

Abstract
The protein tyrosine phosphatases (PTPs) T cell PTP (TCPTP) and PTP1B share a high level of catalytic domain sequence and structural similarity yet display distinct differences in substrate recognition and function. Their noncatalytic domains contribute to substrate selectivity and function by regulating TCPTP nucleocytoplasmic shuttling and targeting PTP1B to the endoplasmic reticulum (ER). The Drosophila TCPTP/PTP1B orthologue PTP61F has two variants with identical catalytic domains that are differentially targeted to the ER and nucleus. Here we demonstrate that the PTP61F variants differ in their ability to negatively regulate insulin signaling in vivo, with the nucleus-localized form (PTP61Fn) being more effective than the ER-localized form (PTP61Fm). We report that PTP61Fm is reliant on the adaptor protein Dock to attenuate insulin signaling in vivo. Also, we show that the PTP61F variants differ in their capacities to regulate growth, with PTP61Fn but not PTP61Fm attenuating cellular proliferation. Furthermore, we generate a mutant lacking both PTP61F variants, which displays a reduction in median life span and a decrease in female fecundity, and show that both variants are required to rescue these mutant phenotypes. Our findings define the role of PTP61F in life span and fecundity and reinforce the importance of subcellular localization in mediating PTP function in vivo.
INTRODUCTION
The phosphorylation of proteins on tyrosine residues is a fundamental aspect of cellular signaling, allowing cells to respond to varied extracellular and intracellular cues. Tyrosine phosphorylation is a reversible dynamic process controlled by the opposing actions of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). PTPs are a large and structurally diverse family of enzymes found in both eukaryotes and prokaryotes (1, 2). In mammals, the PTP superfamily includes 38 classical PTPs that act exclusively on tyrosine phosphorylated substrates (1, 2). PTPs have been implicated in various physiological processes, and aberrations in PTP function have been linked with many human diseases, including immune and neurological disorders, metabolic diseases, and cancer (1, 2).
The prototypic PTP1B and T cell PTP (TCPTP) are two of the most closely related PTPs in the human genome, sharing a high degree of similarity in primary (72% identity/86% similarity) and tertiary structures and virtually indistinguishable active sites (1, 3). In particular, the two PTPs share a second phosphotyrosine-binding pocket adjacent to the active site that allows for the selective recognition of tandem phosphorylated substrates (4, 5) such as the insulin receptor (IR) PTK (5) and Janus-activated PTKs (JAK) (6). However, despite this striking conservation in catalytic domain structure, the two PTPs display exquisite substrate preference in a cellular and biological context (1, 7). For example, PTP1B and TCPTP dephosphorylate different JAK family PTKs (PTP1B dephosphorylates JAK-2 and Tyk2, and TCPTP dephosphorylates JAK-1 and JAK-3) (6, 8) and can differentially contribute to the dephosphorylation of overlapping substrates such as c-Src; PTP1B dephosphorylates the C-terminal Y529 inhibitory site to activate c-Src, whereas TCPTP dephosphorylates the Y416 autophosphorylation site to inactivate c-Src (9–13). Furthermore, PTP1B and TCPTP can act cooperatively to regulate IR β-subunit Y1162/Y1163 phosphorylation to control the intensity and duration of IR activation and signaling, respectively (14), and can also function in concert to regulate JAK/signal transducer and activator of transcription (STAT) signaling (1); for example PTP1B can act at the level of the Y1007/Y1008 phosphorylated JAK2 (8, 15) and TCPTP at the level of the Y705 phosphorylated STAT3 (16) to attenuate leptin signaling. The overall difference in PTP1B versus TCPTP substrate selectivity and function is reflected by the overt phenotypic differences of PTP1B- versus TCPTP-null mice (17–19). Mice that are globally deficient for TCPTP die soon after birth from severe anemia and immune system dysfunction (19), whereas PTP1B knockout mice have a normal life span and exhibit enhanced insulin sensitivity and obesity resistance (17, 18).
The capacity of PTP1B and TCPTP to differentially contribute to cellular signaling has been ascribed to both inherent differences in catalytic domain substrate specificity and differences in their noncatalytic domains that contain distinct protein-protein interaction and subcellular targeting motifs (1). PTP1B is targeted to the endoplasmic reticulum (ER) by a hydrophobic C terminus and accesses substrates at cell-cell junctions and after receptor PTK endocytosis (20–24). The noncatalytic C terminus of PTP1B also contains a proline-rich sequence (PXXP motif) that allows PTP1B to interact with SH3 domains of proteins such as p130cas (25) to influence PTP1B substrate selectivity. On the other hand, TCPTP exists as two forms with distinct subcellular localizations (1). The TCPTP variants arise from alternative splicing to generate a 48-kDa protein (TC48) with a hydrophobic C terminus that is targeted to the ER similarly to PTP1B and a 45-kDa variant (TC45) that lacks the hydrophobic C terminus and is targeted to the nucleus by a bipartite nuclear localization signal (NLS) (1); both TCPTP variants lack the PXXP motif present in the C terminus of PTP1B. Importantly, although its nuclear locale affords TC45 access to nuclear substrates such as STAT-1, -3, -5, and -6 (16, 26–30), TC45 can also exit the nucleus to access substrates such as the IR and JAK-1/3 at the plasma membrane (6, 31–33). TC45 exit from the nucleus occurs in response to varied physiological stimuli, including hormones such as insulin, as well as cellular stresses such as hyperosmotic shock that activate the AMP-activated protein kinase (1, 31–33).
Drosophila melanogaster is an ideal organism to study the regulation and functions of PTPs, as many signaling pathways are highly conserved between mammals and flies (34). In Drosophila, PTP61F is the single orthologue of PTP1B and TCPTP (35). The catalytic domain of PTP61F exhibits approximately 60% similarity to that of PTP1B or TCPTP and includes the second phosphotyrosine-binding pocket (our unpublished data) that allows the recognition of tandem tyrosine phosphorylated substrates. Alternative splicing of the PTP61F message can result in two characterized variants with identical noncatalytic N termini and PTP catalytic domains but differing extreme C termini (Fig. 1): one has a hydrophobic C-terminal tail similar to PTP1B/TC48, is targeted to intracellular membranes including the ER, and will here be referred to as PTP61Fm, and the other lacks the hydrophobic C terminus, is targeted to the nucleus like TC45 by a yet to be defined NLS, and will hereon be referred to as PTP61Fn (35). Like PTP1B, both variants have a proline-rich sequence (five PXXP motifs) in their noncatalytic C-terminal domains (35) that allows for interactions with SH3 domain-bearing proteins such as the adaptor protein Dock (9, 25, 36–38). Like PTP1B and TCPTP, PTP61F has been shown to negatively regulate both IR and JAK/STAT signaling (38–40) as well as to play roles in actin remodeling and organization (41, 42). Moreover, as for PTP1B/TCPTP, PTP61F can recognize the corresponding tandem phosphorylated IRβ Y1162/Y1163 in Drosophila (dIR Y1153/Y1154) (38). In this study, we have compared the roles of PTP61Fm and PTP61Fn in IR regulation and tissue growth and generated PTP61F-deficient flies to assess PTP61F function in vivo. We report that the PTP61F variants can differentially regulate IR signaling and growth and that PTP61F-deficiency results in alterations in life span and female fecundity (reproductive capacity) associated with elevated levels of IR and JAK/STAT signaling.
Fig 1.
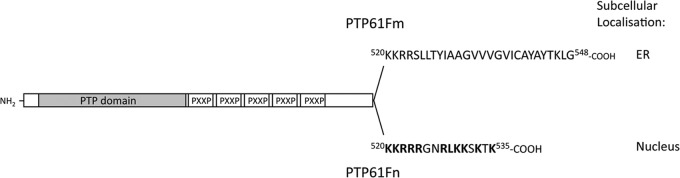
Schematic representation of PTP61Fm and PTP61Fn. The two PTP61F variants are identical except for their extreme C termini. The PTP61Fm hydrophobic C-terminal tail that targets PTP61Fm to membranes, including the ER (35), is shown. Basic residues in the PTP61Fn C terminus shown in bold are thought to constitute a nuclear localization sequence (NLS) targeting PTP61Fn to the nucleus (35). The PTP catalytic domain and the PXXP motifs that are present in both variants and allow interaction with SH3 domain-containing proteins are also highlighted.
MATERIALS AND METHODS
Drosophila stocks.
The w1118 (BL no. 5905) and Df(3L)ED4191/TM2 (BL no. 8049) lines were obtained from Bloomington Stock Center. The upstream activation sequence-RNA interference (UAS-RNAi) line against STAT92E was obtained from the Vienna Drosophila RNAi Center (VDRC no. 43866). The P{RS5}5-HA-2445 strain was obtained from the Szeged Stock Centre. The GMR-Gal4, ey-Gal4, UAS-dp110, UAS-dp110DN, and UAS-IR lines were kindly provided by Helena Richardson (Peter MacCallum Cancer Centre, Australia). The arm-Gal4 and dpp-Gal4 lines were kindly provided by Gary Hime (The University of Melbourne, Australia). The UAS-Dock line was kindly provided by Larry Zipursky (Howard Hughes Medical Institute, University of California, Los Angeles, CA). Stocks were reared on yeasted semolina-syrup medium in 30-ml vials or 250-ml bottles at 22°C in a room with access to natural light. Crosses were performed at 25°C with the exception of crosses involving UAS-dp110, which were performed at 29°C.
Generation of UAS-PTP61F transgenic lines.
Full-length cDNAs for PTP61Fm and PTP61Fn were amplified using reverse transcriptase PCR (RT-PCR) from whole-fly Canton-S mRNA. The fidelity of the coding region was verified by sequencing (Micromon, Monash University). The cDNAs were cloned into the Nmyc-pUAST vector (C. G. Warr, unpublished data) in frame with the initiation codon and three copies of the Myc tag-coding sequence such that the resulting proteins are N-terminally Myc tagged. Transgenic flies were generated via microinjection into w1118 embryos using standard procedures for P-element-mediated transformation. Several independent transgenic lines were generated and analyzed for each UAS construct.
Generation of the PTP61FΔ mutant.
