

Abstract
Regulation of the activity of N-methyl-d-aspartate receptors (NMDARs) at glutamatergic synapses is essential for certain forms of synaptic plasticity underlying learning and memory and is also associated with neurotoxicity and neurodegenerative diseases. In this report, we investigate the role of Src-like adaptor protein (Slap) in NMDA receptor signaling. We present data showing that in dissociated neuronal cultures, activation of ephrin (Eph) receptors by chimeric preclustered eph-Fc ligands leads to recruitment of Slap and NMDA receptors at the sites of Eph receptor activation. Interestingly, our data suggest that prolonged activation of EphA receptors is as efficient in recruiting Slap and NMDA receptors as prolonged activation of EphB receptors. Using established heterologous systems, we examined whether Slap is an integral part of NMDA receptor signaling. Our results showed that Slap does not alter baseline activity of NMDA receptors and does not affect Src-dependent potentiation of NMDA receptor currents in Xenopus oocytes. We also demonstrate that Slap reduces excitotoxic cell death triggered by activation of NMDARs in HEK293 cells. Finally, we present evidence showing reduced levels of NMDA receptors in the presence of Slap occurring in an activity-dependent manner, suggesting that Slap is part of a mechanism that homeostatically modulates the levels of NMDA receptors.
INTRODUCTION
In an effort to identify genes involved in cortical development, we have previously cloned slap, which encodes a Src-like adaptor protein (Slap) (1). slap is expressed strongly in the forebrain, but its function there remains unknown. slap encodes a 34-kDa protein containing SH2 and SH3 domains followed by a unique 104-amino-acid COOH terminal. Similar to results seen with members of the Src family kinases (SFKs), myristoylation at the NH2 terminus targets Slap to cellular membranes (2). Slap has been studied in the immune system, where it inhibits T-cell receptor (TCR) signaling (3) and is involved in the internalization and degradation of the TCRζ subunit (4). Furthermore, Slap abrogates the mitogenic response to platelet-derived growth factors (PDGFs) in NIH 3T3 fibroblasts, antagonizing the mitotic activity of Src kinase (5).
Slap was initially identified in a yeast two-hybrid screen using the cytoplasmic tail of EphrinA2 as bait (6). Ephrin (Eph) receptors constitute the largest known family of receptor tyrosine kinases and, together with their membrane-bound ligands (eph ligands), have been implicated in a variety of patterning events during the development of the central nervous system (7–21). Ephs and their ligands act as contact-dependent adhesive molecules and are implicated in the development and function of synapses. They are divided into two classes (EphA and EphB), based on sequence homologies and binding specificity (22). EphBs have established roles in the formation of synapses, transforming dendritic filopodia to spines and clustering and phosphorylating N-methyl-d-aspartate receptors (NMDARs) (7, 9, 23), and are implicated in synaptic plasticity, modulating NMDAR function (16). Less is known about the role of EphAs, although evidence suggests that they contribute to the induction of synaptogenesis (24), spine morphology, maturation, and collapse (25, 26). EphA signaling alters the stability of synaptic boutons, modulating synaptic plasticity and regulating adhesion to the extracellular matrix (27) and the reorganization of the cytoskeleton (28). Moreover, acute administration of an EphA antagonist in rat hippocampal slices impairs long-term potentiation (LTP), whereas administration of an agonist leads to LTP-like potentiation (29). Finally, EphA4 induces the degradation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors upon chronic elevation of synaptic activity (30).
Here we report that activation of Ephs mediates the recruitment of Slap at neuronal synapses. We show that activation of EphAs is more efficient in recruiting Slap than activation of EphB. Additionally, we show that NMDARs can be also recruited alongside Slap in an EphA-dependent manner. We provide further evidence that Slap offers protection from NMDAR-induced excitotoxicity and modulates NMDAR signaling, by regulating its degradation.
MATERIALS AND METHODS
Synaptosomal preparation and Western blotting.
The synaptosomal preparation was performed according to a previous published method (31). Briefly, 10 brains of adult Sprague-Dawley rats were used and their cerebral cortex was removed. The brains were homogenized in 5 mM HEPES (pH 7.4)–0.32 mM sucrose and subjected to centrifugation (1,500 × g). The pellet was resuspended in 0.32 M sucrose–1 mM NaHCO3 and layered over a sucrose gradient (1.2 M, 1 M, and 0.85 M sucrose) and centrifuged at 70,000 × g. The obtained fractions were removed and recovered by centrifugation. The pellets for each fraction were resuspended in 50 mM HEPES (pH 7.4)–2 mM EDTA. Western blot analyses were performed according to standard procedures. The primary antibodies (Abs) used were rabbit anti-Slap and mouse antisynaptophysin (anti-Syp) (Sigma, United Kingdom; catalog no. S5768). Horseradish peroxidase (HRP)-conjugated secondary anti-rabbit and anti-mouse antibodies were from Jackson ImmunoResearch Laboratories.
Primary hippocampal neuronal cultures.
Primary hippocampal neuronal cultures were prepared from embryonic day 18 (E18) Sprague-Dawley rat brains. Cells (750/mm2; i.e., 100,000/coverslip) were plated on poly-d-lysine (Sigma) (0.1 mg/ml in borate buffer [pH 8.5])-coated glass coverslips. The plating medium was Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS), penicillin-streptomycin (P/S), and 0.5 mM l-glutamine (all from Invitrogen). On the next day, the medium was changed to Neurobasal medium supplemented with B27, penicillin-streptomycin, and 0.5 mM l-glutamine (all from Invitrogen) or with NS21, a homemade supplement prepared as described previously (32). Cultures were routinely used at 14 days in vitro (14DIV).
Plasmids and lentiviral vectors.
pSP64T-derived plasmids containing rat GluN1-1a, GluN2A, and GluN2B subunits were used as oocyte expression vectors and have been described previously (33, 34). An IRAV Mammalian Gene Collection (MGC) mouse cDNA clone containing full-length slap was purchased from RZPD. Full-length Slap cDNA was subcloned into a pSP6T plasmid for cRNA synthesis. Slap was amplified using primers (Invitrogen) TTTCTAGACCACCATGCTCTGCAGGCTTCGA for the 5′ end and AAGTCGACTTAATCTTCAAAGTACTG for the 3′ end and cloned in the pGEM-T Easy vector (Promega). Following confirmation of sequences, Slap was then cloned under the control of the cytomegalovirus (CMV) promoter in the self-inactivating lentiviral plasmid pRRL_scC_IRESeGFP_W using XbaI and SalI restriction enzymes (Promega) (bold and italics in primer sequences) to generate pRRL_scC_Slap_IRESeGFP_W. No untranslated region was inserted between Slap and the internal ribosome entry site (IRES) of the vector (PubMed accession number of vector, pRRLsc_Cslap_eIeGFP_W KC262216). Third-generation, integration-deficient lentiviral vectors (35) were produced as previously described (36). Briefly, transient cotransfection of four plasmids into HEK 293T cells was used. In addition to the pRRL-type transfer plasmid, we used packaging plasmid pMDLg/pRREintD64V (integration deficient through a D64V point mutation), rev plasmid pRSV-rev, and a standard vesicular stomatitis virus G glycoprotein (VSV-G; plasmid pMD2. G) for pseudotyping. Culture supernatant was harvested 48 and 72 h posttransfection, and viral particles were then concentrated by ultracentrifugation, resuspended in DMEM, aliquoted, and frozen. Lentiviral vectors were titrated by quantitative PCR (qPCR) as previously described (36).
Clustering experiments and NMDAR activation.
Recombinant mouse ephA2-Fc, ephB1-Fc, and ephB2-Fc chimeras were purchased from R&D systems (catalog no. 603-A2-200, 437-EB, and 496-EB-200, respectively). A control human IgG, Fc fragment-purified protein, and the anti-Fc (fluorescein isothiocyanate [FITC]-conjugated) antibodies were from Chemicon (catalog no. AG714 and AP113F). Ligands and the control fragment were preclustered with the antibody in full Neurobasal medium for 1 h at room temperature (RT), and the medium in each treatment well was substituted for the medium containing clusters. The final concentration of ligand clusters was 0.03 μg/well and the antibody/ligand ratio 1:50. The cells were incubated at 37°C for either 1 h or 8 h and subsequently fixed with Cytofix/Cytoperm reagent (BD Biosciences) for 12 min.
In a subset of clustering experiments, after incubation of the neurons with the eph-Fc ligands for 8 h, activation medium (AM) was added to the wells containing 100 μM glycine, 100 μM NMDA, and 50 mM KCl. The activation medium was left for 2, 10, or 20 min, and the neurons were subsequently fixed as described above.
Immunocytochemistry.
Coverslips were incubated with different combinations of primary and secondary antibodies and then embedded using ProLong Gold reagent (Invitrogen, United Kingdom). The primary antibodies used were rabbit anti-Slap, rabbit anti-GluN2A/B (Chemicon, United Kingdom; catalog no. AB1548), mouse anti-EphA4 (Invitrogen, United Kingdom; catalog no. 37-1600), mouse antisynaptophysin (Sigma, United Kingdom; catalog no. S5768), rabbit antisynaptophysin (Santa Cruz; catalog no. sc-7568), and mouse anti-Map2 (Sigma, United Kingdom; catalog no. M4403). Fluorescent secondary antibodies (all from Invitrogen) were anti-rabbit antibody–Alexa Fluor 568, anti-mouse antibody–Alexa Fluor 488, and anti-mouse antibody–Alexa Fluor 647. The cells were visualized on a spinning disk confocal system (CARV from Digital Imaging Solutions) with an electron microscopy charge-coupled-device (EM-CCD) camera (Rolera/QI Cam 3500) mounted on an Olympus X71 microscope, using a 100× Fluoplan objective (numerical aperture [NA], 4.2). The microscope confocal system was supported by Image Pro 6.0 software.
