

Abstract
p53 prevents cancer via cell cycle arrest, apoptosis, and the maintenance of genome stability. p53 also regulates energy-generating metabolic pathways such as oxidative phosphorylation (OXPHOS) and glycolysis via transcriptional regulation of SCO2 and TIGAR. SCO2, a cytochrome c oxidase assembly factor, is a metallochaperone which is involved in the biogenesis of cytochrome c oxidase subunit II. Here we have shown that SCO2 functions as an apoptotic protein in tumor xenografts, thus providing an alternative pathway for p53-mediated apoptosis. SCO2 increases the generation of reactive oxygen species (ROS) and induces dissociation of the protein complex between apoptosis signal-regulating kinase 1 (ASK-1) (mitogen-activated protein kinase kinase kinase [MAPKKK]) and its cellular inhibitor, the redox-active protein thioredoxin (Trx). Furthermore, SCO2 induces phosphorylation of ASK-1 at the Thr845 residue, resulting in the activation of the ASK-1 kinase pathway. The phosphorylation of ASK-1 induces the activation of mitogen-activated protein kinase kinases 4 and 7 (MAP2K4/7) and MAP2K3/6, which switches the c-Jun N-terminal protein kinase (JNK)/p38-dependent apoptotic cascades in cancer cells. Exogenous addition of the SCO2 gene to hypoxic cancer cells and hypoxic tumors induces apoptosis and causes significant regression of tumor xenografts. We have thus discovered a novel apoptotic function of SCO2, which activates the ASK-1 kinase pathway in switching “on” an alternate mode of p53-mediated apoptosis. We propose that SCO2 might possess a novel tumor suppressor function via the ROS–ASK-1 kinase pathway and thus could be an important candidate for anticancer gene therapy.
INTRODUCTION
The p53 protein induces apoptosis, cell cycle arrest, senescence, and differentiation, which prevents proliferation of stressed or damaged cells (1). The role of p53 in the regulation of cellular metabolism was recently identified (2, 3). p53 plays an important role in the regulation of energy-generating metabolic pathways that switch from oxidative phosphorylation (OXPHOS) to glycolysis. It inhibits glycolysis and increases OXPHOS by transcriptionally regulating two downstream genes, SCO2 (synthesis of cytochrome c oxidase 2) (3) and a TIGAR (p53-transactivated TP53-induced glycolysis apoptosis regulator) (2). Human SCO2, a novel p53-inducible protein, is a cytochrome c oxidase (COX) assembly protein that participates in the mitochondrial copper pathway, acting downstream of the COX17 protein. Severe cellular copper deficiency is observed in patients with nonfunctional SCO2 protein, and wild-type SCO2 overexpression complements the copper deficiency phenotype (4). SCO2 was shown previously to increase the rate of OXPHOS by stabilization of the COX17 subunit in the cytochrome c oxidase assembly (5).
Reactive oxygen species (ROS) are a toxic by-product of the mitochondrial energy production pathway, OXPHOS, in cancer cells (6). Since p53 increases OXPHOS, it is logical to assume that p53-mediated SCO2 upregulation will eventually lead to accumulation of cellular ROS. ROS play a major role in the progressive accumulation of cellular and tissue damage in neoplastic cells (7). Furthermore, ROS also provide an efficient pathway of eliminating cancerous cells through apoptosis (7). ROS function primarily by altering the status of cellular signaling and functions by activating various mitogen-activated protein (MAP) kinase (MAPK) pathways (8). One of the ROS-activated kinases is apoptosis signal-regulating kinase 1 (ASK-1), which is an extensively characterized mitogen-activated protein kinase kinase kinase (MAPKKK) (8). ASK-1 regulates the c-Jun N-terminal protein kinase (JNK) and p38 MAPK pathways, which play multiple important roles in cellular apoptosis. ASK-1 directly phosphorylates JNK and p38 and activates their respective mitogen-activated protein kinase kinases (MAP2Ks), MAP2K4 (SEK1)/MAP2K7 and MAP2K3/MAP2K6 (9). ASK-1 is activated via phosphorylation, which is initiated by cellular stressors, including ROS, mitochondrial oxidative stress, and endoplasmic reticulum (ER) stress (10, 11). ASK-1 has been shown to induce apoptosis in various cells through mitochondrion-dependent caspase activation and JNK and p38 kinase pathways (12, 13). ASK-1 is regulated in the cellular system by an evolutionarily conserved 12-kDa protein, thioredoxin (Trx). Trx is a redox protein which contains the redox-active-site sequence Trp-Cys-Gly-Pro-Cys-Lys. This sequence has known biological functions related to cell proliferation and apoptosis (14). The reduced form of Trx interacts with the N terminus of ASK-1 both in vitro and in vivo and inhibits its serine-threonine kinase activity (15). Trx is a molecular target of ROS and can be oxidized by various ROS signaling intermediates (16, 17). ROS-mediated oxidation of Trx disrupts the Trx–ASK-1 complex, enabling ASK-1 activation. Since p53 and SCO2 might be involved in ROS generation via high cellular OXPHOS levels, there is a possibility of a strong cellular relationship between SCO2 and the ASK-1 pathway. In this study, we demonstrate that the p53-inducible gene SCO2, which is known to upregulate OXPHOS, plays a major role in p53-mediated apoptosis. SCO2 provides an alternative pathway of cellular apoptosis by linking the role of p53 as a regulator of cellular metabolism and as a tumor suppressor. SCO2 increases ROS production and activates ASK-1 by inducing phosphorylation of ASK-1 at the Thr845 residue and disrupting the binding of ASK-1 with Trx. SCO2-activated ASK-1 shows high kinase activity and induces activation of downstream MAP2Ks, ultimately resulting in the triggering of JNK- and p38-dependent apoptosis. In ASK-1 knockout tumor xenografts, we show that ASK-1 activation is indispensable for SCO2-mediated apoptosis. Furthermore, the exogenous addition of SCO2 cDNA results in regression of the tumor xenografts, indicating that SCO2 is a potential candidate for anticancer gene therapy.
MATERIALS AND METHODS
Cell lines and culture conditions.
KB, MCF-7, HCT p53+/+, HCT p53−/−, A-431, and H1299 cells were obtained from ATCC (Manassas, VA) and the National Center for Cell Sciences (Pune, India) (18, 19). ASK-1−/− HCT p53+/+ and HCT p53−/− cells were prepared as described previously (23). The cells were cultured as monolayers in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) heat-inactivated fetal bovine serum (FBS) and antibiotics and incubated at 37°C in a humidified atmosphere of 95% air and 5% CO2.
Hypoxia exposure.
For hypoxic exposure, cells were placed into a humidified chamber maintained at 1.8% O2 and 5% CO2 (balance N2) for 24 h, as described previously (18, 19, 21). The oxygen level in the chamber was monitored with an oxygen analyzer (Vascular Technology). As a control, cells were cultured in a standard incubator (normoxia; air with 5% CO2) for 24 h.
Chemicals and antibodies.
DMEM, penicillin, streptomycin, FBS, trypsin-EDTA, and other chemicals of culture grade were purchased from Gibco Life Sciences (India). Tamoxifen, cisplatin, N-acetylcysteine (NAC), tumor necrosis factor alpha (TNF-α), propidium iodide (PI), 4′,6-diamidino-2-phenylindole (DAPI), and phalloidin (fluorescein isothiocyanate labeled) were purchased from Sigma (St. Louis, MO). Anti-goat IgG antibodies (Abs) for SCO2 (catalog number sc-49110), Trx (catalog number sc-18215), α-tubulin (catalog number sc-31779), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalog number sc-48166); anti-mouse antibody for p53 (catalog number sc-6243), α-tubulin (catalog number sc-5286), and GAPDH (catalog number sc-137179); and anti-rabbit antibody for α-tubulin (catalog number sc-5546) and GAPDH (catalog number sc-25778) were procured from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-rabbit primary antibodies for ASK-1 (catalog number 3762), phospho-ASK-1 (Thr845) (catalog number 3765), MAP2K4 (catalog number 9152), phospho-MAP2K4 (catalog number 9155), MAP2K7 (catalog number 4172), phospho-MAP2K7 (catalog number 4171), p38 (catalog number 9212), phospho-p38 (catalog number 4631), JNK (catalog number 4672), and phospho-JNK (catalog number 4668) were purchased from Cell Signaling Technology (Beverley, MA). Anti-rabbit IgG antibodies for MAP2K3 (catalog number SAB4300391), phospho-MAP2K3 (catalog number SAB4300119), MAP2K6 (catalog number SAB4300422), and phospho-MAP2K6 (catalog number SAB4300120) were procured from Sigma-Aldrich (St. Louis, MO). Goat anti-rabbit IgG (catalog number sc-2030), anti-goat IgG (catalog number sc-2768), and anti-mouse IgG (catalog number sc-2302) secondary antibodies were procured from Santa Cruz Biotechnology (Santa Cruz, CA).
Transfections.
p53 cDNA, SCO2 cDNA, p53 small interfering RNA (siRNA), and SCO2 siRNA were purchased from Origene (18, 19, 21–25). Cells were split 2 days before transfection at a density of 5 × 105 cells per plate. The siRNAs were transfected into the desired cells as described previously (21), and the in vivo cDNA and siRNA transfections were performed as described previously (18, 21, 23).
Tumor induction.
An 80-μl cell suspension containing 1 × 107 cells was subcutaneously injected into the hind leg of each mouse (18, 19). Tumor volumes were monitored weekly by caliper measurement of length, width, and height and were calculated by using the formula for a semiellipsoid (2πr3/3) (18, 19). After 3 weeks, mice bearing tumors with volumes averaging approximately 200 mm3 were randomized for treatment. Because of the variations in tumors and initial tumor growth as well as the removal of mice for analysis at various time points, the number of mice at each time point varied from experiment to experiment. The number of mice used is reported in the text.
