

Abstract
Mammalian poxviruses, including vaccinia virus (VACV), have evolved multiple mechanisms to evade the host type I interferon (IFN) responses at different levels, with viral proteins targeting IFN induction, signaling, and antiviral effector functions. Avian poxviruses (avipoxviruses), which have been developed as recombinant vaccine vectors for permissive (i.e., poultry) and nonpermissive (i.e., mammals, including humans) species, encode no obvious equivalents of any of these proteins. We show that fowlpox virus (FWPV) fails to induce chicken beta IFN (ChIFN2) and is able to block its induction by transfected poly(I·C), an analog of cytoplasmic double-stranded RNA (dsRNA). A broad-scale loss-of-function genetic screen was used to find FWPV-encoded modulators of poly(I·C)-mediated ChIFN2 induction. It identified fpv012, a member of a family of poxvirus genes highly expanded in the avipoxviruses (31 in FWPV; 51 in canarypox virus [CNPV], representing 15% of the total gene complement), encoding proteins containing N-terminal ankyrin repeats (ANKs) and C-terminal F-box-like motifs. Under ectopic expression, the first ANK of fpv012 is dispensable for inhibitory activity and the CNPV ortholog is also able to inhibit induction of ChIFN2. FWPV defective in fpv012 replicates well in culture and barely induces ChIFN2 during infection, suggesting that other factors are involved in blocking IFN induction and resisting the antiviral effectors. Nevertheless, unlike parental and revertant viruses, the mutants induce moderate levels of expression of interferon-stimulated genes (ISGs), suggesting either that there is sufficient ChIFN2 expression to partially induce the ISGs or the involvement of alternative, IFN-independent pathways that are also normally blocked by fpv012.
INTRODUCTION
The host antiviral type I interferon (IFN) system is targeted by many viruses (1), and poxviruses are no exception. The prototypic, mammalian poxvirus, vaccinia virus (VACV), encodes a number of proteins that have been shown to modulate the IFN system in diverse ways, as reviewed recently (2). Functionally, they can be grouped into those that inhibit induction of beta IFN (IFN-β), such as NF-κB activation suppressor K1 (3) and TBK-1 adaptor binding protein C6 (4); those that inhibit IFN signaling via the Jak/Stat pathway necessary to induce expression of IFN-stimulated genes (ISGs), such as soluble IFN receptor mimic B18 (5, 6) and Stat phosphorylation inhibitor H1 (7); and those that antagonize the activity of the major antiviral ISGs, such as double-stranded RNA (dsRNA)-binding protein E3 (8) and α subunit of eukaryotic initiation factor 2 (eIF-2α) mimic K3 (9).
With the exception of H1, no orthologs (or even any functional equivalents) of these modulators have been described in avian poxviruses. A diverse group of viruses isolated from more than 230 species of birds (10) and infecting only avian species, all avian poxviruses are grouped into the single Avipoxvirus genus of the Chordopoxvirinae subfamily (11), with Fowlpox virus (FWPV) being the type species. Like VACV, FWPV has been developed for use as a live recombinant vaccine vector in both permissive hosts (i.e., poultry) and nonpermissive hosts (i.e., mammals, including humans, in which its replication is abortive) (12–18). Notably, a commercial FWPV recombinant vaccine (TROVAC-H5) expressing the hemagglutinin gene of H5N8 isolate A/turkey/Ireland/1378/83 has become the most extensively used live recombinant virus used in any sector, with some 2 billion doses used to counter highly pathogenic influenza H5N2 in Mexico up to 2005 (19). Another avipoxvirus, canarypox virus (CNPV), which is well diverged from FWPV (20), has been developed extensively for use in nonpermissive mammalian hosts (21–23), with several licensed commercial vaccines available for diseases of livestock and companion animals. CNPV, as ALVAC, was also the recombinant virus in the recent Thai HIV vaccine trial (RV144) that showed marginal indications of potential efficacy (24).
We show that, in chicken cell culture, FWPV fails to induce chicken IFN-2 (ChIFN2), believed to be the chicken equivalent of IFN-β (25, 26), and is able to block its induction by transfected poly(I·C), an analog of cytoplasmic dsRNA. We have used a broad-scale genetic loss-of-function screen involving a library of 48 FWPV in vitro-generated mutants, each defective in a single, nonessential gene, to identify a gene involved in blocking induction of the ChIFN2 promoter mediated by the dsRNA mimic poly(I·C). The screen identified a member of a poxvirus gene family that is far more extensive in avipoxviruses than in mammalian poxviruses and has not been previously associated with IFN modulation.
MATERIALS AND METHODS
Cells and viruses.
Primary chicken embryo fibroblasts (CEFs) produced from specific-pathogen-free (SPF)-quality embryos (10 days old) at the Institute for Animal Health (Compton, Berkshire, United Kingdom) were grown in medium 199 supplemented with 10% tryptone phosphate broth (TPB), 10% newborn bovine serum, nystatin, and penicillin-streptomycin. DF-1 cells (27) obtained from the American Type Culture Collection (ATCC) were maintained in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (Autogen Bioclear) and penicillin-streptomycin.
Attenuated FWPV strain FP9, used throughout this study, and its pathogenic European progenitor, HP1, have been described previously (28). Infectious bursal disease virus strain PBG98, a mild vaccine strain from laboratory stocks, was propagated in CEFs (29).
Plasmids.
The ChIFN2 promoter reporter plasmid (pChIFN2lucter [30]) and the constitutive β-galactosidase reporter plasmid (pJATlacZ [31]) have been described previously. Expression plasmids were constructed by cloning FWPV or CNPV genes into pEFPlink2 or pEFPlink2Flag (32). Sequences were amplified by PCR using oligonucleotides (Table 1) containing BamHI and SpeI (NcoI in the case of fpv213) sites and cloned into pEFPlink2 or pEFPlink2Flag restricted with BamHI and SpeI (NcoI in the case of fpv213). Plasmids were sequenced to ensure that no errors had been introduced and the reading frame was intact.
Table 1.
Primers used for constructing expression clones
| Gene | Vector | Orientationa | Primer sequence (5′–3′) |
|---|---|---|---|
| cnpv030 | pEFPlink2 | Fwd | CCGGATCCTCAGTCACGATGGATTTACC |
| cnpv030 | pEFPlink2 | Rev | CGATACTAGTTTATTTGTGTATATTTAAAGCG |
| fpv012 | pEFPlink2 | Fwd | CCGGATCCATGGATACAGAAATGGACGG |
| fpv012 | pEFPlink2 | Rev | GGTTCCACTAGTTTAACCAGTCTTATTATTAAC |
| fpv012 | pEFPlink2Flag | Fwd | CCGGATCCGATACAGAAATGGACGGTGTCAATAACG |
| fpv012mut1-126 (Mut1) | pEFPlink2Flag | Fwd | CCGGATCCGTAGATTCCTACACTCCGTTAC |
| fpv012mut1-222 (Mut2) | pEFPlink2Flag | Fwd | CCGGATCCACTAATGATGGTTATACGGCTC |
| fpv012mut1-324 (Mut3) | pEFPlink2Flag | Fwd | CCGGATCCAAATACGGTATTACTCCTCTTG |
| fpv012 | pEFPlink2Flag | Rev | CGATACTAGTTTAACCAGTCTTATTATTAACTTTAATAGCG |
| fpv012mut955-996 (Mut4) | pEFPlink2Flag | Rev | CGATACTAGTTTACAATTTTTTTATATTATCGTTATTAAG |
| fpv155 | pEFPlink2Flag | Fwd | CCGGATCCATGTTTAATAGTATGATAACCGG |
| fpv155 | pEFPlink2Flag | Rev | CGATACTAGTTTAGTTATCTGAAAATATTTTATTTATATCC |
| fpv213 | pEFPlink2Flag | Fwd | GCATCCATGGGGGAACGAGTAAAAAAATGTTTTAG |
| fpv213 | pEFPlink2Flag | Rev | CGATGGATCCTTAGCTATCCTGTAAAGGAGATAC |
Fwd, forward; Rev, reverse.
Transfection of cells with poly(I·C) and assay of luciferase reporters.
Chicken DF-1 cells in 12-well plates were transfected with the ChIFN2 promoter reporter pChIFN2lucter (167 ng) and the constitutive reporter plasmid pJATlacZ (167 ng), sometimes additionally with an expression plasmid driving the overexpression of viral proteins or the control empty vector pEFPlink2 (250 ng). Following recovery for 24 h, cells were either left uninfected or infected with infectious bursal disease virus (IBDV) attenuated vaccine PBG98, pathogenic FWPV HP1, or parental or mutant attenuated FWPV FP9 (multiplicity of infection [MOI], 10). Following infection for 4 h, cells, when appropriate, were transfected with poly(I·C) (10 μg/ml) using Polyfect reagent (Qiagen), as described by Childs et al. (30), and incubated for 16 h. Luciferase assays were carried out, and data were normalized using β-galactosidase measurements.