Image analysis.
To establish a suitable approach that would enable us to quantitatively measure the extent of colocalization of two signals in double immunolabeling, we examined an independent biological issue and asked how a colocalization analysis approach could portray the dynamics of synaptic vesicle (SV) regulation by a family of phosphoproteins, the synapsins (Snp). Double immunolabeling was performed in mature hippocampal neurons for synaptophysin (Syp) using a rabbit anti-Syp antibody (Santa Cruz, sc-7568) and Snp using a mouse anti-Snp antibody (Santa Cruz; catalog no. sc-37662) or for Syp using a mouse anti-Syp antibody (Sigma, United Kingdom; catalog no. S5768) and phosphor-Snp (p-Snp) Ser9 using a rabbit antibody (Abcam; catalog no. ab76260) in resting neurons and in neurons depolarized with 55 mM KCl for 5 min. A total of 15 images (5 images from each of three independent experiments) were analyzed using the JaCoP plug-in for ImageJ (37) to calculate the split Manders coefficient and the Pearson's coefficient. To avoid potential digital overlap, all images throughout the article are single-section confocal images.
The main difference between the Manders coefficient and Pearson's coefficient values is that Pearson's correlation describes (or, rather, measures) the relationship between the intensities in two images whereas the Manders overlap coefficient measures the proportion of one signal, signal a (i.e., green), coinciding with the other signal, signal b (i.e., red), even if the intensities in both channels differ. Although the latter measurement is seemingly more accurate because it does not take into consideration the amount of signal in signal b and asks only how much of signal a overlaps signal b, the fact that it does not take into consideration intensity (as long as it is above zero, it is positive) suggests that the background had to be removed from the images. To allow consistency of the removal of the background, two random areas devoid of staining within the images were independently measured on each channel using ImageJ and the average background measurement was removed from the entire single-channel image before analysis. The calculation of the Pearson's coefficient was performed in nonedited images (raw images). To avoid introducing an inconsistent bias into our calculations, no threshold was applied.
The data for the Manders coefficient (see Fig. 2) are the values of the fraction of the green signal (Syp) overlapping the red signal (Snp or p-Snp), and in the remaining figures, we present the values of the fraction of the green signal (i.e., eph-Fc clusters) overlapping the red signal.
Fig 2.
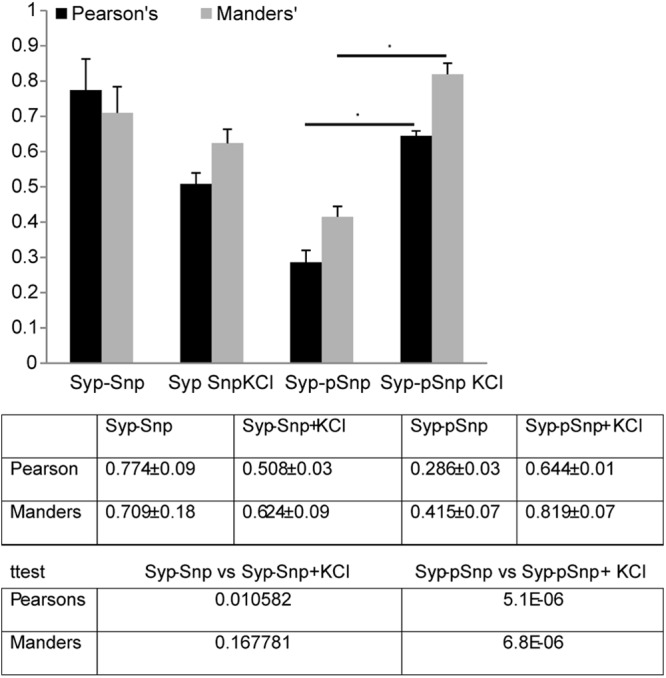
Comparison of colocalization values (obtained using the JaCoP plug-in for ImageJ) of immunolabeling for Syp and either Snp or pSnp in resting mature hippocampal neurons (21DIV) and in neurons treated with 55 mM KCl to induce depolarization, showing that both approaches can be used to describe the dynamics of signaling pathways by immunolabeling.
It should be mentioned that the extent of colocalization obtained by the Pearson's coefficient appeared consistently lower. This is not surprising, since the Pearson's coefficient can be affected by unique signals of both channels that contribute to the measured value, whereas the Manders coefficient provides information only on the extent to which a reference channel overlaps with the other.
In conclusion, it seems that both values can be used to provide meaningful and biologically relevant information. Our results here show that these values produce subtle differences. For this report, we calculated both values and the results produced were similar. In our article, we present the Manders coefficient values since our primary focus was to investigate the ability of ephrin receptors (Eph receptors) visualized by preclustered eph-fc ligands (using a FITC-conjugated Ab) to induce the clustering of candidate molecules and not the behavior or simultaneous correlation of the two molecules.
Recombinant NMDAR and Slap expression in Xenopus oocytes and two-electrode voltage clamp (TEVC) electrophysiology.
cRNA was synthesized from MluI- or NotI-linearized runoff transcripts using an SP6 or T7 Promega RiboMax RNA synthesis kit (Madison, WI). cRNA was synthesized in vitro according to manufacturer's instructions except for the addition of 0.75 mM m7G(5′)ppp(5′)G capping nucleotide (Promega, Madison, WI) and adjusting the GTP concentration to 1.6 mM. cRNA amounts and integrity were estimated by intensity of fluorescence in ethidium bromide-stained agarose gels. GluN1, GluN2, and Slap cRNAs were mixed in a nominal ratio ranging between 1:1 and 1:9, with total injected content fixed at approximately 5 ng per oocyte.
Stage V to VI Xenopus laevis oocytes were obtained from the European Xenopus Resource Centre at the University of Portsmouth. Xenopus oocytes were obtained from Xenopus laevis frogs held under United Kingdom Home Office regulations. Before cRNA injection, the follicular membranes of oocytes were removed mechanically, and after injection, oocytes were incubated at 18°C in separate wells of 24-well plates containing a modified Barth's solution containing (in mM) NaCl (88), KCl (1), NaHCO3 (2.4), MgCl2 (0.82), CaCl2 (0.77), and Tris-Cl (15), adjusted to pH 7.35 with NaOH (Sigma-Aldrich, United Kingdom) and supplemented with 50 IU/ml penicillin and 50 μg/ml streptomycin (Invitrogen, United Kingdom) and 100 μM dl-2-amino-5-phosphonovaleric acid (DL-APV) (Tocris Bioscience, Bristol, United Kingdom). Oocytes were incubated for 24 to 48 h to allow for receptor expression and then stored at 4°C until required for electrophysiological measurements.
TEVC recordings were made, using a Turbo TEC-03 amplifier (npi electronics, Tamm, Germany), from oocytes that were placed in a modified Ringer's solution containing (in mM) NaCl (115), KCl (2.5), HEPES (10), and BaCl2 (1.8), adjusted to pH 7.35 with NaOH (20°C). Chemicals were purchased from Sigma (Poole, United Kingdom). Current and voltage electrodes were made from thin-walled borosilicate glass (catalog no. GC150TF-7.5; Harvard Apparatus, Kent, United Kingdom) using a PC-10 electrode puller (Narashige Instruments, Japan) and were filled with 3 M KCl and possessed resistances of between 0.5 and 1.5 MΩ. Oocytes were voltage clamped at −40 mV. For l-glutamate concentration-response measurements, the recording solution was further supplemented with 50 μM glycine. The application of solutions was controlled manually, and oocytes were perfused under conditions of gravity flow (15 ml/min). Data were filtered at 20 Hz and digitized at 100 Hz (Digidata 1322A; Molecular Devices, Sunnyvale, CA) before being recorded to a computer hard disk. All recordings were acquired using a Windows PC-based program, WinEDR v3.0.6 (John Dempster, University of Strathclyde, United Kingdom). Agonist-containing solutions were applied until a plateau with respect to the agonist-evoked response was achieved.
Concentration-response curves were fitted individually for each oocyte with the Hill equation using Igor Pro 6.12 (Wavemetrics, Tigard, OR) as follows:
where nH is the Hill coefficient, Imax is the maximum current, [A] is the concentration of agonist, and EC50 is the concentration of agonist that produces a half-maximum response. Each data point was then normalized to the fitted maximum of the concentration-response curve. The normalized values were then pooled and means taken for each construct and fitted again with the Hill equation, with the maximum and minimum for each curve being constrained to the asymptote to 1 and 0, respectively.
For the insulin potentiation experiments, modified Ringer's solution was supplemented with 1 μM insulin (Sigma, Poole, United Kingdom). Maximal agonist-evoked inward currents in response to 100 μM glutamate and 50 μM glycine were measured from oocytes expressing NMDAR subunits alone or with Slap. Oocytes were then incubated with Ringer solution–1 μM insulin for 10 min before the agonist-evoked currents were measured again. Values for potentiation in current response were expressed as the percentages of increase in current from each oocyte before and after exposure to insulin.
Cell culture experiments.
HEK2393T cells were cultured in DMEM containing 10% fetal bovine serum, penicillin-streptomycin, and 2 mM l-glutamine (all from Invitrogen). Cells were transiently transfected using Lipofectamine 2000 (Invitrogen). A total of 4 μg of DNA was added to each well of a 6-well plate. Slap in pRRL_scC_Slap_IRESeGFP_W was transfected at 2 μg/well, and 2 μg of pEGFP-C1 was added to keep the total amount of DNA constant. pRK plasmids containing GluN1 or GluN2A cDNAs downstream of a pCMV promoter were transfected at 1 μg/well each, and either 2 μg of pEGFP-C1 was added to keep the total amount of DNA constant (NMDA treatments with or without activation) or 2 μg of Slap–pRRL_scC_Slap_IRESeGFP_W was added in the all-plasmids treatment.
