

Abstract
Broadly neutralizing antibodies PG9 and PG16 effectively neutralize 70 to 80% of circulating HIV-1 isolates. In this study, the neutralization abilities of PG9 and PG16 were further enhanced by bioconjugation with aplaviroc, a small-molecule inhibitor of virus entry into host cells. A novel air-stable diazonium hexafluorophosphate reagent that allows for rapid, tyrosine-selective functionalization of proteins and antibodies under mild conditions was used to prepare a series of aplaviroc-conjugated antibodies, including b12, 2G12, PG9, PG16, and CD4-IgG. The conjugated antibodies blocked HIV-1 entry through two mechanisms: by binding to the virus itself and by blocking the CCR5 receptor on host cells. Chemical modification did not significantly alter the potency of the parent antibodies against nonresistant HIV-1 strains. Conjugation did not alter the pharmacokinetics of a model IgG in blood. The PG9-aplaviroc conjugate was tested against a panel of 117 HIV-1 strains and was found to neutralize 100% of the viruses. PG9-aplaviroc conjugate IC50s were lower than those of PG9 in neutralization studies of 36 of the 117 HIV-1 strains. These results support this new approach to bispecific antibodies and offer a potential new strategy for combining HIV-1 therapies.
INTRODUCTION
Innovative new approaches to HIV-1 prophylaxis and therapy are desperately needed. Despite the successes of highly active antiretroviral therapy (HAART), more than 2 million people die each year and more than 33 million individuals are infected worldwide (http://aids.gov/hiv-aids-basics/hiv-aids-101/global-statistics/). Although HAART is typically effective, it is not without problems, including complicated drug-drug interactions, adherence issues, and a myriad of side effects. The development of potent and broadly acting biologic drugs might offer a solution to some of these problems and complement traditional HAART.
Broadly neutralizing monoclonal antibodies (BNmAbs) that recognize features conserved across clades of HIV are promising starting points for the development of immunotherapeutic agents against HIV-1 (1–8). Several studies have shown that the transfer of sufficient quantities of broadly neutralizing antibodies can achieve sterilizing immunity against intravenous, vaginal, or rectal challenge in macaque models (9, 10). The delivery of broadly neutralizing antibodies using gene-based approaches has also been shown to be effective in animal models (11, 12). Indeed, soon after our discovery of BNmAb b12, we developed protein engineering methods to increase the potency and breadth of neutralization by b12 with the original aim of developing evolved b12 variants for HIV-1 therapy (13, 14). Collectively, these studies suggest that BNmAbs could be effective HIV-1 prophylactic and therapeutic agents. Unfortunately, even the most broadly neutralizing antibody is vulnerable to viral escape, because a single amino acid change on the target protein can alter the binding epitope. If a BNmAb could be modified to inhibit HIV in multiple ways, the evolutionary hurdle for escape would be significantly elevated. Furthermore, by combining multiple inhibitory functions in a single molecule, the regulatory and cost issues for a biologic complement to combinatorial drug therapy might be minimized.
Recently, we developed a new class of therapeutic molecules by demonstrating that catalytic monoclonal antibodies covalently linked to designed ligands possess potent biological activities in a variety of animal models of disease (15–19). Several of these are now in clinical development (20). These studies revealed the many advantages of coupling active small molecules and peptides with antibodies. In contrast to bispecific-antibody approaches based on protein engineering, such as the dual-variable-domain (DVD)-Ig (21) or single-chain variable fragment (scFv)-Ig (22) fusion approaches, among others, laborious protein engineering is not required to endow a second specificity when the desired ligand is chemically coupled to the antibody. Furthermore, expression issues are bypassed, since development of a new cell line is not required.
A promising additional blockade to HIV-1 infection that should complement the targeting of viral proteins is the targeting of host proteins required for viral entry and replication. Unlike viral proteins, host proteins are not under selective pressure to evolve to evade the therapeutic agent. A number of small-molecule inhibitors of the HIV-1 coreceptors CCR5 and CXCR4 have been developed (23, 24), and one CCR5-targeting drug has been approved (25–28). Here we covalently linked a CCR5-targeting small molecule, aplaviroc, to BNmAbs and CD4-IgG. This approach provided rapid access to bispecific proteins with exceptional breadth in their abilities to neutralize diverse isolates of HIV-1.
MATERIALS AND METHODS
Antibodies.
Antibodies b12, 2G12, and DEN3 were provided by Dennis R. Burton (Scripps Research Institute); antibodies PG9 and PG16 were provided by the IAVI; the CD4-IgG2 immunoadhesion protein was obtained from Progenics (PRO542). Antibodies were stored at 4°C. Therapeutic-grade trastuzumab (Genentech) was used without additional purification.
Synthesis of labeling reagents.
The synthesis and characterization of aplaviroc with a linker have been described previously (29).
Antibody labeling procedure.
In a 1.5-ml tube, an antibody solution (99 μl; 1.5 mg/ml in 0.1 M Na2HPO4 [pH 8.0]) and 10 equivalents of 4-formylbenzene diazonium hexafluorophosphate (FBDP; 1 μl; 10 mM solution in CH3CN) were combined (30). The solution was mixed gently and was allowed to react for 30 min at room temperature with intermittent mixing. The solution turned yellow upon completion of the reaction. After 30 min, excess FBDP was removed using a Zeba Spin desalting column (molecular weight cutoff [MWCO], 7,000 [7K]; Pierce), and buffer was exchanged with 0.1 M Na2HPO4 (pH 6.0). Aplaviroc-oxyamine (20 equivalents; 2 μl; 10 mM solution in CH3CN) was added, and the solution was incubated overnight at 4°C. Excess aplaviroc-oxyamine was removed using a Zeba Spin desalting column (MWCO, 7K), and free aplaviroc was removed by using a protein A spin column (GE Healthcare) according to the manufacturer's instructions.
Tryptic digestion and quadrupole time of flight tandem mass spectrometry (Q-TOF MS-MS) characterization of antibody conjugates.