β-Galactosidase assay.
Cell lysate β-galactosidase concentrations were measured by incubation of 10 μl of cell lysate with ortho-nitrophenyl-β-galactoside (50 μl of 0.5 mg ml−1 diluted in 60 mM Na2HPO4·7H2O, 40 mM Na2H2PO4·H2O, 10 mM KCl, 1 mM MgSO4·7H2O, 2.7 ml liter−1 β-mercaptoethanol). The reaction mixture was incubated at 37°C until a yellow coloration appeared, when the A420 was measured using a spectrophotometer.
Analysis of gene expression by RNase protection assay.
Total RNA was prepared from 9-cm-diameter dishes of confluent cells, treated as described above, using the acid phenol method, and analyzed by RNase protection using the method of Zinn et al. (33) as described previously (34). To generate a probe against chicken β-actin (Ch-Act-β), a 158-bp fragment of CEF genomic DNA was amplified by PCR using the primers 5′-CCCCATGGATGATATTGCTGCGC-3′ and 5′-TAATACGACTCACTATAGCTGATGTCTGGGGCGACCCACGA-3′; the underlined region contains the T7 promoter, allowing generation of an RNA probe from the PCR product by transcription with T7 RNA polymerase. To generate a probe for ChIFN2, a PCR product (amplified using the primers 5′-CCCAGATCTCCTCCAGTACAGCCACCACATGGT-3′ and 5′-CCCTCTAGACAGTCACTGGGTGTTGAGAC-3′) was cloned into pCRIIBlunt.Topo (Invitrogen). The orientation of the PCR product within pCRII.Blunt.Topo was deduced by restriction mapping; it was linearized with the restriction enzyme EarI and transcribed using T7 polymerase to generate a 357-bp RNA probe complementary to cellular mRNA.
Construction of insertion-knockout mutant FWPV.
Generation of an FWPV knockout library by insertion of the Escherichia coli xanthine-guanine phosphoribosyl-transferase (gpt) gene was carried out by splice-overlap-extension PCR (SOE-PCR). To construct the knockout FWPV, SOE-PCR (using high-fidelity Taq polymerase) was used to assemble linear recombination templates from three constituent parts: approximately 350-bp PCR fragments of the FWPV genome from either side, fragments i and ii, of the center of the target gene disrupted in the middle by fragment iii, a VACV p7.5 promoter upstream of the gpt gene. For each gene, fragment i was amplified by primers 1 and 2, fragment ii was amplified by primers 5 and 6, and fragment iii was amplified by primers 3 and 4. Primers 2 and 3, as well as primers 4 and 5, had 20 bases of complementary sequence, half from the target gene sequence and half from the p7.5 gpt cassette. Details of the primers used for generation of the library (data not shown) are available upon request. Following the first round of PCR, products were purified using a QIAquick PCR purification kit (Qiagen) and combined into a SOE-PCR in order to amplify a product consisting of the complete gene of interest with gpt inserted into the center of the gene. The resulting PCR product was then transfected into FWPV FP9-infected CEFs, and recombinant viruses were selected for the gpt gene with fresh medium containing mycophenolic acid (25 μg ml−1), xanthine (250 μg ml−1), and hypoxanthine (15 μg ml−1) (MXH). Recovered viruses were bulk passaged three times in CEFs in MXH and then plaque purified thrice. Viral genomic DNA was then extracted and analyzed by PCR to confirm disruption of the target gene and loss of parental virus.
Construction of deletion-knockout mutant FWPV.
An fpv012 deletion mutant, FP9Δ012TD (trans dominant), was generated independently of the FWPV FP9 knockout mutant library by the transient dominant selection (TDS) method of Falkner and Moss (35), as described previously (36). Briefly, two 0.6-kbp regions of the FP9 genome, comprising 500 bp upstream of fpv012 plus 100 bp from the 5′ end of the open reading frame (ORF) (amplicon i) and 100 bp from the 3′ end of fpv012 plus 500 bp downstream (amplicon ii), were amplified by PCR using oligonucleotides 5′-ATCGGGATCCCTTTAGTATTAGTTATTAAACCCGG and 5′-CATTCTGTATTTAACGATGGAATCTACGTTCGGTGTATTAGGATTTACACC for amplicon i and 5′-CCTAATACACCGAACGTAGATTCCATCGTTAAATACAGAATGGTGTTTACTTCC and 5′-ATCGGACGTCCTTAGCAGTGCAGAAGAATTTATC for amplicon ii. The two amplicons were joined by SOE-PCR (deleting about 800 bp from the fpv012 ORF), digested with BamHI and AatII, and then ligated into pGNR (36) to give pGNRdel012. CEFs were infected with FP9 (MOI, 0.1) and then transfected with pGNRdel012 using Lipofectin (Life Technologies), according to the manufacturer's instructions. Twenty-four hours following transfection, the medium was replaced with fresh medium containing MXH, and then infection was allowed to proceed for a further 72 h. Progeny virus was harvested and plaque purified thrice on CEFs in the presence of MXH, and then resolution was accomplished by plaque purification in the absence of MXH. The resolved viruses were tested for loss of resistance to mycophenolic acid, and the genotypes of these gpt-negative viruses were established by PCR (using internal and flanking primers), following DNA isolation from infected cells using Wizard SV genomic DNA purification (Promega). In this way, FP9Δ012TD was isolated.
Construction of knock-in mutant and revertant FWPV.
To construct knock-in viruses expressing tandem affinity-purified (TAP)-tagged fpv012, transient dominant (TD) plasmid pUC13-FL012TAP was constructed in two steps. First, the 400-bp 3′ flanking sequence from fpv012 was amplified by PCR and cloned into the poxvirus transient dominant TAP vector pUC13TAP (from G. Smith), such that the 3′ flanking sequence was downstream of the TAP tag, generating the intermediate plasmid pUC13TAP012-3′. Subsequently, the ORF, minus the stop codon, and 400 bp of upstream sequence were amplified by PCR and cloned into pUC13TAP012-3′ so that the virus ORF was fused in frame with the TAP tag. Constructs were sequenced to ensure that no errors had been introduced. Recombinant TAP-tagged fpv012 virus was generated by transfection of the construct into CEFs infected with FP9Δ012TD. The knock-in mutants were then isolated by the TDS method, as described above.
The TDS method described above was also used to construct a revertant virus from mutant FP9Δ012TD. The fpv012 ORF was amplified, with 500-bp flanking regions, using primers 5′-ATCGGGATCCCTTTAGTATTAGTTATTAAACCCGG and 5′-ATCGGACGTCCTTAGCAGTGCAGAAGAATTTATC, digested with BamHI and AatII, and then ligated into pGNR to generate pGNR012REV. This was used to generate revertant virus 012REV with a reconstituted fpv012 locus by the TDS method described above.
RNA extraction and processing of samples.
RNA was extracted from cells using an RNeasy kit (Qiagen) according to the manufacturer's instructions. On-column DNA digestion was performed using RNase-free DNase (Qiagen) to remove contaminating genomic DNA. RNA samples were quantified using a Nanodrop spectrophotometer (Thermo Scientific) and checked for quality using a 2100 Bioanalyzer (Agilent Technologies). All RNA samples had an RNA integrity number (RIN) of ≥9.6.
RT-PCR and qRT-PCR.
Quantitative real-time reverse transcription-PCR (qRT-PCR) was performed using Mesa Green quantitative PCR (qPCR) MasterMix Plus for SYBR Assay I dTTP (Eurogentec) according to the manufacturer's instructions. A final volume of 10 μl per reaction mixture was used with 1 μl cDNA diluted 1:10 in nuclease-free H2O as the template. Primers (Table 2) were used at a final concentration of 300 nM. qPCR was performed on an ABI-7900HT Fast real-time PCR system (Applied Biosystems) using the following program: 95°C for 5 min; 40 cycles of 95°C for 15 s, 57°C for 20 s, and 72°C for 20 s; 95°C for 15 s; and 60°C for 15 s.
Table 2.