Tumor regression.
The sizes of the tumors were carefully monitored after treatments by using vernier caliper measurements of length, width, and height and were calculated by using the formula for a semiellipsoid (4/3πr3/2), as described previously (18, 19).
Apoptosis in tumor samples.
The cellular apoptosis in tumors was measured by using a Red SR FLIVO in vivo apoptosis staining kit (Immuno Chemistry Technologies USA, Bloomington, MN) with flow cytometric analysis, as described previously (19).
Human apoptosis PCR array.
The Human Apoptosis RT2 Profiler PCR array kit (PAHS-012; SA-Bio Sciences) was used to profile the expression of 84 genes involved in programmed cell death, as described previously (21, 23).
Please refer to the supplemental material for a more details of the materials and methods used.
RESULTS
p53 regulates SCO2 at high doses of UV and tamoxifen.
p53 downregulates glycolysis through TIGAR activation (2, 19) and upregulates OXPHOS through SCO2 (3). We hypothesized that p53 fails to antagonistically regulate glycolysis and OXPHOS under conditions of cellular stress (19). We asked whether p53-mediated regulation of SCO2 is dependent upon the degree of cellular stress. Recently, we have shown that p53 specifically regulates the TIGAR gene in cancer cells exposed to low doses of UV and tamoxifen stress (19), suggesting that p53-mediated regulation of glycolysis works in cells undergoing repairable cellular stress. Thus, it was interesting to observe the cellular conditions under which p53 regulates SCO2. We began by analyzing the expression of SCO2 mRNA using reverse transcriptase PCR (RT-PCR) and quantitative PCR (qPCR). SCO2 mRNA expression (Fig. 1A) was analyzed in KB cells treated with low (25 J/cm2) and high (50 J/cm2) doses of UV (UV-25 and UV-50, respectively) and tamoxifen (10 and 100 nM [Tam-10 and Tam-100, respectively]) in a time-dependent manner. KB cells were treated with UV (25 J/cm2 and 50 J/cm2) for 0 h (control), 12 h, 18 h, and 24 h. Following treatment, the total mRNA of the cancer cells was extracted and used for real-time PCR (quantitative) and RT-PCR analysis. The results of both qPCR and RT-PCR were similar and showed that UV-50 (high-dose) treatment resulted in a progressive increase in the SCO2 mRNA level from 0 h to 24 h, whereas UV-25 (low dose) was unable to induce any significant increase in the SCO2 mRNA level from 0 h to 24 h. Similarly, KB cells were incubated with the two doses of tamoxifen for 12 h, 18 h, or 24 h. The results showed that Tam-100 (high dose) was able to induce maximum expression of the SCO2 mRNA at the 24-h time point. On the other hand, Tam-10 (low dose) had no significant effect on SCO2 mRNA expression. In conclusion, the SCO2 mRNA expression level was at least 10-fold higher at high doses of UV and tamoxifen than at lower doses. Similarly, SCO2 protein expression was analyzed in UV- and tamoxifen-treated KB cells by using quantitative in vivo enzyme-linked immunosorbent assay (ELISA) and Western blot techniques (Fig. 1B). The results showed that only UV-50 and Tam-100 (high doses) were effective in upregulating SCO2 protein expression at the 24-h time point, whereas UV-25 and Tam-10 (low doses) were not effective in inducing the expression of SCO2 protein in KB cells. In conclusion, the SCO2 expression level was 8-fold higher in KB cells treated with low doses of UV and tamoxifen. These data suggested that p53-mediated regulation of SCO2 gene expression might exist only at high doses of cellular stress.
Fig 1.

p53 regulates SCO2 in cancer cells treated with high doses of UV radiation and tamoxifen. (A) KB cells were treated with 25 J/cm2 (UV-25) or 50 J/cm2 (UV-50) of UV light for periods of 0, 12, 18, and 24 h. The expression of SCO2 mRNA was analyzed by using quantitative real-time PCR (graphs) and an RT-PCR assay (agarose gels). Both real-time and RT-PCR assays showed that a low dose of UV exposure (UV-25) was unable to induce SCO2 mRNA expression at 12 h, 18 h, or 24 h of UV exposure (graph, bars 3 to 5; gels, lanes 2 to 4). An asterisk represents a significant difference between bars 5 and 8 (P < 0.032). High doses of UV exposure (UV-50) induced a significant increase in the SCO2 mRNA level (graph, bars 6 to 8; gels, lanes 5 to 7) (n = 5). Similarly, in KB cells treated with 10 nM tamoxifen (low dose) (Tam-10), no significant change in the SCO2 mRNA expression level was observed via both real-time PCR (top, bars 3 to 5) and RT-PCR (bottom, lanes 2 to 4) studies. On the other hand, a high dose of tamoxifen (Tam-100) led to a significant increase in the SCO2 mRNA expression level (graph, bars 6 to 8; gels, lanes 5 to 7); (an asterisk represents a significant difference between bars 5 and 8 [P < 0.048]; n = 5; error bars represent standard deviations determined by analysis of variance). (B) The effect of low and high doses of UV and tamoxifen on SCO2 protein expression was observed by using a quantitative in vivo ELISA (top) and Western blots (bottom). Similar to SCO2 mRNA expression, the SCO2 protein was also upregulated with high doses of UV (UV-50) (an asterisk represents a significant difference between bars 5 and 8 [P < 0.038]) and tamoxifen (Tam-100) (an asterisk represents a significant difference between bars 5 and 8 [P < 0.045]) (n = 5; error bars represent standard deviations determined by analysis of variance). (C) The dose-dependent expression of SCO2 protein was observed in vivo in Swiss albino mice. The skin tissue of Swiss albino mice was subjected to UV and tamoxifen treatment, and the expression of SCO2 protein was measured by Western blotting. High doses of UV (UV-50) and tamoxifen (Tam-100) were again more effective in upregulating SCO2 protein levels in Swiss albino mice (n = 5). (D) The effect of p53-mediatedtranscription at the SCO2 promoter region carrying the p53 RE was observed in KB cells by using a luciferase assay. Results show that p53 initiates transcription at the SCO2 promoter at high doses of UV stress (UV-40 to UV-50); the transcription was p53 dependent, since p53 siRNA abolished the luciferase activity (∗, P < 0.025; n = 7; error bars represent standard deviations determined by analysis of variance). (E) The binding of p53 at its SCO2-RE at chromatin was studied by chromatin immunoprecipitation of UV-treated KB cells. Results showed that p53 shows a 15-fold increase in its binding at its SCO2-RE in UV-50-treated cells compared to untreated cells. Only high doses of UV influence binding of p53 at the SCO-RE (∗, P < 0.031) (n = 5). (F) For quantitative analysis, the results of chromatin immunoprecipitation were repeated by using quantitative ChIP. The results showed similar data, where only high doses of UV induced binding of p53 at the SCO2-RE (∗, P < 0.029; n = 7; error bars represent standard deviations determined by analysis of variance). IC, internal control; PC, positive control.
Next, the role of UV and tamoxifen stress in regulation of SCO2 protein expression was determined by using epithelial tissues of UV- and tamoxifen-treated Swiss albino mice (Fig. 1C). After treatment of the mouse epithelial tissue with UV-25, UV-50, Tam-10, and Tam-100 for 24 h, tissue samples were collected and analyzed for the expression of SCO2 protein by Western blotting. The results showed that the SCO2 expression level was high only at high doses of UV (UV-50) and tamoxifen (Tam-100). Low doses of UV and tamoxifen did not affect SCO2 protein expression. Since SCO2 is an established transcriptional target of p53 (3), it was important to understand the role of p53 in the UV-50- and Tam-100-induced increase in SCO2 mRNA and protein levels. Gene silencing using p53 siRNA showed that the UV-50-induced increase in SCO2 mRNA and protein levels was p53 dependent. p53 gene silencing abolished expression of SCO2 (see Fig. S1 in the supplemental material); however, p53 gene silencing did not alter SCO2 protein expression in UV-25-treated cells. These data suggest that p53 does not transcriptionally regulate SCO2 at low doses of cellular stress. Furthermore, a luciferase assay was conducted to determine p53-mediated transcriptional regulation of the SCO2 gene promoter in KB cells exposed to high doses of UV stress (Fig. 1D). The SCO2 gene promoter region containing the p53 response element (RE) was cloned into a pGL3 vector, and the construct was transfected into KB cells treated with increasing doses of UV irradiation for 24 h. The results showed an increase in p53-dependent luciferase activity only in KB cells that were treated with high doses of UV (Fig. 1D, bars 9 to 12). In the luciferase assay, p53 siRNA was used as the control (Fig. 1D). These data suggested that p53 transcriptionally regulates the SCO2 gene promoter only in cancer cells subjected to high doses of cellular stress. The level of p53-mediated transcription at the SCO2 gene promoter was significantly reduced upon decreasing UV doses, suggesting that p53-mediated transcriptional regulation of the SCO2 gene promoter is abolished at low doses of cellular stress. Since p53 mediates its transcriptional efficiency by directly binding to its response elements in the promoter region of downstream genes, we observed the direct binding of p53 at the SCO2 RE in UV-treated (UV-5 to UV-50) KB cells using chromatin immunoprecipitation (ChIP). Analysis of p53 binding to p53 RE, as described previously by Matoba et al. (3), at the SCO2 promoter by ChIP showed that there was a >15-fold increase in the binding of p53 at its RE in KB cells exposed to a high dose of UV (50 J/cm2) compared to cells treated with a low dose of UV (25 J/cm2) (Fig. 1E). p53 showed very high levels of binding to its RE at the SCO2 promoter in KB cells treated with UV-40, UV-45, and UV-50 (Fig. 1E, lanes 10 to 12). The real-time ChIP experiment showed similar results, where high doses of UV resulted in strong binding of p53 to its response element at the SCO2 promoter (Fig. 1F). These data confirm the fact that p53 regulates the SCO2 gene promoter exclusively in cancer cells subjected to high doses of cellular stress, and at low doses of stress, p53-mediated regulation of SCO2 gene was abolished. Overall, the results established that p53 binds to the SCO2 promoter and activates SCO2 expression at high doses of UV and tamoxifen.