Purified antibody samples (100 μl; 1 mg/ml) were exchanged into 6 M guanidine, 0.1 M Tris (pH 8.0) by using 0.5-ml Zeba Spin desalting columns (MWCO, 7K) according to the manufacturer's instructions. To each sample, 1 M dithiothreitol (DTT) (Fisher) was added to a final concentration of 20 mM. The samples were incubated for 1 h at 37°C with gentle shaking, and a 1 M solution of iodoacetamide (Fisher) was added to each sample to a final concentration of 40 mM. Samples were incubated at room temperature in the dark for 40 min. The alkylation reactions were quenched by adding 1 M DTT to a 40 mM final concentration. Sample buffer was then exchanged for trypsin digestion buffer (50 mM Tris [pH 7.5], 5 mM CaCl2) using 0.5-ml Zeba Spin desalting columns (MWCO, 7K). Trypsin Gold (0.5 mg/ml; Pierce) was dissolved in 50 mM acetic acid. The trypsin solution was added to each sample at an enzyme-to-protein ratio of 1:20 (wt/wt), and samples were incubated at 37°C with shaking at 600 rpm for approximately 18 h. Digestion was stopped by the addition of trifluoroacetic acid (TFA) to approximately 0.1%.
Samples (with 20 μg protein injected) were analyzed using an Agilent 6510 Q-TOF mass spectrometer equipped with a Zorbax SB C18 column (narrow bore; inner diameter, 2.1 mm; length, 150 mm; particle size, 3.5 μm) (Agilent). High-performance liquid chromatography (HPLC) parameters were as follows: flow rate, 0.2 ml/min; a gradient from 0 to 40% mobile phase B over 80 min, followed by a gradient to 0% B from 80 to 90 min. Mobile phase A was 0.05% TFA (vol/vol) in HPLC-grade H2O, 2% acetonitrile (vol/vol), and mobile phase B was 0.04% TFA (vol/vol) in 90% acetonitrile (vol/vol). MS data were collected for 200 to 2,500 m/z and 100 to 2,000 m/z, positive polarity, a gas temperature of 325°C, a nebulizer pressure of 30 lb/in2, and a capillary voltage of 3,500 V. Data were analyzed using MassHunter software (Agilent) and GPMAW software (version 8.20; ChemSW).
Flow cytometry.
Flow cytometry experiments were performed as described previously (29). The cell lines used were A431 cells, which do not express CCR5, and TZM-bl cells, which do express CCR5. In brief, a single-cell suspension was prepared, and cells were washed twice with cold stain buffer (BD Pharmingen) and were pelleted by centrifugation (300 × g). The cell pellet was resuspended in cold stain buffer to a final concentration of 2 × 107 cells/ml. Aliquots of 50 μl were distributed to V-bottom wells of microwell plates (Corning). Primary antibodies were added to a final concentration of 20 μg/ml; each sample was tested in triplicate. The cells were incubated on ice for 1 h, washed twice with stain buffer, and resuspended in 100 μl of stain buffer. A fluorescence-labeled secondary antibody was added at a concentration recommended by manufacturer. The plate was incubated on ice, protected from light, for 1 h. The cells were washed twice with the stain buffer, resuspended in 200 μl of stain buffer, and transferred to filter-top fluorescence-activated cell sorter (FACS) tubes (BD Biosciences) containing 300 μl of buffer (2% fetal bovine serum [FBS] in phosphate-buffered saline [PBS]). Cell counting was performed using a digital LSR II cytometer. Data were analyzed with FlowJo software, version 8.7.1.
Pharmacokinetic study.
The pharmacokinetic experiment was carried out as described previously (19). In brief, all animal experiments were performed according to Division of Animal Resources (TSRI) guidelines following approved protocols. Female athymic nude mice (8 weeks of age) were injected subcutaneously with 100 μg trastuzumab or trastuzumab-aplaviroc in 100 μl sterile PBS (4 animals per group). Blood was collected from tail veins after 5 min, 2 h, 4 h, 8 h, 24 h, 48 h, 72 h, 96 h, 168 h, and 336 h. In order to minimize blood loss, only 10 μl of blood was withdrawn per bleed; blood was diluted 1/5 in PBS containing 1% bovine serum albumin (BSA). Samples were allowed to chill on ice for 20 min. Insoluble components were removed by centrifugation, and samples were stored at −20°C until analysis.
The binding and wash buffer for enzyme-linked immunosorbent assays (ELISA) was PBS containing 1% BSA. Half-area ELISA plates (Corning) were coated with anti-human IgG Fc (Pierce) at 750 ng/well overnight at 4°C. After wells were blocked with 3% BSA, serum samples diluted in binding buffer were added, and samples were incubated for 1 h at 37°C. Trastuzumab and trastuzumab-aplaviroc were detected by incubation with donkey anti-human IgG conjugated with horseradish peroxidase (HRPO) (Jackson ImmunoResearch). To determine the serum dilution at which 80% saturation was reached, the 5-min samples were serially diluted and analyzed; 80% saturation of the ELISA signal at time zero was reached at a dilution of 1/500 for the anti-human IgG. In all subsequent experiments, the optical density (OD) at time zero was set as 100%, and the absorption at all later time points was displayed as a percentage of that at time zero. The resulting curves were fitted using GraphPad Prism one-phase exponential decay.
Virus neutralization assay.
Neutralization assays with single-round infectious pseudovirus were performed as described elsewhere (10) by using U87.CD4.CCR5 target cells obtained from the NIH AIDS Research and Reference Reagent Program (contributed by HongKui Deng and Dan Littman). Briefly, 1 × 104 target cells in a volume of 100 μl were seeded into wells of 96-well plates (Corning) and were incubated overnight at 37°C under 5% CO2. After overnight incubation, the medium was removed, and 50 μl of fresh medium was added to each well. Serially diluted samples (50 μl) were transferred to plated target cells and were incubated for 1 h at 37°C. An equal volume of virus previously determined to yield 2 × 105 relative light units (RLU) was added (100 μl), and plates were incubated for an additional 72 h. To measure luciferase activity, the medium was removed, the wells were washed once with Ca2+- and Mg2+-free phosphate-buffered saline, and 50 μl of an appropriately diluted luciferase cell culture lysis reagent (Promega) was added and mixed by pipetting vigorously up and down. Aliquots (20 μl) were transferred to opaque 96-well assay plates (Corning), and luciferase activity was measured on a luminometer (EG&G Berthold LB96V; Perkin-Elmer) using a luciferase assay substrate (Promega). The percentage of virus neutralization at a given antibody concentration was determined by calculating the reduction in luciferase activity in the presence of an antibody (relative to that in virus-only wells).
High-throughput virus neutralization assay.
High-throughput screening of the PG9-aplaviroc conjugate was performed in the laboratory of Michael Seaman, Harvard Medical School. The highest concentration of PG9-aplaviroc tested was 50 μg/ml; seven 5-fold dilutions were also tested in duplicate wells. The highest concentration of aplaviroc tested was 100 nM.