Primers used to quantify gene expression in real-time qRT-PCR
| Gene producta or gene | GenBank accession no. | Sequence (5′–3′) |
|
|---|---|---|---|
| Forward PCR primer | Reverse PCR primer | ||
| ChGAPDH | NM_204305.1 | GGCACTGTCAAGGCTGAGAA | TGCATCTGCCCATTTGATGT |
| ChIFN2 | GU119897.1 | CAAGGCACGCGCTCCCAGAG | TGTGCCGTAGGAAGTTGTGGATGG |
| ChMX1 | HM775376.1 | GACCAGTACCGCGGACGGGA | CGCAACAGCTGGCTCCTCCA |
| ChZC3HAV1 | NM_001012938.1 | TCGGCGCCTCTCTACGCCAT | TCAGTCCACTGGCCGTGGTCA |
| ChIFIH1/MDA5 | AB371640.1 | TTGCTATGGTGCAGGCCCGG | TGGCGGCATCTCTTGGACACG |
| ChIFIT5 | NW_003763812.1 | TGACCAGCCAAGGGGATGGCT | AGAGGGATTTGGGAGTGCTTCAGC |
| ChISG12-2 | NM_001001296.4 | TGACCAGAACGTCCACAAAGCCG | ACCTGCTCCTGGACCGATGCTT |
| fpv012 | AJ581527 | CACCCTCATCTAACAAAACA | CGCTAAAACGGGCAATATAA |
| fpv094 | AJ581527 | TATAATGAATGGCGCTGTGT | GTTTTGCTATCTTGGCTGT |
| fpv100 | AJ581527 | GTGTTCACGCCAAAGTAG | AGTAGGTTCTTCGTCAGATG |
| fpv126 | AJ581527 | AACAACGAACAAATACCC | AATCCAAGTAGCATATCAAG |
| fpv165 | AJ581527 | CCCCAAACGGTTAAAAACTACACG | ACGTATTCGTGTCCGTAATCGT |
| fpv168 | AJ581527 | ACCTCAAACAACCTCATC | GTTAATACTTGTGACTGCTG |
| fpv176 | AJ581527 | ACGTGTCCCTTTACCTCC | TTCCTTGCCATCTACGCC |
Ch, chicken.
The qRT-PCR primer pairs (Table 2) were validated by generating standard curves using PCR products corresponding to each gene. A 10-fold dilution series was made for each PCR product, and 1 μl was used with the Mesa Green qPCR master mix. Threshold cycle (CT) values were analyzed using SDS v2.3 software (Applied Biosystems). The slopes of the standard curves were used to identify the amplification efficiencies (E) of the qRT-PCR primer pairs, using the equation 10(−1/slope) − 1. Only qRT-PCR primer pairs with efficiencies of 90 to 110% were used further. The linear correlation coefficient (R2) was used to assess the linearity of the standard curve. Standard curves with R2 values of >0.985 were used.
Data were analyzed using SDS v2.3 and RQ Manager v1.2 software (Applied Biosystems). All target gene expression levels were calculated relative to the expression levels of the reference gene, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which were shown to remain constant over 24 h in uninfected and FP9-infected cells, and the target gene expression level in control CEFs, using the comparative CT method (also referred to as the 2−ΔΔCT method).
RESULTS
Fowlpox virus fails to induce ChIFN2 in chicken DF-1 cells.
Infection of immortalized chicken fibroblast DF-1 cells with FWPV FP9 led to no detectable induction in the expression of ChIFN2 mRNA, as determined by RNase protection assay. In contrast, infection with the chicken birnavirus infectious bursal disease virus (IBDV) attenuated vaccine strain PBG98 (29) showed clear induction of ChIFN2 (Fig. 1A).
Fig 1.

Induction of the ChIFN2 promoter upon transfection with poly(I·C) and/or infection with viruses. (A) RNase protection analysis of expression of ChIFN2 in DF-1 cells upon virus infection. DF-1 cells, mock treated or infected with virus (FWPV strain FP9 or IBDV strain PBG98) for the times stated, were analyzed for expression of ChIFN2 by RNase protection using 10 μg RNA. The levels of control chicken β-actin are also shown. Gels were exposed (exp) for 18 h or 72 h. (B to D) Luciferase reporter analysis of expression of ChIFN2 in DF-1 cells. DF-1 cells were transfected with the ChIFN2 promoter luciferase reporter plasmid (pChIFN2lucter) and plasmid pJATlacZ, constitutively expressing β-galactosidase from the rat β-actin promoter (31). Following recovery for 24 h, cells were either left uninfected or infected (at an MOI of 10) with poxviruses (attenuated FWPV FP9 or its progenitor, HP1) or attenuated IBDV vaccine strain PBG98. Following infection for 4 h, cells were either left untreated or transfected with poly(I·C) (10 μg ml−1) and incubated for 16 h. Luciferase expression values were normalized to those of β-galactosidase. ChIFN2 expression levels were compared to the level of the uninduced control to calculate the fold induction. (B) Induction of the ChIFN2 promoter by transfected poly(I·C) as a positive control or by infection alone. (C) Modulation of induction of the ChIFN2 promoter mediated by transfected poly(I·C) following infection with FWPV (FP9 or HP1) or IBDV PBG98. (D and E) Modulation of induction of the ChIFN2 promoter mediated by virus infection or by transfected poly(I·C) following infection with FWPV FP9 or IBDV PBG98. Samples from the same experiment were split for luciferase assay (D) and for qRT-PCR (E). ChIFN2 expression levels were calculated relative to those of GAPDH and the untreated control. (F) The same samples used for panels D and E were tested for expression of early (fpv094) and late (fpv176) genes by qRT-PCR. Their expression was normalized against that of GAPDH and is presented relative to that in the FP9 sample.
To test whether the failure of FWPV to induce ChIFN2 was reflected by a failure to activate the ChIFN2 promoter, the effects of infection on expression of a luciferase reporter gene under the control of the ChIFN2 promoter (30) transfected into chicken DF-1 cells were examined. Compared to poly(I·C) transfection or IBDV infection, FWPV infection did not induce the ChIFN2 promoter (Fig. 1B and D). This effect was seen whether the highly passaged and attenuated FP9 strain of FWPV or its pathogenic precursor strain (HP1) was used (Fig. 1B), despite the fact that the former has undergone numerous mutations, including the loss of 22 kbp of genomic sequence (28).
FWPV actively blocks dsRNA induction of the ChIFN2 promoter.
We next examined whether the failure of FWPV to activate the ChIFN2 promoter represented a failure to generate a pathogen-associated molecular pattern or the production of an FWPV-encoded inhibitor of IFN induction. Figures 1C and D show that FWPV-infected DF-1 cells exhibited a block to subsequent induction of the ChIFN2 promoter by transfected poly(I·C). In contrast, not only did the positive control IBDV PBG98 fail to block poly(I·C)-mediated induction of the promoter, but also it actually enhanced the induction. The FWPV-mediated block to induction of the ChIFN2 promoter upon virus infection alone or after subsequent poly(I·C) transfection was confirmed by qRT-PCR in parallel with luciferase reporter assays (Fig. 1D and E). Infection of appropriate samples with FWPV was demonstrated by qRT-PCR for FWPV early and late genes (Fig. 1F). These data indicated that FWPV encodes an inhibitor of transfected poly(I·C)-mediated ChIFN2 induction.
Identification of a knockout mutant of FWPV partially defective in the ability to block induction of ChIFN2.
A loss-of-function approach was adopted to identify an FWPV gene(s) that blocked the induction of ChIFN2 by poly(I·C). To facilitate this, a panel of FWPV nonessential gene gpt-insertion-knockout mutants (48 single genes and 1 double gene; Table 3), was screened by luciferase reporter assay for complete or partial loss of the ability of FP9 to block induction of ChIFN2 by transfected poly(I·C).
Table 3.
FWPV FP9 gene knockouts screened for loss of ability to block poly(I·C) induction of ChIFN2luc reportera
| Targeted FWPV FP9 geneb |
|---|
| 006 (C4/C10L) |
| 010 (SERPIN) |
| 011 (SNAP) |
| 012 (ANK) |
| 014 (ANK) |
| 016 (IFN-γ-binding protein) |
| 017 (V Ig domain) |
| 018 (ANK) |
| 020 (C4/C10L) |
| 021 (GPCR) |
| 022 (ANK) |
| 023 (ANK) |
| 024 (ANK) |
| 026 (ANK) |
| 027 (GPCR) |
| 034 (ANK) |
| 040 (SERPIN) |
| 044 (SERPIN) |
| 046 (A44L; steroid DH) |
| 047 (semaphorin; A39L) |
| 048 (ELOVL4) |
| 055 (V Ig domain) |
| 063 (unknown) |
| 075 (N1R/p28) |
| 076 (β-NGF) |
| 080 (TGF-β) |
| 092 (E11L) |
| 097/098 (VARV B22R) |
| 099 (VARV B22R) |
| 105 (F15L) |
| 107 (VARV B22R) |
| 109 (F12L) |
| 119 (G6R) |
| 122 (VARV B22R) |
| 150 (N1R/p28) |
| 155 (N1R/p28) |
| 213 (unknown) |
| 216 (ANK) |
| 217 (NPHV protein) |
| 219 (ANK) |
| 224 (ANK) |
| 226 (B1R; S/T kinase) |
| 227 (ANK) |
| 228 (ANK) |
| 229 (A47L?) |
| 230 (ANK) |
| 231 (ANK) |
| 232 (ANK) |
| 011/033 (SNAP double K/O) |
Gene assignments are as described previously (28).