Cells were left to express for ca. 22 h before being treated for 10 min with PSS (140 mM NaCl, 1.4 mM CaCl2, 5.4 mM KCl, 1.2 mM NaH2PO4, 21 mM glucose, 26 mM NaHCO3, pH 7.4) with or without the addition of 1 mM NMDA and 50 μM glycine (both from Sigma) (NMDA activation treatment). Bortezomib was used as a proteasome inhibitor at a final concentration of 0.01 mg/ml (Millenium Science). Following treatment, cells either were used to calculate cell death or were lysed for protein extraction.
Proteins were extracted in 25 mM HEPES (pH 7.5)–150 mM NaCl–1% NP-40–10 mM MgCl2–1 mM EDTA–2% glycerol supplemented with protease inhibitors (Roche). The protein concentration of the protein extract was determined by Bradford's protein assay, and equal amounts were loaded on a 10% precast gel (Invitrogen) for the detection of GluN1 using Western blot analysis. For protein normalization, a background band of 14 kDa detected by the antibody was used and the relative amount of GluN1 was calculated against this band using the Gel Analyzer tool from ImageJ.
Cell death experiments were performed largely as described previously (38). Transfected and activated cells from different treatments were incubated in fresh media for 6 h prior to assessing cell death using the trypan blue (Sigma) assay, where the exclusion of the reagent from living cells was used to distinguish between dead and living cells in both the attached and the floating cells. Cell counts on a hemocytometer were performed either after addition of trypan blue to media from each well or after trypsinizing the cells in the well. All data points represent the combined results for floating and attached cells for each treatment and correspond to the means ± standard errors of the means (SEM) of the results from three experiments each performed in triplicate.
Statistical analysis.
All data in this study are reported as means ± SEM. Statistical analyses were performed by a t test (two-tailed) using SPSS 15.0. Statistical significance was set at P = 0.05.
RESULTS
Subcellular localization of Slap.
To study the function of Slap in the central nervous system (CNS), we first raised an antibody specific for Slap using a unique peptide scanning the last 16 amino acids of the rodent sequence to immunize rabbits. The specificity of the affinity-purified serum from immunized rabbits and its potential use in Western blotting and immunolabeling were examined in HEK293T cells transduced with a nonintegrating lentiviral vector expressing Slap and enhanced green fluorescent protein (eGFP) from a bicistronic mRNA. Western blot analysis of HEK293T cells revealed that the antibody recognized a specific band only in cells transduced with the Slap expression vector, verifying its specificity and its appropriateness for detection of Slap by Western blot analysis (Fig. 1a). Furthermore, Slap immunofluorescence labeling of HEK293T cells produced a visible signal only in cells transduced with the lentiviral vector expressing Slap and not in those transduced with eGFP-only vector, suggesting that the antibody was suitable for immunolabeling as well (Fig. 1b and c). Next, we used our antibody to examine the subcellular distribution of Slap by Western blotting of different subcellular fractions from rat adult brain homogenates. A major band of ∼34 kDa, the expected size of Slap, was detected in most fractions, with a prominent band detected at the synaptosomal fraction. This distribution closely followed the distribution of the synaptic marker synaptophysin (Syp), indicating that Slap is present at neuronal synapses (Fig. 1f). To corroborate the subcellular distribution of Slap, mature 21DIV dissociated hippocampal neurons were doubly labeled for Slap and neuronal markers. Double labeling for Slap and the presynaptic marker Syp showed that Slap was juxtaposed to Syp, confirming our previous observations that Slap is enriched at neuronal synapses and, specifically, at the postsynaptic terminal (Fig. 1d). Furthermore, double labeling for Slap and the postsynaptic marker PSD95 confirmed that, indeed, Slap could be detected at postsynaptic sites.
Fig 1.

Subcellular distribution of Slap. Cell lysates from HEK293 cells transduced with a nonintegrating lentiviral vector expressing Slap and eGFP from a bicistronic mRNA were used to examine the specificity of a Slap polyclonal antibody. (a) Western blots reveal a major band of 34 kDa in cells transduced with the Slap-expressing vector that was absent from cells transduced with the control, eGFP-only lentiviral vector. (b and c) Immunolabeling for Slap shows that specific labeling is evident only in cells transduced with the Slap-expressing vector (b) and not with the control vector (c). (d and e) Representative images from dissociated 21DIV hippocampal neurons labeled for Syp and PSD95 (green in panels d and e, respectively) and Slap (red) show that Slap is localized at the postsynaptic terminal. A higher magnification image of the boxed area in panel d (below the main image) shows that Slap is juxtaposed to Syp labeling (d) and colocalizes with PSD95 (e). (f) Western blots of subcellular fractions from adult rat brain homogenate show that Slap is enriched at synaptic contacts. Bar, 10 μm.
Colocalization analysis.
For the purposes of measuring the dynamics of the colocalization between Ephs and candidate molecules, we first established an approach that would allow us to measure the extent of colocalization between two signals. To this end, we examined an independent biological issue and asked if a colocalization analysis approach could portray the dynamics of synaptic vesicle (SV) regulation by a family of phosphoproteins, the synapsins (39). Synapsins are present at the presynaptic terminal, and their role is to cluster SVs by binding to them and to cytoskeletal elements. Synapsins are phosphorylated in response to various signals (chemical or electrical). A key phosphorylation site is Ser9 (site 1). Site 1 is the target of protein kinase A (PKA) and Ca2+/calmodulin-dependent protein kinases I and IV, and under basal conditions, small amounts of Snp are phosphorylated at this site. A depolarization-induced, Ca++ influx increases the phosphorylation levels at site 1. Phosphorylation at this site is transient, and dephosphorylation is mediated by protein phosphatase 2A. Upon phosphorylation, the affinity of Snp to actin and SVs decreases, allowing more SV to participate in neurotransmitter release.
We performed double immunolabeling in mature hippocampal neurons for synaptophysin (Syp) and synapsin (Snp) or for Syp and phosphor-synapsin (p-Snp Ser9) in resting neurons and in neurons depolarized with 55 mM KCl for 5 min, and single-section confocal images were analyzed using the JaCoP plug-in for ImageJ (37) to calculate the split Manders coefficient and the Pearson's coefficient (Fig. 2). According to the literature, the extents of colocalization between Syp and Snp in resting neurons and in depolarized neurons would be similar. In contrast, we expected a significant increase in the colocalization between Syp and p-Snp in depolarized neurons compared to resting neurons. Our results showed that for the Manders coefficient, depolarization of neurons did not reveal a significant difference between Syp and Snp in the colocalization, whereas calculation of the Pearson's coefficient showed a marginal decrease in the extent of colocalization between Syp and Snp (Fig. 2). In contrast, measuring the extent of colocalization between Syp and p-Snp showed that depolarization of neurons resulted in a significant increase in the amount of p-Snp at synaptic contacts marked by Syp that was reflected by both values.
The issue now would be which of the two values should be used. We believe that the answer depends on the focus of the experiment. If the focus is evaluate the behavior or correlation of both candidate molecules, then the Pearson's coefficient seems more appropriate, since it provides an overall estimate of the extent of colocalization of both molecules. Furthermore, images can be analyzed without threshold or removal of background, provided that the immunolabeling experiments are performed with Abs that produce a clean signal. On the other hand, if the focus is to evaluate the response of a candidate molecule (X) in relation to another (Y), then the Manders coefficient seems more accurate, since it provides an estimate of the extent to which X overlaps Y without taking into consideration the overall dispersion of Y. The drawback of using the Manders coefficient is that background has to be removed in each channel independently. We calculated both values in all our experiments, and the results produced were similar. We present, though, the Manders coefficient values, since our primary focus was to investigate the ability of ephrin (Eph) receptors to induce the clustering of candidate molecules and not the dynamics of the two molecules independently.
Slap and Eph receptors in dissociated hippocampal neurons.
Next, we explored the possibility that Slap functions downstream of Ephs. Although it has been shown that Slap interacts with the intracellular domain of EphA2, this receptor is not expressed in the hippocampus or the cortex (40). However, other EphAs, such as EphA4, that share significant homology with EphA2 in their intracellular domain are abundantly expressed in the CA1 region of the hippocampus (40), where Slap is also expressed (1). Dissociated hippocampal neurons were used to assess a possible association between Slap and EphA receptors. First, we examined the presence of EphAs in our neuronal preparation, as reports of their expression in dissociated hippocampal neurons are controversial (5, 25, 41, 42). The presence of EphA4 in our culture system and its possible association with Slap and/or NMDARs was confirmed by double immunolabeling of fixed hippocampal neurons. We focused our efforts on 14DIV hippocampal neurons to examine the possible role of Slap downstream of Eph signaling during synaptogenesis, since 14DIV hippocampal neurons have relatively few mature dendritic spines and their synaptic connections are still developing (43, 44). Double immunolabeling for EphA4 and Slap or NMDARs showed that EphA4 was indeed expressed in fixed hippocampal neurons. Moreover, although colocalization between EphA4 and Slap was evident, we could not detect significant colocalization between EphA4 and NMDARs (Fig. 3a and b). To quantify the dynamics of this colocalization, we calculated the extent of colocalization using the JaCoP plug-in for ImageJ (37) and compared the extents to which EphA4 labeling overlaps Slap or GluN2A/B labeling. Our results showed that the colocalization between EphA4 and Slap was significantly higher than that between EphA4 and NMDAR (Fig. 3g) (M = 0.316 ± 0.028 and 0.152 ± 0.04, respectively; t test, P = 0.0038; n = 6), confirming our observation.