RESULTS
Model trastuzumab-aplaviroc conjugate.
In order to independently assess the activity of aplaviroc conjugated to a carrier IgG without anti-HIV-1 activity, trastuzumab, an anti-Her2 monoclonal antibody (MAb), was used as a model human IgG for chemical modification and testing. Bioconjugation conditions were selected on the basis of our previous study (30) and were further optimized here (for full optimization details, see the tables in section S3 in the supplemental material). The two-step conjugation methodology involved modification of the IgG with FBDP (4-formylbenzene diazonium hexafluorophosphate) followed by ligation of the antibody to aplaviroc-oxyamine (Fig. 1). The trastuzumab-aplaviroc conjugate was subjected to extensive purification, including gel filtration and protein A purification, to ensure complete removal of unreacted aplaviroc.
Fig 1.
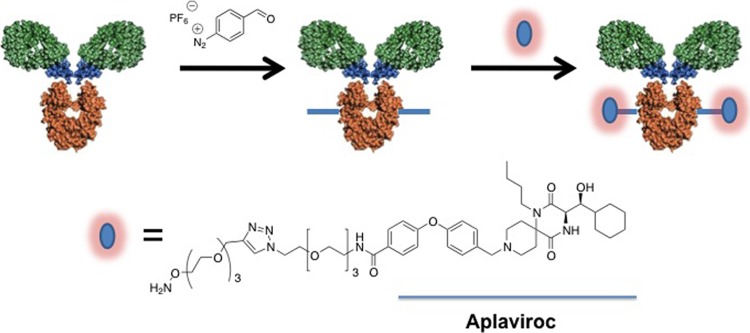
Schematic representation of the chemical modification of an antibody with a diazonium hexafluorophosphate (FBDP) reagent. The antibody is first modified with the FBDP reagent at a surface-exposed tyrosine residue(s), introducing an aldehyde tag onto the antibody. In the second step, chemoselective conjugation of the aldehyde with oxyamine is performed, resulting in the introduction of the aplaviroc moiety onto the surface of the antibody.
Characterization of trastuzumab-aplaviroc by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry revealed the incorporation of an average of 1 aplaviroc moiety per IgG molecule (see the supplemental material). Tryptic digestion and MS-MS characterization of the trastuzumab-aplaviroc indicated that the diazonium modification occurred preferentially at the conserved surface-exposed tyrosine in the Fc domain at position 319 by Kabat numbering. Some minor modification was also observed in the heavy chain of Fab (see the supplemental material). This observation is in agreement with previous studies that demonstrated preferential diazene formation with the most accessible tyrosines (22, 31, 32). When trastuzumab that was not modified with FBDP was incubated with aplaviroc-oxyamine, no conjugation of aplaviroc-oxyamine occurred, as shown by MALDI-TOF mass spectrometry. This sample had no activity in the HIV neutralization assays, indicating that the purification protocol used removed unconjugated aplaviroc-oxyamine.
Flow cytometry demonstrated that trastuzumab-aplaviroc bound to Her2-positive A431 cells and, to a lesser extent, to CCR5-positive TZM-bl cells (Fig. 2A). The parent antibody, trastuzumab, also bound to TZM-bl cells, due to low-level expression of Her2; however, mean fluorescence intensity was considerably higher for trastuzumab-aplaviroc bound to TZM-bl cells than for the parent antibody, indicating that the CCR5 binding activity of aplaviroc was maintained following conjugation.
Fig 2.

Evaluation of the model antibody conjugate trastuzumab-aplaviroc. (A) Flow cytometry analysis of binding to CCR5-positive TZM-bl cells and CCR5-negative A431 cells. The potent CCR5 binding antibody (Ab) 2D7 served as a positive control in TZM-bl cell assays. MFI, mean fluorescence intensity. (B) Neutralization of HIV-1 JR-FL and YU2 by trastuzumab and the trastuzumab-aplaviroc conjugate. (C) Pharmacokinetic profiles of trastuzumab and the trastuzumab-aplaviroc conjugate in athymic mice (4 mice per experimental group). All experimental data represent two or more independent experiments performed in triplicate. Error bars represent standard deviations.
The anti-HIV-1 activity of trastuzumab-aplaviroc was assessed by neutralization assays with a single round of infectious pseudovirus. Trastuzumab-aplaviroc neutralized HIV-1 strains JR-FL and YU2 with 50% inhibitory concentrations (IC50s) of 2.3 nM and 4.9 nM, respectively; unmodified trastuzumab showed no neutralizing activity (Fig. 2B). The potent CCR5 binding antibody 2D7 neutralized HIV-1 strains JR-FL and YU2 with IC50s of 0.2 nM and 0.1 nM, respectively. To ensure the CCR5-based mechanism of trastuzumab-aplaviroc, it was also tested for neutralization of CXCR4-tropic HIV HXB2 and was found to be inactive (see the supplemental material).
Modification of the antibody occurred primarily at Tyr 319 in the CH2 region of the heavy chain. Because the in vivo half-lives (t1/2) of antibodies are mediated primarily by contacts in the CH2 region of the IgG with the neonatal Fc receptor FcRn, we evaluated the half-lives of the parent and the conjugate in mice. The trastuzumab-aplaviroc conjugate had pharmacokinetic properties similar to those of the unmodified parent antibody (Fig. 2C). The half-life of trastuzumab in athymic nude mice was 115 h, whereas the half-life of the trastuzumab-aplaviroc conjugate was 168 h. The half-life of aplaviroc in mice is 30 min (33). Therefore, conjugation of aplaviroc with trastuzumab dramatically extended the half-life of aplaviroc in vivo and did not negatively impact the half-life of the scaffold antibody.
Conjugation of aplaviroc to broadly neutralizing monoclonal antibodies and CD4-IgG.
BNmAbs b12, 2G12, PG9, and PG16 and the immunoadhesin protein CD4-IgG were conjugated with aplaviroc, and the conjugates were characterized by MALDI-TOF mass spectrometry. We observed the incorporation of 0.5 to 2 aplaviroc moieties per protein molecule (Table 1). Tryptic digestion and MS-MS analysis of conjugates indicated that the heavy-chain constant region Tyr 319 was the primary site of chemical modification in each BNmAb, although minor modification sites were also identified (see the supplemental material). No modifications were observed in the Fv regions of the antibodies.
Table 1.