Gene prediction and/or VACV ortholog. SERPIN, serine proteinase inhibitor; GPCR, G protein-coupled receptor; β-NGF, beta nerve growth factor; TGF-β, transforming growth factor β; VARV, variola virus; NPHV, nuclear polyhedrosis virus; S/T kinase, serine/threonine protein kinase; SNAP, soluble NSF attachment protein; V Ig domain, V-type immunoglobulin domain; steroid DH, hydroxysteroid dehydrogenase; ELOVL4, elongation of very-long-chain fatty acids protein 4; K/O, knockout.
One gpt-insertion-knockout mutant consistently demonstrated higher levels of transfected poly(I·C)-induced luciferase activity than the parental FP9 strain and the other mutants (Fig. 2A). This mutant, FP9_012::gpt, had a copy of the gpt gene inserted in the center of fpv012. An independent deletion mutant, FP9Δ012TD, generated using the transient dominant method (35, 36), displayed the same phenotype (Fig. 2B). Multistep growth curves of the replication of parental FWPV FP9 and FP9Δ012TD in CEFs, infected at an MOI of 0.01, showed no significantly adverse effect of the fpv012 disruption on the production of intra- and extracellular virus up to 72 h postinfection (hpi) (Fig. 3).
Fig 2.
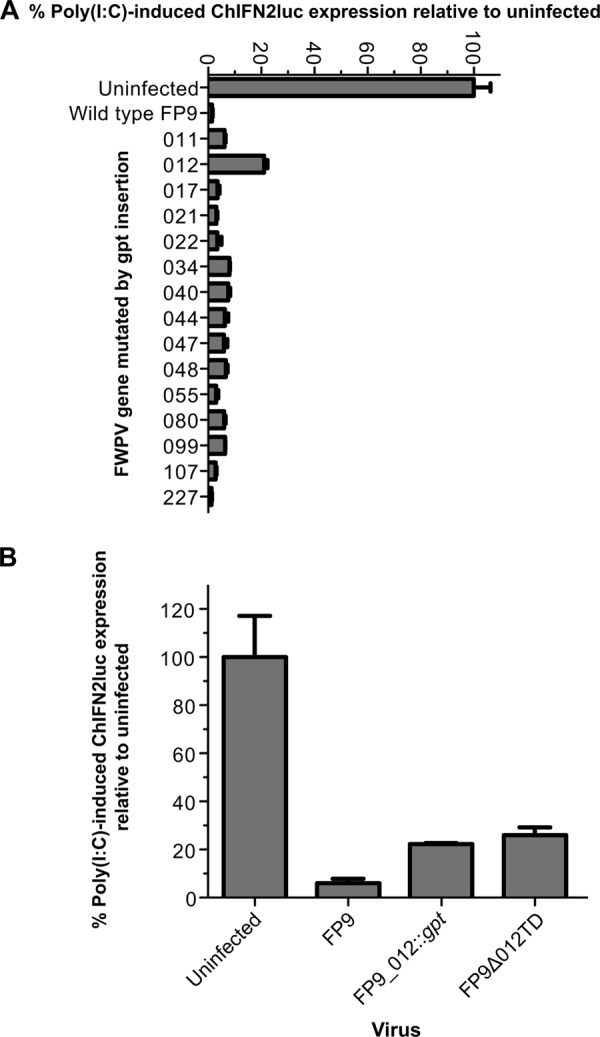
Screening of FWPV FP9 mutants for reduced ability to block poly(I·C)-mediated induction of the ChIFN2 promoter. (A) Chicken DF1 cells were transfected with the ChIFN2 promoter reporter (pChIFN2lucter) and the constitutive lacZ reporter plasmid pJATlacZ. Following recovery for 24 h, cells were either left uninfected or infected with parental FWPV FP9 or single-gene mutants of FP9 at an MOI of 10. Following infection for 4 h, cells were either left untreated or transfected with poly(I·C) (10 μg ml−1) and incubated for 16 h. Luciferase assays were carried out, and data were normalized using β-galactosidase measurements. The level of induction for each sample was compared to that for the uninfected, poly(I·C)-treated control to calculate percent induction. Results show the mean (n = 3) + SD. (A) Results of an experiment screening 15 single-gene insertion mutants; (B) blocking of induction of the ChIFN2 promoter by two independently isolated fpv012 mutants created by two different methods (insertion of the gpt selection cassette in FP9_012::gpt or trans-dominant selection-mediated deletion in FP9Δ012TD) is compared with that observed for parental strain FP9.
Fig 3.
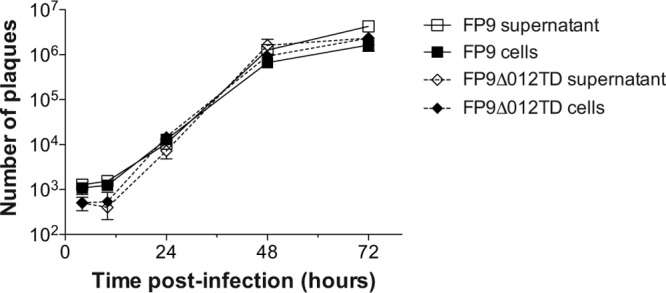
Multistep growth kinetics of the parental FWPV FP9 strain and transient dominant selection deletion mutant FP9Δ012TD. CEFs were infected with FP9 or FP9Δ012TD at an MOI of 0.01. Extracellular (supernatant) and intracellular (cells) samples were harvested at 2, 10, 24, 48, and 72 h postinfection. Samples were freeze-thawed three times, and the virus titer was determined in triplicate using plaque assay.
Expression kinetics of fpv012.
Analysis of the kinetics of expression of fpv012 by qRT-PCR revealed that it was expressed early during FWPV infection, but only weakly so (Fig. 4A). The levels of fpv012 RNA, relative to those of GAPDH, were below those observed for control genes fpv094 (an ortholog of VACV E9L encoding DNA polymerase) and fpv100 (an ortholog of VACV E4L encoding RNA polymerase subunit RPO30). Both of the VACV orthologs showed early expression kinetics (37).
Fig 4.

qRT-PCR analysis of FWPV gene expression by parental, 012 mutant, and revertant viruses. Expression of mRNA specific for fpv012 was assayed by qRT-PCR, using as controls the FWPV genes fpv094 (ortholog of VACV E9L; DNA polymerase), fpv100 (ortholog of VACV E4L; RNA polymerase subunit RPO30), fpv165 (ortholog of VACV A2L; VLTF-3 late transcription factor), fpv168 (core protein with a molecular weight of 39,000 [39K] [68, 69]; ortholog of VACV A4L), and fpv176 (ortholog of VACV A12L). Expression was standardized against that for chicken GAPDH. Graphs show the results for parental strain FP9 (A), FP9Δ012TD (B), and revertant (C) viruses from two experiments (mean ± SD). (D) Expression of TAP-tagged fpv012 (with TAP-tagged fpv014 as a control) inserted back into the native locus in FWPV FP9 under the control of its cognate promoter in infected CEF and DF-1 cells. TAP-tagged proteins were detected by immunoblotting of SDS-polyacrylamide gels with anti-FLAG antibody (Sigma) and anti-mouse secondary antibody (LI-COR) per the manufacturers' protocols. The immunoblots were imaged using a LI-COR Odyssey infrared imaging system. Samples were obtained at 24 hpi (MOI, 3). Molecular mass markers (M) are shown, as are the predicted (Pred.) masses of TAP-tagged fpv012 (41 kDa) and TAP-tagged fpv014 (53 kDa).
As expected, no fpv012 expression was detected in cells infected with the fpv012 deletion mutant FP9Δ012TD (Fig. 4B), but it was evident in cells infected with the revertant (Fig. 4C). Analysis of the same data plotted on a linear scale (data not shown) revealed that in cells infected with the mutant, there appeared to be a slight delay (1.5 to 2.5 h) in the kinetics of expression of late FWPV genes (fpv165, fpv168, and fpv176), with half-maximal expression at 12 hpi rather than 10 hpi. Expression of the early genes (fpv094 and fpv100) appeared to be delayed by 3 to 4 h in FP9Δ012TD (with half-maximal expression at 7 hpi rather than 3 to 4 hpi).