SCO2 induces p53 downstream apoptosis by generating ROS.
The SCO2 protein is an active member of the electron transport chain in mitochondria; thus, we studied whether SCO2 protein upregulation results in ROS synthesis. The effect of SCO2 on alterations of cellular ROS content was observed both in vitro and in vivo (Fig. 2A and B). SCO2 protein was ectopically overexpressed in p53 wild-type cells (KB, MCF-7, and HCT p53+/+), p53 mutant cells (A-431), and p53 null cells (H1299 and HCT p53−/−) (Fig. 2A). The results showed a >5-fold increase in the ROS content of the SCO2-overexpressing cells (Fig. 2A, bars 7 to 12) in comparison to the control cells (bars 1 to 6). Tamoxifen treatment served as a positive control (Fig. 2A, bars 13 to 18). Similarly, the effect of SCO2 in generating ROS was observed for the p53 mutant (A-431), p53 null (HCT p53−/−), and p53 wild-type (MCF-7 and HCT p53+/+) tumor xenografts (Fig. 2B). SCO2 cDNA was overexpressed in tumor tissue by using an in vivo transfection kit from Altogen Biosystems (18, 19). Results showed that ectopic expression of SCO2 cDNA resulted in a significant increase in the ROS content in the tumor xenografts (Fig. 2B, bars 5 to 8); tamoxifen treatment served as the positive control. ROS are important signaling molecules which regulate cellular apoptosis via p53-dependent and -independent pathways (18). To counterestablish the role of SCO2 in cellular generation of ROS, we observed whether SCO2 gene silencing in tamoxifen-treated cancer cells resulted in downregulation of cellular ROS (see Fig. S2 in the supplemental material). Tam-100-treated KB and MCF-7 cells showed significant increases in cellular ROS content (see Fig. S2, bar 2, in the supplemental material). Upon silencing of the SCO2 gene in Tam-100-treated KB and MCF-7 cells, there was a 4-fold decrease in the cellular ROS content (see Fig. S2, bar 3). Similarly, p53 gene silencing in Tam-100-treated KB and MCF-7 cells resulted in a 5-fold decrease in cellular ROS (see Fig. S2, bar 4). Since SCO2 was efficient in inducing a significant increase in ROS content, we determined if SCO2 overexpression in cancer cells and tumor xenografts could result in cellular apoptosis (Fig. 2C and D). p53 wild-type cells (KB, MCF-7, and HCT p53+/+), p53 mutant cells (A-431), and p53 null cells (H1299) were ectopically expressed with SCO2 cDNA (Fig. 2C, bars 7 to 12), and cellular apoptosis was compared with that of control cells (bars 1 to 6), using annexin V staining and flow cytometry. The results showed a significant increase in the apoptotic fraction of the SCO2-overexpressing cancer cells, irrespective of the status of p53 gene expression. Next, we observed the role of SCO2-induced ROS in SCO2-mediated apoptosis by quenching cellular ROS via N-acetyl cysteine (NAC) (Fig. 2C, bars 13 to 18). The results showed that ROS quenching via NAC abolished SCO2-mediated apoptosis in cancer cells, suggesting that SCO2 induces ROS-mediated apoptosis. Similar results were obtained by using terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining, confirming that SCO2 expression induces cellular apoptosis that is abolished upon ROS quenching via NAC (see Fig. S3 in the supplemental material) The role of SCO2 in inducing apoptosis in tumor tissue was also determined by measuring the apoptotic fraction in p53 mutant (A-431), p53 null (HCT p53−/−), and p53 wild-type (MCF-7 and HCT p53+/+) tumor xenografts by using the SR FLIVO in vivo apoptosis kit (19) (Fig. 2D). FLIVO was injected intravenously into nude mice bearing tumors post-therapy. The tissue was extracted, and the cells were trypsinized for flow cytometric analysis of cellular apoptosis. The results showed significant increases in the apoptotic fraction of the SCO2-transfected tumor xenografts in comparison with control tumors (Fig. 2D, bars 5 to 8). Interestingly, SCO2-induced apoptosis was reversed upon ROS quenching via NAC (Fig. 2D, bars 9 to 12). These data established that SCO2 induces ROS-mediated apoptosis in cancer cells and tumor xenografts. Next, we determined the effect of SCO2 overexpression in cancer cells and tumor xenografts on the cleavage of the poly(ADP-ribose) polymerase (PARP) (a cellular marker of apoptosis) (Fig. 2E and F). A-431, HCT p53+/+, and HCT p53−/− cells were ectopically expressed with SCO2 cDNA in the presence and absence of NAC. The results showed cleavage of PARP in cancer cells overexpressing SCO2 protein (Fig. 2E, lane 2). However, SCO2-mediated PARP cleavage was abolished upon ROS quenching (Fig. 2E, lane 3). Tamoxifen treatment served as a positive control. Similarly, in HCT p53+/+ and HCT p53−/− tumor xenografts (Fig. 2F), the ectopic expression of SCO2 resulted in PARP cleavage, which was abolished upon ROS quenching. These data provide further evidence that SCO2 induces ROS-mediated apoptosis.
Fig 2.

SCO2 induces p53 downstream apoptosis by generating ROS. (A) The effect of SCO2 on cellular ROS was observed in p53 wild-type cells (KB, MCF-7, and HCT p53+/+), p53 mutant cells (A-431), and p53 null cells (H1299). SCO2-overexpressing cells (bars 7 to 12) showed a >5-fold increase in the ROS content in comparison with control cells (bars 1 to 6). Tamoxifen treatment served as a positive control (an asterisk represents a significant difference between control and SCO2-transfected groups [P < 0.029]; n = 7; error bars represent standard deviations determined by analysis of variance). MFI, mean fluorescence intensity. (B) The effect of SCO2 on cellular ROS was also analyzed in tumor xenografts. p53 mutant (A-431), p53 null (HCT p53−/−), and p53 wild-type (MCF-7 and HCT p53+/+) tumor xenografts were ectopically expressed with SCO2 cDNA, which resulted in a 4-fold increase in ROS content (bars 5 to 8). Tamoxifen treatment served as a positive control (∗, P < 0.045; n = 7; error bars represent standard deviations determined by analysis of variance). (C) The effect of SCO2 overexpression on cellular apoptosis was observed in p53 wild-type cells (KB, MCF-7, and HCT p53+/+), p53 mutant cells (A-431), and p53 null cells (H1299) by annexin V staining and flow cytometry. A significant increase in the apoptotic fraction of SCO2-overexpressing cells was seen irrespective of the p53 gene status (bars 7 to 12). ROS quenching via NAC abolished SCO2-mediated apoptosis (bars 13 to 18). Untreated cells served as negative controls, while tamoxifen served as a positive control (an asterisk represents a significant difference between control and SCO2-treated cells and between SCO2-treated and ROS-quenched cells [P < 0.021 and P < 0.025]; n = 7; error bars represent standard deviations determined by analysis of variance). (D) The role of SCO2 in inducing apoptosis was observed in p53 mutant (A-431), p53 null (HCT p53−/−), and p53 wild-type (MCF-7 and HCT p53+/+) tumor xenografts by using a FLIVO in vivo apoptosis kit (23). A significant increase in the apoptotic fraction of SCO2-transfected xenografts was seen in comparison with control tumors (bars 5 to 8), and ROS quenching via NAC suppressed SCO2-induced apoptosis (∗, P < 0.034 and P < 0.039; n = 7; error bars represent standard deviations determined by analysis of variance). (E) The effect of SCO2 overexpression on cleavage of PARP was observed in vitro for A-431, HCT p53+/+, and HCT p53−/− cancer cells. SCO2 cDNA was expressed ectopically in cancer cells in the presence and absence of NAC. Results show that ectopic expression of SCO2 resulted in cleavage of PARP in all the cancer cell lines (lane 2). Quenching of ROS via NAC abolished PARP cleavage (lane 3). Tamoxifen treatment served as a positive control and cleaved PARP (n = 5). (F) The effect of exogenous addition of SCO2 on PARP cleavage in p53+/+ and HCT p53−/− tumor xenografts was observed in the presence and absence of NAC. Ectopic expression of SCO2 resulted in PARP cleavage (lane 2). NAC abolished PARP cleavage (lane 3). Untreated cells and tamoxifen-treated cells served as controls (n = 4). (G) The potential of SCO2 to induce tumor regression was observed in A-431, MCF-7, HCT p53+/+, and HCT p53−/− tumor xenografts. Results show that exogenous addition of SCO2 cDNA led to a significant reduction in the size of tumor xenografts in 4 weeks. Combinatorial therapy of SCO2 and cisplatin and of SCO2 and tamoxifen caused a >85% regression of the tumor in 4 weeks. Untreated tumors served as controls; tamoxifen treatment served as a positive control (n = 10; error bars represent standard deviations determined by analysis of variance).