Neutralization of HIV-1 JR-FL and YU2 by aplaviroc-conjugated BNmAbs and CD4-Ig
| Antibodya | Avg no. of aplaviroc moieties per IgG molecule | IC50 (nM) for: |
|
|---|---|---|---|
| JR-FL | YU2 | ||
| b12-apl | 0.5 | 0.1 | 0.1 |
| b12 | 0 | 0.3 | 46.7 |
| 2G12-apl | 2 | 0.3 | 25.5 |
| 2G12 | 0 | 0.8 | >100 |
| PG9-apl | 1 | 11.6 | 1.2 |
| PG9 | 0 | >100 | 22.6 |
| PG16-apl | 2 | 5.9 | 0.4 |
| PG16 | 0 | >100 | 1.5 |
| CD4-IgG–apl | 1 | 0.6 | 0.6 |
| CD4-IgG | 0 | 0.6 | 0.7 |
| Aplaviroc | 0.95 | 0.86 | |
| 2D7 (positive control) | 0 | 0.2 | 0.1 |
| DEN3 (negative control) | 0 | >1,000 | >1,000 |
apl, aplaviroc.
HIV-1 neutralization studies showed that the potencies of BNmAb conjugates against HIV-1 isolates JR-FL and YU2 were significantly higher than those of the unconjugated parental antibodies or the CD4 fusion protein (Table 1; Fig. 3). Improvements in potency over the parent antibody ranged from ∼3-fold for 2G12-aplaviroc against the JR-FL isolate to >400-fold for b12-aplaviroc against the YU2 isolate. Differences between conjugates and parent antibodies were smallest for the most potent of the parental antibodies. In the neutralization assay with the JR-FL isolate, PG9 and PG16 were inactive except as aplaviroc conjugates. CD4-IgG potently neutralizes both JR-FL and YU2. Conjugation of aplaviroc to CD4-IgG did not have a notable effect on the neutralization of these viruses, since the immunoadhesin protein itself can neutralize JR-FL and YU2 more potently than aplaviroc alone. To determine whether aplaviroc conjugation can have a positive effect on the activity of this protein, we additionally assayed the clade A variant 92RW 020.5, a strain relatively resistant to neutralization by CD4-IgG. Against this strain, the conjugation resulted in a 20-fold improvement in the IC50. CD4-IgG neutralized the 92RW pseudovirus with an IC50 of 10 nM, whereas CD4-IgG–aplaviroc neutralized this isolate with an IC50 of 0.5 nM. Significantly, no evidence of enhanced infection was noted with any conjugated protein studied.
Fig 3.

Evaluation of the neutralization abilities of antibody conjugates against HIV-1 strains JR-FL and YU2 and of the CD4-IgG2–aplaviroc conjugate against HIV-1 strains JR-FL, YU2, and 92RW. HIV-1 JR-FL is resistant to neutralization by parent antibodies PG9 and PG16. The bottom right graph shows results for controls only: 2D7, positive-control MAb; DEN3, negative-control MAb; aplaviroc, small-molecule positive control.
We also studied the neutralization activity of a 1:1 molar mixture of aplaviroc and PG9 against HIV-1 strains JR-FL and YU2, and we observed a potency equal to that of aplaviroc. The in vitro assay, however, does not allow us to assess the potential clinical benefit of the extended half-life of the antibody-aplaviroc conjugate relative to the 1:1 mixture of two agents (vide infra).
Neutralization of HIV-1 strains by PG9-aplaviroc.
The PG9 conjugate was chosen for more in-depth study because the parent antibody, PG9, is representative of a recently described group of BNmAbs that demonstrate tremendous breadth of neutralization activity. ELISA studies of PG9 and PG9-aplaviroc performed using gp120 from HIV strain 16055, a clade C virus, indicated no loss of binding activity for PG9 following conjugation. The neutralizing ability of PG9-aplaviroc was assessed against a panel of 117 pseudoviruses representing major circulating HIV-1 subtypes (Table 2; see also section S5 in the supplemental material). PG9 neutralized 101 of the 117 viruses tested with IC50s below 50 μg/ml. The 16 viruses that were not well neutralized by PG9 were neutralized by PG9-aplaviroc with a mean IC50 of 6 μg/ml (Table 3). PG9-aplaviroc neutralized all 117 of the subtypes with IC50s below 17 μg/ml and with a mean IC50 28 μg/ml (see the supplemental material). PG9-aplaviroc neutralized 94.9% of the 117 viruses with IC80s of <50 μg/ml; for 22 of the pseudovirus strains neutralized by the PG9-aplaviroc conjugate, the IC80 of the parent antibody, PG9, was above 50 μg/ml (Table 4). Unlike the parent antibody, the conjugate neutralized all clade B and C viruses, including transmitted/founder viruses. Clade C is predominant in Southern and East Africa, India, and Nepal and is responsible for about half of worldwide infections. Clade B is the major cause of infection in the Americas, Europe, Japan, and Australia. Thus, conjugation to aplaviroc significantly improved the breadth of neutralization ability.
Table 2.
Summary of neutralization data for a panel of 117 HIV-1 strains
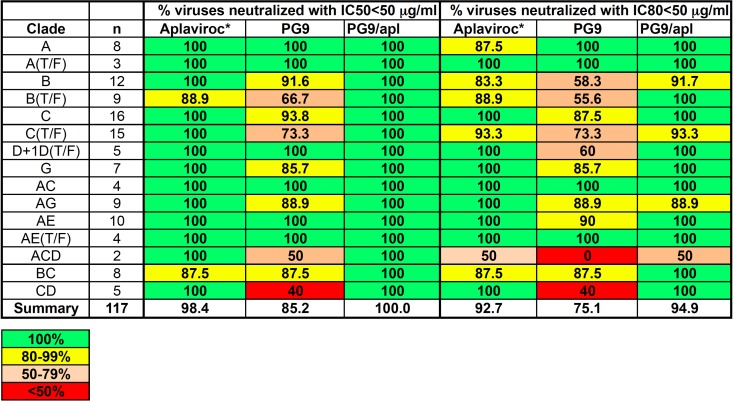
a Aplaviroc neutralization with IC50s of <100 nM and IC80s of <100 nM.
Table 3.