Antibodies for fpv012 are unavailable, but a recombinant knock-in virus with a TAP-tagged version of fpv012 inserted into the native locus under the control of the resident promoter was constructed, allowing demonstration of the expression of the tagged protein by Western blotting of lysates with anti-FLAG antibodies (Fig. 4D).
Expression of ISGs is elevated in CEFs infected with the fpv012 deletion mutant.
Complex viruses often encode multiple antagonists of IFN-mediated responses, targeting the IFN induction, signaling, and antiviral activities of IFN-stimulated genes (ISGs). Releasing the viral block to induction of ChIFN2 might not, therefore, lead to expression of ISGs if robust blocks to IFN signaling remain intact. To investigate whether this might be the case for FWPV, CEFs were infected with parental strain FP9 or the fpv012 deletion mutant, and the induction of selected ISGs by virus infection alone was analyzed by qRT-PCR (Table 2). Significant, moderate-level induction of Mx1 by infection with the fpv012 deletion mutant alone and lower-level (but highly significant) induction of ZC3HAV1 were observed (Fig. 5A), but no significant induction of mda-5/IFIH1 or ChIFN2 was observed. No significant induction of any of the ISGs or ChIFN2 by virus infection alone was observed for parental FP9. Expression of Mx1, but not ZC3HAV1, induced by the mutant alone appeared to be the highest at 8 hpi. The experiment (Fig. 5B) was therefore repeated to include an earlier time point (4 hpi), additional ISGs (IFIT5 and ISG12-2), and the revertant virus. This confirmed that the fpv012 deletion mutant is phenotypically distinct from parental FP9 and the revertant in terms of ISG expression following virus infection alone (Fig. 5B). It is clear that Mx1 shows a biphasic response with an immediate peak of expression at 4 hpi, a trough at 8 to 16 hpi, and then increased expression at 24 hpi. A similar profile is shared by IFIT5, though it showed much higher induction at 24 hpi. Another ISG (ISG12-2) showed a different profile, with no immediate peak at 4 hpi but increased expression at 16 and especially 24 hpi. This profile actually resembles that for ChIFN2, which showed trace induction (2-fold) at 24 hpi.
Fig 5.

qRT-PCR analysis of IFN-β and ISG mRNA expression after infection with the FP9 parental, fpv012 deletion mutant, or revertant strain. CEFs were infected (MOI, 5) with the parental FP9, Δ012TD, or 012REV (panel B only) strain and harvested for RNA extraction at 0, 4 (panel B only), 8, 16, and 24 h postinfection. Total RNA was extracted from samples, cDNA was synthesized, and qRT-PCR was performed in triplicate to quantify relative expression kinetics. (A and B) Results from 2 different experiments, using completely different batches of CEFs. Bar graphs show the results from three experiments (mean ± SD), each performed with a different batch of CEFs. Asterisks indicate probability (two-way analysis of variance, followed by the Bonferroni post hoc test): *, P < 0.05; **, P < 0.01; ****, P < 0.0001. Data were normalized against those for chicken GAPDH and are presented relative to mock-infected (A) or parental FP9-infected (B) cells.
Ectopic expression of fpv012 blocks induction of ChIFN2.
Following transfection of the pEFPlink2-derived expression plasmids, ectopic expression of native or N-terminally FLAG-tagged fpv012 but not of control fpv155 (N1R/p28-like) or fpv213 in the absence of other FWPV proteins blocked poly(I·C)-mediated induction of the ChIFN2 promoter in DF-1 cells (Fig. 6B), offering the opportunity for ready analysis of its activity.
Fig 6.

Domain structure and deletion mutant analysis of fpv012. (A) Domain structure of fpv012 showing N-terminal ankyrin repeats (ANK) as well as the C-terminal F-box motif and the larger, encompassing PRANC domain. Structures are to scale. (B) Schematic illustrating the position of deletions of fpv012 in pEF012Flag mutants 1 to 4 (Mut1 to Mut4, respectively, with deletion of amino acids 1 to 42, 1 to 74, 1 to 108, and 319 to 331, respectively) with comparison of the ability of the FLAG-tagged parental (pEF012Flag) and mutant (pEF012FlagMut1 to -4) forms of fpv012 to block poly(I·C)-mediated induction of the ChIFN2 promoter. Empty pEFPlink2 vector, vectors expressing native and FLAG-tagged fpv012 (pEF012 and pEF012FLAG, respectively), or vectors expressing other FWPV genes (fpv155 or fpv213 [pEF155 and pEF213], respectively) served as controls. The ability of the CNPV ortholog of fpv012 (CNPV030) expressed from pEFPlink2 (pEFCNPV030) to block the ChIFN2 promoter was also assayed.
The gene for the IFN modulator fpv012 is a member of the ANK/PRANC poxvirus gene family.
The IFN modulator identified by this study, fpv012, is encoded by a member of the largest gene family in poxviruses (Fig. 6A), representing those proteins containing multiple copies of the ankyrin repeat (ANK; InterPro IPR002110). The majority of poxviral ANK proteins, including fpv012, appear to be of a particular type, ANK/PRANC, with an N-terminal domain (InterPro IPR020683) containing multiple ANKs (4 in the case of fpv012) and a C-terminal F-box-like motif in what has been described by Mercer and colleagues (38) to be a PRANC domain (pox protein repeats of ankyrin—C-terminal; InterPro IPR018272). Deletion analysis was therefore used to define whether both of these domains were required for activity.
Inhibition of the ChIFN2 promoter is not dependent on fpv012 ankyrin repeat 1.
In an attempt to analyze the relative importance of the ANK domain and the C-terminal F-box-like motif in the ability of fpv012 to inhibit the induction of ChIFN2 by poly(I·C), a panel of fpv012 variants with domain deletions (Fig. 6B) was generated and examined in transient transfections. Unfortunately, the levels of expression of wild-type and mutant fpv012 from the constructs were too low to allow investigation of expression and/or stability of the mutated proteins by Western blot analysis using anti-FLAG antibody. Nevertheless, the first ANK appears to be dispensable for the inhibitory activity of fpv012, with mutant 1 (Mut1) displaying as much inhibitory activity as full-length or N-terminally FLAG-tagged fpv012 (Fig. 6B). Removal of additional ANKs from the N terminus (in Mut2 and Mut3) or deletion of just 13 amino acids from the C terminus (Mut4) downstream of the acknowledged F-box motif (38) completely abrogated the inhibitory effect (Fig. 6B), but we cannot eliminate the possibility that the loss of inhibitory activity is due to reduced stability of the truncated proteins.
cnpv030, the canarypox virus ortholog of fpv012, also inhibits poly(I·C) induction of the ChIFN2 promoter.
Although both CNPV and FWPV are members of the Avipoxvirus genus, they are considerably diverged, they are found in different major clades of the genus (39, 70), and they display significant differences in gene complement (20, 40). Comparisons between ANK proteins can be problematic, but extensive phylogenetic analysis (41) revealed that fpv012 is most similar (45% amino acid identity) to an ANK protein, cnpv030, from CNPV. Genes fpv012 and cnpv030 appear to be in relatively syntenic, yet diverged, locations within their respective genomes (Fig. 7), in that cnpv030 is flanked by orthologs of fpv011 (cnpv025) and fpv016 (cnpv032). Moreover, cnpv030 expressed ectopically appeared to be as effective as fpv012 at blocking the transfected poly(I·C)-mediated induction of the ChIFN2 promoter (Fig. 6B), indicating that the possession of genes for modulating the induction of the avian equivalent of IFN-β is probably conserved across the Avipoxvirus genus.
Fig 7.

Comparative genomic organization of the fpv010 to fpv016 loci in FWPV and CNPV. Genes conserved between FWPV and CNPV are shown as filled boxes; nonconserved genes are shown as open blocks. The arrowhead at the end of each block shows the direction of transcription. Reading frames (RF) are indicated. Short and long dashed lines correlate to the left and right ends of the genes, respectively, in the two avipoxviral genomes. The drawing is to scale.
DISCUSSION
Although poxviruses have double-stranded DNA genomes, they have long been known to produce dsRNA by production of convergent, heterogeneous-length, complementary late transcripts (42). Indeed, a VACV mutant temperature sensitive in transcription factor A18 produces more dsRNA, which leads to increased activation of 2′,5′-oligoadenylate synthetase (OAS) (43). VACV E3 has long been known to play a major role in resisting the antiviral effects of IFN, sequestering dsRNA and thereby preventing allosteric activation of the dsRNA-dependent protein kinase (protein kinase R [PKR]) and OAS (44). More recently, E3 was also shown to inhibit activation of IRF3 (45), mediated via PKR and IPS-1 (46), thereby interfering with induction of IFN-β. Although avipoxviruses lack an ortholog of E3, parental strain FP9 is able to block transfected poly(I·C)-mediated induction of ChIFN2 (Fig. 1), so we sought to identify the proteins involved.