Fig 3.

Expression of EphA4 in dissociated hippocampal neurons. (a and b) Representative images from double-immunolabeling experiments of fixed neurons for EphA4 (green) and Slap (a) or GluN2A/B (red) (b). High-magnification images of boxed areas (below the respective images) show colocalization between EphA4 and Slap (the arrow indicates an example of colocalization between EphA4 and Slap) but not with GluN2A/B. (c and d) Double labeling with preclustered ephA2-Fc (green) and Slap (c) or GluN2A/B (d). High-magnification images of boxed areas (below the respective images) show the more pronounced colocalization between ephA2-Fc and Slap (arrow) compared to GluN2A/B. (e and f) Double labeling with preclustered ephB2-Fc (green) and Slap (e) or GluN2A/B (f). High-magnification images of boxed areas (below the respective images) show the more pronounced colocalization between ephB2-Fc and GluN2A/B (arrow) compared to Slap. (g) Values for quantification of colocalization using the Manders coefficient (M) were 0.316 ± 0.03 and 0.152 ± 0.04 (n = 6) for EphA4 with Slap and NMDARs, respectively, 0.377 ± 0.03 and 0.099 ± 0.05 (n = 5) for ephA2-Fc with Slap and NMDARs, respectively, and 0.178 ± 0.03 and 0.38 ± 0.02 (n = 6) for ephB2 with Slap and NMDARs, respectively. *, P value ≤ 0.005 with t test. Bar, 10 μm.
Having established the presence of at least one EphA in our cultures, we then investigated if Slap is associated preferentially with EphAs or if it is associated with EphBs as well. To label different classes of Eph receptors, we employed an indirect labeling approach using preclustered chimeric eph-Fc ligands that have been extensively used to study Ephs both in vivo and in vitro (7, 16, 25, 45). Fixed 14DIV hippocampal neurons were double labeled for Ephs, using different preclustered eph-Fc ligands (to enable visualization, a FITC anti-Fc antibody was used to cluster eph-Fc ligands) and Slap or NMDAR. Similar to our previous results, colocalization between Slap and preclustered ephA2-Fc was more pronounced than that seen with NMDARs (Fig. 3c, d, and g) (M = 0.377 ± 0.03 and 0.099 ± 0.05, respectively; t test, P = 0.0002; n = 10). In contrast, colocalization between preclustered ephB-Fc and Slap was significantly lower than that seen with NMDARs (Fig. 3e, f, and g) (M = 0.178 ± 0.03 and 0.380 ± 0.02, respectively; t test, P = 0.0005; n = 6). These data are in agreement with previously published work that supports the idea of a specific association between NMDARs and EphBs (7). Furthermore, our data suggest that in dissociated hippocampal neurons, Slap preferentially (although not extensively) associates with EphA receptors.
Activation of Eph receptors.
Since the ligand for EphA4 is found in glia cells (25), it is likely that, even though EphA4 was detected in our neuronal cultures, it might not be activated, as we did not employ neuron-glia cell cocultures. We next sought to investigate whether activation of Ephs could induce further recruitment of Slap. Since it has been shown that only membrane-bound or Fc-clustered ligands are capable of activating the receptor in vitro whereas soluble monomeric ligands that bind the receptor do not induce receptor autophosphorylation and activation (46), activation of Ephs was induced by incubation of neurons with preclustered ephA2-Fc, ephB1-Fc, or ephB2-Fc fusion ligands for 1 h and 8 h prior to fixation. We first examined the promiscuity of binding between Ephs and ephs by means of exposure to preclustered ligands for 1 h and 8 h prior to fixation and subsequent colabeling for EphA4 (Fig. 4). Based on the reported binding promiscuity between ephBs and EphA4, we used preclustered ephB1-Fc for its specificity in binding EphB receptors and ephB2-Fc as a control for its reported binding to EphA4 (47, 48).
Fig 4.

Binding promiscuity between preclustered ephB-Fc ligands and EphA4 in live neurons. (a to f) Representative images showing colocalization between eph-Fc ligands (green) and EphA4 (red) in 14DIV hippocampal neurons incubated with preclustered ephA2-Fc (a and b), ephB1-Fc (c and d), and ephB2-Fc (e and f) for 1 h and 8 h, respectively. High-magnification images of boxed areas (below the respective images) show a high degree of colocalization between EphA4 and all eph-Fc. (g) Values of quantification of colocalization (M) were 0.698 ± 0.05 at 1 h and 0.857 ± 0.04 at 8 h between EphA4 and ephA2-Fc, 0.47 ± 0.04 at 1 h and 0.889 ± 0.01 at 8 h between EphA4 and ephB2-Fc (t test = 8.5E-8), and 0.725 ± 0.03 at 1 h and 0.894 ± 0.02 at 8 h between EphA4 and ephB1-Fc (t test = 0.001). *, P value of 0.001 with t test. n ≥ 3 independent experiments for each set of conditions. Bar, 10 μm.
Labeling for EphA4 in neurons exposed to ephA2-Fc showed extensive colocalization between them at both time points (Fig. 4a and b). Surprisingly, labeling for EphA4 in neurons exposed to both ephB-Fc ligands showed extensive colocalization between ephB-Fc and EphA4 that appeared more prominent after 8 h (Fig. 4c to f). Quantification of the colocalization between EphA4 and eph-Fc ligands confirmed our observations (Fig. 4g). The extent of colocalization between EphA4 and ephA2-Fc was significant at both time points examined (M = 0.698 ± 0.05 at 1 h and 0.857 ± 0.04 at 8 h; n = 3). Interestingly, after 1 h of incubation, although the extent of colocalization between EphA4 and ephB1-Fc was similar to that seen with ephA2-Fc (M = 0.725 ± 0.03; n = 5), the extent of colocalization between EphA4 and ephB2-Fc was significantly lower than that seen with ephA2-Fc (M = 0.47 ± 0.04; n = 5; P = 0.0025 with t test). After 8 h of activation, though, the extents of colocalization between EphA4 and all preclustered eph-Fc ligands were increased at similar levels (M = 0.857 ± 0.04 for ephA2 [P = 0.0123 with t test], 0.889 ± 0.01 for ephB2 [P = 8.5E-8 with t test], and 0.894 ± 0.01 for ephB1 [P = 0.001 with t test]; n = 10 for ephB-Fc).
These results are seemingly different from those of previous reports where no interaction between ephB1 and EphA4 was identified (47, 48). It should be mentioned, though, that our experiments assessed only the extent of colocalization between EphA4 and the preclustered ligands and not the ability of the preclustered ligands to activate the receptor. Nevertheless, since our data showed reduced colocalization between EphA4 and ephB2-Fc compared to ephB1-Fc after 1 h of exposure, to differentiate between signaling cascades involving EphBs and EphAs at the early time point, we present here our data from the experiments in which ephB2-Fc was used to activate EphB receptors.
Recruitment of Slap and NMDARs upon Eph activation.
Having established the association between Slap and Ephs in fixed neurons, we next investigated if activating Ephs could induce recruitment of Slap.
14DIV hippocampal neurons were incubated with preclustered eph-Fc ligands for 1 h and 8 h to activate their respective receptors prior to fixation (Fig. 5). Immunolabeling revealed that Slap was partially colocalized with ephA2-Fc and ephB2-Fc in neurons after 1 h of exposure to the ligands (Fig. 5a and c). However, in neurons incubated with preclustered eph-Fc ligands for 8 h, Slap was detected with the vast majority of preclustered ephA2-Fc compared to a partial overlap of ephB2-Fc (Fig. 5b and d). Quantification of colocalization using the Manders coefficient showed colocalization between ephA2-Fc and Slap after 1 h of incubation (MP = 0.375 ± 0.04). After 8 h of incubation with the ligand, a significant increase in colocalization between ephA2-Fc and Slap was detected (M = 0.809 ± 0.04; n = 10; P = 0.00005 with t test). In contrast, there was little colocalization between ephB2-Fc and Slap after 1 h of incubation, with a less significant increase after 8 h of incubation with the ligand (M = 0.26 ± 0.03 at 1 h and 0.578 ± 0.03 at 8 h; n = 10; P = 0.0004 with t test). These data suggest that prolonged incubation of neurons with preclustered ephA2-Fc can trigger a delayed but highly efficient recruitment of Slap compared to prolonged incubation with preclustered ephB2-Fc (Fig. 5e) (P = 0.0008 with t test).
Fig 5.

Recruitment of Slap upon Eph activation. Representative images show colocalization between preclustered eph-Fc ligands (green) and Slap (red). (a to d) 14DIV hippocampal neurons were incubated with ephA2-Fc (a and b) and ephB2-Fc (c and d) for 1 h and 8 h, respectively. High-magnification images of boxed areas (below the respective images) show in detail that extensive colocalization was detected only between ephA2-Fc and Slap upon 8 h of incubation (b). (e) Values of quantification of colocalization (M) were 0.375 ± 0.04 at 1 h and 0.809 ± 0.04 at 8 h (n = 10) between ephA2-Fc and Slap and 0.26 ± 0.03 at 1 h and 0.578 ± 0.03 at 8 h (n = 10) between ephB2-Fc and Slap, confirming our observation. *, P value ≤ 0.0004 with t test. Bar, 10 μm.