PCR analysis was used to confirm that the {RS5}5-HA-2445 P element was inserted 66 bp upstream of the start codon of PTP61Fm and PTP61Fn. The P element was excised using the transposase source Δ2-3, and 327 stable excision lines were obtained for analysis. Of these, PTP61FΔ was confirmed by sequencing to have a 2,098-bp deletion mutation extending 970 bp upstream and 1,128 bp downstream of the P element insertion site, removing the first exon of PTP61Fm and PTP61Fn.
Protein extraction and immunoblot analysis.
Ten Drosophila heads or 20 pairs of ovaries were homogenized in lysis buffer (50 mM HEPES [pH 7.4], 1% [vol/vol] Triton X-100, 1% [vol/vol] sodium deoxycholate, 0.1% [vol/vol] SDS, 150 mM NaCl, 10% [vol/vol] glycerol, 1.5 mM MgCl2, 1 mM EGTA, 100 mM sodium fluoride, 5 μg/ml aprotinin [Sigma], 5 μg/ml leupeptin [Invitrogen], 1 μg/ml pepstatin A [Sigma], 1 mM benzamadine, 1 mM NaN3). Samples were incubated on ice for 45 min and clarified by centrifugation (16,000 × g, 15 min, 4°C) and equal amounts of protein were resolved by SDS-PAGE and transferred onto Immobilon-P (Millipore) for immunoblotting with the following antibodies: 1:1,000 anti-phosphorylated IRβ Y1162/Y1163 (Biosource), 1:4,000 anti-phosphorylated STAT92E Y704 serum (rabbit polyclonal raised to the peptide CVLDPVTG(pY)VKST), 1:1,000 anti-c-Myc (Sigma), and 1:7,000 antiactin (Neomarkers). Immunoblots were developed using enhanced chemiluminescence (Amersham) and medical X-ray film (AGFA).
Immunostaining.
Dissected tissue was fixed in 4% paraformaldehyde (PFA)–phosphate-buffered saline (PBS) for 30 min and washed in PBS-T (0.3 [vol/vol] Triton X-100–PBS) before being placed in 1% (wt/vol) bovine serum albumin [BSA]–PBS to block nonspecific binding. Primary antibodies were diluted in 1% (wt/vol) BSA–PBS to the following concentrations before overnight incubation at 4°C: anti-elav (DSHB), 1:50; anti-cleaved caspase-3 (Cell Signaling), 1:200; anti-vasa (DSHB), 1:50; anti-fasIII (DSHB), 1:50. After washing in PBS-T, the sample was incubated with the secondary antibody (anti-mouse antibody–Alexa Fluor 488 or anti-rabbit antibody–Alexa Fluor 568) diluted to 1:250 in 1% (wt/vol) BSA–PBS for 1 h at room temperature. Samples were washed in PBS-T and mounted in VectaShield mounting medium (Vector Laboratories, Inc.) and visualized using a Leica DMLB compound microscope or a Nikon C1 confocal microscope (Monash Microimaging, Monash University). Images were processed using LeicaIM50 imaging software, NIS Elements (Monash Microimaging, Monash University), or Image J.
BrdU staining.
Tissue was incubated with 500 μg/ml of BrdU (Sigma) for 60 min at 25°C while shaking, followed by washing with PBS to remove excess BrdU, and then fixed in 4% (wt/vol) PFA–PBS for 1 h at room temperature while shaking. Samples were washed three times quickly in PBS-T followed by 2 10-min washes in PBS and subjected to DNase treatment (85% PBS, 10% DNase buffer [Promega], 5% DNase I [1 U/1 μl stock; Promega]) at 37°C for 1.5 h while shaking. Samples were blocked in 1% (wt/vol) BSA–PBS for 1 h at room temperature. Following blocking, the samples were incubated with the primary antibody (1:50 mouse anti-BrdU [BD Biosciences] diluted in PBS–1% [wt/vol] BSA) overnight at 4°C while shaking followed by 3 20-min washes in PBS-T and then incubated overnight with the secondary antibody (1:250 anti-mouse antibody–Alexa Fluor 488 [Molecular Probes] diluted in 1% [wt/vol] BSA–PBS). Samples were washed with PBS-T and visualized using a Leica DMLB compound microscope. Images were processed using LeicaIM50 imaging software.
TUNEL staining.
Egg lays from PTP61FΔ homozygous and heterozygous flies were performed over 4 h to control density of progeny. Forty-four virgin female PTP61FΔ homozygous and heterozygous Drosophila flies were collected and were mated in separate vials to w1118 males (11 females/vial). Ovaries were dissected and subjected to terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) staining using an in situ cell detection kit, TMR Red (Roche), as per the manufacturer's protocol. Ovaries were visualized using a Leica DMLB compound microscope and processed using Leica IM50 imaging software.
Reverse transcriptase PCR.
mRNA was extracted from 10 adult Drosophila flies, and reverse transcriptase (RT) reactions were performed with Superscript III (Invitrogen) as per the manufacturer's instructions. PCR was performed using primers complementary to a 300-bp cDNA region (genomic DNA [gDNA]; 4,200 bp) within the first and second exons of PTP61Fm and PTP61Fn (forward primer, 5′-GTCCAGCTCTAGTTCTCCTC-3′; reverse primer, 5′-ACATCCCGATAGCGATTCAG-3′). The primers used for the positive control for the cDNA preparation were for the Synaptotagmin gene (forward primer, 5′-CGGATCCCTATGTCAAGGTG-3′; reverse primer, 5′-TCTGGTCGTGCTTCGAGAAG-3′). These primers also flanked an intron to distinguish between the presence of cDNA (200-bp product) and gDNA (300-bp product).
Light and scanning electron microscopy.
Drosophila flies were collected 3 to 5 days posteclosion. Light microscopy was performed at a magnification of ×110 on an Olympus XZS16 dissecting microscope, and images were processed using Leica IM50 imaging software. To prepare samples for scanning electron microscopy, 10 adult flies per sample were fixed overnight in 5% gluteraldehyde-Superfix (with 0.025× volume detergent) solution at 4°C. The samples were washed 3 times for 10 min in 0.1 M sodium phosphate buffer and then postfixed in 1% osmium tetroxide in 0.1 M sodium phosphate buffer for 1 h. Samples were washed 3 times for 7 min in 0.1 M sodium phosphate buffer and were incubated overnight at 4°C in the final wash. Samples were dehydrated with increasing concentrations of ethanol (EtOH) at room temperature: 50% (vol/vol) EtOH for 10 min, 70% (vol/vol) EtOH for 10 min, 90% (vol/vol) EtOH for 10 min, 100% (vol/vol) EtOH for 30 min (twice), and absolute dry EtOH for 30 min (twice). Samples were then dried in hexamethyldisilazane (HMDS) at room temperature in the following ratios: 1 part HMDS with 2 parts dry ethanol for 5 min, 2 parts HMDS and 1 part dry ethanol for 5 min, and straight HMDS for 5 min. The dried samples were mounted onto stainless steel stubs on double-sided tape and gold plated in a Polaron Sputter coater in an argon atmosphere. Scanning electron microscopy was performed on a Hitachi S570 scanning electron microscope at 15 kV and magnification of ×110 (Monash MicroImaging, Monash University). Images were captured using Spectrum NT software.
Wing growth experiments.
Wing centroid size was calculated using IMP Morphogenetic software (provided by Monash University and generated by F. J. Rohlf, http://www.canisius.edu/∼sheets/morphsoft.html). Wing growth experiments using dpp-Gal4 were performed as described in reference 43. For determination of wing cell size and number, dissected wings were placed in PBS-T in an Eppendorf tube for 90 min and mounted on a microscope slide in a solution of 80% (vol/vol) glycerol–PBS under a coverslip. Images were captured using an Olympus BX51 compound microscope using OlyVIA software and Olympus VS-ASW virtual slide system software (Monash Microimaging, Monash University). Determination of wing cell size and cell number was performed using Image J Software. Statistical analysis was performed using the two-tailed Student t test.
Longevity assays.
Drosophila stocks used in the life span experiments were maintained at 25°C for 2 generations. Fifteen females and 10 males were mated in bottles at 25°C for 24 h to generate the progeny with the genotypes required for the life span analysis. Progeny were collected within 24 h of eclosion. For each genotype, 3 to 5 vials containing 10 females and 10 males were set up for life span analysis and were maintained at 25°C throughout the experiment. Flies were tipped into fresh vials every 2 to 3 days, and the number and sex of dead flies were determined. Data were analyzed using GraphPad Prism 5 Software, and statistical analysis was performed using the log rank test (df = 1).
Fecundity assays.
Single-pair matings were performed between virgin female and male Drosophila flies between 2 and 5 days old (n = 10 per genotype). Flies were stored in the dark at 25°C for 2 days to acclimatize. Following this, the number of eggs laid per female fly was determined every 24 h for 5 days. Statistical analysis was performed using a two-tailed Student t test.
RESULTS
Regulation of insulin receptor signaling by PTP61Fm and PTP61Fn.
Previously we have reported that the Drosophila IR can serve as a substrate for PTP61F in vitro and that the ER-targeted form of PTP61F can attenuate IR signaling in the developing Drosophila eye (38). Overexpression of the IR during Drosophila eye development causes pronounced overgrowth associated with significantly increased cellular proliferation and hypertrophy (44). As a first step toward assessing the biological roles of the nucleus- versus ER-targeted forms of PTP61F, we compared their capacity to suppress IR signaling in the developing Drosophila eye (Fig. 2A). We generated transgenic flies carrying the PTP61Fm versus PTP61Fn coding sequences under the control of the UAS regulatory sequence and tagged at the N terminus with a myc epitope tag, and we induced expression in the developing eye with GMR-Gal4. Although the transgene expression of PTP61Fm was greater than of that PTP61Fn (Fig. 2B), we found that PTP61Fn (but not PTP61Fm) overexpression alone caused a disrupted bristle pattern and mild rough-eye phenotype (Fig. 2A), consistent with the two variants having the capacity to exert differential effects in vivo. IR overexpression using GMR-Gal4 resulted in severe eye overgrowth, with substantial outgrowth and disorganization, as reflected by loss of ommatidial structure and bristle pattern (Fig. 2A). As reported previously (38), PTP61Fm coexpression attenuated, but did not prevent, the IR-mediated eye overgrowth (Fig. 2A). Strikingly, coexpression of the nucleus-targeted PTP61Fn prevented the overgrowth, with any remaining eye roughness being comparable to that seen with PTP61Fn expression alone (Fig. 2A). These results provide evidence for the nucleus- and ER-targeted PTP61F variants exerting differential effects on IR signaling in vivo.