Since SCO2 was able to induce apoptosis in the tumor xenografts, the role of SCO2 as an anticancer gene therapy molecule was observed by analyzing its potential to induce tumor regression. A-431, MCF-7, HCT p53+/+ and HCT p53−/− tumor xenografts were grown on the hind leg of nude mice to an approximate volume of 2 cm3 (Fig. 2G). The tumors were treated with tamoxifen as a positive control (Fig. 2G). Tamoxifen treatment was able to induce tumor regression after 4 weeks of the treatment. Interestingly, SCO2 gene therapy conducted by exogenous addition of SCO2 cDNA induced a significant reduction in the size of the tumor xenografts (Fig. 2G). The effect of SCO2 gene therapy in combination with the known anticancer drugs cisplatin and tamoxifen was observed. The combinatorial therapy of SCO2 plus cisplatin and SCO2 plus tamoxifen resulted in more than 85% tumor regression after 4 weeks of treatment (Fig. 2G). The untreated tumors served as controls. These data suggested that SCO2 can be a potential candidate for anticancer gene therapy. We next studied the role of ROS in SCO2-mediated tumor regression. A-431, MCF-7, HCT p53+/+, and HCT p53−/− tumor xenografts were ectopically expressed with SCO2 cDNA, and SCO2-mediated tumor regression was observed for a period of 4 weeks. SCO2 addition resulted in a consistent regression of these tumor xenografts (see Fig. S4, black line, in the supplemental material). However, upon quenching of ROS in these tumor xenografts using NAC, SCO2-mediated tumor regression was abolished, and the tumors showed moderate growth from 0 to 4 weeks (see Fig. S4, red line, in the supplemental material). Untreated tumors served as controls (see Fig. S4, green line, in the supplemental material). These data suggested that SCO2-mediated tumor regression was ROS dependent, and ROS quenching abolished SCO2-mediated gene therapy.
Our study established SCO2 as an apoptotic protein which has the potential to function as an anticancer gene therapy molecule. In order to understand SCO2-induced apoptosis in detail, we determined the activation and suppression of a key set of 84 genes (19) (PAHS-012; SA-Bio Sciences) involved in both induction and inhibition of apoptosis. The mRNA expression analysis of these genes was conducted with p53 wild-type cells (KB, MCF-7, and HCT p53+/+), p53 mutant cells (A-431), and p53 null cells (H1299) (Fig. 3). In the experimental setup, these cells were overexpressed with the apoptotic SCO2 cDNA. The results were consistent in all cell lines and showed that SCO2 overexpression resulted in the downregulation of genes involved in apoptosis inhibition and upregulated genes involved in apoptosis induction (Fig. 3, lanes 13, 17, 19, 20, 23, and 24). Upon ectopic expression of SCO2 cDNA in cancer cells, where ROS was quenched through NAC treatment, the apoptotic gene expression level was altered to be consistently low (Fig. 3, lanes 1 to 4, 6, and 8). Treatment with tamoxifen, used as the positive control, resulted in upregulation of genes involved in apoptosis (Fig. 3, lanes 14 to 16, 18, 21, and 22). Untreated cancer cells served as controls (Fig. 3, lanes 5, 7, and 9 to 12). Furthermore, the expression of these 84 genes was observed in A-431, MCF-7, HCT p53+/+, and HCT p53−/− tumor xenografts (Fig. 4). The results showed that SCO2 ectopic expression resulted in the upregulation of genes involved in apoptosis and downregulation of genes involved in the inhibition of apoptosis (Fig. 4, lanes 1, 6, 7, and 9). Furthermore, ROS quenching via NAC resulted in reversal of this gene expression pattern (Fig. 4, lanes 2 and 12 to 14). Tamoxifen treatment was used as a positive control (Fig. 4, lanes 3 to 5 and 8), and untreated tumors served as a negative control (Fig. 4, lanes 10, 11, 15, and 16). The results suggest that SCO2 induces apoptosis and tumor regression in the xenograft model via upregulation of ROS and alters the expression of genes involved in apoptosis.
Fig 3.

SCO2 induces expression of apoptotic genes in cancer cells. A PCR gene array for genes involved in the regulation of cellular apoptosis was conducted by using SCO2 cDNA-transfected p53 wild-type cells (KB, MCF-7, and HCT p53+/+), p53 mutant cells (A-431), and p53 null cells (HCT p53−/− and H1299). Overexpression of SCO2 resulted in the downregulation of genes inhibiting apoptosis and the upregulation of proapoptotic genes (lanes 13, 17, 19, 20, 23, and 24). ROS quenching in SCO2-overexpressing cell resulted in inhibition of apoptotic genes and increases in the expression levels of antiapoptotic genes (lanes 1 to 4, 6, and 8). Untreated cells were used as controls (lanes 5, 7, and 9 to 12). Tamoxifen was used as a positive control, which led to upregulation of genes involved in apoptosis (lanes 14 to 16, 18, 21, and 22) (n = 10).
Fig 4.

SCO2 induces expression of apoptotic genes in tumor xenografts. A PCR gene array for genes involved in regulation of cellular apoptosis was conducted by using SCO2 cDNA-transfected A-431, MCF-7, HCT p53+/+, and HCT p53−/− tumor xenografts. Overexpression of SCO2 resulted in the downregulation of genes inhibiting apoptosis and the upregulation of proapoptotic genes (lanes 1, 6, 7, and 9). ROS quenching in SCO2-overexpressing cell resulted in inhibition of apoptotic gens and increases in the expression levels of antiapoptotic genes (lanes 2 and 12 to 14). Untreated cells were used as controls (lanes 10, 11, 15, and 16). Tamoxifen treatment was used as a positive control, which led to the upregulation of genes involved in apoptosis (lanes 3 to 5 and 8) (n = 5).
SCO2 inhibits the ASK-1–Trx complex and phosphorylates ASK-1 at the Thr845 residue.
We further studied the mechanism of SCO2-mediated apoptosis in cancer cells. ASK-1 is a MAP kinase kinase kinase (MAPKKK) known to activate SEK1-JNK and MAP2K3/MAP2K6-p38 signaling cascades (9). ASK-1 is a pivotal component in the mechanism of inflammation, stress, and ROS-induced apoptosis (26). SCO2 induces ROS, which is a prerequisite for the higher kinase activity of ASK-1. Thus, we sought to analyze the role of ASK-1 in SCO2-mediated apoptosis. The role of SCO2 and SCO2-induced ROS in regulating the cellular expression of ASK-1 protein was observed in HCT p53+/+ cells (Fig. 5A) along with HCT p53+/+ and HCT p53−/− tumor xenografts (Fig. 5B). Results showed that ectopic expression of SCO2 cDNA (1 μg) resulted in a significant upregulation of the ASK-1 protein level (Fig. 5A, lane 2). Interestingly, SCO2 cDNA expression in HCT cells, where ROS was quenched through NAC treatment, showed no increase in ASK-1 protein levels (Fig. 5A, lane 3). Tamoxifen, which is known to increase the ASK-1 protein level (27), served as a positive control (Fig. 5A, lane 4). To further demonstrate the role of SCO2 in ASK-1 upregulation, the SCO2 gene was silenced by using SCO2 siRNA, which resulted in a significant decrease in the ASK-1 expression level (Fig. 5A, lane 5). ASK-1 cDNA transfection was used as a positive control (Fig. 5A, lane 6). The observed effect of SCO2-mediated ASK-1 upregulation was also seen in HCT p53+/+ and HCT p53−/− tumor xenografts (Fig. 5B). Western blot analysis showed that the ectopic expression of SCO2 cDNA in tumor tissue resulted in a significant increase in the ASK-1 expression level (Fig. 5B, lane 2). ROS quenching via NAC reversed the SCO2-induced ASK-1 upregulation (Fig. 5B, lane 3). ASK-1 cDNA transfection in tumor tissue served as the positive control (Fig. 5B, lane 4). The data suggest that SCO2 induces upregulation of ASK-1 under both in vitro and in vivo conditions. Furthermore, the data suggested that SCO2-mediated ASK-1 upregulation is dependent upon SCO2-mediated increases in levels of cellular ROS. The results also hint that SCO2-mediated apoptosis might be due to activation of the ASK-1 pathway. Next, we analyzed whether the increase in the expression level of ASK-1 protein was linked to the increase in ASK-1 kinase activity (Fig. 5C). ASK-1 kinase activity was observed in HCT p53+/+ cells, using maltose-binding protein as a substrate (28). TNF-α, which is known to activate ASK-1 kinase activity, was used as a positive control (27). Untreated cells showed minimal ASK-1 kinase activity (Fig. 5C, bar 1). In order to establish a positive control, HCT cells were ectopically expressed with ASK-1 cDNA and simultaneously treated with TNF-α, which resulted in a significant increase in the ASK-1 kinase activity (Fig. 5C, bar 2); these data served as our positive control. TNF-α-induced ASK-1 kinase activity was reversed upon ROS quenching through NAC treatment (bar 3). These data served as a negative control. The role of SCO2 in regulating ASK-1 kinase activity was determined by exogenously transfecting SCO2 cDNA (1 μg) into the HCT cells. This resulted in a significant increase in ASK-1 kinase activity, suggesting that SCO2 activated the ASK-1 pathway by regulating both its cellular protein level and its kinase activity (Fig. 5C, bar 4). These data clearly showed that the SCO2 protein was important in the regulation of ASK-1 kinase activity in cancer cells.
Fig 5.