Having established that activation of EphAs is more efficient in recruiting Slap than activation of EphBs, we next investigated the dynamics of NMDAR's recruitment upon Eph signaling. Although it has been shown that activation of EphBs recruits both NMDA and AMPA receptors (9, 16, 23), there is limited information regarding EphAs. 14DIV hippocampal neurons were incubated with preclustered ephA2-Fc or ephB2-Fc as described previously. Immunolabeling of treated neurons for NMDARs showed that prolonged exposure of dissociated neurons to both ligands induced colocalization between them and NMDARs (Fig. 6a to d). Unexpectedly, though, we noticed that incubation of neurons with preclustered ephA2-Fc for 8 h induced colocalization of NMDARs with the majority of the clustered ligand. We next quantified the dynamics of this colocalization (Fig. 6e). Our results showed that although colocalization between ephA2-Fc and NMDARs after 1 h of activation was near background levels (M = 0.193 ± 0.02; n = 10), it was significantly enhanced after 8 h of incubation (M = 0.701 ± 0.04; n = 10; P = 3E-8 with t test), suggesting that recruitment of NMDARs requires prolonged exposure of neurons to ephA2-Fc. On the other hand, the extent of colocalization between NMDARs and ephB2-Fc was significant at both time points and did not change dramatically over time (M = 0. 3711 ± 0.04 at 1 h and 0.615 ± 0.09 at 8 h; n = 5; P = 0.0026 with t test). These data indicate that ephB2-Fc is more efficient in recruiting NMDARs at early time points (P = 0.0056 with t test) but ephA2-Fc is as efficient as ephB2-Fc after 8 h of activation.
Fig 6.

Recruitment of NMDARs upon Eph activation. Representative images show colocalization between preclustered eph-Fc ligands (green) and GluN2A/B (red). (a to d) 14DIV hippocampal neurons were incubated with ephA2-Fc (a and b) and ephB2-Fc (c and d) for 1 h and 8 h. High-magnification images of boxed areas (below the respective images) show in detail colocalization between ephB2-Fc and GluN2A/B under all conditions (c and d) and colocalization between ephA2-Fc and GluN2A/B only after 8 h of incubation (a and b). (e) Values of quantification of colocalization (M) were 0.193 ± 0.02 at 1 h and 0.701 ± 0.04 at 8 h (n = 10) between ephA2-Fc and NMDARs and 0.371 ± 0.04 at 1 h and 0.615 ± 0.09 at 8 h (n = 5) between ephB2-Fc and NMDARs. *, P value ≤ 0.0056 with t test. (f) Triple labeling with ephA2-Fc (green), GluN2A/B, and Syp. A high-magnification image of boxed area (below the main image) shows in detail that ephA2-Fc clusters are juxtaposed to Syp labeling. Bar, 10 μm.
It has been reported that activation of EphA4 in young immature neurons results in the internalization of the receptor/ligand complex whereas in mature neurons it induces an increased colocalization with the postsynaptic marker PSD-95 (49). To confirm these findings and to see if the colocalization between ephA2 and NMDARs we observed was synaptic, neurons exposed to ephA2-Fc for 8 h were triple labeled for NMDAR and Syp, a presynaptic marker (Fig. 6f). Similar to previous reports (30, 41), our results showed that ephA2-Fc labeling was partially detected at synapses. Interestingly, colocalization between NMDARs and ephA2-Fc was not restricted solely to synapses but was more widespread, suggesting that activation of EphAs recruits NMDA receptors independently of synaptic contacts.
Slap does not interfere with the activity of NMDARs.
Our results so far showed that activation of Ephs recruits both NMDARs and Slap. Since the role of Slap in the CNS has not been studied, we next examined if it is involved in modulating the activity of NMDARs. To do so, we used established heterologous systems that offer the advantage of directly assessing the possible cross talk between Slap and NMDARs independently of synaptic complexity.
First, we examined whether Slap interferes with the activation of NMDARs in Xenopus oocytes injected with in vitro-transcribed cRNAs expressing GluN1 and GluN2B or GluN2A in the presence or absence of a cRNA expressing Slap. The presence of Slap at the membrane of the oocytes was detected by immunolabeling of injected oocytes (Fig. 7a), and the expression of NMDARs was confirmed by measuring agonist-evoked inward currents from oocytes expressing recombinant NMDARs (Fig. 7b to e). We first examined if Slap had an effect on the baseline activity of NMDARs by comparing maximal agonist inward currents in response to glutamate and glycine in oocytes expressing recombinant NMDARs alone and subsequently in oocytes expressing both NMDARs and Slap. Our results showed that the NMDAR inward currents from GluN1/N2B (Fig. 6b and c) and GluN1/N2A (Fig. 7d and e) receptors in the absence or presence of Slap were almost identical. Furthermore, we did not observe any major alterations in glutamate EC50 values in the presence of Slap compared to those reported previously for wild-type (WT) recombinant NMDARs expressed in Xenopus oocytes (33). Our results show that Slap does not affect activation of NMDARs. Additionally, Slap does not preferentially interfere with either of the two major GluN2 subtypes expressed in hippocampal neurons.
Fig 7.

Slap does not affect NMDAR activity. (a) Expression of Slap in Xenopus oocytes visualized by immunolabeling. Panels b and d show examples of traces of cumulative glutamate concentration-response curves recorded from Xenopus oocytes expressing recombinant GluN1/GluN2B (b) and GluN1/GluN2A (d) NMDA receptors. (c and d) NMDAR currents in Xenopus oocytes expressing GluN1/GluN2B (c) or GluN1/GluN2A (d) did not change in the presence of Slap (GluN1/GluN2B mean maximal current = 0.28 ± 0.1 μA [n = 6], GluN1/GluN2BSlap mean maximal current = 0.37 ± 0.1 μA [n = 6], GluN1/GluN2A mean maximal current = 1.1 ± 0.4 μA [n = 6], and GluN1/GluN2ASlap mean maximal current = 2 ± 0.5 μA [n = 6]). No change was found in the mean glutamate EC50 values for GluN2A or GluN2B containing NMDARs in the absence and presence of Slap. (c) Mean GluN1/GluN2B EC50 = 1.35 ± 0.04 μM (n = 6; filled circles); mean GluN1/GluN2BSlap EC50 = 1.06 ± 0.06 μM (n = 6; filled squares). (e) Mean GluN1/GluN2A EC50 = 3.5 ± 0.7 μM (n = 6; filled circles); GluN1/GluN2ASlap EC50 = 2.1 ± 0.1 μM (n = 6; filled squares). (f) Mean values of potentiation of NMDA currents by SFKs in 100 μM glutamate–50 μM glycine (31.4% ± 3.6%) (n = 10; −Slap are not affected by Slap (36.9% ± 9.3%) (n = 11; +Slap coexpressed in Xenopus oocytes. Data represent sample traces of inward currents recorded before (black line) and after (gray line) insulin treatment coexpressed with Slap (i) and in WT GluN1/GluN2B NMDARs without Slap (ii).
It has been demonstrated that in cortical neurons, activation of EphBs leads to tyrosine phosphorylation of NMDARs through activation of the Src family kinases (SFKs) (7, 16). Since Slap has been shown to antagonize the activity of SFKs (3) and is recruited alongside NMDARs upon Eph signaling, we next investigated whether it has a role in modulating NMDAR activity by regulating its phosphorylation by SFKs. To induce SFK-dependent phosphorylation of NMDARs, we exposed injected Xenopus oocytes to 1 μM insulin for 10 min. This treatment has been shown to enhance NMDAR currents in a SFK-dependent manner (50, 51). NMDAR currents were measured initially in oocytes prior to insulin exposure and were then compared to the currents produced after the exposure to insulin. Our results showed that exposure to insulin for 10 min potentiated NMDAR currents (Fig. 7f). We then repeated the experiment with oocytes coinjected with Slap and recombinant NMDARs and saw little change in the percentage of potentiation of agonist-evoked current response following insulin exposure compared to the results seen with recombinant NMDARs in the absence of Slap (Fig. 7f) (31.4% ± 3.6% [n = 10] and 36.9% ± 9.3% [n = 11], respectively). Therefore, our results showed that Slap did not affect the enhanced response of NMDARs to insulin, suggesting that it is not directly involved in the regulation of NMDARs by SFKs.
Activity-dependent regulation of NMDARs.
We next investigated if Slap has a role downstream of NMDAR signaling. Since a link between Slap and cell death has been previously reported (52), we examined if Slap exerts a protective effect upon NMDAR-dependent, glutamate-induced excitotoxicity (53). HEK293T cells expressing NMDARs have been used to study modulation of NMDAR currents (54, 55) and NMDAR-mediated excitotoxicity (38, 56). We adopted a similar approach and compared the amounts of cell death induced by a NMDAR activation medium (containing 1 mM NMDA and 50 μM glycine) in HEK293T cells transiently expressing GluN1/GluN2A subunits alone or in combination with Slap. Transfected cells were incubated with the NMDAR activation medium (AM) for 10 min and were left to recover for 6 h prior to measurement of cell death (38). Our results (Fig. 8a) showed increased cell death in cells expressing NMDARs and treated with AM (34% ± 2.76%) compared to control, nonactivated cells (11% ± 3.2%; P = 0.0001) or to cells expressing Slap alone and treated with AM (17% ± 1.21%). Interestingly, when Slap was expressed alongside NMDAR, it significantly (20% ± 3.02%; P = 0.0014; n = 3) prohibited cell death upon the activation of the receptor, indicating that coexpression of Slap protects cells from NMDAR-induced cell death.
Fig 8.

