr4, ensuring efficient transmission of H3K9 methylation. RNAi defects are proposed to prolong replisome stalling, leading to an alternative pathway of replication fork restart requiring homology-directed repair (HDR) (22). This pathway engages a replisome that lacks the associated Rik1 complex, leading to the loss of histone modifications required for heterochromatic gene silencing (Fig. 6). Aberrant repair via this alternative pathway may underlie the formation of isochromosomes, which account for ∼50% of spontaneous gross chromosomal rearrangements (GCRs) in fission yeast (50). Rad3 suppresses these GCRs, supporting a model in which they originate from a failure to stabilize stalled replication forks near centromeres. Mutants lacking Brc1 have increased spontaneous foci of Rad52 (Rad22) HDR protein, and there is a severe synergistic growth defect when brc1Δ and rad22Δ mutations are combined, indicating that stalled replication forks are more likely to be restarted via an HDR pathway in brc1Δ cells (16). Incorporating the data presented in this study, the most parsimonious model is that Brc1 stabilizes replication forks stalled in pericentromeric heterochromatin, favoring the restart pathway in which the replisome maintains its association with the Rik1 complex (Fig. 6).
Fig 6.

Model for Brc1 stabilizing replication forks in pericentromeric heterochromatin. Collisions between the replisome and RNA Pol II trigger γH2A formation by Rad3 (not shown), resulting in recruitment of Brc1. Stabilization of the replication fork by Brc1 favors RNAi-mediated disassociation of RNA Pol II from the chromosome, allowing fork restart by the Rik1-associated replisome (22). Loss of Brc1 destabilizes the fork and increases the frequency of HDR-mediated fork restart by a reassembled replisome that is not associated with the Rik1 complex. See the text for additional description.
ACKNOWLEDGMENTS
We thank Robin Allshire and Shiv Grewal for kindly providing strains and members of the Scripps Cell Cycle Group for discussion.
This research was supported by NIH grants GM59447 and CA77325 awarded to P.R.
Footnotes
Published ahead of print 28 January 2013
REFERENCES
- 1. Harper JW, Elledge SJ. 2007. The DNA damage response: ten years after. Mol. Cell 28:739–745 [DOI] [PubMed] [Google Scholar]
- 2. Musacchio A, Salmon ED. 2007. The spindle-assembly checkpoint in space and time. Nat. Rev. Mol. Cell Biol. 8:379–393 [DOI] [PubMed] [Google Scholar]
- 3. Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461:1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SI. 2005. Comprehensive analysis of heterochromatin- and RNAi-mediated epigenetic control of the fission yeast genome. Nat. Genet. 37:809–819 [DOI] [PubMed] [Google Scholar]
- 5. Almeida R, Allshire RC. 2005. RNA silencing and genome regulation. Trends Cell Biol. 15:251–258 [DOI] [PubMed] [Google Scholar]
- 6. Pidoux AL, Allshire RC. 2004. Kinetochore and heterochromatin domains of the fission yeast centromere. Chromosome Res. 12:521–534 [DOI] [PubMed] [Google Scholar]
- 7. Bernard P, Hardwick K, Javerzat JP. 1998. Fission yeast bub1 is a mitotic centromere protein essential for the spindle checkpoint and the preservation of correct ploidy through mitosis. J. Cell Biol. 143:1775–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kawashima SA, Yamagishi Y, Honda T, Ishiguro K, Watanabe Y. 2010. Phosphorylation of H2A by Bub1 prevents chromosomal instability through localizing shugoshin. Science 327:172–177 [DOI] [PubMed] [Google Scholar]
- 9. Cimprich KA, Cortez D. 2008. ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9:616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Branzei D, Foiani M. 2010. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11:208–219 [DOI] [PubMed] [Google Scholar]
- 11. Dickey JS, Redon CE, Nakamura AJ, Baird BJ, Sedelnikova OA, Bonner WM. 2009. H2AX: functional roles and potential applications. Chromosoma 118:683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stucki M, Jackson SP. 2006. gammaH2AX and MDC1: anchoring the DNA-damage-response machinery to broken chromosomes. DNA Repair 5:534–543 [DOI] [PubMed] [Google Scholar]