SCO2 induces dissociation of the ASK-1–Trx complex. (A) The role of SCO2 and SCO2-induced ROS in the regulation of expression of ASK-1 protein in HCT (p53+/+) cells was studied. Untreated cells were used as controls (lane 1). HCT cells ectopically expressed with SCO2 cDNA (1 μg) resulted in a significant upregulation of the ASK-1 protein (lane 2). ROS quenching by NAC treatment resulted in a significant decrease in the ASK-1 protein level (lane 3). Tamoxifen was used as a positive control (lane 4). Silencing of the SCO2 gene by SCO2 siRNA reversed the tamoxifen-induced increase in the ASK-1 protein expression level (lane 5). ASK-1 cDNA transfection was used as a positive control (lane 6) (n = 5). (B) The role of SCO2 and SCO2-induced ROS in expression of ASK-1 protein was observed by using HCT p53+/+ and HCT p53−/− tumor xenografts. Western blot analysis showed increased ASK-1 protein expression levels in SCO2-overexpressed tumors (lane 2). ROS quenching via NAC reversed SCO2-induced ASK-1 upregulation (lane 3). Transfected ASK-1 cDNA in tissue was used as a positive control (lane 4) (n = 4). (C) The role of SCO2 in the regulation of ASK-1 kinase activity was studied in HCT p53+/+ cells, using MBP as a substrate. TNF-α, which is a known activator of ASK-1 kinase activity, was used as a positive control. The level of ASK-1 kinase activity was very low in the untreated cells (bar 1), and treatment with TNF-α significantly increased the ASK-1 kinase activity (bar 2). ROS quenching through NAC reversed the TNF-α-induced increase in the ASK-1 kinase activity (bar 3). The exogenous addition of SCO2 cDNA (1 μg) in HCT cells resulted in a significant increase in ASK-1 kinase activity (bar 4), which was reversed upon ROS quenching (bar 5) (n = 5; an asterisk represents a significant difference between bars 4 and 5 [P < 0.029]; error bars represent standard deviations determined by analysis of variance). (D) The role of SCO2 in inducing the dissociation of the ASK-1–Trx complex was studied by using coimmunoprecipitation in HCT p53+/+ cells. ASK-1 protein was pulled down by using anti-ASK-1 Ab, and Western blots were developed by using anti-ASK-1 and anti-Trx Abs. In HCT p53+/+ cells, ASK-1 bound to its inhibitor Trx (lane 2). TNF-α treatment broke the interaction between ASK-1 and Trx (lane 3), and ROS quenching restored their interaction (lane 4). Exogenous addition of SCO2 cDNA (1 μg) resulted in dissociation of ASK-1 from the Trx protein (lane 5), and ROS quenching restored their interaction (lane 6) (n = 4). (E) Exogenous addition of SCO2 results in dissociation of the ASK-1–Trx complex in HCT p53+/+ and HCT p53−/− tumor xenografts. Exogenous addition of SCO2 cDNA resulted in ASK-1–Trx protein dissociation (lane 4). ROS quenching by NAC reversed the dissociation (lane 5). Input, IPP with no Ab. Untreated cells were used as controls (n = 4). (F) The role of SCO2 in inducing the phosphorylation of ASK-1 at the Thr845 residue was studied by using Western blotting of HCT p53+/+ cells. Control HCT cells showed minimal Thr845 phosphorylation (lane 1). Exogenous addition of SCO2 cDNA resulted in a significant increase in ASK-1 Thr845 residue phosphorylation (lane 2), and ROS quenching abolished this phosphorylation (lane 3). TNF-α also induced phosphorylation, which was reversed upon ROS quenching (lanes 5 and 6). Tamoxifen induced phosphorylation (lane 7), and SCO2 siRNA abolished it (lane 8) (n = 5). (G) Similarly, SCO2 was observed to induce the phosphorylation of ASK-1 at the Thr845 residue in HCT p53+/+ and HCT p53−/− tumor xenografts (lane 2), and ROS quenching reversed it (lane 3). Tamoxifen treatment was used as a positive control (n = 4).
ASK-1 exists in an inactive state in cancer cells, as a protein complex with its cellular inhibitor Trx (29). We analyzed whether SCO2 activates ASK-1 by regulating its protein-protein interaction with Trx. Previously, it was shown that Trx is highly susceptible to ROS-induced oxidation, and ROS rapidly induces the translocation of Trx into the nucleus (30). ROS generated by cytokines or stress may oxidize and consequently remove Trx from preexisting Trx–ASK-1 complexes, leading to the activation of ASK-1 and subsequent ASK-1-dependent apoptotic signaling cascades. The dissociation of ASK-1 from the ASK-1–Trx complex indicates the activity status of the ASK-1 protein. In order to study the role of SCO2 in the regulation of the ASK-1–Trx complex, coimmunoprecipitation was conducted in HCT p53+/+ cells (Fig. 5D) along with HCT p53+/+ and HCT p53−/− tumor xenografts (Fig. 5E). TNF-α, which is known to break the ASK-1–Trx complex, was used as a positive control, whereas tamoxifen treatment served as a positive control for the tumor xenografts. The treatment of HCT cells with TNF-α led to the dissociation of the ASK-1–Trx complex (Fig. 5D, lane 3). ROS quenching by NAC treatment reversed the effect of TNF-α on the ASK-1 and Trx interaction; these data served as our negative control (Fig. 5D, lane 4). To establish the role of SCO2 in regulation of the ASK-1 and Trx complex, SCO2 cDNA (1 μg) was exogenously added to HCT cells, which resulted in the dissociation of ASK-1 from its complex with the Trx protein (Fig. 5D, lane 5). ROS quenching in SCO2-transfected cells reversed SCO2-induced dissociation of the ASK-1–Trx complex, suggesting that this activity of SCO2 was via the activation of cellular ROS (Fig. 5D, lane 6). Similar results were observed for the HCT p53+/+ and HCT p53−/− tumor xenografts, where exogenous addition of SCO2 cDNA resulted in dissociation of the ASK-1 protein from its cellular inhibitor Trx (Fig. 5E, lane 4). ROS quenching again led to a reversal of this SCO2-induced effect (Fig. 5E, lane 5); tamoxifen treatment served as the positive control.
ASK-1 kinase activity is tightly regulated by events such as protein-protein interactions, oligomerization, and phosphorylation. It was previously shown that ASK-1 can be phosphorylated at several sites which either up- or downregulate ASK-1 kinase activity. The Thr845 residue of ASK-1 has been demonstrated to be an autophosphorylation site responsible for increasing its kinase activity (31). Since the phosphorylation of the Thr845 residue of ASK-1 has an important role in regulation of its kinase activity, we sought to determine the role of the SCO2 protein in the regulation of phosphorylation of ASK-1 at the Thr845 residue. Western blot analyses were conducted with HCT p53+/+ cells (Fig. 5F) along with HCT p53+/+ and HCT p53−/− tumor xenografts (Fig. 5G) and antibody against the Thr845 residue of ASK-1. The results showed minimal phosphorylation of the Thr845 residue in control HCT cells (Fig. 5F, lane 1). The exogenous addition of SCO2 cDNA led to a significant increase in ASK-1 Thr845 residue phosphorylation (Fig. 5F, lane 2), which was reversed upon ROS quenching (lane 3). The data suggest that the SCO2 protein regulates the phosphorylation of ASK-1 at the Thr845 residue via generation of cellular ROS. TNF-α and tamoxifen served as positive controls for these experiments (Fig. 5F, lanes 4 to 8). Similar effects of the ectopic expression of the SCO2 gene on the HCT p53+/+ and HCT p53−/− tumor xenografts were observed (Fig. 5G, lanes 2 and 3). These data show that SCO2 induces overall activation of ASK-1 MAPKKK by increasing its cellular level, increasing its kinase activity, inducing its dissociation from its inhibitor (Trx), and increasing the phosphorylation at the ASK-1 Thr845 residue.
SCO2-activated ASK-1 induces MAP2K4/7 and MAP2K3/6 kinases and the JNK and p38 apoptotic cascade.
Upon activation, MAPKKK (ASK-1) phosphorylates and activates MAPKKs like MAP2K4, MAP2K7, MAP2K3, and MAP2K6, which then phosphorylates and activates MAPKs. Two mammalian MAPKs, c-Jun N-terminal protein kinase (JNK) and p38 MAPK, are known to be activated by various environmental stresses and regulate diverse cellular functions, including cytokine production, differentiation, and apoptosis (32–39). Since SCO2 induced the activation of ASK-1, we sought to find if the SCO2-mediated apoptosis is via the MAPKKK kinase cascade. ASK-1 activates MAP2Ks by phosphorylating them; thus, we analyzed the expression of phosphorylated MAP2K4, MAP2K7, MAP2K3, and MAP2K6 and total expression of MAP2K4, MAP2K7, MAP2K3, and MAP2K6 in SCO2-overexpressing HCT p53+/+ cells and HCT p53+/+ tumor xenografts (Fig. 6A and B). We used an in vivo ELISA to quantify the altered levels of both total and phosphorylated forms of MAP2K3, MAP2K6, MAP2K4, MAP2K7, p38, and JNK. Untreated cells were used as controls (Fig. 6A), which showed low expression levels of MAP2Ks and the phosphorylated forms of these kinases (bar 1). The effect of the ectopic expression of SCO2 cDNA was observed in HCT p53+/+ cells, and the results showed significant increases in levels of both the total expression of the MAP2Ks and the expression of their phosphorylated forms (Fig. 6A, bar 2). The role of SCO2-induced ROS in the expression of these MAP2Ks was analyzed by quenching ROS in SCO2-transfected cells. The results showed significant decreases in the total expression level of these MAP2Ks and also the expression level of their phosphorylated forms (Fig. 6A, bar 3). Treatment with tamoxifen was used as a positive control, which showed a significant increase in the expression levels of both the total and phosphorylated forms of these kinases (Fig. 6A, bar 4). These data show that SCO2 not only induces the activity of MAPKKK but also ensures the progression of the ASK-1 kinase cascade pathway by helping to induce phosphorylation of its downstream MAP2Ks. The results were repeated with HCT p53+/+ tumor xenografts, where we found that the ectopic expression of SCO2 cDNA in these tumors led to significant increases in the total expression levels of MAP2K4, MAP2K7, MAP2K3, and MAP2K6 and the expression levels of the phosphorylated forms of these MAP2Ks (Fig. 6B, bar 2). The quenching of ROS via NAC treatment in these tumors led to significant decreases in the SCO2-induced expression levels of MAP2Ks and the phospho-MAP2Ks (Fig. 6B, bar 3); tamoxifen served as the positive control.