Fig 2.

Differential regulation of IR signaling by PTP61Fm and PTP61Fn. (A) Scanning electron micrographs of a control Drosophila eye (GMR-Gal4/+) and those overexpressing IR alone using GMR-Gal4 (GMR>IR) and either N-terminal myc-tagged PTP61Fm (GMR>PTP61Fm) or PTP61Fn (GMR>PTP61Fn) alone using GMR-Gal4 or IR and either N-terminal myc-tagged PTP61Fm (GMR>IR+PTP61Fm) or PTP61Fn (GMR>IR+PTP61Fn) using GMR-Gal4. PTP61Fm attenuates and PTP61Fn almost completely suppresses the eye overgrowth induced by IR overexpression. Results shown are representative of two independent experiments (n = 4 or 5 per genotype). (B) The UAS-myc-PTP61Fm transgene is expressed at a higher level than UAS-myc-PTP61Fn. Protein extracts from the heads of GMR>PTP61Fm or GMR>PTP61Fn flies were resolved by SDS-PAGE and immunoblotted with anti-myc and antiactin. Results shown are representative of two independent experiments. (C) PTP61Fn suppresses IR phosphorylation level to that seen in controls. Protein extracts from the heads of GMR>IR or GMR>IR+PTP61Fn flies or from UAS-IR+PTP61Fn control flies were resolved by SDS-PAGE and immunoblotted with antibodies to the Y1553/Y1554 phosphorylated IR (p-IR) and actin. (D) PTP61Fm does not by itself suppress IR phosphorylation but does so in combination with Dock. Protein extracts from the heads of GMR>IR or GMR>IR+PTP61Fm flies or those overexpressing IR plus PTP61Fm and Dock (GMR>IR+PTP61Fm+Dock), or Dock alone (GMR>Dock) or from UAS-IR+PTP61Fn+Dock control flies were resolved by SDS-PAGE and immunoblotted with antibodies to the Y1553/Y1554 phosphorylated IR (p-IR) and actin. (E) Scanning electron micrographs show that compared to a control eye (GMR-Gal4/+), overexpression of the catalytic subunit of PI3K (dp110) using GMR-Gal4 (GMR>p110) results in significant eye overgrowth and this is not suppressed by coexpression of PTP61Fn (GMR>p110+PTP61Fn). Results shown are representative of two independent experiments (n = 4 or 5 per genotype).
Since PTP61Fn is targeted to the nucleus, we asked whether PTP61Fn might exert its effects at the level of the IR, as reported for its mammalian counterpart, TC45, which can exit the nucleus in response to insulin to dephosphorylate the IR (31, 32), or otherwise whether PTP61Fn might act downstream of the IR on a nuclear substrate to attenuate the eye overgrowth. We have reported previously that PTP61F can dephosphorylate the Drosophila IR Y1553/Y1554 activation loop tandem phosphorylation site that is required for IR activation (38). Accordingly, we assessed the status of IR Y1553/Y1554 phosphorylation in Drosophila eye homogenates from GMR-Gal4; UAS-IR transgenic flies with and without PTP61F coexpression; we compared the effects of PTP61Fm and PTP61Fn on IR phosphorylation. Consistent with the prevention of IR-driven eye overgrowth, we found that PTP61Fn suppressed IR phosphorylation to the level seen in controls (Fig. 2C). In contrast, PTP61Fm expression had no overt effect on IR phosphorylation (Fig. 2D), in keeping with the modest effects on eye overgrowth (Fig. 2A). Previously, we have reported that the adaptor protein Dock associates with PTP61Fm and recruits PTP61Fm to the IR and that this is required for effective IR dephosphorylation in vitro and for the attenuation of IR-mediated eye overgrowth in vivo (38). Consistent with this, we found that the coexpression of PTP61Fm with Dock allowed for the effective suppression of IR phosphorylation (Fig. 2D). These results suggest that both PTP61F variants can dephosphorylate the IR in vivo, albeit in a distinct manner, with PTP61Fm but not PTP61Fn being reliant on Dock.
To exclude PTP61Fn also acting downstream of the IR to suppress IR-driven eye overgrowth, we next asked if PTP61Fn could suppress the eye overgrowth that is induced by overexpressed phosphatidylinositol 3-kinase (PI3K). PI3K is activated downstream of IR and IRS-1/2/3/4 (Chico in flies) at the plasma membrane and mediates many of the metabolic and mitogenic effects of IR signaling (45, 46). GMR-Gal4-driven expression of the wild-type catalytic subunit of PI3K (UAS-dp110) induced a significant eye overgrowth phenotype (Fig. 2E), although not as severe as that induced by IR overexpression. However, PTP61Fn coexpression had no overt effect on the rough eye phenotype mediated by dp110 (Fig. 2E). These results are consistent with PTP61Fn exerting its effects upstream of PI3K at the level of the IR, as has been shown for its mammalian counterpart, TC45, which dephosphorylates the IR at the plasma membrane (31).
Regulation of growth by PTP61Fm and PTP61Fn.
The IR and the pathways it controls play an important role in organismal growth in both mammals and flies (47–49). In flies, mutations in genes encoding negative regulators such as phosphatase and tensin homologue (PTEN) and tuberous sclerosis 1/2 (Tsc1/2) enhance IR signaling and promote overgrowth (43, 50–53), whereas suppressing IR signaling by inactivating positive components of IR signaling using loss-of-function mutations (e.g., chico, dp110, or dAkt) (45, 46), expression of dominant negatives (e.g., dominant negative dp110) (51), or the overexpression of negative regulators such as PTEN, Tsc1/2, or Susi (43, 50, 52, 54) decreases cell size and cell number and overall tissue and organism size. To examine whether the nucleus- and ER-targeted PTP61F variants could affect tissue growth, we overexpressed PTP61Fm versus PTP61Fn in the Drosophila wing using dpp-Gal4, which drives expression in the wing imaginal disc between the developing third and fourth longitudinal wing veins (Fig. 3A) (43). This approach has been used previously to characterize the effects of dp110 and dPTEN on growth (43, 51). Overexpression of PTP61Fn with dpp-Gal4 was sufficient to significantly reduce growth to approximately 70% of that in controls (Fig. 3B). However, overexpression of PTP61Fm had no significant effect, despite being expressed at higher levels than PTP61Fn (Fig. 2B). PTP61Fm had no effect even when the transgene copy number was increased to two (Fig. 3B). The decrease in growth caused by PTP61Fn overexpression was attributable to a decrease both in cell size and in cell number (Table 1). Thus, these results are consistent with the nucleus-targeted PTP61Fn playing an important role in growth, in line with the regulation of IR signaling.
Fig 3.
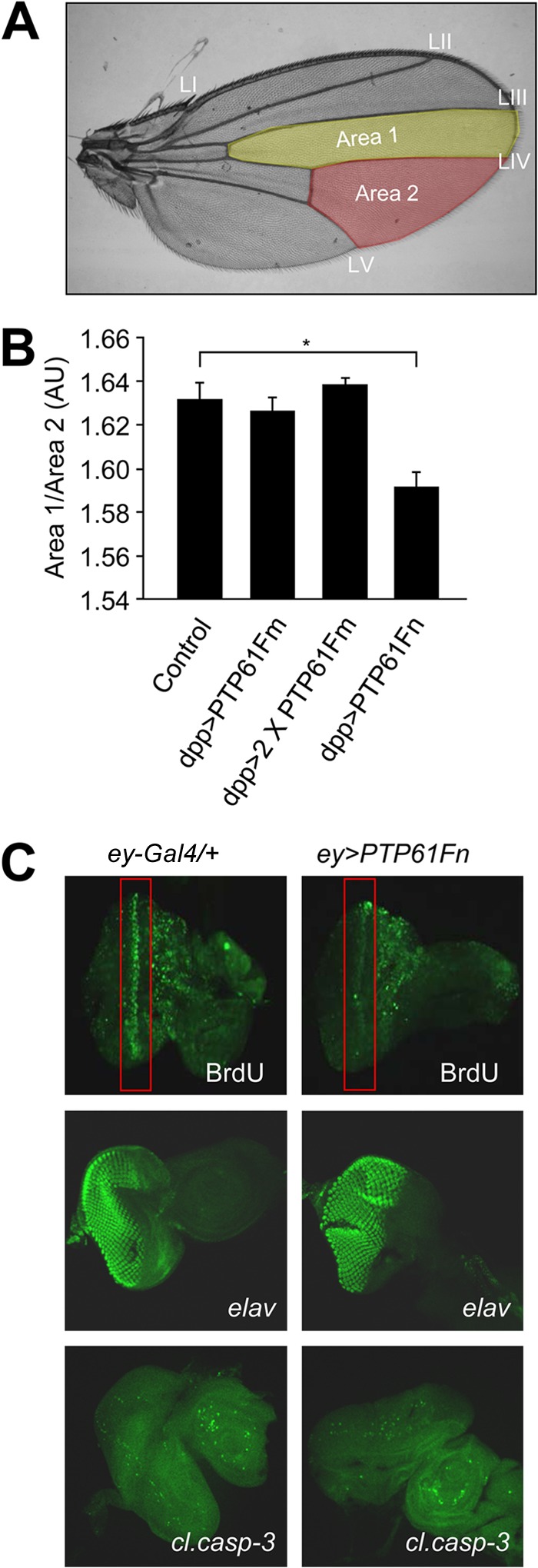
PTP61Fn is a negative regulator of growth and cellular proliferation. (A) The dpp-Gal4 driver was used to overexpress PTP61Fm versus PTP61Fn in the region of the Drosophila wing between longitudinal wing veins LIII and LIV bordered by the posterior cross vein and wing margin (area 1, yellow highlighting). The effect on growth was assessed by comparing the area of this region to the area of a region within the same wing where the dpp-Gal4 driver was not expressed, between wing veins LIV and LV (area 2, red highlighting). (B) Overexpression of PTP61Fn in the Drosophila wing significantly reduced growth (P = 0.02) compared to control (dpp-Gal4/+); however, overexpression of 1 or 2 copies of PTP61Fm had no significant effect. Results are means ± standard errors of the means (SEM) for n = 6 per genotype. (C) Immunostaining of Drosophila third-instar larval eye imaginal discs overexpressing PTP61Fn under ey-Gal4 and control flies (ey-Gal4/+), showing that PTP61Fn overexpression reduces cell proliferation as assessed by BrDu incorporation, without affecting cell differentiation (anti-elav) or apoptosis (anti-cleaved caspase-3 [cl.casp-3]).