Summary of the pseudoviruses neutralized by PG9-aplaviroc with IC50s of <50 μg/ml that are not neutralized by PG9 (IC50s, >50 μg/ml)
| Virus IDa | Cladeb | IC50 titer in TZM-bl cells (μg/ml)c |
|
|---|---|---|---|
| PG9 | PG9-aplaviroc | ||
| QH0692.42 | B | >50 | 10.817 |
| 1054_07_TC4_1499 | B (T/F) | >50 | 6.916 |
| 6244_13_B5_4576 | B (T/F) | >50 | 7.674 |
| 62357_14_D3_4589 | B (T/F) | >50 | 4.305 |
| ZM214M.PL15 | C | >50 | 2.136 |
| Ce1086_B2 | C (T/F) | >50 | 1.183 |
| Ce2010_F5 | C (T/F) | >50 | 11.145 |
| 246F C1G | C (T/F) | >50 | 1.262 |
| 7030102001E5(Rev-) | C (T/F) | >50 | 6.763 |
| CNE30 | BC | >50 | 9.645 |
| T251-18 | CRF02_AG | >50 | 16.982 |
| X2088_c9 | G | >50 | 10.453 |
| 6480.v4.c25 | CD | >50 | 1.476 |
| 6952.v1.c20 | CD | >50 | 1.666 |
| 6811.v7.c18 | CD | >50 | 5.080 |
| 0815.v3.c3 | ACD | >50 | 1.399 |
ID, identification.
T/F, transmitted/founder virus.
The PG9 data are historical data for this 117-virus panel bridged for the current experiment with a set of representative viruses. For PG9-aplaviroc, the median IC50 titer in TZM-bl cells is 5.921 μg/ml, and the mean ± standard deviation is 6.181 ± 4.703 μg/ml.
Table 4.
Summary of the pseudoviruses neutralized by PG9-aplaviroc with IC80s of <50 μg/ml that are not neutralized by PG9 (IC80s, >50 μg/ml)
| Virus IDa | Cladeb | IC80 titer in TZM-bl cells (μg/ml)c |
|
|---|---|---|---|
| PG9 | PG9-aplaviroc | ||
| WEAU_d15_410_5017 | B (T/F) | >50 | 19.772 |
| HIV-16845-2.22 | C | >50 | 18.415 |
| 620345.c01 | CRF01_AE | >50 | 14.409 |
| 0815.v3.c3 | ACD | >50 | 7.406 |
| 6480.v4.c25 | CD | >50 | 8.097 |
| 6952.v1.c20 | CD | >50 | 11.544 |
| 6811.v7.c18 | CD | >50 | 24.897 |
| X2088_c9 | G | >50 | 30.361 |
| 3016.v5.c45 | D | >50 | 1.571 |
| 191821_E6_1 | D (T/F) | >50 | 0.976 |
| 246F C1G | C (T/F) | >50 | 3.500 |
| 7030102001E5(Rev-) | C (T/F) | >50 | 29.579 |
| CNE30 | BC | >50 | 41.902 |
| Ce1086_B2 | C (T/F) | >50 | 4.069 |
| ZM214M.PL15 | C | >50 | 18.933 |
| PVO.4 | B | >50 | 8.239 |
| TRO.11 | B | >50 | 35.584 |
| RHPA4259.7 | B | >50 | 7.092 |
| THRO4156.18 | B | >50 | 20.071 |
| 1054_07_TC4_1499 | B (T/F) | >50 | 36.094 |
| 6244_13_B5_4576 | B (T/F) | >50 | 28.640 |
| 62357_14_D3_4589 | B (T/F) | >50 | 17.254 |
ID, identification.
T/F, transmitted/founder virus.
The PG9 data are historical data for this 117-virus panel bridged for the current experiment with a set of representative viruses. For PG9-aplaviroc, the median IC80 titer in TZM-bl cells is 17.834 μg/ml, and the mean ± standard deviation is 17.655 ± 12.160 μg/ml.
The PG9-aplaviroc conjugate demonstrated a dramatic improvement over the parent antibody in the maximum percentage of inhibition (MPI) across the 117-virus panel (Table 5; see also the supplemental material). PG9 is sensitive to the glycosylation profile of a virus and cannot completely neutralize certain viral strains even at high antibody concentrations (34). The PG9-aplaviroc conjugate did not display this glycan sensitivity and was found to be more potent than native PG9 when MPIs were compared. PG9 and PG9-aplaviroc neutralized 40 and 84 of the 117 viral isolates tested at MPIs of >99.5%, respectively.
Table 5.
Summary of MPIsa for PG9 and PG9-aplaviroc
| Cladeb | No. of viruses tested | No. of viruses neutralized with an MPI of 100% |
Avg MPI |
||
|---|---|---|---|---|---|
| PG9 | PG9-aplaviroc | PG9 | PG9-aplaviroc | ||
| A | 8 | 3 | 6 | 97 | 99 |
| A (T/F) | 3 | 3 | 3 | 100 | 100 |
| B | 12 | 1 | 4 | 77 | 95 |
| B (T/F) | 9 | 0 | 2 | 63 | 94 |
| C | 16 | 7 | 12 | 88 | 99 |
| C (T/F) | 15 | 10 | 13 | 76 | 97 |
| D and D (T/F) | 5 | 1 | 3 | 83 | 99 |
| G | 7 | 1 | 4 | 85 | 98 |
| AC | 4 | 1 | 3 | 98 | 100 |
| AG | 9 | 3 | 7 | 92 | 97 |
| AE | 10 | 5 | 7 | 96 | 99 |
| AE (T/F) | 4 | 0 | 2 | 93 | 100 |
| ACD | 2 | 0 | 0 | 29 | 84 |
| BC | 8 | 5 | 6 | 89 | 98 |
| CD | 5 | 0 | 2 | 47 | 97 |
MPIs of >99.5% were rounded up to 100% by the analysis software.
T/F, transmitted/founder virus.
DISCUSSION
The success of HAART is predicated on the fact that although HIV-1 is a hypermutating virus, a therapy consisting of a combination of inhibitors targeting three viral enzymes—reverse transcriptase, protease, and integrase—presents a stringent barrier to viral replication. HAART often reduces viral replication to undetectable levels in patients. HAART does not, however, present an insurmountable barrier to viral replication, and drug resistance can evolve rapidly when patient compliance is poor. Two key factors that contribute to drug compliance failure are frequency of administration, which has been reduced to once daily for some HAART regimens, and adverse side effects, often driven by metabolic toxicity. Both of these limitations might be addressed by the development of a HAART equivalent based on protein drugs. For example, antibodies have long in vivo half-lives. Native human IgG1 has a t1/2 of 23 days in humans, and engineered variants of the Fc regions of antibodies promise t1/2s of >6 months (35, 36). The metabolic liability of HIV-1 small-molecule drugs is a particular challenge, because the drugs are dosed chronically at very high compound loading levels due to their short pharmacokinetic profiles. The metabolism of antibodies does not create new active metabolites.