Fig 6.

(A) The role of SCO2 in inducing the activation of ASK-1 downstream kinases was studied in HCT p53+/+ cells. The ratio of the phosphorylated and total cellular levels of MAP2Ks, JNK, and p38 in SCO2-overexpressing HCT p53+/+ cells was obtained by an in vivo ELISA. Untreated control cells showed low-level expression of total MAP2Ks and phosphorylated MAP2Ks (bar 1). Ectopic expression of SCO2 cDNA resulted in significant increases in the expression levels of total MAP2Ks and their phosphorylated forms (bar 2). ROS quenching by NAC resulted in significant decreases in the expression levels of both forms in SCO2-transfected cells (bar 3). Tamoxifen treatment was used as a positive control (bar 4) (error bars represent standard deviations determined by analysis of variance; n = 7). (B) The ratio of the phosphorylated and total cellular levels of MAP2Ks, JNK, and p38 in SCO2-transfected HCT p53+/+ tumor xenografts was determined by an in vivo ELISA. Untreated control cells showed low-level expression of total MAP2Ks and phosphorylated MAP2Ks (bar 1). Ectopic expression of SCO2 cDNA resulted in significant increases in the expression levels of total MAP2Ks and their phosphorylated forms (bar 2). ROS quenching via NAC resulted in significant decreases in the expression levels of both forms in SCO2-transfected cells (bar 3). Tamoxifen treatment was used as a positive control (bar 4) (error bars represent standard deviations determined by analysis of variance; n = 5). O.D, optical density.
MAP2Ks are known to activate MAP kinases by inducing their phosphorylation. The MAP kinases finally evoke apoptotic signals, pushing the cell toward the apoptotic mode of cell death. Two known mammalian MAPKs, JNK and p38 MAPK, are activated by the ASK-1 pathway and are required for ASK-1-mediated apoptosis. In order to study the apoptotic cascade activated by SCO2, we analyzed the protein levels and phosphorylation of JNK and p38 MAPK in HCT p53+/+ cells and in HCT p53+/+ tumor xenografts. The ectopic expression of SCO2 cDNA in HCT p53+/+ cells led to significant increases in the expression levels of p38, JNK, and the phosphorylated forms of JNK and p38 (Fig. 6A, bar 2). ROS quenching led to a reversal of the SCO2-induced increases in the expression levels of p38, JNK, phospho-p38, and phospho-JNK (Fig. 6A, bar 3). Tamoxifen (100 nM)-treated HCT p53+/+ cells were used as a positive control, which showed increases in the JNK and p38 protein levels (Fig. 6A, lane 4). Similar results were observed for HCT p53+/+ tumor xenografts, where ectopic expression of SCO2 resulted in a significant increase in the expression levels of p38, JNK, phospho-p38, and phospho-JNK. ROS quenching resulted in a reversal of this increase (Fig. 6B, bars 2 and 3). The results of the in vivo ELISA were repeated by Western blotting for both HCT p53+/+ cells (see Fig. S5a in the supplemental material) and HCT p53+/+ xenografts (see Fig. S5b). The results showed the SCO2-induced changes in the levels of the total and phosphorylated forms of the above-mentioned kinases. Both systems showed similar results of SCO2-induced activation of the MAP2Ks JNK and p38. Furthermore, the effect of SCO2 protein on the percent change in the phosphorylated MAP2Ks/total MAP2Ks and the phosphorylated MAPKs/total MAPKs is represented in Fig. 7A.
Fig 7.

(A) The percent change in the ratios of phosphorylated (P) to total (T) MAP2K3 (M2K3), M2K6, M2K4, M2K7, JNK, and p38 was obtained for HCT p53+/+ cells (error bars represent standard deviations determined by analysis of variance; n = 7). (B) The change in the ratio of the above-mentioned proteins in HCT p53+/+ tumors was determined (error bars represent standard deviations determined by analysis of variance; n = 5).
SCO2 induces ASK-1-dependent apoptosis in hypoxic tumors.
Hypoxia is a unique condition where cells may exist in a quiescent state (40). Some cells, under hypoxic conditions, are resistant to apoptosis (41) and undergo high rates of aerobic and anaerobic glycolysis (42). The role of SCO2 and SCO2-induced ASK-1 in generating the apoptotic signals in hypoxic cancer cells and regression of the hypoxic tumors was realized. In order to understand the role of ASK-1 in SCO2-induced effects on apoptosis in hypoxic cancer cells, the ASK-1 gene was knocked down in HCT p53+/+ and HCT p53−/− cell lines, and the cell lines were confirmed for the successful knockdown of the gene by Western blot analysis using anti-p53 and anti-ASK-1 Abs (Fig. 8A); GAPDH was used as a loading control. We cultured HCT p53+/+, HCT p53−/−, HCT p53+/+ ASK-1−/−, and HCT p53−/− ASK-1−/− cell lines in 1.8% O2 to provide hypoxic exposure, as described previously (18, 19, 21). The cultured cells, which were ectopically expressed with SCO2 cDNA (Fig. 8B), showed a significant increase in the level of SCO2-mediated apoptosis in both normoxic and hypoxic cancer cells (lanes 3 and 6). Tamoxifen was not efficient in inducing apoptosis in hypoxic cancer cells (Fig. 8B, bars 2 and 5) (18, 21). Instead, SCO2 anticancer gene therapy was able to induce a higher fraction of apoptosis than tamoxifen treatment. The most interesting observation was that SCO2-induced apoptosis was absent in the ASK-1 knockdown normoxic and hypoxic cancer cells. These data established that SCO2-induced ASK-1 signaling is indispensable for SCO2-induced apoptosis. Next, we established the role of ASK-1 in SCO2-induced apoptosis in the in vivo system. HCT p53+/+, HCT p53−/−, HCT p53+/+ ASK-1−/−, and HCT p53−/− ASK-1−/− tumor xenografts were induced in the hind leg of nude mice and allowed to grow to a volume of around 3 cm3 (Fig. 8C). The tumors were ectopically expressed with SCO2 cDNA by using an in vivo transfection kit, as described previously (18, 19). The results showed that SCO2 induced tumor regression only in ASK-1+/+ tumors (Fig. 8C, graphs 1 and 2), and SCO2-induced tumor regression was missing in ASK-1−/− tumor xenografts (Fig. 8C, graphs 3 and 4). Furthermore, we measured the SCO2-induced apoptotic fraction in ASK-1+/+ and ASK-1−/− tumors upon SCO2 gene therapy. The results showed that SCO2 induced significant apoptosis in the ASK-1+/+ tumors (Fig. 8D, bars 1 and 2). However, SCO2-induced apoptosis was absent in the ASK-1−/− tumors (Fig. 8D, bars 3 and 4). The role of ASK-1 in the profile of SCO2-induced expression of the apoptotic genes was also observed in SCO2-treated HCT p53+/+, HCT p53−/−, HCT p53+/+ ASK-1−/−, and HCT p53−/− ASK-1−/− tumor xenografts (Fig. 9). The apoptotic gene PCR array showed that SCO2 induced apoptosis only in ASK-1+/+ tumors (Fig. 9, lanes 1 and 2). ASK-1 knockdown led to a reversal in the expression of the apoptotic gene in the tumors that were ectopically expressed with SCO2 cDNA. These results suggested that ASK-1 is indispensable for SCO2-mediated apoptosis and tumor regression.
Fig 8.

SCO2-mediated apoptosis is ASK-1 dependent. (A) Two ASK-1 knockout cell lines were prepared from HCT p53+/+ and HCT p53−/− cells. The cell lines were validated for the presence of ASK-1 and p53 proteins by Western blotting. The results show that ASK-1 protein was missing from the HCT p53+/+ ASK-1−/− and HCT p53−/− ASK-1−/− cell lines (n = 4). (B) SCO2-induced apoptosis was observed in normoxic and hypoxic HCT p53+/+ ASK-1−/− and HCT p53−/− ASK-1−/− cell lines. Both tamoxifen and SCO2 induced significant apoptosis in normoxic cancer cells. However, SCO2-induced apoptosis was absent in ASK-1−/− cells (bar 3). Tamoxifen was not able to induce a high apoptotic fraction in the hypoxic cancer cells (bar 5). SCO2 induced high-level apoptosis in hypoxic cancer cells with the ASK-1+/+ genotype. SCO2-induced apoptosis was absent in ASK-1−/− cells (bar 6) (n = 7; error bars represent standard deviations determined by analysis of variance; an asterisk represents a significant difference in apoptosis between ASK-1+/+ and ASK-1−/− cells [P < 0.02]). (C) The role of ASK-1 in SCO2-induced tumors was studied by using tumor regression in SCO2-transfected HCT p53+/+ ASK-1−/− and HCT p53−/− ASK-1−/− tumor xenografts. Results show that SCO2 induced apoptosis in ASK-1+/+ tumors (graphs 1 and 2) but did not induce tumor regression in ASK-1−/− knockdown tumor xenografts (graphs 3 and 4). Untreated tumors and tumors where ROS was quenched by NAC served as controls (error bars represent standard deviations determined by analysis of variance; n = 5). (D) The role of ASK-1 in SCO2-mediated apoptosis in tumor xenografts was also observed in ASK-1 knockout tumor xenografts by using a FLIVO in vivo apoptosis kit. Results show significant decreases in the apoptotic fraction in ASK-1−/− tumor xenografts (bars 3 and 4) (a red asterisk represents a significant difference in SCO2-induced apoptosis between the HCT p53+/+ and HCT p53+/+ ASK-1−/− tumors [P < 0.04]; a blue asterisk represents a significant difference in SCO2-induced apoptosis between the HCT p53−/− and HCT p53−/− ASK-1−/− tumors [P < 0.043]; error bars represent standard deviations determined by analysis of variance; n = 7).