- 13. Nakamura TM, Du LL, Redon C, Russell P. 2004. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol. Cell. Biol. 24:6215–6230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kilkenny ML, Dore AS, Roe SM, Nestoras K, Ho JC, Watts FZ, Pearl LH. 2008. Structural and functional analysis of the Crb2-BRCT2 domain reveals distinct roles in checkpoint signaling and DNA damage repair. Genes Dev. 22:2034–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sofueva S, Du LL, Limbo O, Williams JS, Russell P. 2010. BRCT domain interactions with phospho-histone H2A target Crb2 to chromatin at double-strand breaks and maintain the DNA damage checkpoint. Mol. Cell. Biol. 30:4732–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. 2010. gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 29:1136–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee KM, Nizza S, Hayes T, Bass KL, Irmisch A, Murray JM, O'Connell MJ. 2007. Brc1-mediated rescue of Smc5/6 deficiency: requirement for multiple nucleases and a novel Rad18 function. Genetics 175:1585–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X, Liu K, Li F, Wang J, Huang H, Wu J, Shi Y. 2012. Structure of C-terminal tandem BRCT repeats of Rtt107 protein reveals critical role in interaction with phosphorylated histone H2A during DNA damage repair. J. Biol. Chem. 287:9137–9146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yan W, Shao Z, Li F, Niu L, Shi Y, Teng M, Li X. 2011. Structural basis of gammaH2AX recognition by human PTIP BRCT5-BRCT6 domains in the DNA damage response pathway. FEBS Lett. 585:3874–3879 [DOI] [PubMed] [Google Scholar]
- 20. Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, Durocher D. 2010. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat. Struct. Mol. Biol. 17:299–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rozenzhak S, Mejia-Ramirez E, Williams JS, Schaffer L, Hammond JA, Head SR, Russell P. 2010. Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-phase in fission yeast. PLoS Genet. 6:e1001032 doi:10.1371/journal.pgen.1001032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, de Castro E, Marin L, Chang AY, Goto D, Cande WZ, Antequera F, Arcangioli B, Martienssen RA. 2011. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature 479:135–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li F, Martienssen R, Cande WZ. 2011. Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature 475:244–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Forsburg SL, Rhind N. 2006. Basic methods for fission yeast. Yeast 23:173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakagawa H, Lee JK, Hurwitz J, Allshire RC, Nakayama J, Grewal SI, Tanaka K, Murakami Y. 2002. Fission yeast CENP-B homologs nucleate centromeric heterochromatin by promoting heterochromatin-specific histone tail modifications. Genes Dev. 16:1766–1778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Halic M, Moazed D. 2010. Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell 140:504–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li F, Goto DB, Zaratiegui M, Tang X, Martienssen R, Cande WZ. 2005. Two novel proteins, dos1 and dos2, interact with rik1 to regulate heterochromatic RNA interference and histone modification. Curr. Biol. 15:1448–1457 [DOI] [PubMed] [Google Scholar]
- 28. Yamane K, Mizuguchi T, Cui B, Zofall M, Noma K, Grewal SI. 2011. Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol. Cell 41:56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rougemaille M, Shankar S, Braun S, Rowley M, Madhani HD. 2008. Ers1, a rapidly diverging protein essential for RNA interference-dependent heterochromatic silencing in Schizosaccharomyces pombe. J. Biol. Chem. 283:25770–25773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rodriguez-Gabriel MA, Watt S, Bahler J, Russell P. 2006. Upf1, an RNA helicase required for nonsense-mediated mRNA decay, modulates the transcriptional response to oxidative stress in fission yeast. Mol. Cell. Biol. 26:6347–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pebernard S, Wohlschlegel J, McDonald WH, Yates JR, III, Boddy MN. 2006. The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol. Cell. Biol. 26:1617–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Partridge JF, Borgstrom B, Allshire RC. 2000. Distinct protein interaction domains and protein spreading in a complex centromere. Genes Dev. 14:783–791 [PMC free article] [PubMed] [Google Scholar]