Both the high genetic variability and the diversity of HIV-1 pose challenges to the creation of a HAART therapy based on protein therapeutic agents such as antibodies. The extracellular targets on which this approach focuses are the HIV-1 envelope and the host cell receptors CD4, CCR5, and CXCR4. Despite the high genetic variability and preexisting diversity of the HIV envelope protein, a number of BNmAbs that possess exceptional anti-HIV-1 activity and breadth have been characterized (1–8). Targeting the host cell receptors CD4, CCR5, and CXCR4 has the advantage of genetic stability but could have deleterious effects if the therapeutic agent negatively impacts the host cells. Certain therapeutic antibodies targeting CD4 and CCR5 are in clinical studies and appear to possess promising safety profiles; however, none are approved drugs (37–39).
The road to an approved drug cocktail is long and requires independent testing and approval of each species. One approach to streamlining this process is to construct single entities that possess multiple activities. The first step toward this goal is the creation of bispecific-antibody therapeutic agents. To address this challenge, modified antibody formats (e.g., knobs into holes [40], IgG-scFv [41], and DVD-Ig [21]) have been created using protein engineering methods. Often these approaches suffer from protein instability and low expression yields, although the recently described peptide fusion approach overcomes many of these problems (42). The approach used here involves chemical conjugation of a peptide or small molecule to a monoclonal antibody that possesses a desired specificity. The conjugation is performed using a methodology that allows both the MAb and the programming agent to bind their respective targets.
Here we conjugated the CCR5-specific drug aplaviroc to several broadly neutralizing antibodies and CD4-IgG. Aplaviroc was withdrawn from clinical study following the observation of idiosyncratic hepatotoxicity in phase IIb clinical trials (43). In humans, aplaviroc has a half-life of approximately 3 h, making high doses an unavoidable requirement. We hypothesized that in the context of an antibody conjugate, the half-life of aplaviroc would be considerably extended. To test this, we prepared a trastuzumab-aplaviroc conjugate through a tyrosine-selective reaction. The plasma half-life of the trastuzumab-aplaviroc conjugate in mice was significantly longer (168 h) than that of aplaviroc alone (30 min). It has been shown previously that chemical programming of an antibody resulted in a 1,000-fold reduction of the amount of a small molecule required for therapeutic effect (17). Reduction of the effective dose will likely mitigate the metabolic toxicity observed with anti-HIV drugs such as aplaviroc, although this remains to be demonstrated. The trastuzumab-aplaviroc conjugate was able to recognize the CCR5 receptor on the surfaces of CCR5-expressing cells and efficiently blocked HIV entry at low nanomolar concentrations. Thus, attributes of both the parent antibody and the small molecule were maintained in the context of the conjugate. Success with aplaviroc suggests that clinically approved CCR5 inhibitors, such as maraviroc, can potentially benefit from evaluation as BNmAb conjugates. However, maraviroc derivatives that are amenable to linkage to proteins or long-lived carriers have not yet been described.
We prepared aplaviroc conjugates with the broadly neutralizing antibodies b12, 2G12, PG9, PG16, and CD4-IgG. The incorporation of aplaviroc significantly improved the neutralization abilities of BNmAbs for strains resistant to or weakly neutralized by the parent antibodies. The antiviral activity of PG9-aplaviroc was analyzed against a panel of 117 different viral strains. The conjugate neutralized the activities of 100% of viral strains with IC50s lower than 50 μg/ml. To the best of our knowledge, none of the currently known BNmAbs neutralize all of these 117 cross-clade pseudoviruses with IC50s lower than 50 μg/ml. Notably, compared to parent PG9-aplaviroc had lower IC50s against 13 out of 32 tested transmitted/founder viruses of different clades and was more potent than any of the parent molecules alone against 10 of these viruses (see the supplemental material). PG9-aplaviroc also displayed MPIs significantly improved over those of the parent antibody.
The median IC50 of the PG9-aplaviroc conjugate against the panel was 2.76 nM, whereas that of the parent antibody, PG9, was 0.77 nM. On a panel of 76 of 117 viruses, PG9-aplaviroc had higher IC50s than the naked PG9 antibody (see section S5 in the supplemental material), with median IC50s of 0.674 nM for PG9 and 1.737 nM for PG9-aplaviroc. Notably, aplaviroc itself was not very potent on this set of the viruses, with a median IC50 of 4.6 nM. We hypothesize that during the neutralization assay there is competition between binding to CCR5 and binding to the HIV-1 envelope, and thus, the conjugate is not always used in the most efficient neutralization pathway. Since these two mechanisms of neutralization are in competition when the conjugate is assayed against viruses that are very sensitive to PG9 neutralization, some reduction in potency against these isolates is expected.
Combining potent molecules that prevent HIV entry through different mechanisms into a single multifunctional molecule may create an insurmountable evolutionary challenge limiting the development of resistance. This hypothesis is supported by a recent report by Zhou et al. showing that HIV escape mutants selected by exposure to entry inhibitors were much more sensitive to BNmAbs than the original virus (44). The chemical approach to the synthesis of multispecific BNmAbs described here will allow ready access to various combinations of neutralizing antibodies and host-protecting small-molecule drugs that bind to the receptors crucial for HIV entry (CCR5, CXCR4, CD4). Intelligent design of the linker architecture will allow the preparation of tri- and tetraspecific antibody conjugates.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grants AI095038 (C.F.B.), AI33292 (D.R.B.), AI100663 (D.R.B.), and IAVI (D.R.B.).
We thank B. Hahn, F. McCutchan, G. Shaw, D. Montefiori, M. Thomson, J. Overbaugh, R. Swanstrom, L. Morris, J. Kim, L. Zhang, D. Ellensberger, and C. Williamson for contributing the HIV-1 envelope plasmids used in the extended neutralization panel.
Footnotes
Published ahead of print 20 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03146-12.