Fig 9.

SCO2-induced expression of apoptotic genes is absent in ASK-1 knockdown tumors. A PCR gene array for genes involved in the regulation of cellular apoptosis was conducted by using SCO2 cDNA-transfected HCT p53+/+, HCT p53−/−, HCT p53+/+ ASK-1−/−, and HCT p53−/− ASK-1−/− tumor xenografts. SCO2 transfection in the HCT p53+/+ and HCT p53−/− tumors resulted in significant increases in apoptotic gene expression levels (lanes 1 and 2). The effect of SCO2 on the expression of apoptotic genes was abolished in HCT p53+/+ ASK-1−/− and HCT p53−/− ASK-1−/− tumor xenografts (lanes 4 and 6). NAC- and SCO2-treated tumors (lanes 8 and 10 to 12) and untreated tumors (lanes 3, 5, 7, and 9) were used as controls (n = 5).
DISCUSSION
The direct regulation of cellular metabolism by p53 has important implications for an understanding of tumor growth and development. The tumor suppressor activity of p53 lies in its ability to induce cell cycle arrest and apoptosis in oncogenic cells. It is not known how the role of p53 as a regulator of cellular glycolysis (2) and OXPHOS (3) is synchronized with its tumor suppressor function. It was proposed previously that, through regulation of OXPHOS via transcriptional control of SCO2, p53 antagonizes tumorigenesis by decreasing cellular dependence on oxygen, potentially permitting growth in more hypoxic environments (25). However, no mechanism in support of this hypothesis is available (2). In this study, we have correlated two diverse functions of p53, the regulation of metabolism and tumor suppressor activity. We have shown that p53 upregulates SCO2, which may provide an alternative pathway of p53-dependent apoptosis. The p53-mediated upregulation of SCO2 is observed only in cells that are under conditions of high cellular stress. Chromatin immunoprecipitation and luciferase assays confirm that p53 binds to the SCO2 promoter in cells that are exposed to high doses of UV radiation and tamoxifen. It was thus concluded that p53-mediated apoptosis is executed via upregulation of SCO2, OXPHOS, ROS production, and activation of the ASK-1 pathway. Our conclusion is further substantiated by the fact that p53 was previously shown to activate the SCO2 promoter, thus increasing both mRNA and protein levels (3).
SCO2 plays a major role in copper homeostasis. Most SCO2 patients carry an E140K missense mutation on one allele adjacent to the conserved CXXXC motif, which regulates the efficiency of SCO2 to bind to copper and function as a redox protein (43, 44). Our study shows that SCO2 induces cellular apoptosis by activating the ASK-1 pathway of JNK and p38 activation. Also, we have shown that the exogenous addition of SCO2 cDNA to cancer cells shows a significant increase in the kinase activity of the ASK-1 protein. We propose that p53-mediated SCO2 overexpression might be involved in the generation of cellular ROS. We observed that SCO2 could induce up to a 6-fold increase in the level of cellular ROS, and SCO2-mediated ROS generation could be due to alterations in mitochondrial signaling or due to an imbalance of copper homeostasis. The transcriptional regulation of SCO2 by p53 might also be responsible for the p53-mediated regulation of mitochondrial signals and cellular thiol-disulfide oxidoreductase reactions that are required for oxidation of the copper-binding cysteine amino acids in the mitochondrial proteins (44).
SCO2 induces the dissociation of the interaction of ASK-1 from its physiological inhibitor Trx and induces phosphorylation of ASK-1 at its Thr845 residue, leading to its activation. In cancer cells, overexpression of SCO2 is linked to the upregulation and phosphorylation of MAP2K4/7, MAP2K3/6, JNK, and p38 through the activation of ASK-1. ASK-1 was shown previously to activate p38 and JNK via activating the MAP2Ks MAP2K4/MAP2K7 and MAP2K3/MAP2K6 (9). ASK-1 is activated by a variety of stresses, including calcium influx, endoplasmic reticulum (ER) stress, lipopolysaccharide (LPS), tumor necrosis factor, and, especially, ROS (10), resulting in the activation of ASK-1 through Thr845 phosphorylation (45, 46). ASK-1 plays pivotal roles in a wide variety of cellular responses, such as cell differentiation, apoptosis, and the immune response, with special focus on oxidative stress-induced apoptosis (47). Activation of ASK-1 can selectively activate JNK and p38 MAP kinases, leading to apoptosis. It was also shown that ASK-1 activity can be regulated by a number of ASK1-interacting proteins. Among them, thioredoxin (Trx) and 14-3-3 can directly bind to ASK-1, leading to inhibition of ASK-1 activity (45). We have shown that SCO2 inhibits the protein-protein interaction between ASK-1 and Trx in a ROS-dependent manner. Exogenous addition of SCO2 protein abolishes ASK-1–Trx interactions in cancer cells, thus rendering ASK-1 free and active to participate in cellular signaling. SCO2-mediated phosphorylation of the Thr845 residue of ASK-1 was thus a crucial event in its activation.
Ectopic addition of SCO2 cDNA to cancer cells and tumor xenografts induced ASK-1-mediated cellular apoptosis. SCO2 addition led to cleavage of PARP, and annexin V staining showed that SCO2 cDNA induced significant apoptosis in cancer cells and in chemoresistant hypoxic cancer cells, respectively. SCO2 induced the ASK-1 cascade, as p38 and JNK downstream of ASK-1 were both upregulated and phosphorylated. These data established SCO2 as a key protein involved in the p53 apoptotic response to cellular stress. The mechanism of SCO2-mediated cell death in tumor xenografts was similar to the one observed for cancer cells. In the human colon cancer cell lines DLD1 and SW480, overexpression of SCO2 protein increased OXPHOS even in the presence of p53 mutations, suggesting that p53-mediated regulation of OXPHOS is via SCO2 (3). In the HCT116 human colon cancer cell line, deficiency in p53 caused low-level expression of SCO2, resulting in lower OXPHOS levels, which were balanced by the increase in glycolysis (20). This suggests that the downregulation of p53-dependent regulation of SCO2 impairs the mitochondrial respiratory chain, causing a shift of ATP production from OXPHOS to glycolysis. Hypoxic cancer cells show chemoresistance and render p53 in a transcriptionally inactive form, resulting in the lack of apoptosis in hypoxic tumors (21, 23, 40). Interestingly, exogenous addition of SCO2 in hypoxic tumors led to SCO2-mediated apoptosis and tumor regression, suggesting that SCO2 is active under conditions of hypoxia but is not expressed in these cells, possibly due to a lack of transcriptional activity of p53 under conditions of hypoxia. Based on the results of the present study, we proposed a model to explain the mechanism of SCO2-mediated apoptosis in cancer cells (Fig. 10).
Fig 10.
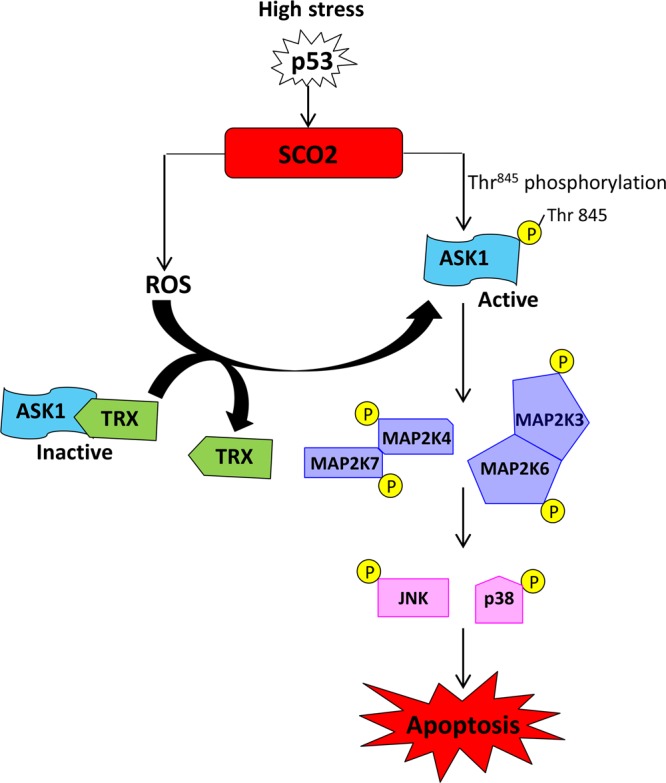
Model depicting the mechanism of SCO2-induced apoptosis in cancer cells and tumor xenografts via the activation of the ROS and ASK-1 signaling cascade.
In conclusion, we have identified a novel apoptotic function of SCO2, which activates p53-mediated apoptosis via an alternate pathway. This novel role of SCO2 may find relevance in SCO2-mediated gene therapy in tumor regression protocols. We propose that SCO2 might be a novel tumor suppressor that could function in a ROS–ASK-1 signal pathway, and this novel role of SCO2 may find relevance in SCO2-mediated gene therapy in tumor regression protocols.
Supplementary Material
ACKNOWLEDGMENTS
We thank the University Grant Commission, India, Capacity Build Up Fund (to U.P.), Indian Council of Medical Research (grant to A.A.M.), and NIH (RO1 grant NIH EB004031 to P.K.).
We thank Muzzammill Sayyid and Shally Awasthi (faculty in charge, Research Cell, CSMMU) for their contributions.
We declare no conflict of interest.
E.M. participated in the conceptualization of the study, conducted the majority of the experiments, analyzed the data, interpreted the results, and prepared the manuscript. R.G. helped in conceptualization, conducted some key experiments, helped to analyze the data, interpreted the results, and equally participated in the preparation of the manuscript. P.K. helped in the fine-tuning of the experimental design, interpretation, and preparation of the manuscript. M.B. provided technical editing of the manuscript. A.A.M. provided the design of the study. U.P. provided the design of the study and manuscript.