Slap is downstream of NMDAR signaling. (a) Activation of NMDAR in HEK293 cells significantly (34% ± 2.76%) increases cell death compared to that seen with nonactivated cells expressing NMDARs (11% ± 3.2%; P = 0.0001) or Slap alone (17% ± 1.21%). Coexpression of Slap prevents NMDA-induced cell death (20% ± 3.02%; *, P value of 0.0014; n = 3), and the results are comparable to the cell death seen in cells expressing Slap alone (P = 0.02; n = 3). (b) Comparison of the normalized ratios of GluN1 levels (from HEK293 cells transfected with GluN1/GluN2A and activated for 10 min [NMDAR act] showed that activation of NMDAR in HEK293 cells coexpressing Slap significantly reduces the levels of GluN1 (P = 4E-5). This reduction is inhibited in the presence of proteasome inhibitors (P = 0.007; n = 4). The inset shows a representative gel. (c to f) Activation of NMDARs for 10 min (c and e) and 20 min (d and f) in hippocampal neurons exposed to preclustered ephA2-Fc for 8 h and labeled for ephA2-Fc clusters (green), Slap (red in panels c and d), or GluN2A/B (red in panels e and f) and Syp (blue). High-magnification images of boxed areas (below each image, respectively) show in detail that the colocalization between Slap and ephA2-Fc clusters was not affected upon NMDAR activation for 10 and 20 min (c and d, respectively). In contrast, NMDARs could not be detected within ephA2-Fc clusters after 10 and 20 min of activation but could be detected in other parts of the coverslip (e and f, respectively). **, P ≤ 0.007. Bar, 10 μm.
We next investigated the possible mechanism for this protective function against NMDAR-induced apoptosis. It has been established that neuronal activity homeostatically reduces the levels of synaptic NMDARs (57–59). A likely mechanism proposed for homeostatic plasticity is proteasome-dependent degradation of glutamate receptors, since proteasome inhibitors block the removal of GluN1 from the postsynaptic density (PSD) (59). Since Slap interacts with c-Cbl, a ubiquitin ligase responsible for the degradation of Eph and TCR (4, 60–63), we examined if Slap was also involved in the degradation of NMDARs. We focused on HEK293T cells and compared the levels of GluN1 in cells transfected with plasmids expressing GluN1/GluN2A subunits alone or in combination with Slap (Fig. 8b). Our results showed that expression of Slap did not alter the expression levels of NMDARs. Next, we incubated cells with the NMDAR activation medium for 10 min prior to lysis to activate the receptors. Our results showed that coexpression of Slap under these conditions induced a 20% reduction of GluN1 levels. Furthermore, this reduction was abrogated in cells preincubated with 10 μg/ml of the proteasome inhibitor bortezomib (P = 0.007; n = 4), suggesting that expression of Slap induced the degradation of NMDARs in an activity-dependent manner. The next step was to examine this effect of Slap in neurons. Hippocampal neurons were incubated with epha2-Fc for 8 h to induce coclustering of NMDARs and Slap. NMDARs were then activated for 10 min and 20 min using an NMDAR activation medium (containing 100 μM glycine, 100 μM NMDA, and 50 mM KCl) prior to fixation. To assess the success of EphA activation, triple labeling for ephA2-Fc clusters with Syp and Slap was performed. Our results showed high colocalization between Slap and ephA2-Fc throughout the activation time points (Fig. 8c and d). We then looked at the colocalization of NMDA receptors. In contrast to the extensive colocalization between NMDARs and ephA2-Fc clusters consistently observed in our previous experiments, we could not detect NMDAR labeling along ephA2-Fc clusters after incubation with the activation medium (Fig. 8e and f). Extrasynaptic as well as synaptic labeling of NMDAR beyond ephA2-Fc clusters was evident at the same coverslips, suggesting that upon activation, NMDARs were downregulated or preferentially removed from the ephA2-Fc clusters where Slap was also present.
DISCUSSION
slap encodes a SH2-SH3 adapter protein, and although Slap expression in the brain has been previously reported (1), its function there remains unknown. Here we present data showing that Slap is recruited to synapses and is downstream of NMDARs.
Synaptic localization of Slap.
First, we looked into the cellular distribution of Slap. Western blot analyses of subcellular fractions from adult rat brain extracts and immunolabeling in dissociated hippocampal neurons showed that Slap was enriched at the postsynaptic terminals. We then investigated the possible mechanism by which Slap is recruited there. Since Slap was initially identified as a binding partner of EphA2 (6), we first examined their possible association in dissociated neurons by looking into the extent of colocalization of different classes of Ephs with Slap and comparing it to the results seen with NMDARs that have been associated with EphB signaling (7). We confirmed the association between EphBs and NMDARs and further revealed that Slap was preferentially localized with EphAs.
Cross talk between EphAs and EphBs.
We then examined if Slap could be actively recruited by Eph signaling, looking at the temporal dynamics of the localization of Slap upon activation of Ephs in neuronal cultures. First, we optimized our conditions, performing a series of control experiments to assess the reported binding promiscuity of eph/Eph interactions (47, 48). Interestingly, our data revealed extensive colocalization between preclustered ephB-Fcs and EphA4, with colocalization between ephB1-Fc and EphA4 more pronounced than that seen with the more promiscuous ephB2-Fc. Although this colocalization could be seen as an indication of binding promiscuity between the ligands and the receptor, it should be emphasized that our experiments were not designed to ascertain interactions of different classes of Ephs with their ligands. What is more, the lack of significant levels of colocalization between the promiscuous ephB2-Fc and EphA4 after 1 h of incubation suggests that a direct cross-interaction between ephBs and EphA4 is not a likely explanation for our results.
Subsequently, we performed our experiments using both ephB1-Fc and ephB2-Fc, and the results obtained with both ligands were identical. Here we present the data obtained using ephB2-Fc, since its colocalization with EphA4 after 1 h of incubation was not as pronounced as that seen with ephB1-Fc.
Recruitment of Slap upon Eph signaling.
Next, we showed that activation of Ephs in cultured neurons enhanced Slap's colocalization with each preclustered eph-Fc, with sustained exposure to ephA2-Fc being more efficient than exposure to ephB2-Fc. We also showed that, interestingly, sustained exposure of hippocampal neurons to ephA2-Fc was as efficient as exposure to ephB2-Fc in recruiting NMDARs. Although our focus was to examine the extent to which activation of Eph receptors differentially recruits Slap, our results showed that, as with EphB, activation of EphA recruits NMDARs, contributing to synaptic development. Furthermore, our data provide support to a hypothesis proposed by Henkemeyer et al., who suggested that in vivo, EphAs can partially compensate for the loss of EphBs, since hippocampal neurons lacking EphB expression fail to form dendritic spines in vitro but not in vivo (9). Finally, the different dynamics of recruitment of Slap by ephA2-Fc and ephB2-Fc suggest that the colocalization between ephB-Fc ligands and EphA4 we observed previously is not indicative of a direct interaction between them, since such an interaction would activate the receptor inducing the recruitment of Slap in a manner similar to that seen with ephA2-Fc. An alternative explanation for the observed colocalization could be that activation of EphBs induces the recruitment of EphA4, probably via a signaling cascade that allows for a cross talk between different Ephs. Although further experiments should be performed to unambiguously demonstrate this hypothesis, the data presented here indicate that activation of EphBs can induce the recruitment of EphAs and that subsequent activation of EphA4 (i.e., from ephA3 present on glia cells) would further stabilize synaptic contacts, aiding in the recruitment of molecules such as NMDARs and Slap.
Slap does not interfere with NMDAR activity.
Having established the recruitment of Slap by Eph signaling, we next focused on its possible role. Since Slap and NMDARs can be present at the same sites, we examined if Slap has an effect downstream of NMDAR signaling. Activation of NMDARs at synaptic contacts has pleiotropic effects, and assessing the role of Slap in such a complex background without any prior information on its function would be challenging. To circumvent this, we used established heterologous systems to determine if Slap is involved in any aspects of NMDAR signaling.
First, we asked if Slap interferes directly with the activation of NMDARs or with signals that modulate its activity. We compared NMDA currents in Xenopus oocytes expressing NMDARs in the presence or absence of Slap. Our results showed that coexpression of Slap did not affect the activity of NMDARs. We next investigated if Slap is associated with signaling mechanisms that modulate the activation of NMDARs. It has been shown, for instance, that Ephs bind to SFKs through their yeast extract-peptone-dextrose (YEPD) cytoplasmic motifs (64) and that members of the SFKs have a significant role in modulating NMDAR activity by enhancing its function (16, 65). Since it has been shown that in NIH 3T3 fibroblasts, Slap antagonizes the activity of SFKs (3), we investigated if Slap affects the modulation of NMDAR activation by SFKs. Coexpression of Slap in Xenopus oocytes had no effect on SFK-dependent potentiation of NMDA currents induced by insulin, suggesting that it is not involved in the modulation of NMDARs by SFKs.
Slap protects against NMDAR-induced excitotoxicity.
Next, we examined if Slap was downstream of NMDAR signaling. Aberrant activation of NMDARs has been linked to excitotoxicity, and the mechanisms behind it are complex, with several pathways implicated (66). Since Slap has been shown to protect against apoptosis in osteoclasts (52), we examined if Slap affects NMDAR-induced excitotoxic cell death in HEK293T cells, which have been extensively used to study the mechanisms of NMDAR-subtype-specific excitotoxicity (67). Our data showed that expression of Slap conferred significant protection against NMDAR-dependent excitotoxic cell death.
Slap induces NMDAR degradation.