REFERENCES
- 1. Binley JM, Wrin T, Korber B, Zwick MB, Wang M, Chappey C, Stiegler G, Kunert R, Zolla-Pazner S, Katinger H, Petropoulos CJ, Burton DR. 2004. Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J. Virol. 78:13232–13252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Diskin R, Scheid JF, Marcovecchio PM, West AP, Jr, Klein F, Gao H, Gnanapragasam PN, Abadir A, Seaman MS, Nussenzweig MC, Bjorkman PJ. 2011. Increasing the potency and breadth of an HIV antibody by using structure-based rational design. Science 334:1289–1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Falkowska E, Ramos A, Feng Y, Zhou T, Moquin S, Walker LM, Wu X, Seaman MS, Wrin T, Kwong PD, Wyatt RT, Mascola JR, Poignard P, Burton DR. 2012. PGV04, an HIV-1 gp120 CD4 binding site antibody, is broad and potent in neutralization but does not induce conformational changes characteristic of CD4. J. Virol. 86:4394–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Scheid JF, Mouquet H, Ueberheide B, Diskin R, Klein F, Oliveira TY, Pietzsch J, Fenyo D, Abadir A, Velinzon K, Hurley A, Myung S, Boulad F, Poignard P, Burton DR, Pereyra F, Ho DD, Walker BD, Seaman MS, Bjorkman PJ, Chait BT, Nussenzweig MC. 2011. Sequence and structural convergence of broad and potent HIV antibodies that mimic CD4 binding. Science 333:1633–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Walker LM, Huber M, Doores KJ, Falkowska E, Pejchal R, Julien JP, Wang SK, Ramos A, Chan-Hui PY, Moyle M, Mitcham JL, Hammond PW, Olsen OA, Phung P, Fling S, Wong CH, Phogat S, Wrin T, Simek MD, Koff WC, Wilson IA, Burton DR, Poignard P. 2011. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477:466–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walker LM, Phogat SK, Chan-Hui PY, Wagner D, Phung P, Goss JL, Wrin T, Simek MD, Fling S, Mitcham JL, Lehrman JK, Priddy FH, Olsen OA, Frey SM, Hammond PW, Kaminsky S, Zamb T, Moyle M, Koff WC, Poignard P, Burton DR. 2009. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326:285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wu X, Yang ZY, Li Y, Hogerkorp CM, Schief WR, Seaman MS, Zhou T, Schmidt SD, Wu L, Xu L, Longo NS, McKee K, O'Dell S, Louder MK, Wycuff DL, Feng Y, Nason M, Doria-Rose N, Connors M, Kwong PD, Roederer M, Wyatt RT, Nabel GJ, Mascola JR. 2010. Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329:856–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu X, Zhou T, Zhu J, Zhang B, Georgiev I, Wang C, Chen X, Longo NS, Louder M, McKee K, O'Dell S, Perfetto S, Schmidt SD, Shi W, Wu L, Yang Y, Yang ZY, Yang Z, Zhang Z, Bonsignori M, Crump JA, Kapiga SH, Sam NE, Haynes BF, Simek M, Burton DR, Koff WC, Doria-Rose NA, Connors M, Mullikin JC, Nabel GJ, Roederer M, Shapiro L, Kwong PD, Mascola JR. 2011. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333:1593–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hessell AJ, Hangartner L, Hunter M, Havenith CE, Beurskens FJ, Bakker JM, Lanigan CM, Landucci G, Forthal DN, Parren PW, Marx PA, Burton DR. 2007. Fc receptor but not complement binding is important in antibody protection against HIV. Nature 449:101–104 [DOI] [PubMed] [Google Scholar]
- 10. Parren PW, Marx PA, Hessell AJ, Luckay A, Harouse J, Cheng-Mayer C, Moore JP, Burton DR. 2001. Antibody protects macaques against vaginal challenge with a pathogenic R5 simian/human immunodeficiency virus at serum levels giving complete neutralization in vitro. J. Virol. 75:8340–8347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. 2012. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 481:81–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson PR, Schnepp BC, Zhang J, Connell MJ, Greene SM, Yuste E, Desrosiers RC, Clark KR. 2009. Vector-mediated gene transfer engenders long-lived neutralizing activity and protection against SIV infection in monkeys. Nat. Med. 15:901–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saphire EO, Parren PW, Barbas CF, III, Burton DR, Wilson IA. 2001. Crystallization and preliminary structure determination of an intact human immunoglobulin, b12: an antibody that broadly neutralizes primary isolates of HIV-1. Acta Crystallogr. D Biol. Crystallogr. 57:168–171 [DOI] [PubMed] [Google Scholar]
- 14. Zwick MB, Bonnycastle LL, Menendez A, Irving MB, Barbas CF, III, Parren PW, Burton DR, Scott JK. 2001. Identification and characterization of a peptide that specifically binds the human, broadly neutralizing anti-human immunodeficiency virus type 1 antibody b12. J. Virol. 75:6692–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gavrilyuk JI, Wuellner U, Barbas CF., III 2009. Beta-lactam-based approach for the chemical programming of aldolase antibody 38C2. Bioorg. Med. Chem. Lett. 19:1421–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gavrilyuk JI, Wuellner U, Salahuddin S, Goswami RK, Sinha SC, Barbas CF., III 2009. An efficient chemical approach to bispecific antibodies and antibodies of high valency. Bioorg. Med. Chem. Lett. 19:3716–3720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Popkov M, Rader C, Gonzalez B, Sinha SC, Barbas CF., III 2006. Small molecule drug activity in melanoma models may be dramatically enhanced with an antibody effector. Int. J. Cancer 119:1194–1207 [DOI] [PubMed] [Google Scholar]
- 18. Rader C, Sinha SC, Popkov M, Lerner RA, Barbas CF., III 2003. Chemically programmed monoclonal antibodies for cancer therapy: adaptor immunotherapy based on a covalent antibody catalyst. Proc. Natl. Acad. Sci. U. S. A. 100:5396–5400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wuellner U, Gavrilyuk JI, Barbas CF., III 2010. Expanding the concept of chemically programmable antibodies to RNA aptamers: chemically programmed biotherapeutics. Angew. Chem. Int. Ed. Engl. 49:5934–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doppalapudi VR, Tryder N, Li LN, Aja T, Griffith D, Liao FF, Roxas G, Ramprasad MP, Bradshaw C, Barbas CF., III 2007. Chemically programmed antibodies: endothelin receptor targeting CovX-Bodies. Bioorg. Med. Chem. Lett. 17:501–506 [DOI] [PubMed] [Google Scholar]