Table 1.
Overexpression of PTP61Fn decreases cell size and number in the adult winga
| Characteristic | Value for: |
P value | |
|---|---|---|---|
| Control (dpp-Gal4/+) flies | dpp>PTP61Fn mutant flies | ||
| No. of wings | 4 | 5 | |
| Area 1 (105 μm2) | 3.18 ± 0.05 | 2.29 ± 0.05 | |
| Area 2 (105 μm2) | 3.32 ± 0.05 | 3.41 ± 0.02 | |
| Area ratio (area 1/area 2) | 0.96 ± 0.02 | 0.67 ± 0.015 | |
| Percent difference in area ratio versus controlb | 0 ± 1 | −30.2 ± 0.75 | |
| Cell density of area 1 (10−3 cells/μm2)c | 5.6 ± 0.3 | 6.9 ± 0.2 | |
| No. of cells in area 1d | 1778 ± 83 | 1566 ± 44 | 0.047 |
| Cell area in area 1 (μm2)e | 180 ± 10 | 146 ± 3 | 0.01 |
| Cell density of area 2 (10−3 cells/μm2)c | 5.5 ± 0.2 | 5.9 ± 0.1 | |
| No. of cells in area 2d | 1826 ± 42 | 2015 ± 52 | 0.085 |
| Cell area in area 2 (μm2)e | 182 ± 6 | 170 ± 4 | 0.11 |
PTP61Fn was overexpressed in a segment of the wing using dpp-Gal4 (area 1), and the effect on cell size and cell number compared to a control region (area 2) was analyzed. Overexpression of PTP61Fn (dpp>PTP61Fn) caused a 30% reduction in growth within area 1 compared to control due to a reduction in both cell size and cell number. No significant difference was observed in cell size or number in area 2 between genotypes. Results shown are means ± standard errors of the means (SEM).
Generated by comparing the experimental area ratio with the control area ratio.
Determined by counting the wing hairs (each representing a single cell) in a 200- by 100-μm rectangle. For area 1, the rectangle was located near the wing margin with the long edge of the rectangle parallel to LIII. For area 2, the rectangle was parallel to LIV near the wing margin.
Generated by multiplying the size of the area with the cell density of the area.
Reciprocal of cell density.
We then asked whether the decreased growth accompanying PTP61Fn overexpression was due to decreased cellular proliferation or to increased apoptosis and/or whether PTP61Fn overexpression affected cellular differentiation. To this end, PTP61Fn was overexpressed in the third-instar larval eye-antennal imaginal disc using ey-Gal4 (55). BrdU labeling of PTP61Fn-overexpressing imaginal discs demonstrated a clear reduction in cellular proliferation compared to controls (Fig. 3C). Cellular differentiation and apoptosis were examined using the neuronal marker anti-elav (50) and the apoptotic cell marker anti-cleaved-caspase-3 (56), respectively. No significant differences in cellular differentiation or apoptosis due to PTP61Fn overexpression were observed (Fig. 3C). Therefore, these results indicate that PTP61Fn suppresses wing growth by attenuating cellular proliferation.
Normal growth but altered life span and female fecundity in PTP61FΔ mutant flies.
To further delineate the roles of the nucleus- and ER-targeted forms of PTP61F in vivo, we generated a loss-of-function mutant with the ultimate aim of performing rescue experiments with the PTP61F variants. We used imprecise P-element excision to generate a deletion mutation, PTP61FΔ, lacking the expression of PTP61Fm and PTP61Fn. The P element in the {RS5}5-HA-2445 strain, which is inserted 66 bp upstream of the start codon utilized by PTP61Fm and PTP61Fn, was excised, and a 2,098-bp deletion mutation was recovered. PCR and sequencing showed that the deletion extended 970 bp upstream from the insertion site and 1,128 bp downstream and removed the first exon utilized by PTP61Fm and PTP61Fn (see Fig. S1A in the supplemental material); this deletion included the promoter and start codon of PTP61Fm and PTP61Fn. RT-PCR confirmed the deletion and lack of expression of PTP61Fm and PTP61Fn in the homozygous PTP61FΔ mutants, although PTP61Fm and PTP61Fn expression was readily detected in w1118 control flies and in flies in which a precise excision of the P element had occurred (see Fig. S1B and C in the supplemental material). Despite the overt effects of PTP61Fn overexpression on wing size, we found that homozygous PTP61FΔ flies were viable and of normal size, as measured by wing centroid size (see Fig. S2 in the supplemental material).
Given the capacity of PTP61F to regulate IR signaling, we asked whether other known life history traits controlled by IR signaling might be altered in PTP61FΔ flies. Decreased IR signaling has been linked with increased longevity in flies (57–61), thus raising the possibility that heightened IR signaling in PTP61FΔ mutants conversely decreases longevity. Accordingly, we assessed the life span of PTP61FΔ mutant flies. We found that PTP61FΔ female flies exhibited a significant reduction in life span as assessed by comparing survival curves using the log rank test (P < 0.001) (Fig. 4); similar results were seen in males (data not shown). The decrease in life span mapped to the PTP61F region as it was also observed in transheterozygotes for the PTP61FΔ allele and a genomic deficiency uncovering PTP61F (see Fig. S3 in the supplemental material; data not shown).
Fig 4.
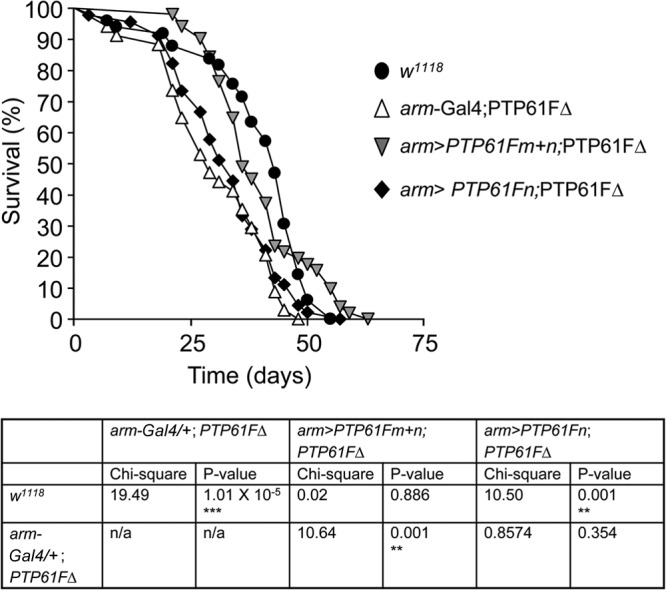
PTP61FΔ mutant flies have a decreased life span. The life span of PTP61FΔ (arm-Gal4/+; PTP61FΔ) female flies is significantly reduced compared to that of w1118 female controls (P < 0.0001). Constitutive overexpression of PTP61Fm and PTP61Fn in the PTP61FΔ background using arm-Gal4 (arm>PTP61Fm+n; PTP61FΔ) is sufficient to rescue the life span defect of PTP61FΔ mutants; however, overexpression of PTP61Fn alone using arm-Gal4 (arm>PTP61Fn; PTP61FΔ) does not rescue the life span defect. n = 30 to 50. Statistical analysis was performed using the log rank test.
Since altered IR signaling has also been linked with the regulation of reproduction and fertility (57, 58, 61, 62), we next assessed PTP61FΔ fertility. Although no defect in male fertility was observed, female fecundity (number of eggs laid per fly per day) was significantly reduced in PTP61FΔ flies (Fig. 5A). To define the nature of the decrease in PTP61FΔ female fecundity, we examined the ovaries for defects in egg chamber development. Ovarian somatic and germ line cells were visualized using the markers fasciclin III and vasa, respectively (63–66). The number and morphology of somatic cells surrounding the developing egg chambers in PTP61FΔ mutant ovaries appeared to be similar to those of w1118 control flies (data not shown). However, compared with ovaries from the w1118 control strain, PTP61FΔ ovaries lacked late-stage egg chambers, particularly those at stage 10 or later, and had an accumulation of germ line stem cells and/or early-stage egg chambers (Fig. 5B). In addition, ovaries from PTP61FΔ females showed an approximate 8-fold increase in the number of apoptotic pre-stage 10 egg chambers (as assessed by TUNEL staining) compared to PTP61FΔ heterozygotes (Fig. 5C). These results indicate that PTP61F deficiency increases pre-stage 10 egg chamber apoptosis, resulting in a lack of mature oocytes and a consequent reduction in female fecundity. Taken together, these results show that PTP61FΔ mutant flies exhibit a phenotype consistent with heightened IR signaling.
Fig 5.