The genetic strategy employed identified fpv012, a member of a large avipoxvirus gene family encoding ANK proteins, most of which have C-terminal F-box motifs in so-called PRANC domains. Extensive passage (more than 430 times) of pathogenic FWPV HP1 through CEF culture by Mayr and Malicki, with concomitant attenuation (47), led to the loss or disruption in FP9 of 12 of the 31 FWPV ANK genes, but fpv012 was not affected (28).
Many studies with mammalian poxviruses have demonstrated the role of multiple viral modulators in controlling host IFN responses at various levels. Data shown in Fig. 2 indicate that fpv012 is responsible for blocking about 20% of expression from the ChIFN2 promoter induced by transfected poly(I·C), suggesting that the deletion mutant still has another mechanism(s) to control induction of ChIFN2. The additional mechanisms for controlling ChIFN2 induction appear to be sufficient, even in the absence of fpv012, to control the bulk of the residual ChIFN2 induction stimulated by virus infection, as infection alone by the fpv012 deletion mutant virus stimulated only a trace increase (not more than 2-fold at 24 hpi) in induction of ChIFN2 over that observed for FP9 infection alone (Fig. 5). Nevertheless, infection alone with the fpv012 deletion mutant, but not with FP9, led to significant, moderate-level induction of the ISGs Mx1, ISG12-2, and especially IFIT5 and lower-level (but highly significant) induction of ZC3HAV1 (Fig. 5A and B). Several studies have described induction of such IFN-induced downstream effectors by avian viruses in avian systems (48, 49). Increased expression of the effectors in cells infected by the fpv012 deletion virus might be attributable to the induced trace-level expression of ChIFN2. We cannot exclude the possible involvement of alternative, IFN-independent pathways in induction of the ISG, especially given the different kinetic expression profiles observed for the various ISGs, although it should be stressed that any such pathways are also clearly normally blocked by fpv012. Clarifying this issue will need extensive study of the chicken innate responses, which remain relatively poorly characterized, but fpv012 should prove a useful tool in those studies.
Despite moderate-level induction of ISGs and some delay in expression of viral genes, the fpv012 deletion virus did not appear to be significantly compromised in its replicative capacity in tissue culture (Fig. 3). This indicates that virus mechanisms for subverting the antiviral effectors must exist in FWPV and must remain intact in the fpv012 deletion mutant. We have identified fpv014 to be a contributor to chicken type I IFN resistance in VACV modified virus Ankara (MVA)/FWPV chimeras (see the accompanying paper by Buttigieg et al. [50]), but, on the basis of the VACV paradigm, it is likely that more genes are involved.
Ectopic expression of fpv012 leads to inhibition of transfected poly(I·C)-mediated induction of the ChIFN2 promoter (Fig. 6). This phenotype offered the ready opportunity to analyze the viral determinants of modulation. Unfortunately, the levels of expression of wild-type and mutant fpv012 from the constructs were too low to allow investigation of the expression and/or stability of the mutated proteins by Western blot analysis using anti-FLAG antibody. Nevertheless, the data shown in Fig. 6 demonstrate that the first ANK is dispensable for the inhibitory activity of fpv012. Although removal of further ANKs or just 12 residues from the C terminus of fpv012 disrupted its function when expressed ectopically (Fig. 6B), we were not able to confirm the stability of the truncated protein, so detailed mutagenic analysis of fpv012 must await an extensive study using expression of mutated FLAG-tagged proteins expressed from knock-in virus mutants.
In the accompanying paper (50), we demonstrated interaction between the fpv014 ANK/PRANC protein involved in ChIFN1 (chicken IFN-α) resistance and proteins of the SCF (Skp1, Cullin-1, F-box) ubiquitin ligase complex, as has been observed for a number of mammalian poxvirus ANK proteins (51–55). It is postulated (38) that such interactions allow ANK/PRANC proteins to act as adaptors targeting for ubiquitination (and probable proteasomal degradation) ligands captured by the N-terminal ANK domains. Despite considerable effort, probably because of the low levels of expression of fpv012 in the various systems, we have so far been unable to identify interaction of fpv012 with the SCF complex. We have also been unable to identify any cellular proteins captured by the N-terminal ANK domain of fpv012, but ligands for this family of proteins have generally proved elusive. Thus far, the only ligands identified are Akt, for myxoma virus MT-5 (56), and NF-κB, for variola virus G1 (57) and its cowpox virus (CPXV) ortholog CPXV006 (58), as well as for CPXV CP77, encoded by CPXV025 (52).
It is not clear why so many ANK proteins are encoded by the avipoxviruses. The difference in numbers of ANK protein genes carried by relatively closely related viruses (e.g., 31 in FWPV and 51 in CNPV) suggests that gene expansion involves duplication and subsequent evolution. Such expansion, driven by IFN, has recently been demonstrated (using deep sequencing) for VACV in cell culture (59). In a genetic background defective for E3L, the copy number of the K3L gene was observed to expand, allowing it to mutate and evolve so that it was more effective at inhibiting PKR. The authors coined the term “viral gene accordions” for this mechanism, as selection of an advantageous allele allowed subsequent contraction in copy number (59). They also recognized that the expansion of the ANK genes in avipoxviruses is a “particularly clear example of such adaptive gene expansions” and, moreover, that it represents “an exceptional example” whereby multiple functional variants have been generated so that the accordion has not collapsed to a single copy. Our demonstration that fpv012 and cnpv030, which are an orthologous pair (41), share the ability to block induction of avian IFN-β provides a strong indication that ANK gene expansion in the avipoxviruses predates speciation of FWPV and CNPV, particularly as fpv012 and cnpv030 are far from basal to the avipoxvirus ANK phylogenetic tree (see the supplemental figure of Sonnberg et al. [41]).
Expansion of such a large complement of related genes to the extent seen for this viral gene accordion might initially have facilitated high-level expression of inhibitors of a limited number of targets by a gene dosage effect (59). Subsequent selection and evolution might have allowed the targeting of a limited number of host factors in a redundant manner, as seen for binding of NF-κB by both CPXV006 and CPXV025 (52, 58). Ultimately, the accordion might have become capable of targeting multiple, distinct, cellular proteins. Our demonstration of different functions for two avipoxvirus ANK proteins (fpv012 here and fpv014 in the accompanying paper [50]), both of which fall in the same larger phylogenetic cluster of avipoxvirus ANK genes (see the supplemental figure of Sonnberg et al. [41]), hints at a complex and highly dynamic evolutionary picture. There is clearly wide scope for evolutionary selection of functions for the avipoxvirus ANK genes. It is notable that avipoxviruses lack equivalents of mammalian poxvirus genes encoding a family of related proteins with structural homology to cellular Bcl-2 (note that fpv039, an antiapoptotic, Bcl-2-like FWPV protein [60], appears to be unrelated [61]). In VACV, these proteins (A46, A52, B14, C1, C6, C16/B22, K7, and N1 [61–67]) target host factors involved in immunomodulation and the control of apoptosis. It is likely that these genes represent another viral gene accordion. It will be interesting to discover whether the avipoxvirus ANK protein family has evolved to recognize a range of targets similar to that of the VACV Bcl-2-like proteins.
ACKNOWLEDGMENTS
We thank Geoffrey Smith (University of Cambridge) for pUC13TAP. We acknowledge the financial support of the Biotechnology and Biological Sciences Research Council (BBSRC) with funding to M.A.S. (grants BBS/B/00115/2, BB/E009956/1, and BB/G018545/1) and S.G. (grants BBS/B/00344 and BB/G018332/1).