- 21. Wu C, Ying H, Grinnell C, Bryant S, Miller R, Clabbers A, Bose S, McCarthy D, Zhu RR, Santora L, Davis-Taber R, Kunes Y, Fung E, Schwartz A, Sakorafas P, Gu J, Tarcsa E, Murtaza A, Ghayur T. 2007. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 25:1290–1297 [DOI] [PubMed] [Google Scholar]
- 22. Kovacs EW, Hooker JM, Romanini DW, Holder PG, Berry KE, Francis MB. 2007. Dual-surface-modified bacteriophage MS2 as an ideal scaffold for a viral capsid-based drug delivery system. Bioconjug. Chem. 18:1140–1147 [DOI] [PubMed] [Google Scholar]
- 23. Chen W, Zhan P, De Clercq E, Liu X. 2012. Recent progress in small molecule CCR5 antagonists as potential HIV-1 entry inhibitors. Curr. Pharm. Des. 18:100–112 [DOI] [PubMed] [Google Scholar]
- 24. Liu T, Weng Z, Dong X, Hu Y. 2010. Recent advances in the development of small-molecule CCR5 inhibitors for HIV. Mini Rev. Med. Chem. 10:1277–1292 [DOI] [PubMed] [Google Scholar]
- 25. Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, Mori J, Rickett G, Smith-Burchnell C, Napier C, Webster R, Armour D, Price D, Stammen B, Wood A, Perros M. 2005. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob. Agents Chemother. 49:4721–4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hunt JS, Romanelli F. 2009. Maraviroc, a CCR5 coreceptor antagonist that blocks entry of human immunodeficiency virus type 1. Pharmacotherapy 29:295–304 [DOI] [PubMed] [Google Scholar]
- 27. Kromdijk W, Huitema AD, Mulder JW. 2010. Treatment of HIV infection with the CCR5 antagonist maraviroc. Expert Opin. Pharmacother. 11:1215–1223 [DOI] [PubMed] [Google Scholar]
- 28. Lorenzen T. 2010. Profile of maraviroc: a CCR5 antagonist in the management of treatment-experienced HIV patients. HIV AIDS (Auckl.) 2:151–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gavrilyuk J, Uehara H, Otsubo N, Hessell A, Burton DR, Barbas CF., III 2010. Potent inhibition of HIV-1 entry with a chemically programmed antibody aided by an efficient organocatalytic synthesis. Chembiochem 11:2113–2118 [DOI] [PubMed] [Google Scholar]
- 30. Gavrilyuk J, Ban H, Nagano M, Hakamata W, Barbas CF., III 2012. Formylbenzene diazonium hexafluorophosphate reagent for tyrosine-selective modification of proteins and the introduction of a bioorthogonal aldehyde. Bioconjug. Chem. 23:2321–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hooker JM, Kovacs EW, Francis MB. 2004. Interior surface modification of bacteriophage MS2. J. Am. Chem. Soc. 126:3718–3719 [DOI] [PubMed] [Google Scholar]
- 32. Schlick TL, Ding Z, Kovacs EW, Francis MB. 2005. Dual-surface modification of the tobacco mosaic virus. J. Am. Chem. Soc. 127:3718–3723 [DOI] [PubMed] [Google Scholar]
- 33. Nakata H, Maeda K, Miyakawa T, Shibayama S, Matsuo M, Takaoka Y, Ito M, Koyanagi Y, Mitsuya H. 2005. Potent anti-R5 human immunodeficiency virus type 1 effects of a CCR5 antagonist, AK602/ONO4128/GW873140, in a novel human peripheral blood mononuclear cell nonobese diabetic-SCID, interleukin-2 receptor gamma-chain-knocked-out AIDS mouse model. J. Virol. 79:2087–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Doores KJ, Burton DR. 2010. Variable loop glycan dependency of the broad and potent HIV-1-neutralizing antibodies PG9 and PG16. J. Virol. 84:10510–10521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hinton PR, Xiong JM, Johlfs MG, Tang MT, Keller S, Tsurushita N. 2006. An engineered human IgG1 antibody with longer serum half-life. J. Immunol. 176:346–356 [DOI] [PubMed] [Google Scholar]
- 36. Igawa T, Tsunoda H, Kuramochi T, Sampei Z, Ishii S, Hattori K. 2011. Engineering the variable region of therapeutic IgG antibodies. MAbs 3:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bruno CJ, Jacobson JM. 2010. Ibalizumab: an anti-CD4 monoclonal antibody for the treatment of HIV-1 infection. J. Antimicrob. Chemother. 65:1839–1841 [DOI] [PubMed] [Google Scholar]
- 38. Song R, Franco D, Kao CY, Yu F, Huang Y, Ho DD. 2010. Epitope mapping of ibalizumab, a humanized anti-CD4 monoclonal antibody with anti-HIV-1 activity in infected patients. J. Virol. 84:6935–6942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tenorio AR. 2011. The monoclonal CCR5 antibody PRO-140: the promise of once-weekly HIV therapy. Curr. HIV/AIDS Rep. 8:1–3 [DOI] [PubMed] [Google Scholar]
- 40. Ridgway JB, Presta LG, Carter P. 1996. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 9:617–621 [DOI] [PubMed] [Google Scholar]
- 41. Bostrom J, Yu SF, Kan D, Appleton BA, Lee CV, Billeci K, Man W, Peale F, Ross S, Wiesmann C, Fuh G. 2009. Variants of the antibody herceptin that interact with HER2 and VEGF at the antigen binding site. Science 323:1610–1614 [DOI] [PubMed] [Google Scholar]
- 42. LaFleur DW, Abramyan D, Kanakaraj P, Smith RG, Shah RR, Wang G, Yao X-T, Kankanala S, Boyd E, Zaritskaya L, Nam V, Puffer BA, Buasen P, Kaithamana S, Burnette AF, Krishnamurthy R, Patel D, Roschke VV, Kiener P, Hilbert DM, Barbas CF., III 2013. Monoclonal antibody therapeutics with up to five specificities: functional enhancement through fusion of target-specific peptides. mAbs 5:208–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nichols WG, Steel HM, Bonny T, Adkison K, Curtis L, Millard J, Kabeya K, Clumeck N. 2008. Hepatotoxicity observed in clinical trials of aplaviroc (GW873140). Antimicrob. Agents Chemother. 52:858–865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou N, Fan L, Ho HT, Nowicka-Sans B, Sun Y, Zhu Y, Hu Y, McAuliffe B, Rose B, Fang H, Wang T, Kadow J, Krystal M, Alexander L, Colonno R, Lin PF. 2010. Increased sensitivity of HIV variants selected by attachment inhibitors to broadly neutralizing antibodies. Virology 402:256–261 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.