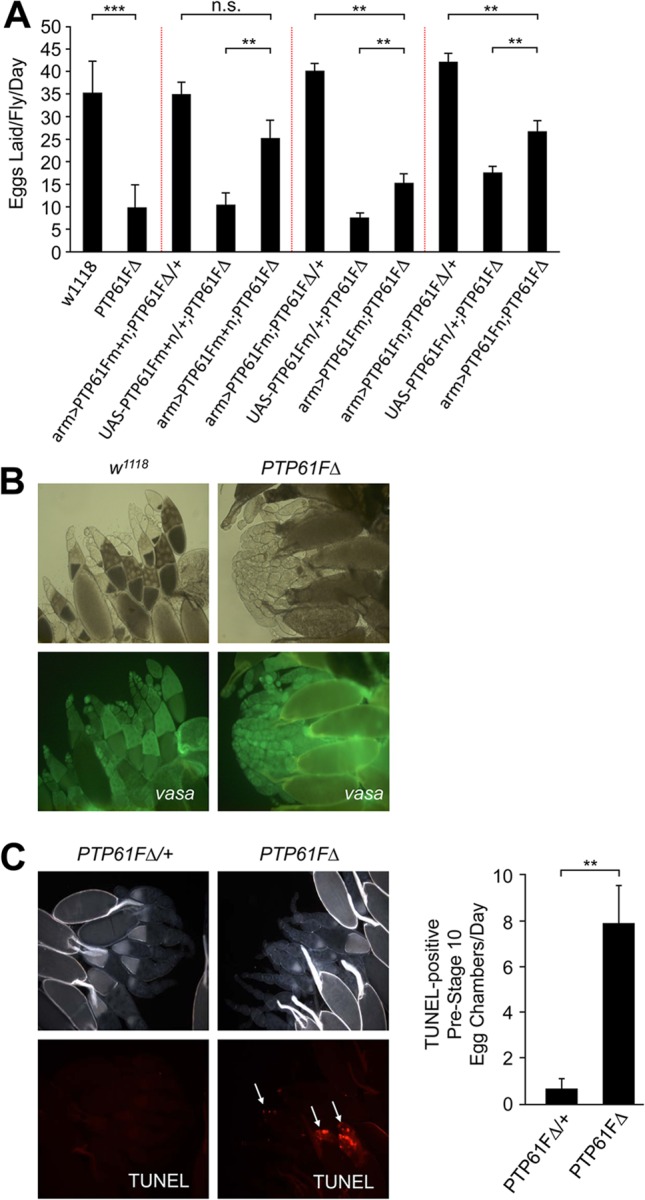
PTP61FΔ mutant flies have reduced female fecundity due to increased apoptosis in developing egg chambers. (A) Fecundity assays were performed using single-pair matings of females of the indicated genotype. Fecundity assays separated by dashed lines were performed independently of each other. PTP61FΔ females have significantly decreased fecundity compared to w1118 female controls (P = 9 × 10−5). When overexpressed together using arm-Gal4, PTP61Fm and PTP61Fn (arm>PTP61Fm+n) significantly rescued the fecundity defect of the PTP61FΔ mutant. PTP61Fm+n restored fecundity to a level not significantly different from that of control PTP61FΔ heterozygotes overexpressing PTP61Fm+n (P = 0.07). Overexpression of PTP61Fm or PTP61Fn individually in the PTP61FΔ mutant background resulted in only a partial rescue of fecundity, which was statistically different from what was observed in controls overexpressing the PTPs in the heterozygote background. Results are means ± standard errors of the means (SEM) for n = 8 to 10. **, P < 0.01; ***, P < 0.001. (B) Ovaries from w1118 and PTP61FΔ females were visualized under bright field and also immunostained with anti-vasa antibodies to identify germ line cells in the ovary. Ovaries from PTP61FΔ mutant females lack later-stage egg chambers (post-stage 10) compared to controls. Results shown are representative of 3 experiments. (C) TUNEL staining of egg chambers (magnification, ×100) from PTP61FΔ homozygous and heterozygous female ovaries demonstrated that in the PTP61FΔ mutants there was an approximate 8-fold increase in the number of apoptotic pre-stage 10 egg chambers compared to PTP61FΔ heterozygotes. Results are means ± SEM for n = 8. **, P < 0.01.
Regulation of life span and female fecundity by PTP61Fm and PTP61Fn.
Having established that PTP61F deficiency decreases life span and fecundity, we next determined the roles of the nucleus- and ER-targeted forms of PTP61F in these specific traits. First, we confirmed that the decrease in PTP61FΔ life span was due to loss of PTP61F by performing rescue experiments. We initially attempted the rescue experiments using the constitutive driver actin-Gal4 to drive expression of PTP61Fm and/or PTP61Fn; however, while overexpressing PTP61Fm using actin-Gal4 gave viable flies, overexpressing PTP61Fn with actin-Gal4 resulted in lethality (data not shown). We thus used the weaker constitutive driver arm-Gal4 for experiments involving PTP61Fn, as driving PTP61Fn with arm-Gal4 gave viable flies. Expression of both PTP61Fm and PTP61Fn using arm-Gal4 in the PTP61FΔ background restored life span to levels in controls (as assessed by comparing survival curves using the log rank test) (P = 0.89) (Fig. 4; male life span data not shown), confirming that the decrease in PTP61FΔ flies' life span was indeed due to loss of PTP61F. Next, we determined the roles of the individual PTP61F isoforms. Although the combined expression of PTP61Fm and PTP61Fn rescued the decreased longevity evident in the PTP61FΔ flies, this was not the case when the PTP61F variants were expressed individually. Overexpression of PTP61Fn alone did not increase life span to control levels (P = 0.001), and the life span of these flies was not significantly different from that of PTP61FΔ flies (P = 0.354) (Fig. 4; male life span data not shown). PTP61Fm overexpression with the stronger actin-Gal4 driver also did not increase life span (data not shown). Hence, these results argue for both PTP61Fm and PTP61Fn being required for the maintenance of life span.
Next, we assessed the individual effects of PTP61Fm and PTP61Fn on female fecundity. As for life span, we confirmed that the decrease in female fecundity in PTP61FΔ could be largely rescued by the expression of both PTP61Fm and PTP61Fn using arm-Gal4, with no statistical difference evident between arm-Gal4; UAS-PTP61Fm+n; PTP61FΔ/+ and arm-Gal4; UAS-PTP61Fm+n; PTP61FΔ flies (Fig. 5A). Furthermore, as for life span, we found that both variants were required for the rescue of female fecundity, with the expression of either PTP61Fm or PTP61Fn alone using arm-Gal4 resulting only in a partial rescue (Fig. 5A). Taken together, these results point toward a lack of functional redundancy between the PTP61F variants in distinct biological processes.
The role of IR and STAT92E signaling in the PTP61FΔ female fecundity defect.
The PTP61FΔ female fecundity phenotype could be the result of deregulated IR signaling, since insulin-like peptides and the IR have been implicated in the regulation of germ line stem cell division, cyst development, and progression through oogenesis (62). However, in addition to regulating IR signaling, previous studies have established the capacity of PTP61F (39, 40) and its mammalian counterparts, PTP1B and TCPTP, to regulate JAK/STAT signaling (6, 8). The JAK/STAT pathway in flies (Hop/STAT92E [67, 68]) is involved in the regulation of germ line and stem cell number through interactions with dpp signaling, and increases in JAK/STAT signaling promote germ line tumors (69, 70). Therefore, to explore the molecular basis for the fecundity defect in PTP61FΔ female flies, we first assessed the activation status of these pathways in ovaries using antibodies to the Y1553/Y1554 phosphorylated IR and the Y711 phosphorylated STAT92E. Ovaries from control (w1118) and heterozygous versus homozygous PTP61FΔ flies were processed for immunoblot analysis. We found striking increases in IR and STAT92E phosphorylation in homozygous mutant ovaries, consistent with the IR and STAT92E serving as substrates for PTP61F (Fig. 6A). Interestingly, STAT92E but not IR phosphorylation was increased in the ovaries of flies that were heterozygous for PTP61FΔ (Fig. 6A), highlighting the sensitivity of the Hop/STAT92E pathway to PTP61F deficiency.
Fig 6.
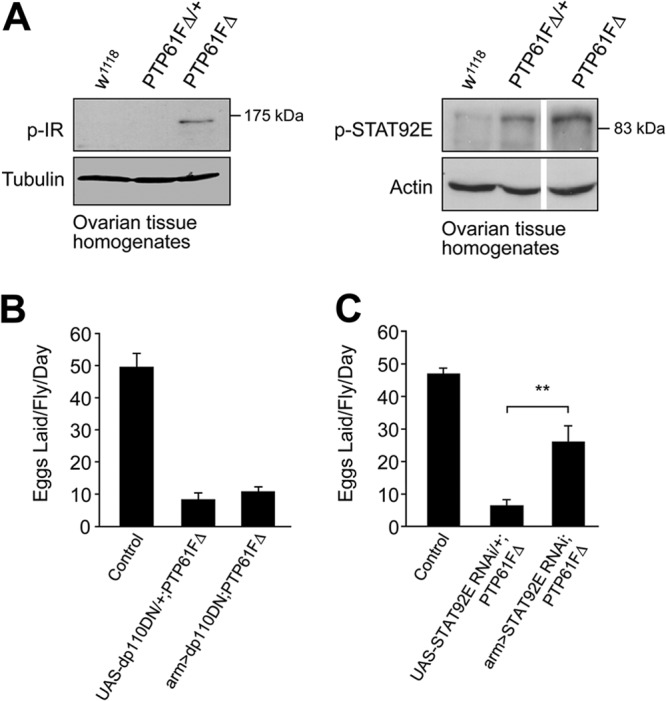
Heightened STAT92E signaling contributes to the female fecundity defect in PTP61FΔ mutant flies. (A) Protein extracts from ovarian tissue were immunoblotted with antibodies against the phosphorylated and activated forms of STAT (p-STAT92E) and the IR (p-IR). p-IR is increased in the ovaries of PTP61FΔ homozygous mutants. p-STAT92E is increased in both PTP61FΔ homozygotes and heterozygotes. Results shown are representative of at least 2 or 3 independent experiments. (B and C) Fecundity assays were performed using single-pair matings of females of the indicated genotype with w1118 males. Results are means ± SEM; n = 8 to 10. **, P < 0.01. (B) Reducing PI3K signaling in the PTP61FΔ mutant background by overexpressing dominant negative dp110 (dp110DN) using the arm-Gal4 driver did not restore fecundity (P = 0.36). Control, arm>dp110DN; PTP61FΔ/+. (C) Suppression of STAT92E in the PTP61FΔ homozygous background using in vivo RNAi (STATRNAi) resulted in a partial rescue of fecundity. Control, arm>STATRNAi; PTP61FΔ/+.
Next, we investigated the relative contributions of the heightened IR versus STAT92E signaling to the fecundity defect in PTP61FΔ female flies. To investigate the contributions of IR activation to the fecundity defect, we suppressed IR signaling in homozygous PTP61FΔ mutants by overexpressing a dominant negative form of PI3K (dp110DN) (51) with arm-Gal4. Although dp110DN effectively suppressed IR-driven eye overgrowth when expressed using the GMR-Gal4 driver (see Fig. S4 in the supplemental material), it did not improve fecundity (number of eggs laid per fly per day) in PTP61FΔ flies (Fig. 6B). Therefore, these results point to PI3K-independent or alternate pathways contributing to the fecundity defect in PTP61FΔ mutants. To determine whether the heightened STAT92E phosphorylation might contribute to the fecundity defect, we suppressed the Hop/STAT92E pathway by knocking down the expression of STAT92E by RNA interference. The arm-Gal4 driver was used to express a UAS-STAT92E RNAi construct in the PTP61FΔ background. We found that suppression of STAT92E in the PTP61FΔ mutant background resulted in a significant, albeit partial rescue of fecundity (Fig. 6C). Therefore, these results argue for heightened STAT92E signaling being at least an important contributing factor to the fecundity defect associated with PTP61F-deficiency.