- 33. Grewal SI, Elgin SC. 2007. Transcription and RNA interference in the formation of heterochromatin. Nature 447:399–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ekwall K, Javerzat JP, Lorentz A, Schmidt H, Cranston G, Allshire R. 1995. The chromodomain protein Swi6: a key component at fission yeast centromeres. Science 269:1429–1431 [DOI] [PubMed] [Google Scholar]
- 35. Nonaka N, Kitajima T, Yokobayashi S, Xiao G, Yamamoto M, Grewal SI, Watanabe Y. 2002. Recruitment of cohesin to heterochromatic regions by Swi6/HP1 in fission yeast. Nat. Cell Biol. 4:89–93 [DOI] [PubMed] [Google Scholar]
- 36. Bernard P, Maure JF, Partridge JF, Genier S, Javerzat JP, Allshire RC. 2001. Requirement of heterochromatin for cohesion at centromeres. Science 294:2539–2542 [DOI] [PubMed] [Google Scholar]
- 37. Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ. 1999. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol. Biol. Cell 10:2905–2918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hammet A, Magill C, Heierhorst J, Jackson SP. 2007. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 8:851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Du LL, Nakamura TM, Russell P. 2006. Histone modification-dependent and -independent pathways for recruitment of checkpoint protein Crb2 to double-strand breaks. Genes Dev. 20:1583–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Braun S, Garcia JF, Rowley M, Rougemaille M, Shankar S, Madhani HD. 2011. The Cul4-Ddb1(Cdt)(2) ubiquitin ligase inhibits invasion of a boundary-associated antisilencing factor into heterochromatin. Cell 144:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pebernard S, Schaffer L, Campbell D, Head SR, Boddy MN. 2008. Localization of Smc5/6 to centromeres and telomeres requires heterochromatin and SUMO, respectively. EMBO J. 27:3011–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Scholes DT, Banerjee M, Bowen B, Curcio MJ. 2001. Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics 159:1449–1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Brady TL, Schmidt CL, Voytas DF. 2008. Targeting integration of the Saccharomyces Ty5 retrotransposon. Methods Mol. Biol. 435:153–163 [DOI] [PubMed] [Google Scholar]
- 44. Qian Z, Huang H, Hong JY, Burck CL, Johnston SD, Berman J, Carol A, Liebman SW. 1998. Yeast Ty1 retrotransposition is stimulated by a synergistic interaction between mutations in chromatin assembly factor I and histone regulatory proteins. Mol. Cell. Biol. 18:4783–4792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Houben A, Schubert I. 2003. DNA and proteins of plant centromeres. Curr. Opin. Plant Biol. 6:554–560 [DOI] [PubMed] [Google Scholar]
- 46. Andrulis ED, Zappulla DC, Alexieva-Botcheva K, Evangelista C, Sternglanz R. 2004. One-hybrid screens at the Saccharomyces cerevisiae HMR locus identify novel transcriptional silencing factors. Genetics 166:631–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zappulla DC, Maharaj AS, Connelly JJ, Jockusch RA, Sternglanz R. 2006. Rtt107/Esc4 binds silent chromatin and DNA repair proteins using different BRCT motifs. BMC Mol. Biol. 7:40 doi:10.1186/1471-2199-7-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fernandez-Capetillo O, Mahadevaiah SK, Celeste A, Romanienko PJ, Camerini-Otero RD, Bonner WM, Manova K, Burgoyne P, Nussenzweig A. 2003. H2AX is required for chromatin remodeling and inactivation of sex chromosomes in male mouse meiosis. Dev. Cell 4:497–508 [DOI] [PubMed] [Google Scholar]
- 49. Ichijima Y, Ichijima M, Lou Z, Nussenzweig A, Camerini-Otero RD, Chen J, Andreassen PR, Namekawa SH. 2011. MDC1 directs chromosome-wide silencing of the sex chromosomes in male germ cells. Genes Dev. 25:959–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Nakamura K, Okamoto A, Katou Y, Yadani C, Shitanda T, Kaweeteerawat C, Takahashi TS, Itoh T, Shirahige K, Masukata H, Nakagawa T. 2008. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 27:3036–3046 [DOI] [PMC free article] [PubMed] [Google Scholar]