We then looked into a possible mechanism by which Slap interferes with NMDAR activation. A key aspect of modulating receptor activity is the degradation of receptor molecules. It has been shown that Slap interacts with c-Cbl, a ubiquitin ligase, and that this interaction leads to the ubiquitination and degradation of TCR and BCR signaling complexes (4, 62, 63). Furthermore, c-Cbl is recruited to Eph-ephrin complexes, contributing to termination of the signal by degradation of the receptor (60, 61). Thus, we investigated if the antiexcitotoxic effect of Slap could be mediated, at least in part, by degradation of NMDARs. We compared the expression levels of GluN1 in HEK293T cells in the presence or absence of Slap. We show that although coexpression of Slap with NMDARs does not affect the levels of GluN1, activation of the receptors results in a proteasome- and Slap-dependent reduction of GluN1 levels. We also provide evidence that a similar mechanism is present in neurons. Activation of NMDARs in neurons preincubated with ephA2-Fc to induce coclustering of Slap and NMDARs results in a severe reduction of NMDAR immunoreactivity along ephA-Fc clusters, although overall labeling of NMDARs on the same coverslip appeared unaffected. These data show that Slap is involved in activity-dependent NMDAR degradation, suggesting that it might be also involved in regulating homeostatic plasticity. Interestingly, the involvement of EphAs in homeostatic plasticity has been recently demonstrated. Activation of EphA4 induced downregulation of AMPA receptors without affecting the levels of NMDARs (5). Similarly, we found no evidence that activation of EphAs affect the levels of NMDARs. In contrast, prolonged activation of EphAs induced increased levels of NMDARs at the sites of activation. However, subsequent activation of NMDARs resulted in their rapid removal from EphA clusters, suggesting an activity-dependent downregulation of NMDARs. Even though the role of Slap and the precise mechanisms in this downregulation are yet to be elucidated, the known biochemical function of Slap in T cells and our data in HEK293T cells suggest that it has a key role in mediating the downregulation of NMDARs.
Concluding remarks.
Taken together, our results reveal that Slap is recruited to synaptic contacts through signaling cascades activated by Eph receptors and is involved in synaptic development and homeostasis. It can protect cells from aberrant NMDAR activation, and it regulates the levels of NMDARs in an activity- and proteasome-dependent manner. A significant number of proteins undergo regulated, proteasome-dependent degradation under conditions of homeostatic plasticity (59). The identity and the role of the ubiquitin ligases involved, their substrate specificity, and the mechanisms controlling their function are still under investigation. The implication of Slap in activity-dependent degradation of NMDARs is a significant addition to our knowledge and can be used as a starting point to shed more light on the regulation of synaptic scaling by identifying, for instance, the ubiquitin ligases involved as well as the signaling cascades that activate them.
ACKNOWLEDGMENTS
P.A. is grateful for funding from the Biotechnology and Biological Sciences Research Council (United Kingdom). P.E.C. was funded by the Royal Society and a SouthWest London Academic Network studentship to S.S.S. R.J.Y.-M. acknowledges support from the 7th EU Framework Programme (PERSIST project, grant agreement 222878).
H.H.A.-R. is on leave from Department of Biology, Faculty of Science, Al-Mustansiriya University, Baghdad, Iraq.
P.A. conceived the project and interpreted data. S.S. performed experiments and contributed to the interpretation. P.E.C. performed the oocyte electrophysiology experiments, S.S.S. assisted with the recombinant NMDAR expression experiments, and H.H.A.-R. and R.J.Y.-M. provided the lentiviral expertise and vectors; V.T. contributed to materials and the conception of the project. P.A. drafted the manuscript, and all authors contributed to the draft and approved the final manuscript.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 4 February 2013
REFERENCES
- 1.Alifragis P, Molnar Z, Parnavelas JG. 2003. Restricted expression of Slap-1 in the rodent cerebral cortex. Gene Expr. Patterns 3:437–440 [DOI] [PubMed] [Google Scholar]
- 2.Manes G, Bello P, Roche S. 2000. Slap negatively regulates Src mitogenic function but does not revert Src-induced cell morphology changes. Mol. Cell. Biol. 20:3396–3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sosinowski T, Pandey A, Dixit VM, Weiss A. 2000. Src-like adaptor protein (SLAP) is a negative regulator of T cell receptor signaling. J. Exp. Med. 191:463–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Myers MD, Dragone LL, Weiss A. 2005. Src-like adaptor protein down-regulates T cell receptor (TCR)-CD3 expression by targeting TCRzeta for degradation. J. Cell Biol. 170:285–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fu AK, Hung KW, Fu WY, Shen C, Chen Y, Xia J, Lai KO, Ip NY. 2011. APC(Cdh1) mediates EphA4-dependent downregulation of AMPA receptors in homeostatic plasticity. Nat. Neurosci. 14:181–189 [DOI] [PubMed] [Google Scholar]
- 6.Pandey A, Duan H, Dixit VM. 1995. Characterization of a novel Src-like adapter protein that associates with the Eck receptor tyrosine kinase. J. Biol. Chem. 270:19201–19204 [DOI] [PubMed] [Google Scholar]
- 7.Dalva MB, Takasu MA, Lin MZ, Shamah SM, Hu L, Gale NW, Greenberg ME. 2000. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell 103:945–956 [DOI] [PubMed] [Google Scholar]
- 8.Irie F, Yamaguchi Y. 2004. EPHB receptor signaling in dendritic spine development. Front. Biosci. 9:1365–1373 [DOI] [PubMed] [Google Scholar]
- 9.Henkemeyer M, Itkis OS, Ngo M, Hickmott PW, Ethell IM. 2003. Multiple EphB receptor tyrosine kinases shape dendritic spines in the hippocampus. J. Cell Biol. 163:1313–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murai KK, Pasquale EB. 2004. Eph receptors, ephrins, and synaptic function. Neuroscientist 10:304–314 [DOI] [PubMed] [Google Scholar]
- 11.Gerlai R. 2001. Eph receptors and neural plasticity. Nat. Rev. Neurosci. 2:205–209 [DOI] [PubMed] [Google Scholar]
- 12.Henderson JT, Georgiou J, Jia Z, Robertson J, Elowe S, Roder JC, Pawson T. 2001. The receptor tyrosine kinase EphB2 regulates NMDA-dependent synaptic function. Neuron 32:1041–1056 [DOI] [PubMed] [Google Scholar]
- 13.Contractor A, Rogers C, Maron C, Henkemeyer M, Swanson GT, Heinemann SF. 2002. Trans-synaptic Eph receptor-ephrin signaling in hippocampal mossy fiber LTP. Science 296:1864–1869 [DOI] [PubMed] [Google Scholar]
- 14.Grunwald IC, Korte M, Adelmann G, Plueck A, Kullander K, Adams RH, Frotscher M, Bonhoeffer T, Klein R. 2004. Hippocampal plasticity requires postsynaptic ephrinBs. Nat. Neurosci. 7:33–40 [DOI] [PubMed] [Google Scholar]
- 15.Calò L, Cinque C, Patane M, Schillaci D, Battaglia G, Melchiorri D, Nicoletti F, Bruno V. 2006. Interaction between ephrins/Eph receptors and excitatory amino acid receptors: possible relevance in the regulation of synaptic plasticity and in the pathophysiology of neuronal degeneration. J. Neurochem. 98:1–10 [DOI] [PubMed] [Google Scholar]
- 16.Takasu MA, Dalva MB, Zigmond RE, Greenberg ME. 2002. Modulation of NMDA receptor-dependent calcium influx and gene expression through EphB receptors. Science 295:491–495 [DOI] [PubMed] [Google Scholar]
- 17.Knöll B, Drescher U. 2002. Ephrin-As as receptors in topographic projections. Trends Neurosci. 25:145–149 [DOI] [PubMed] [Google Scholar]
- 18.Dickson BJ. 2002. Molecular mechanisms of axon guidance. Science 298:1959–1964 [DOI] [PubMed] [Google Scholar]
- 19.Klein R. 2004. Eph/ephrin signaling in morphogenesis, neural development and plasticity. Curr. Opin. Cell Biol. 16:580–589 [DOI] [PubMed] [Google Scholar]
- 20.Martínez A, Soriano E. 2005. Functions of ephrin/Eph interactions in the development of the nervous system: emphasis on the hippocampal system. Brain Res. Brain Res. Rev. 49:211–226 [DOI] [PubMed] [Google Scholar]
- 21.Pasquale EB. 2005. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 6:462–475 [DOI] [PubMed] [Google Scholar]
- 22.Pasquale EB. 1997. The Eph family of receptors. Curr. Opin. Cell Biol. 9:608–615 [DOI] [PubMed] [Google Scholar]
- 23.Kayser MS, Nolt MJ, Dalva MB. 2008. EphB receptors couple dendritic filopodia motility to synapse formation. Neuron 59:56–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Akaneya Y, Sohya K, Kitamura A, Kimura F, Washburn C, Zhou R, Ninan I, Tsumoto T, Ziff EB. 2010. Ephrin-A5 and EphA5 interaction induces synaptogenesis during early hippocampal development. PLoS One 5:e12486 doi:10.1371/journal.pone.0012486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murai KK, Nguyen LN, Irie F, Yamaguchi Y, Pasquale EB. 2003. Control of hippocampal dendritic spine morphology through ephrin-A3/EphA4 signaling. Nat. Neurosci. 6:153–160 [DOI] [PubMed] [Google Scholar]