DISCUSSION
There have been numerous studies highlighting the exquisite specificity of even closely related phosphatases, such as PTP1B and TCPTP or SHP-1 and SHP-2, in varied cellular and biological contexts (reviewed in reference 1). In particular, early studies using chimeric PTP1B/TCPTP or SHP-1/SHP-2 proteins (33, 71) established the importance of inherent catalytic domain specificity to PTP function. Here, we have taken advantage of the Drosophila PTP61F nucleus- and ER-targeted variants that are identical in all but their extreme C termini to reinforce the importance of subcellular localization and protein-protein interactions to PTP function in vivo. In addition, we have defined PTP61F's role in life span and fecundity and the regulation of IR and STAT92E signaling.
Using tissue-specific overexpression approaches, we have shown that the nucleus- and ER-targeted PTP61F variants have differential effects on IR signaling. Despite its nuclear locale, PTP61Fn was considerably more effective than the ER-localized PTP61Fm in reversing the eye overgrowth associated with IR overexpression. In keeping with the effect on eye overgrowth, we found that PTP61Fn, but not PTP61Fm, could attenuate IR Y1553/Y1554 phosphorylation (as measured in eye homogenates). As for the IR-induced eye overgrowth, we found that PTP61Fn was also more effective in suppressing Drosophila wing growth. Although overexpression of PTP61Fm had no overt effect on wing growth, PTP61Fn caused a 30% reduction due to decreases in both cell size and number. This is similar to findings for another negative regulator of insulin signaling, PTEN; overexpression of PTEN in the Drosophila wing causes a 25% reduction in wing growth (43). The effects of PTEN on wing growth have been attributed to the attenuation of IR-induced PI3K signaling (43). Although we cannot exclude PTP61Fn acting on other pathways to attenuate wing growth, the parallels with PTEN suggest that the actions of PTP61Fn on growth control might also be mediated via the attenuation of IR signaling.
The stark difference in PTP61Fn- versus PTP61Fm-mediated IR dephosphorylation (despite PTP61Fm being overexpressed at higher levels than PTP61Fn in our transgenic flies) was surprising given that the mammalian ER-targeted PTP1B is more prominent in IR regulation in insulin-responsive tissues in mice than the PTP61Fn orthologue TC45 (7, 17, 18, 27, 72–74). One possibility is that the IR is more accessible to the PTP61Fn variant, which presumably exits the nucleus like TC45 (14, 31, 32) to dephosphorylate the IR. Although we have not directly assessed PTP61Fn localization in response to insulin stimulation, our results are entirely consistent with PTP61Fn acting upstream of PI3K at the level of the IR, since PTP61Fn did not attenuate the eye overgrowth induced by overexpressed PI3K but did attenuate IR Y1553/Y1554 phosphorylation. On the other hand, the ER-targeted PTP61Fm may access the IR after endocytosis or at cell-cell adhesions as reported for PTP1B and its actions on receptor PTKs such as ErbB1 (20–22, 24, 75). Another, but not mutually exclusive, possibility is that the ER-targeted PTP61Fm requires an associated protein to efficiently dephosphorylate the IR, and this may be limited in the context of dPTP61F overexpression in the eye. Previously we have reported that the Drosophila adaptor protein Dock forms a stable complex with both the IR and PTP61Fm and that Dock enhances the PTP61Fm-mediated suppression of IR-mediated eye hypertrophy (38). In this study, we have shown that although PTP61Fm on its own does not attenuate IR signaling (as assessed in the Drosophila eye homogenates), coexpression with Dock allows for almost complete suppression of IR Y1553/Y1554 phosphorylation. Therefore, PTP61Fm but not PTP61Fn is reliant on Dock to dephosphorylate the IR in vivo, consistent with IR accessibility differing between the PTP61F variants and the ER-targeted PTP61F having a more restricted access to the IR. This may also be pertinent to IR regulation and glucose homeostasis in mammals. PTP1B can interact with the Dock homolog Nck-1 to attenuate insulin signaling in HEK293 cells (38). Moreover, Nck-1 knockdown enhances IRS-1 tyrosine phosphorylation in HepG2 cells (76), whereas Nck-1 deficiency in obese mice fed a high-fat diet improves glucose tolerance and insulin sensitivity (76). However, it remains to be established if Nck-1 is required for PTP1B-mediated IR regulation in vivo.
To further explore the role of PTP61F in vivo, we generated PTP61F mutant flies that lacked both PTP61Fm and PTP61Fn. Although we found that PTP61F overexpression attenuated wing growth and IR-induced eye hypertrophy, no overt difference in growth was evident in PTP61FΔ flies. In contrast, flies homozygous for a hypomorphic PTEN allele (77) or for a loss-of-function mutation in susi, a negative regulator of PI3K signaling that binds to the PI3K regulatory subunit (54), exhibit a 50% or 27% increase, respectively, in body weight. Although PTP61Fm and PTP61Fn are the only characterized isoforms of PTP61F, three additional transcripts have been predicted based on genomic data (Flybase). Of these, PTP61F-RD and PTP61F-RE would encode ER-targeted N-terminally truncated proteins lacking PTP catalytic domain elements essential for proper folding and activity. In keeping with this, our preliminary studies have confirmed that the PTP domain encoded by PTP61F-RD is catalytically inactive (unpublished observations). The third potential transcript, PTP61F-RC, would produce catalytically active protein that would be nucleus targeted and differ from PTP61Fn only in its extreme N terminus as a result of alternate exon usage. The P-element excision event resulting in the generation of the PTP61FΔ flies did not disrupt the exon encoding this distinct PTP61F N terminus (our unpublished data), which is ∼70 kb upstream, or any of the predicted exons common to all transcripts (see Fig. S2A in the supplemental material). Therefore, if PTP61F-RC is transcribed, it might compensate for PTP61Fn deficiency in PTP61FΔ flies to ameliorate any effect on growth. However, it is also possible that other PTPs or other negative regulators compensate for PTP61F deficiency.
Although we noted no overt effect on growth, PTP61F deficiency resulted in significantly diminished life span and decreased female fecundity. Importantly, both PTP61Fm and PTP61Fn were required to rescue the loss of function mutant phenotypes, consistent with the ER- and nucleus-targeted PTP61F variants acting together in the control of signaling and biological responses such that the reconstitution of one variant alone may not be sufficient to restore signaling to normal levels and correct the phenotype. This is reminiscent of the capacity of PTP1B and TCPTP to act cooperatively in the attenuation of signaling emanating from the IR receptor (14), as well as in the control of JAK/STAT signaling, both in the context of interleukin-4 stimulation in vitro (78) and leptin stimulation in vivo (16). In Drosophila, both IR and Hop/STAT92E signaling have been implicated in the control of female fertility (58, 61, 62, 69, 70). IR signaling is also a well-established regulator of Drosophila life span (57–61). In addition, both increased and decreased JNK signaling can affect life span (79–82). It is not known if PTP61F can regulate JNK in flies, but TCPTP has been shown to regulate JNK signaling in response to epidermal growth factor (EGF) stimulation (83) and both PTP1B and TCPTP regulate SFKs (9–13). In Drosophila, Src has been shown to increase JNK activation (84–86), raising the possibility that activation of the Src/JNK pathway by PTP61F-deficiency contributes to the life span defect.
It is important to note that although our studies are consistent with the PTP61F variants acting in concert in the regulation of cellular signaling, as reported previously for PTP1B and TCPTP (1, 7), they do not exclude PTP61Fm and PTP61Fn acting in different signaling pathways and cell types and/or tissues in mediating their effects on life span and fecundity. During embryogenesis, PTP61F transcripts are expressed in a tissue-specific manner, with PTP61Fm in the mesoderm/neuroblast layers during germ band extension and PTP61Fn in the nervous system (87). Thus, future studies should directly determine the capacity of the PTP61F variants to cooperate in a cell type-specific manner in ameliorating the life span and fecundity defects in PTP61F mutant flies.
The female fecundity defect in PTP61FΔ flies was due to increased pre-stage 10 egg chamber apoptosis, resulting in a lack of mature oocytes and a corresponding increase in germ line stem cells and/or early stage egg chambers. Upregulation of JAK/STAT signaling in the stem cell niche has been shown to cause ovarian germ line tumors due to increased stem cell numbers (69, 70). RNA interference-mediated suppression of STAT92E in the PTP61FΔ background resulted in a partial rescue of fecundity, and elevated STAT92E phosphorylation was evident in PTP61FΔ heterozygote and homozygous mutant ovaries, consistent with deregulated Hop/STAT92E signaling contributing to the fecundity defect. However, the elevated STAT92E phosphorylation in the PTP61FΔ heterozygotes in the absence of fecundity defects suggests that this alone is not causing the fecundity defect in homozygotes and that the deregulation of additional pathways may be occurring. IR signaling has also been shown to contribute to fecundity by affecting vitellogenesis and germ line stem cell division (58, 61, 62). Although IR activation was increased in homozygous (but not heterozygous) PTP61FΔ ovaries, suppression of IR signaling by the overexpression of a dominant negative PI3K did not improve fecundity. Although this suggests that elevated IR-mediated PI3K signaling is not a key cause of the fecundity defect in PTP61FΔ mutant flies, it does not preclude other deregulated IR pathways contributing to the fecundity defect. Furthermore, it is possible that an altogether different pathway such as the Src/JNK pathway discussed earlier may be altered to function together with STAT92E to cause the defects in fecundity. Even though further studies are needed to dissect the molecular mechanisms underlying the perturbations in life span and fecundity in PTP61FΔ flies, the results are nonetheless consistent with PTP61F deficiency enhancing IR and STAT92E signaling and both PTP61Fm and PTP61Fn acting together in the regulation of biological responses.
The results of this study have for the first time assessed the impact of PTP61F deficiency on the regulation of fundamental biological processes and directly compared the contributions of the differentially targeted PTP61F variants in such processes in vivo. The findings highlight the complexities of PTK regulation by PTPs and the capacity of adaptor proteins to contribute to PTP substrate selectivity and function in a biological context. Moreover, the PTP61F mutant flies generated in this study provide a tractable biological system by which to further dissect the importance of both catalytic and noncatalytic domain interactions in PTP substrate specificity and functions.