Footnotes
Published ahead of print 20 February 2013
REFERENCES
- 1. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 2. Perdiguero B, Esteban M. 2009. The interferon system and vaccinia virus evasion mechanisms. J. Interferon Cytokine Res. 29:581–598 [DOI] [PubMed] [Google Scholar]
- 3. Shisler JL, Jin XL. 2004. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J. Virol. 78:3553–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Unterholzner L, Sumner RP, Baran M, Ren H, Mansur DS, Bourke NM, Randow F, Smith GL, Bowie AG. 2011. Vaccinia virus protein C6 is a virulence factor that binds TBK-1 adaptor proteins and inhibits activation of IRF3 and IRF7. PLoS Pathog. 7:e1002247 doi:10.1371/journal.ppat.1002247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Colamonici OR, Domanski P, Sweitzer SM, Larner A, Buller RM. 1995. Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon alpha transmembrane signaling. J. Biol. Chem. 270:15974–15978 [DOI] [PubMed] [Google Scholar]
- 6. Symons JA, Alcami A, Smith GL. 1995. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81:551–560 [DOI] [PubMed] [Google Scholar]
- 7. Najarro P, Traktman P, Lewis JA. 2001. Vaccinia virus blocks gamma interferon signal transduction: viral VH1 phosphatase reverses Stat1 activation. J. Virol. 75:3185–3196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chang HW, Watson JC, Jacobs BL. 1992. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. U. S. A. 89:4825–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharp TV, Witzel JE, Jagus R. 1997. Homologous regions of the alpha subunit of eukaryotic translational initiation factor 2 (eIF2alpha) and the vaccinia virus K3L gene product interact with the same domain within the dsRNA-activated protein kinase (PKR). Eur. J. Biochem. 250:85–91 [DOI] [PubMed] [Google Scholar]
- 10. Bolte AL, Meurer J, Kaleta EF. 1999. Avian host spectrum of avipoxviruses. Avian Pathol. 28:415–432 [DOI] [PubMed] [Google Scholar]
- 11. Mercer AA, Buller RM, McInnes CJ, Upton C, Lefkowitz EJ, McFadden DG, Damon IK, Skinner MA, Moyer RW. 2011. Poxviridae, p 291–310 In King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ. (ed), Virus taxonomy: classification and nomenclature of viruses: Ninth Report of the International Committee on Taxonomy of Viruses Elsevier, Oxford, United Kingdom [Google Scholar]
- 12. Chen HY, Yang MF, Cui BA, Cui P, Sheng M, Chen G, Wang SJ, Geng JW. 2010. Construction and immunogenicity of a recombinant fowlpox vaccine coexpressing S1 glycoprotein of infectious bronchitis virus and chicken IL-18. Vaccine 28:8112–8119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Johnson DI, Vagnozzi A, Dorea F, Riblet SM, Mundt A, Zavala G, Garcia M. 2010. Protection against infectious laryngotracheitis by in ovo vaccination with commercially available viral vector recombinant vaccines. Avian Dis. 54:1251–1259 [DOI] [PubMed] [Google Scholar]
- 14. Rautenschlein S, Sharma JM, Winslow BJ, McMillen J, Junker D, Cochran M. 1999. Embryo vaccination of turkeys against Newcastle disease infection with recombinant fowlpox virus constructs containing interferons as adjuvants. Vaccine 18:426–433 [DOI] [PubMed] [Google Scholar]
- 15. Coupar BE, Purcell DF, Thomson SA, Ramshaw IA, Kent SJ, Boyle DB. 2006. Fowlpox virus vaccines for HIV and SHIV clinical and pre-clinical trials. Vaccine 24:1378–1388 [DOI] [PubMed] [Google Scholar]
- 16. Jager E, Karbach J, Gnjatic S, Neumann A, Bender A, Valmori D, Ayyoub M, Ritter E, Ritter G, Jager D, Panicali D, Hoffman E, Pan L, Oettgen H, Old LJ, Knuth A. 2006. Recombinant vaccinia/fowlpox NY-ESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc. Natl. Acad. Sci. U. S. A. 103:14453–14458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Radaelli A, Bonduelle O, Beggio P, Mahe B, Pozzi E, Elli V, Paganini M, Zanotto C, De Giuli Morghen C, Combadiere B. 2007. Prime-boost immunization with DNA, recombinant fowlpox virus and VLP(SHIV) elicit both neutralizing antibodies and IFNgamma-producing T cells against the HIV-envelope protein in mice that control env-bearing tumour cells. Vaccine 25:2128–2138 [DOI] [PubMed] [Google Scholar]
- 18. Webster DP, Dunachie S, McConkey S, Poulton I, Moore AC, Walther M, Laidlaw SM, Peto T, Skinner MA, Gilbert SC, Hill AV. 2006. Safety of recombinant fowlpox strain FP9 and modified vaccinia virus Ankara vaccines against liver-stage P. falciparum malaria in non-immune volunteers. Vaccine 24:3026–3034 [DOI] [PubMed] [Google Scholar]
- 19. Bublot M, Pritchard N, Swayne DE, Selleck P, Karaca K, Suarez DL, Audonnet JC, Mickle TR. 2006. Development and use of fowlpox vectored vaccines for avian influenza. Ann. N. Y. Acad. Sci. 1081:193–201 [DOI] [PubMed] [Google Scholar]
- 20. Tulman ER, Afonso CL, Lu Z, Zsak L, Kutish GF, Rock DL. 2004. The genome of canarypox virus. J. Virol. 78:353–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karaca K, Bowen R, Austgen LE, Teehee M, Siger L, Grosenbaugh D, Loosemore L, Audonnet JC, Nordgren R, Minke JM. 2005. Recombinant canarypox vectored West Nile virus (WNV) vaccine protects dogs and cats against a mosquito WNV challenge. Vaccine 23:3808–3813 [DOI] [PubMed] [Google Scholar]
- 22. Soboll G, Hussey SB, Minke JM, Landolt GA, Hunter JS, Jagannatha S, Lunn DP. 2010. Onset and duration of immunity to equine influenza virus resulting from canarypox-vectored (ALVAC) vaccination. Vet. Immunol. Immunopathol. 135:100–107 [DOI] [PubMed] [Google Scholar]
- 23. Welter J, Taylor J, Tartaglia J, Paoletti E, Stephensen CB. 1999. Mucosal vaccination with recombinant poxvirus vaccines protects ferrets against symptomatic CDV infection. Vaccine 17:308–318 [DOI] [PubMed] [Google Scholar]
- 24. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220 [DOI] [PubMed] [Google Scholar]
- 25. Sick C, Schultz U, Munster U, Meier J, Kaspers B, Staeheli P. 1998. Promoter structures and differential responses to viral and nonviral inducers of chicken type I interferon genes. J. Biol. Chem. 273:9749–9754 [DOI] [PubMed] [Google Scholar]
- 26. Sick C, Schultz U, Staeheli P. 1996. A family of genes coding for two serologically distinct chicken interferons. J. Biol. Chem. 271:7635–7639 [DOI] [PubMed] [Google Scholar]
- 27. Himly M, Foster DN, Bottoli I, Iacovoni JS, Vogt PK. 1998. The DF-1 chicken fibroblast cell line: transformation induced by diverse oncogenes and cell death resulting from infection by avian leukosis viruses. Virology 248:295–304 [DOI] [PubMed] [Google Scholar]
- 28. Laidlaw SM, Skinner MA. 2004. Comparison of the genome sequence of FP9, an attenuated, tissue culture-adapted European strain of fowlpox virus, with those of virulent American and European viruses. J. Gen. Virol. 85:305–322 [DOI] [PubMed] [Google Scholar]
- 29. Brown MD, Skinner MA. 1996. Coding sequences of both genome segments of a European ‘very virulent’ infectious bursal disease virus. Virus Res. 40:1–15 [DOI] [PubMed] [Google Scholar]
- 30. Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, Randall R, Goodbourn S. 2007. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology 359:190–200 [DOI] [PubMed] [Google Scholar]
- 31. Masson N, Ellis M, Goodbourn S, Lee KA. 1992. Cyclic AMP response element-binding protein and the catalytic subunit of protein kinase A are present in F9 embryonal carcinoma cells but are unable to activate the somatostatin promoter. Mol. Cell. Biol. 12:1096–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Marais R, Light Y, Paterson HF, Marshall CJ. 1995. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 14:3136–3145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zinn K, DiMaio D, Maniatis T. 1983. Identification of two distinct regulatory regions adjacent to the human beta-interferon gene. Cell 34:865–879 [DOI] [PubMed] [Google Scholar]
- 34. Goodbourn S, Burstein H, Maniatis T. 1986. The human beta-interferon gene enhancer is under negative control. Cell 45:601–610 [DOI] [PubMed] [Google Scholar]
- 35. Falkner FG, Moss B. 1990. Transient dominant selection of recombinant vaccinia viruses. J. Virol. 64:3108–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Laidlaw SM, Anwar MA, Thomas W, Green P, Shaw K, Skinner MA. 1998. Fowlpox virus encodes nonessential homologs of cellular alpha-SNAP, PC-1, and an orphan human homolog of a secreted nematode protein. J. Virol. 72:6742–6751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B. 2010. Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:11513–11518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mercer AA, Fleming SB, Ueda N. 2005. F-box-like domains are present in most poxvirus ankyrin repeat proteins. Virus Genes 31:127–133 [DOI] [PubMed] [Google Scholar]