Footnotes
Published ahead of print 14 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.06798-11.
REFERENCES
- 1. Vogelstein B, Lane D, Levine AJ. 2000. Surfing the p53 network. Nature 408:307–310 [DOI] [PubMed] [Google Scholar]
- 2. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. 2006. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126:107–120 [DOI] [PubMed] [Google Scholar]
- 3. Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. 2006. p53 regulates mitochondrial respiration. Science 312:1650–1653 [DOI] [PubMed] [Google Scholar]
- 4. Stiburek L, Vesela K, Hansikova H, Hulkova H, Zeman J. 2009. Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am. J. Physiol. Cell Physiol. 296:C1218–C1226 doi:10.1152/ajpcell.00564.2008 [DOI] [PubMed] [Google Scholar]
- 5. Yang H, Brosel S, Acin-Perez R, Slavkovich V, Nishino I, Khan R, Goldberg IJ, Graziano J, Manfredi G, Schon EA. 2010. Analysis of mouse models of cytochrome c oxidase deficiency owing to mutations in Sco2. Hum. Mol. Genet. 19:170–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kokoszka JE, Coskun P, Esposito LA, Wallace DC. 2001. Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc. Natl. Acad. Sci. U. S. A. 98:2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G. 2004. AIF deficiency compromises oxidative phosphorylation. EMBO J. 23:4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Takeda K, Matsuzawa A, Nishitoh H, Ichijo H. 2003. Roles of MAPKKK ASK1 in stress-induced cell death. Cell Struct. Funct. 28:23–29 [DOI] [PubMed] [Google Scholar]
- 9. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. 1997. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 275:90–94 [DOI] [PubMed] [Google Scholar]
- 10. Matsuzawa A, Saegusa K, Noguchi T, Sadamitsu C, Nishitoh H, Nagai S, Koyasu S, Matsumoto K, Takeda K, Ichijo H. 2005. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat. Immunol. 6:587–592 [DOI] [PubMed] [Google Scholar]
- 11. Takeda K, Matsuzawa A, Nishitoh H, Tobiume K, Kishida S, Ninomiya-Tsuji J, Matsumoto K, Ichijo H. 2004. Involvement of ASK1 in Ca2+-induced p38 MAP kinase activation. EMBO Rep. 5:161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hatai T, Matsuzawa A, Inoshita S, Mochida Y, Kuroda T, Sakamaki K, Kuida K, Yonehara S, Ichijo H, Takeda K. 2000. Execution of apoptosis signal-regulating kinase 1 (ASK1)-induced apoptosis by the mitochondria-dependent caspase activation. J. Biol. Chem. 275:26576–26581 [DOI] [PubMed] [Google Scholar]
- 13. Kanamoto T, Mota M, Takeda K, Rubin LL, Miyazono K, Ichijo H, Bazenet CE. 2000. Role of apoptosis signal-regulating kinase in regulation of the c-Jun N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol. Cell. Biol. 20:196–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nakamura H, Nakamura K, Yodoi J. 1997. Redox regulation of cellular activation. Annu. Rev. Immunol. 15:351–369 [DOI] [PubMed] [Google Scholar]
- 15. Nonn L, Williams RR, Erickson RP, Powis G. 2003. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol. Cell. Biol. 23:916–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, Goldschmidt-Clermont PJ. 1997. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275:1649–1652 [DOI] [PubMed] [Google Scholar]
- 17. Jacobson MD. 1996. Reactive oxygen species and programmed cell death. Trends Biochem. Sci. 21:83–86 [PubMed] [Google Scholar]
- 18. Gogna R, Madan E, Kuppusamy P, Pati U. 2012. Chaperoning of mutant p53 protein by wild-type p53 protein causes hypoxic tumor regression. J. Biol. Chem. 287:2907–2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Madan E, Gogna R, Kuppusamy P, Bhatt M, Pati U, Mahdi AA. 2012. TIGAR induces p53-mediated cell-cycle arrest by regulation of RB-E2F1 complex. Br. J. Cancer 107:516–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. 2007. A pivotal role for p53: balancing aerobic respiration and glycolysis. J. Bioenerg. Biomembr. 39:243–246 [DOI] [PubMed] [Google Scholar]
- 21. Gogna R, Madan E, Kuppusamy P, Pati U. 2012. Re-oxygenation causes hypoxic tumor regression through restoration of p53 wild-type conformation and post-translational modifications. Cell Death Dis. 3:e286 doi:10.1038/cddis.2012.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gogna R, Madan E, Keppler B, Pati U. 2012. Gallium compound GaQ(3)-induced Ca(2+) signalling triggers p53-dependent and -independent apoptosis in cancer cells. Br. J. Pharmacol. 166:617–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gogna R, Madan E, Kuppusamy P, Pati U. 2012. Reactive oxygen species-mediated p53 core-domain modifications determine apoptotic or necrotic death in cancer cells. Antioxid. Redox Signal. 16:400–412 [DOI] [PubMed] [Google Scholar]
- 24. Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. 2011. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget 2:948–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Madan E, Gogna R, Pati U. 2012. p53 Ser15 phosphorylation disrupts the p53-RPA70 complex and induces RPA70-mediated DNA repair in hypoxia. Biochem. J. 443:811–820 [DOI] [PubMed] [Google Scholar]
- 26. Tobiume K, Inage T, Takeda K, Enomoto S, Miyazono K, Ichijo H. 1997. Molecular cloning and characterization of the mouse apoptosis signal-regulating kinase 1. Biochem. Biophys. Res. Commun. 239:905–910 [DOI] [PubMed] [Google Scholar]
- 27. Won M, Park KA, Byun HS, Sohn KC, Kim YR, Jeon J, Hong JH, Park J, Seok JH, Kim JM, Yoon WH, Jang IS, Shen HM, Liu ZG, Hur GM. 2010. Novel anti-apoptotic mechanism of A20 through targeting ASK1 to suppress TNF-induced JNK activation. Cell Death Differ. 17:1830–1841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. 1998. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 17:2596–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hsieh CC, Papaconstantinou J. 2006. Thioredoxin-ASK1 complex levels regulate ROS-mediated p38 MAPK pathway activity in livers of aged and long-lived Snell dwarf mice. FASEB J. 20:259–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanaka T, Nishiyama Y, Okada K, Hirota K, Matsui M, Yodoi J, Hiai H, Toyokuni S. 1997. Induction and nuclear translocation of thioredoxin by oxidative damage in the mouse kidney: independence of tubular necrosis and sulfhydryl depletion. Lab. Invest. 77:145–155 [PubMed] [Google Scholar]
- 31. Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. 2001. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2:222–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Davis RJ. 2000. Signal transduction by the JNK group of MAP kinases. Cell 103:239–252 [DOI] [PubMed] [Google Scholar]
- 33. Ichijo H. 1999. From receptors to stress-activated MAP kinases. Oncogene 18:6087–6093 [DOI] [PubMed] [Google Scholar]
- 34. Kyriakis JM, Avruch J. 1996. Protein kinase cascades activated by stress and inflammatory cytokines. Bioessays 18:567–577 [DOI] [PubMed] [Google Scholar]
- 35. Matsuzawa A, Ichijo H. 2001. Molecular mechanisms of the decision between life and death: regulation of apoptosis by apoptosis signal-regulating kinase 1. J. Biochem. 130:1–8 [DOI] [PubMed] [Google Scholar]
- 36. Nishida E, Gotoh Y. 1993. The MAP kinase cascade is essential for diverse signal transduction pathways. Trends Biochem. Sci. 18:128–131 [DOI] [PubMed] [Google Scholar]
- 37. Ono K, Han J. 2000. The p38 signal transduction pathway: activation and function. Cell. Signal. 12:1–13 [DOI] [PubMed] [Google Scholar]
- 38. Widmann C, Gibson S, Jarpe MB, Johnson GL. 1999. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79:143–180 [DOI] [PubMed] [Google Scholar]
- 39. Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. 1995. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331 [DOI] [PubMed] [Google Scholar]
- 40. Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. 2001. Hypoxia inhibits G1/S transition through regulation of p27 expression. J. Biol. Chem. 276:7919–7926 [DOI] [PubMed] [Google Scholar]
- 41. Kunz M, Ibrahim SM. 2003. Molecular responses to hypoxia in tumor cells. Mol. Cancer 2:23 doi:10.1186/1476-4598-2-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bristow RG, Hill RP. 2008. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 8:180–192 [DOI] [PubMed] [Google Scholar]
- 43. Hervouet E, Cizkova A, Demont J, Vojtiskova A, Pecina P, Franssen-van Hal NL, Keijer J, Simonnet H, Ivanek R, Kmoch S, Godinot C, Houstek J. 2008. HIF and reactive oxygen species regulate oxidative phosphorylation in cancer. Carcinogenesis 29:1528–1537 [DOI] [PubMed] [Google Scholar]
- 44. Leary SC, Sasarman F, Nishimura T, Shoubridge EA. 2009. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 18:2230–2240 [DOI] [PubMed] [Google Scholar]
- 45. Saito J, Toriumi S, Awano K, Ichijo H, Sasaki K, Kobayashi T, Tamura S. 2007. Regulation of apoptosis signal-regulating kinase 1 by protein phosphatase 2Cepsilon. Biochem. J. 405:591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tobiume K, Saitoh M, Ichijo H. 2002. Activation of apoptosis signal-regulating kinase 1 by the stress-induced activating phosphorylation of pre-formed oligomer. J. Cell. Physiol. 191:95–104 [DOI] [PubMed] [Google Scholar]
- 47. Matsuzawa A, Ichijo H. 2008. Redox control of cell fate by MAP kinase: physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 1780:1325–1336 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.