Supplementary Material
ACKNOWLEDGMENTS
We thank Helena Richardson, Gary Hime, and Larry Zipursky for provision of Drosophila stocks, the Australian Drosophila Biomedical Research Facility (OzDros) for importation of all fly strains, and Monash Microimaging for assistance with microscopy.
B.J.B., T.-C.M., R.B., T.T., and C.G.W. conceived and designed the experiments; B.J.B. and T.K.J. performed the experiments; B.J.B., T.K.J., T.T., and C.G.W. analyzed the data; and B.J.B., T.T., and C.G.W. wrote the paper.
This study was funded by the National Health and Medical Research Council (NHMRC) of Australia to T.T. , the Australian Research Council to T.T. and C.G.W., and Taiwan's National Science Council (98-2311-B-001-019-MY3) to T.-C.M.; T.T. is an NHMRC Research Fellow.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 22 January 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01411-12.
REFERENCES
- 1. Tiganis T, Bennett AM. 2007. Protein tyrosine phosphatase function: the substrate perspective. Biochem. J. 402:1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tonks NK. 2006. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7:833–846 [DOI] [PubMed] [Google Scholar]
- 3. Andersen JN, Mortensen OH, Peters GH, Drake PG, Iversen LF, Olsen OH, Jansen PG, Andersen HS, Tonks NK, Moller NP. 2001. Structural and evolutionary relationships among protein tyrosine phosphatase domains. Mol. Cell. Biol. 21:7117–7136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iversen LF, Moller KB, Pedersen AK, Peters GH, Petersen AS, Andersen HS, Branner S, Mortensen SB, Moller NP. 2002. Structure determination of T cell protein tyrosine phosphatase. J. Biol. Chem. 277:19982–19990 [DOI] [PubMed] [Google Scholar]
- 5. Salmeen A, Andersen JN, Myers MP, Tonks NK, Barford D. 2000. Molecular basis for recognition and dephosphorylation of the activation segment of the insulin receptor by protein tyrosine phosphatase 1B. Mol. Cell 6:1401–1412 [DOI] [PubMed] [Google Scholar]
- 6. Simoncic PD, Lee-Loy A, Barber DL, Tremblay ML, McGlade CJ. 2002. The T cell protein tyrosine phosphatase is a negative regulator of janus family kinases 1 and 3. Curr. Biol. 12:446–453 [DOI] [PubMed] [Google Scholar]
- 7. Tiganis T. 2013. PTP1B and TCPTP—nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 280:445–458 [DOI] [PubMed] [Google Scholar]
- 8. Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, Salmeen A, Barford D, Tonks NK. 2001. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J. Biol. Chem. 276:47771–47774 [DOI] [PubMed] [Google Scholar]
- 9. Dadke S, Chernoff J. 2003. Protein-tyrosine phosphatase 1B mediates the effects of insulin on the actin cytoskeleton in immortalized fibroblasts. J. Biol. Chem. 278:40607–40611 [DOI] [PubMed] [Google Scholar]
- 10. Egan C, Pang A, Durda D, Cheng HC, Wang JH, Fujita DJ. 1999. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene 18:1227–1237 [DOI] [PubMed] [Google Scholar]
- 11. Kant S, Swat W, Zhang S, Zhang ZY, Neel BG, Flavell RA, Davis RJ. 2011. TNF-stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway. Genes Dev. 25:2069–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liang F, Lee SY, Liang J, Lawrence DS, Zhang ZY. 2005. The role of protein-tyrosine phosphatase 1B in integrin signaling. J. Biol. Chem. 280:24857–24863 [DOI] [PubMed] [Google Scholar]
- 13. van Vliet C, Bukczynska PE, Puryer MA, Sadek CM, Shields BJ, Tremblay ML, Tiganis T. 2005. Selective regulation of tumor necrosis factor-induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat. Immunol. 6:253–260 [DOI] [PubMed] [Google Scholar]
- 14. Galic S, Hauser C, Kahn BB, Haj FG, Neel BG, Tonks NK, Tiganis T. 2005. Coordinated regulation of insulin signaling by the protein tyrosine phosphatases PTP1B and TCPTP. Mol. Cell. Biol. 25:819–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML. 2002. Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev. Cell 2:497–503 [DOI] [PubMed] [Google Scholar]
- 16. Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C, Sims NA, Bence KK, Zhang S, Zhang ZY, Kahn BB, Neel BG, Andrews ZB, Cowley MA, Tiganis T. 2011. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab. 14:684–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP. 1999. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283:1544–1548 [DOI] [PubMed] [Google Scholar]
- 18. Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB. 2000. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol. Cell. Biol. 20:5479–5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. You-Ten KE, Muise ES, Itie A, Michaliszyn E, Wagner J, Jothy S, Lapp WS, Tremblay ML. 1997. Impaired bone marrow microenvironment and immune function in T cell protein tyrosine phosphatase-deficient mice. J. Exp. Med. 186:683–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Eden ER, White IJ, Tsapara A, Futter CE. 2010. Membrane contacts between endosomes and ER provide sites for PTP1B-epidermal growth factor receptor interaction. Nat. Cell Biol. 12:267–272 [DOI] [PubMed] [Google Scholar]
- 21. Haj FG, Sabet O, Kinkhabwala A, Wimmer-Kleikamp S, Roukos V, Han HM, Grabenbauer M, Bierbaum M, Antony C, Neel BG, Bastiaens PI. 2012. Regulation of signaling at regions of cell-cell contact by endoplasmic reticulum-bound protein-tyrosine phosphatase 1B. PLoS One 7:e36633 doi:10.1371/journal.pone.0036633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haj FG, Verveer PJ, Squire A, Neel BG, Bastiaens PI. 2002. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science 295:1708–1711 [DOI] [PubMed] [Google Scholar]
- 23. Monteleone MC, Gonzalez Wusener AE, Burdisso JE, Conde C, Caceres A, Arregui CO. 2012. ER-bound protein tyrosine phosphatase PTP1B interacts with Src at the plasma membrane/substrate interface. PLoS One 7:e38948 doi:10.1371/journal.pone.0038948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nievergall E, Janes PW, Stegmayer C, Vail ME, Haj FG, Teng SW, Neel BG, Bastiaens PI, Lackmann M. 2010. PTP1B regulates Eph receptor function and trafficking. J. Cell Biol. 191:1189–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu F, Hill DE, Chernoff J. 1996. Direct binding of the proline-rich region of protein tyrosine phosphatase 1B to the Src homology 3 domain of p130(Cas). J. Biol. Chem. 271:31290–31295 [DOI] [PubMed] [Google Scholar]
- 26. Aoki N, Matsuda T. 2002. A nuclear protein tyrosine phosphatase TC-PTP is a potential negative regulator of the PRL-mediated signaling pathway: dephosphorylation and deactivation of signal transducer and activator of transcription 5a and 5b by TC-PTP in nucleus. Mol. Endocrinol. 16:58–69 [DOI] [PubMed] [Google Scholar]
- 27. Fukushima A, Loh K, Galic S, Fam B, Shields B, Wiede F, Tremblay ML, Watt MJ, Andrikopoulos S, Tiganis T. 2010. T-cell protein tyrosine phosphatase attenuates STAT3 and insulin signaling in the liver to regulate gluconeogenesis. Diabetes 59:1906–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lu X, Chen J, Sasmono RT, Hsi ED, Sarosiek KA, Tiganis T, Lossos IS. 2007. T-cell protein tyrosine phosphatase, distinctively expressed in activated-B-cell-like diffuse large B-cell lymphomas, is the nuclear phosphatase of STAT6. Mol. Cell. Biol. 27:2166–2179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. ten Hoeve J, Ibarra-Sanchez MJ, Fu Y, Zhu W, Tremblay M, David M, Shuai K. 2002. Identification of a nuclear Stat1 protein tyrosine phosphatase. Mol. Cell. Biol. 22:5662–5668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamamoto T, Sekine Y, Kashima K, Kubota A, Sato N, Aoki N, Matsuda T. 2002. The nuclear isoform of protein-tyrosine phosphatase TC-PTP regulates interleukin-6-mediated signaling pathway through STAT3 dephosphorylation. Biochem. Biophys. Res. Commun. 297:811–817 [DOI] [PubMed] [Google Scholar]
- 31. Galic S, Klingler-Hoffmann M, Fodero-Tavoletti MT, Puryer MA, Meng TC, Tonks NK, Tiganis T. 2003. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol. Cell. Biol. 23:2096–2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lam MH, Michell BJ, Fodero-Tavoletti MT, Kemp BE, Tonks NK, Tiganis T. 2001. Cellular stress regulates the nucleocytoplasmic distribution of the protein tyrosine phosphatase TCPTP. J. Biol. Chem. 276:37700–37707 [DOI] [PubMed] [Google Scholar]
- 33. Tiganis T, Bennett AM, Ravichandran KS, Tonks NK. 1998. Epidermal growth factor receptor and the adaptor protein p52Shc are specific substrates of T-cell protein tyrosine phosphatase. Mol. Cell. Biol. 18:1622–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bier E. 2005. Drosophila, the golden bug, emerges as a tool for human genetics. Nat. Rev. Genet. 6:9–23 [DOI] [PubMed] [Google Scholar]
- 35. McLaughlin S, Dixon JE. 1993. Alternative splicing gives rise to a nuclear protein tyrosine phosphatase in Drosophila. J. Biol. Chem. 268:6839–6842 [PubMed] [Google Scholar]
- 36. Clemens JC, Ursuliak Z, Clemens KK, Price JV, Dixon JE. 1996. A Drosophila protein-tyrosine phosphatase associates with an adapter protein required for axonal guidance. J. Biol. Chem. 271:17002–17005 [DOI] [PubMed] [Google Scholar]
- 37. Liu F, Sells MA, Chernoff J. 1998. Protein tyrosine phosphatase 1B negatively regulates integrin signaling. Curr. Biol. 8:173–176 [DOI] [PubMed] [Google Scholar]
- 38. Wu CL, Buszard B, Teng CH, Chen WL, Warr CG, Tiganis T, Meng TC. 2011. Dock/Nck facilitates PTP61F/PTP1B regulation of insulin signalling. Biochem. J. 439:151–159 [DOI] [PubMed] [Google Scholar]
- 39. Baeg GH, Zhou R, Perrimon N. 2005. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 19:1861–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muller P, Kuttenkeuler D, Gesellchen V, Zeidler MP, Boutros M. 2005. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 436:871–875 [DOI] [PubMed] [Google Scholar]