- 26.Carmona MA, Murai KK, Wang L, Roberts AJ, Pasquale EB. 2009. Glial ephrin-A3 regulates hippocampal dendritic spine morphology and glutamate transport. Proc. Natl. Acad. Sci. U. S. A. 106:12524–12529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davy A, Gale NW, Murray EW, Klinghoffer RA, Soriano P, Feuerstein C, Robbins SM. 1999. Compartmentalized signaling by GPI-anchored ephrin-A5 requires the Fyn tyrosine kinase to regulate cellular adhesion. Genes Dev. 13:3125–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Meima L, Kljavin IJ, Moran P, Shih A, Winslow JW, Caras IW. 1997. AL-1-induced growth cone collapse of rat cortical neurons is correlated with REK7 expression and rearrangement of the actin cytoskeleton. Eur. J. Neurosci. 9:177–188 [DOI] [PubMed] [Google Scholar]
- 29.Gao WQ, Shinsky N, Armanini MP, Moran P, Zheng JL, Mendoza-Ramirez JL, Phillips HS, Winslow JW, Caras IW. 1998. Regulation of hippocampal synaptic plasticity by the tyrosine kinase receptor, REK7/EphA5, and its ligand, AL-1/Ephrin-A5. Mol. Cell. Neurosci. 11:247–259 [DOI] [PubMed] [Google Scholar]
- 30.Waxman SJ, Kukanich B, Milligan M, Beard WL, Davis EG. 2012. Pharmacokinetics of concurrently administered intravenous lidocaine and flunixin in healthy horses. J. Vet. Pharmacol. Ther. 35:413–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajós F. 1975. An improved method for the preparation of synaptosomal fractions in high purity. Brain Res. 93:485–489 [DOI] [PubMed] [Google Scholar]
- 32.Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. 2008. NS21: re-defined and modified supplement B27 for neuronal cultures. J. Neurosci. Methods 171:239–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Erreger K, Geballe MT, Kristensen A, Chen PE, Hansen KB, Lee CJ, Yuan H, Le P, Lyuboslavsky PN, Micale N, Jorgensen L, Clausen RP, Wyllie DJ, Snyder JP, Traynelis SF. 2007. Subunit-specific agonist activity at NR2A-, NR2B-, NR2C-, and NR2D-containing N-methyl-D-aspartate glutamate receptors. Mol. Pharmacol. 72:907–920 [DOI] [PubMed] [Google Scholar]
- 34.Chen PE, Geballe MT, Katz E, Erreger K, Livesey MR, O'Toole KK, Le P, Lee CJ, Snyder JP, Traynelis SF, Wyllie DJ. 2008. Modulation of glycine potency in rat recombinant NMDA receptors containing chimeric NR2A/2D subunits expressed in Xenopus laevis oocytes. J. Physiol. 586:227–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wanisch K, Yanez-Munoz RJ. 2009. Integration-deficient lentiviral vectors: a slow coming of age. Mol. Ther. 17:1316–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yáñez-Muñoz RJ, Balaggan KS, MacNeil A, Howe SJ, Schmidt M, Smith AJ, Buch P, MacLaren RE, Anderson PN, Barker SE, Duran Y, Bartholomae C, von Kalle C, Heckenlively JR, Kinnon C, Ali RR, Thrasher AJ. 2006. Effective gene therapy with nonintegrating lentiviral vectors. Nat. Med. 12:348–353 [DOI] [PubMed] [Google Scholar]
- 37.Bolte S, Cordelieres FP. 2006. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 224:213–232 [DOI] [PubMed] [Google Scholar]
- 38.Wagey R, Hu J, Pelech SL, Raymond LA, Krieger C. 2001. Modulation of NMDA-mediated excitotoxicity by protein kinase C. J. Neurochem. 78:715–726 [DOI] [PubMed] [Google Scholar]
- 39.Cesca F, Baldelli P, Valtorta F, Benfenati F. 2010. The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol. 91:313–348 [DOI] [PubMed] [Google Scholar]
- 40.Yun ME, Johnson RR, Antic A, Donoghue MJ. 2003. EphA family gene expression in the developing mouse neocortex: regional patterns reveal intrinsic programs and extrinsic influence. J. Comp. Neurol. 456:203–216 [DOI] [PubMed] [Google Scholar]
- 41.Fu AK, Ip NY. 2007. Cyclin-dependent kinase 5 links extracellular cues to actin cytoskeleton during dendritic spine development. Cell Adh. Migr. 1:110–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu CT, Tran T, Sretavan D. 2010. Axonal/glial upregulation of EphB/ephrin-B signaling in mouse experimental ocular hypertension. Invest. Ophthalmol. Vis. Sci. 51:991–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Brien RJ, Mammen AL, Blackshaw S, Ehlers MD, Rothstein JD, Huganir RL. 1997. The development of excitatory synapses in cultured spinal neurons. J. Neurosci. 17:7339–7350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papa M, Bundman MC, Greenberger V, Segal M. 1995. Morphological analysis of dendritic spine development in primary cultures of hippocampal neurons. J. Neurosci. 15:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gerlai R, Shinsky N, Shih A, Williams P, Winer J, Armanini M, Cairns B, Winslow J, Gao W, Phillips HS. 1999. Regulation of learning by EphA receptors: a protein targeting study. J. Neurosci. 19:9538–9549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T, Goldfarb M, Yancopoulos GD. 1994. Ligands for EPH-related receptor tyrosine kinases that require membrane attachment or clustering for activity. Science 266:816–819 [DOI] [PubMed] [Google Scholar]
- 47.Pasquale EB. 2004. Eph-ephrin promiscuity is now crystal clear. Nat. Neurosci. 7:417–418 [DOI] [PubMed] [Google Scholar]
- 48.Flanagan JG, Vanderhaeghen P. 1998. The ephrins and Eph receptors in neural development. Annu. Rev. Neurosci. 21:309–345 [DOI] [PubMed] [Google Scholar]
- 49.Cowan CW, Shao YR, Sahin M, Shamah SM, Lin MZ, Greer PL, Gao S, Griffith EC, Brugge JS, Greenberg ME. 2005. Vav family GEFs link activated Ephs to endocytosis and axon guidance. Neuron 46:205–217 [DOI] [PubMed] [Google Scholar]
- 50.Chen C, Leonard JP. 1996. Protein tyrosine kinase-mediated potentiation of currents from cloned NMDA receptors. J. Neurochem. 67:194–200 [DOI] [PubMed] [Google Scholar]
- 51.Zheng F, Gingrich MB, Traynelis SF, Conn PJ. 1998. Tyrosine kinase potentiates NMDA receptor currents by reducing tonic zinc inhibition. Nat. Neurosci. 1:185–191 [DOI] [PubMed] [Google Scholar]
- 52.Kim HJ, Zou W, Ito Y, Kim SY, Chappel J, Ross FP, Teitelbaum SL. 2010. Src-like adaptor protein regulates osteoclast generation and survival. J. Cell. Biochem. 110:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lau A, Tymianski M. 2010. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460:525–542 [DOI] [PubMed] [Google Scholar]
- 54.Raymond LA, Tingley WG, Blackstone CD, Roche KW, Huganir RL. 1994. Glutamate receptor modulation by protein phosphorylation. J. Physiol. Paris 88:181–192 [DOI] [PubMed] [Google Scholar]
- 55.Grant ER, Bacskai BJ, Anegawa NJ, Pleasure DE, Lynch DR. 1998. Opposing contributions of NR1 and NR2 to protein kinase C modulation of NMDA receptors. J. Neurochem. 71:1471–1481 [DOI] [PubMed] [Google Scholar]
- 56.Raymond LA, Moshaver A, Tingley WG, Huganir RL. 1996. Glutamate receptor ion channel properties predict vulnerability to cytotoxicity in a transfected nonneuronal cell line. Mol. Cell. Neurosci. 7:102–115 [DOI] [PubMed] [Google Scholar]
- 57.Watt AJ, van Rossum MC, MacLeod KM, Nelson SB, Turrigiano GG. 2000. Activity coregulates quantal AMPA and NMDA currents at neocortical synapses. Neuron 26:659–670 [DOI] [PubMed] [Google Scholar]
- 58.Kato A, Rouach N, Nicoll RA, Bredt DS. 2005. Activity-dependent NMDA receptor degradation mediated by retrotranslocation and ubiquitination. Proc. Natl. Acad. Sci. U. S. A. 102:5600–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ehlers MD. 2003. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 6:231–242 [DOI] [PubMed] [Google Scholar]
- 60.Wang Y, Ota S, Kataoka H, Kanamori M, Li Z, Band H, Tanaka M, Sugimura H. 2002. Negative regulation of EphA2 receptor by Cbl. Biochem. Biophys. Res. Commun. 296:214–220 [DOI] [PubMed] [Google Scholar]
- 61.Walker-Daniels J, Riese DJ, Kinch MS. 2002. c-Cbl-dependent EphA2 protein degradation is induced by ligand binding. Mol. Cancer Res. 1:79–87 [PubMed] [Google Scholar]
- 62.Dragone LL, Myers MD, White C, Gadwal S, Sosinowski T, Gu H, Weiss A. 2006. Src-like adaptor protein (SLAP) regulates B cell receptor levels in a c-Cbl-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 103:18202–18207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dragone LL, Shaw LA, Myers MD, Weiss A. 2009. SLAP, a regulator of immunoreceptor ubiquitination, signaling, and trafficking. Immunol. Rev. 232:218–228 [DOI] [PubMed] [Google Scholar]
- 64.Kalo MS, Pasquale EB. 1999. Signal transfer by eph receptors. Cell Tissue Res. 298:1–9 [PubMed] [Google Scholar]
- 65.Wang YT, Salter MW. 1994. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature 369:233–235 [DOI] [PubMed] [Google Scholar]
- 66.Mulvey CS, Zhang K, Liu WH, Waxman DJ, Bigio IJ. 2011. Wavelength-dependent backscattering measurements for quantitative monitoring of apoptosis, Part 1: early and late spectral changes are indicative of the presence of apoptosis in cell cultures. J. Biomed. Opt. 16:117001 doi:10.1117/1.3644389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Arunachalam S, Waxman SR. 2011. Grammatical form and semantic context in verb learning. Lang. Learn. Dev. 7:169–184 [DOI] [PMC free article] [PubMed] [Google Scholar]