- 39. Jarmin S, Manvell R, Gough RE, Laidlaw SM, Skinner MA. 2006. Avipoxvirus phylogenetics: identification of a PCR length polymorphism that discriminates between the two major clades. J. Gen. Virol. 87:2191–2201 [DOI] [PubMed] [Google Scholar]
- 40. Afonso CL, Tulman ER, Lu Z, Zsak L, Kutish GF, Rock DL. 2000. The genome of fowlpox virus. J. Virol. 74:3815–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sonnberg S, Fleming SB, Mercer AA. 2011. Phylogenetic analysis of the large family of poxvirus ankyrin-repeat proteins reveals orthologue groups within and across chordopoxvirus genera. J. Gen. Virol. 92:2596–2607 [DOI] [PubMed] [Google Scholar]
- 42. Colby C, Duesberg PH. 1969. Double-stranded RNA in vaccinia virus infected cells. Nature 222:940–944 [DOI] [PubMed] [Google Scholar]
- 43. Bayliss CD, Condit RC. 1993. Temperature-sensitive mutants in the vaccinia virus A18R gene increase double-stranded RNA synthesis as a result of aberrant viral transcription. Virology 194:254–262 [DOI] [PubMed] [Google Scholar]
- 44. Watson JC, Chang HW, Jacobs BL. 1991. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 185:206–216 [DOI] [PubMed] [Google Scholar]
- 45. Smith EJ, Marie I, Prakash A, Garcia-Sastre A, Levy DE. 2001. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by vaccinia virus E3L protein. J. Biol. Chem. 276:8951–8957 [DOI] [PubMed] [Google Scholar]
- 46. Zhang P, Samuel CE. 2008. Induction of protein kinase PKR-dependent activation of interferon regulatory factor 3 by vaccinia virus occurs through adapter IPS-1 signaling. J. Biol. Chem. 283:34580–34587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mayr A, Malicki K. 1966. Attenuierung von virulentem Hühnerpockenvirus in Zellkulturen und Eigenschaften des attenuierten Virus. Zentralbl. Veterinärmed. B 13:1–13 [PubMed] [Google Scholar]
- 48. Rue CA, Susta L, Cornax I, Brown CC, Kapczynski DR, Suarez DL, King DJ, Miller PJ, Afonso CL. 2011. Virulent Newcastle disease virus elicits a strong innate immune response in chickens. J. Gen. Virol. 92:931–939 [DOI] [PubMed] [Google Scholar]
- 49. Uchida Y, Watanabe C, Takemae N, Hayashi T, Oka T, Ito T, Saito T. 2012. Identification of host genes linked with the survivability of chickens infected with recombinant viruses possessing H5N1 surface antigens from a highly pathogenic avian influenza virus. J. Virol. 86:2686–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buttigieg K, Laidlaw SM, Ross C, Davies M, Goodbourn S, Skinner MA. 2013. Genetic screen of a library of chimeric poxviruses identifies an ankyrin repeat protein involved in resistance to the avian type I interferon response. J. Virol. 87:5028–5040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Blanie S, Gelfi J, Bertagnoli S, Camus-Bouclainville C. 2010. MNF, an ankyrin repeat protein of myxoma virus, is part of a native cellular SCF complex during viral infection. Virol. J. 7:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chang SJ, Hsiao JC, Sonnberg S, Chiang CT, Yang MH, Tzou DL, Mercer AA, Chang W. 2009. Poxvirus host range protein CP77 contains an F-box-like domain that is necessary to suppress NF-kappaB activation by tumor necrosis factor alpha but is independent of its host range function. J. Virol. 83:4140–4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sperling KM, Schwantes A, Schnierle BS, Sutter G. 2008. The highly conserved orthopoxvirus 68k ankyrin-like protein is part of a cellular SCF ubiquitin ligase complex. Virology 374:234–239 [DOI] [PubMed] [Google Scholar]
- 54. van Buuren N, Couturier B, Xiong Y, Barry M. 2008. Ectromelia virus encodes a novel family of F-box proteins that interact with the SCF complex. J. Virol. 82:9917–9927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Werden SJ, Lanchbury J, Shattuck D, Neff C, Dufford M, McFadden G. 2009. The myxoma virus m-t5 ankyrin repeat host range protein is a novel adaptor that coordinately links the cellular signaling pathways mediated by Akt and Skp1 in virus-infected cells. J. Virol. 83:12068–12083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Werden SJ, Barrett JW, Wang G, Stanford MM, McFadden G. 2007. M-T5, the ankyrin repeat, host range protein of myxoma virus, activates Akt and can be functionally replaced by cellular PIKE-A. J. Virol. 81:2340–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mohamed MR, Rahman MM, Lanchbury JS, Shattuck D, Neff C, Dufford M, van Buuren N, Fagan K, Barry M, Smith S, Damon I, McFadden G. 2009. Proteomic screening of variola virus reveals a unique NF-kappaB inhibitor that is highly conserved among pathogenic orthopoxviruses. Proc. Natl. Acad. Sci. U. S. A. 106:9045–9050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Mohamed MR, Rahman MM, Rice A, Moyer RW, Werden SJ, McFadden G. 2009. Cowpox virus expresses a novel ankyrin repeat NF-kappaB inhibitor that controls inflammatory cell influx into virus-infected tissues and is critical for virus pathogenesis. J. Virol. 83:9223–9236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Elde NC, Child SJ, Eickbush MT, Kitzman JO, Rogers KS, Shendure J, Geballe AP, Malik HS. 2012. Poxviruses deploy genomic accordions to adapt rapidly against host antiviral defenses. Cell 150:831–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Banadyga L, Gerig J, Stewart T, Barry M. 2007. Fowlpox virus encodes a Bcl-2 homologue that protects cells from apoptotic death through interaction with the proapoptotic protein Bak. J. Virol. 81:11032–11045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gonzalez JM, Esteban M. 2010. A poxvirus Bcl-2-like gene family involved in regulation of host immune response: sequence similarity and evolutionary history. Virol. J. 7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Aoyagi M, Zhai D, Jin C, Aleshin AE, Stec B, Reed JC, Liddington RC. 2007. Vaccinia virus N1L protein resembles a B cell lymphoma-2 (Bcl-2) family protein. Protein Sci. 16:118–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Benfield CT, Mansur DS, McCoy LE, Ferguson BJ, Bahar MW, Oldring AP, Grimes JM, Stuart DI, Graham SC, Smith GL. 2011. Mapping the I{kappa}B kinase beta (ikk{beta})-binding interface of the B14 protein, a vaccinia virus inhibitor of IKK{beta}-mediated activation of nuclear factor kappaB. J. Biol. Chem. 286:20727–20735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cooray S, Bahar MW, Abrescia NG, McVey CE, Bartlett NW, Chen RA, Stuart DI, Grimes JM, Smith GL. 2007. Functional and structural studies of the vaccinia virus virulence factor N1 reveal a Bcl-2-like anti-apoptotic protein. J. Gen. Virol. 88:1656–1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Graham SC, Bahar MW, Cooray S, Chen RA, Whalen DM, Abrescia NG, Alderton D, Owens RJ, Stuart DI, Smith GL, Grimes JM. 2008. Vaccinia virus proteins A52 and B14 share a Bcl-2-like fold but have evolved to inhibit NF-kappaB rather than apoptosis. PLoS Pathog. 4:e1000128 doi:10.1371/journal.ppat.1000128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jacobs N, Bartlett NW, Clark RH, Smith GL. 2008. Vaccinia virus lacking the Bcl-2-like protein N1 induces a stronger natural killer cell response to infection. J. Gen. Virol. 89:2877–2881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kalverda AP, Thompson GS, Vogel A, Schroder M, Bowie AG, Khan AR, Homans SW. 2009. Poxvirus K7 protein adopts a Bcl-2 fold: biochemical mapping of its interactions with human DEAD box RNA helicase DDX3. J. Mol. Biol. 385:843–853 [DOI] [PubMed] [Google Scholar]
- 68. Boulanger D, Green P, Jones B, Henriquet G, Hunt LG, Laidlaw SM, Monaghan P, Skinner MA. 2002. Identification and characterization of three immunodominant structural proteins of fowlpox virus. J. Virol. 76:9844–9855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Boulanger D, Green P, Smith T, Czerny CP, Skinner MA. 1998. The 131-amino-acid repeat region of the essential 39-kilodalton core protein of fowlpox virus FP9, equivalent to vaccinia virus A4L protein, is nonessential and highly immunogenic. J. Virol. 72:170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gyuranecz M, Foster JT, Dán A, Ip HS, Egstad KF, Parker PG, Higashiguchi JM, Skinner MA, Höfle U, Kreizinger Z, Dorrestein GM, Solt S, Sós E, Kim YJ, Uhart M, Pereda A, González-Hein G, Hidalgo H, Blanco J-M, Erdélyic K. 2013. Worldwide phylogenetic relationship of avian poxviruses. J. Virol. 87:4938–4951 [DOI] [PMC free article] [PubMed] [Google Scholar]