- 41. Chang YC, Lin SY, Liang SY, Pan KT, Chou CC, Chen CH, Liao CL, Khoo KH, Meng TC. 2008. Tyrosine phosphoproteomics and identification of substrates of protein tyrosine phosphatase dPTP61F in Drosophila S2 cells by mass spectrometry-based substrate trapping strategy. J. Proteome Res. 7:1055–1066 [DOI] [PubMed] [Google Scholar]
- 42. Ku HY, Wu CL, Rabinow L, Chen GC, Meng TC. 2009. Organization of F-actin via concerted regulation of Kette by PTP61F and dAbl. Mol. Cell. Biol. 29:3623–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goberdhan DCI, Paricio N, Goodman E, Mlodzik M, Wilson C. 1999. Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev. 13:3244–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brogiolo W, Stocker H, Ikeya T, Rintelen F, Fernandez R, Hafen E. 2001. An evolutionarily conserved function of the Drosophila insulin receptor and insulin-like peptides in growth control. Curr. Biol. 11:213–221 [DOI] [PubMed] [Google Scholar]
- 45. Bohni R, Riesgo-Escovar J, Oldham S, Brogiolo W, Stocker H, Andruss B, Beckingham K, Hafen E. 1999. Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 97:865–875 [DOI] [PubMed] [Google Scholar]
- 46. Scanga S, Ruel L, Binari R, Snow B, Stambolic V, Bouchard D, Peters M, Calvieri B, Mak T, Woodget J, Manoukian A. 2000. The conserved PI3K/PTEN/Akt signaling pathway regulates both cell size and survival in Drosophila. Oncogene 19:3971–3977 [DOI] [PubMed] [Google Scholar]
- 47. Barbieri M, Bonafe M, Franceschi C, Paolisso G. 2003. Insulin/IGF-1-signaling pathway: an evolutionarily conserved mechanism of longevity from yeast to humans. Am. J. Physiol. Endocrinol. Metab. 285:E1064–E1071 [DOI] [PubMed] [Google Scholar]
- 48. Nijhout HF. 2003. The control of growth. Development 130:5863–5867 [DOI] [PubMed] [Google Scholar]
- 49. Teleman AA. 2010. Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 425:13–26 [DOI] [PubMed] [Google Scholar]
- 50. Huang H, Potter C, Tao W, Li DM, Brogiolo W, Hafen E, Sun H, Xu T. 1999. PTEN affects cell size, cell proliferation and apoptosis during Drosophila eye development. Development 126:5365–5372 [DOI] [PubMed] [Google Scholar]
- 51. Leevers SJ, Weinkove D, MacDougall LK, Hafen E, Waterfield MD. 1996. The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. EMBO J. 15:6584–6594 [PMC free article] [PubMed] [Google Scholar]
- 52. Potter C, Huang H, Xu T. 2001. Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 105:357–368 [DOI] [PubMed] [Google Scholar]
- 53. Tapon N, Ito N, Dickson BJ, Treisman JE, Hariharan IK. 2001. The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 105:345–355 [DOI] [PubMed] [Google Scholar]
- 54. Wittwer F, Jaquenoud M, Brogiolo W, Zarske M, Wustemann P, Fernandez R, Stocker H, Wymann MP, Hafen E. 2005. Susi, a negative regulator of Drosophila PI3-kinase. Dev. Cell 8:817–827 [DOI] [PubMed] [Google Scholar]
- 55. Halder G, Callaerts P, Flister S, Walldorf U, Kloter U, Gehring WJ. 1998. Eyeless initiates the expression of both sine oculis and eyes absent during Drosophila compound eye development. Development 125:2181–2191 [DOI] [PubMed] [Google Scholar]
- 56. Fan Y, Bergmann A. 2010. The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 17:534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Broughton SJ, Piper MD, Ikeya T, Bass TM, Jacobson J, Driege Y, Martinez P, Hafen E, Withers DJ, Leevers SJ, Partridge L. 2005. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. U. S. A. 102:3105–3110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Clancy D, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. 2001. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science 292:104–106 [DOI] [PubMed] [Google Scholar]
- 59. Giannakou ME, Goss M, Junger M, Hafen E, Leevers SJ, Partridge L. 2004. Long-lived Drosophila with over-expressed dFOXO in adult fat body. Science 305:361. [DOI] [PubMed] [Google Scholar]
- 60. Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M. 2004. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429:562–566 [DOI] [PubMed] [Google Scholar]
- 61. Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo R. 2001. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science 292:107–110 [DOI] [PubMed] [Google Scholar]
- 62. LaFever L, Drummond-Barbosa D. 2005. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science 309:1071–1073 [DOI] [PubMed] [Google Scholar]
- 63. McGregor JR, Xi R, Harrison DA. 2002. Jak signalling is somatically required for follicle cell differentiation in Drosophila. Development 129:705–717 [DOI] [PubMed] [Google Scholar]
- 64. Shen R, Weng C, Yu J, Xie T. 2009. eIF4A controls germline stem cell self-renewal by directly inhibiting BAM function in the Drosophila ovary. Proc. Natl. Acad. Sci. U. S. A. 106:11623–11628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tootle TL, Spradling AC. 2008. Drosophila Pxt: a cyclooxygenase-like facilitator of follicle maturation. Development 135:839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wang X, Pan L, Wang S, Zhou J, McDowell W, Park J, Haug J, Staehling K, Tang H, Xie T. 2011. Histone H3K9 trimethylase eggless controls germline stem cell maintenance and differentiation. PLoS Genet. 7:e1002426 doi:10.1371/journal.pgen.1002426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Binari R, Perrimon N. 1994. Stripe-specific regulation of pair-rule genes by hopscotch, a putative Jak family tyrosine kinase in Drosophila. Genes Dev. 8:300–312 [DOI] [PubMed] [Google Scholar]
- 68. Hou SX, Perrimon N. 1997. The JAK-STAT pathway in Drosophila. Trends Genet. 13:105–110 [DOI] [PubMed] [Google Scholar]
- 69. Lopez-Onieva L, Fernandez-Minan A, Gonzalez-Reyes A. 2008. Jak/Stat signalling in niche support cells regulates dpp transcription to control germline stem cell maintenance in the Drosophila ovary. Development 135:533–540 [DOI] [PubMed] [Google Scholar]
- 70. Wang L, Li Z, Cai Y. 2008. The JAK/STAT pathway positively regulates DPP signaling in the Drosophila germline stem cell niche. J. Cell Biol. 180:721–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. O'Reilly AM, Neel BG. 1998. Structural determinants of SHP-2 function and specificity in Xenopus mesoderm induction. Mol. Cell. Biol. 18:161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Delibegovic M, Bence KK, Mody N, Hong EG, Ko HJ, Kim JK, Kahn BB, Neel BG. 2007. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol. Cell. Biol. 27:7727–7734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong EG, Cho YR, Kim JK, Kahn BB, Neel BG, Bence KK. 2009. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced ER stress. Diabetes 58:590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Loh K, Merry TL, Galic S, Wu BJ, Watt MJ, Zhang S, Zhang ZY, Neel BG, Tiganis T. 2012. T cell protein tyrosine phosphatase (TCPTP) deficiency in muscle does not alter insulin signalling and glucose homeostasis in mice. Diabetologia 55:468–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Romsicki Y, Reece M, Gauthier JY, Asante-Appiah E, Kennedy BP. 2004. Protein tyrosine phosphatase-1B dephosphorylation of the insulin receptor occurs in a perinuclear endosome compartment in human embryonic kidney 293 cells. J. Biol. Chem. 279:12868–12875 [DOI] [PubMed] [Google Scholar]
- 76. Latreille M, Laberge MK, Bourret G, Yamani L, Larose L. 2011. Deletion of Nck1 attenuates hepatic ER stress signaling and improves glucose tolerance and insulin signaling in liver of obese mice. Am. J. Physiol. Endocrinol. Metab. 300:E423–E434 [DOI] [PubMed] [Google Scholar]
- 77. Oldham S, Stocker H, Laffargue M, Wittwer F, Wymann MP, Hafen E. 2002. The Drosophila insulin/IGF receptor controls growth and size by modulating PtdInsP3 levels. Development 129:4103–4109 [DOI] [PubMed] [Google Scholar]
- 78. Lu X, Malumbres R, Shields B, Jiang X, Sarosiek KA, Natkunam Y, Tiganis T, Lossos IS. 2008. PTP1B is a negative regulator of interleukin 4-induced STAT6 signaling. Blood 112:4098–4108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Biteau B, Karpac J, Supoyo S, Degennaro M, Lehmann R, Jasper H. 2010. Lifespan extension by preserving proliferative homeostasis in Drosophila. PLoS Genet. 6:e1001159 doi:10.1371/journal.pgen.1001159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Deng Y, Ren X, Yang L, Lin Y, Wu X. 2003. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 115:61–70 [DOI] [PubMed] [Google Scholar]
- 81. Uhlirova M, Jasper H, Bohmann D. 2005. Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc. Natl. Acad. Sci. U. S. A. 102:13123–13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vidal M, Warner S, Read R, Cagan RL. 2007. Differing Src signaling levels have distinct outcomes in Drosophila. Cancer Res. 67:10278–10285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tiganis T, Kemp BE, Tonks NK. 1999. The protein tyrosine phosphatase TCPTP regulates epidermal growth factor receptor-mediated and phosphatidylinositol 3-kinase-dependent signalling. J. Biol. Chem. 274:27768–27775 [DOI] [PubMed] [Google Scholar]
- 84. Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M. 2002. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21:3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Tateno M. 2000. Regulation of JNK by Src during Drosophila development. Science 287:324–327 [DOI] [PubMed] [Google Scholar]
- 86. Williams MJ. 2009. The c-src homologue Src64B is sufficient to activate the Drosophila cellular immune response. J. Innate Immun. 1:335–339 [DOI] [PubMed] [Google Scholar]
- 87. Ursuliak Z, Clemens JC, Dixon JE, Price JV. 1997. Differential accumulation of DPTP61F alternative transcripts: regulation of a protein tyrosine phosphatase by segmentation genes. Mech. Dev. 65:19–30 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.