

Abstract
The tegument proteins encoded by ORF11 and ORF9 of varicella-zoster virus (VZV) are conserved among all alphaherpesvirus. We previously demonstrated that the ORF9 gene is essential, whereas ORF11 is dispensable in vitro but its deletion severely impairs VZV infection of skin xenografts in the SCID mouse model in vivo. Here we report that ORF11 protein interacts with ORF9 protein in infected cells as well as in the absence of other viral proteins, and we have mapped the ORF11 protein domain involved in their interaction. Although ORF11 is an RNA binding protein, the interaction between ORF11 and ORF9 proteins was not mediated by RNA or DNA bridging. VZV recombinants with mutations preventing ORF11 protein binding to ORF9 protein had no effect on 6-day growth kinetics based on plaque numbers, but plaque sizes were reduced in vitro. However, disruption of the ORF11 and ORF9 protein interaction was associated with failure to replicate in skin xenografts in vivo. Further, we demonstrate that in the absence of their interaction, the ORF9 protein displays an identical cellular localization, accumulating in the trans-Golgi region, whereas the ORF11 protein exhibits aberrant localization, dispersing throughout the cytoplasm. Overall, our observations suggest that while complete tegument assembly may not be necessary for VZV replication in vitro, the interaction between the ORF11 and ORF9 proteins appears to be critical for the proper localization of ORF11 protein to the assembly complex and for production of infectious virus during VZV pathogenesis in skin.
INTRODUCTION
Varicella-zoster virus (VZV) is a human alphaherpesvirus that causes varicella (chicken pox) and zoster (shingles) (1, 2). VZV virions, like those of all herpesviruses, contain a capsid that harbors the linear double-stranded DNA genome. The capsid is surrounded by the tegument, a proteinaceous layer located between the capsid and the host-derived outer lipid membrane envelope that expresses the viral glycoproteins which are required for cell entry. The tegument of alphaherpesviruses is predicted to consist of more than 20 virus-encoded proteins (3, 4). These tegument proteins have broad functions, including the regulation of viral and host cell gene expression during replication, transport of virions to and from the nucleus by recruitment of cellular molecular motors during entry and egress, and assembly of the infectious virus particle (5). VZV tegument proteins that have been identified experimentally include the immediate-early regulatory proteins IE4, IE62, and IE63; the open reading frame 47 (ORF47) and ORF66 viral kinases; and proteins encoded by ORF9, -10, and -11 (6–10). VZV ORF9 to ORF12 belong to a gene cluster that is conserved in the alphaherpesviruses, which is designated UL46-UL49 in the other members of the subfamily.
The VZV ORF11 gene is a homologue of UL47 (11). Deleting UL47 from herpes simplex virus 1 (HSV-1), pseudorabies virus (PRV), and Marek's disease virus (MDV) resulted in delayed replication compared to that of the parent virus (12–15). In contrast, deleting the ORF11 gene from the VZV parent OKA (pOka) genome did not affect growth kinetics (7), indicating that ORF11 functions are dispensable in vitro, as was confirmed subsequently (16). However, replication of the ORF11 deletion mutant was dramatically impaired when human skin xenografts were infected in vivo using our SCID mouse model of VZV pathogenesis (7). Thus, ORF11 is a determinant of VZV virulence in differentiated epidermal cells in their tissue microenvironment in vivo.
The VZV ORF9 gene is a homologue of UL49, which encodes the HSV-1 protein VP22. However, ORF9 is essential for VZV replication, whereas VP22 is dispensable, and ORF9 protein localizes almost exclusively to the cytoplasm, while VP22 traffics between the cytoplasm and nucleus (17–21). ORF9 protein is a major component of the VZV tegument and interacts with IE62, which is the predominant VZV transactivating protein; ORF9 also binds to glycoprotein E (gE), an essential and abundant envelope glycoprotein, and to the cellular protein tubulin (7, 17). Our ultrastructural analysis showed that both ORF9 and gE are present in the trans-Golgi region, where secondary envelopment occurs in VZV-infected cells. Other ORF9 functions have not been defined.
Most recently, we showed that ORF11 protein is an RNA binding protein with the capacity to bind to both viral and cellular mRNAs (22). Nevertheless, an ORF11 truncation mutant in which RNA binding was eliminated was as infectious as the parent virus, pOka, in skin xenografts, indicating that the ORF11 gene product is not involved in RNA biogenesis during VZV infection and that the RNA binding capacity of ORF11 does not explain why this protein is required for virulence in skin. The purpose of this study was to further investigate the functions of ORF11 and ORF9 proteins in order to better understand their roles in the pathogenesis of VZV skin infection.
MATERIALS AND METHODS
Recombinant viruses.
The complete genome of VZV pOka is contained in four overlapping SuperCos1 cosmid vectors (Stratagene, La Jolla, CA) designated pvFsp73 (nucleotides [nt] 1 to 33128), pvSpe14 (nt 21795 to 61868), pvPme2 (nt 53755 to 96035), and pvSpe23 (nt 94055 to 125124) (23). ORF11 (nt 13589 to 15970 of pOka VZV genome) is in the pvFsp73 cosmid. Three PCR fragments containing the sequence upstream of the SpeI site at nt 12565 to 14298, 15233, or 15886 were synthesized with the same upper primer (5′-GTTCCGTTCGATTGCCTAGTCCAC-3′) and different lower primers (5′-CGCTAACATAGAGCCTGTTAGGGT-3′, 5′-CAACATTCCTCCACCAAGAGTAAC-3′, and 5′-TAGGTCCACGGAGGGAATGGGCTG-3′). The fourth PCR amplified the regions from either the ORF11 stop codon or nt 15925 to a PacI site with different upstream primers (5′-TAAGGGTTGTGATTTTTTTCATTAG-3′ or 5′-GATTGGAAAAGCGTAGCCATGC-3′) and the same lower primer (5′-TACGGCTGATCAGACACACTCGATG-3′). The pvFsp73 cosmid was the template for synthesizing these PCR products. The resulting PCR products were digested with SpeI or PacI and inserted by triple ligation into a pT7Blue-3 plasmid (New England BioLabs, Beverly, MA) which contained a 6.7-kb NheI-NheI fragment corresponding to VZV genome nt 12339 to 19129 and was digested by SpeI and PacI. The three NheI-NheI fragments containing mutated ORF11 genes were used to replace the NheI fragment in the pvFsp73 cosmid. Recombinant viruses were isolated from human melanoma cells transfected with the mutated pvFsp73 cosmid and three intact cosmids, pvSpe14, pvPme2, and pvSpe23. Mutations were confirmed by sequencing. Viruses were propagated in human embryonic lung fibroblasts (HELF) for infection of skin xenografts in SCID mice, and the replication kinetics and peak titers of the recombinant viruses were assessed by infectious-focus assay (24).
Construction of plasmids and transfection.
Full-length ORF9 protein tagged with histidine (ORF9-His) was generated by PCR with the upper primer 5′-CTACGAAGCTTACCATGGCATCTTCCGACGGTG-3′ and lower primers 5′-CTACGCTCGAGCTAATGATGATGATGATGATGTTTTCGCGCATCAGTTCTTG-3′. Eight PCR fragments, including ORF11 253, ORF11 548, ORF11 600, ORF11 667, ORF11 701, ORF11 750, ORF11 766, and ORF11 780, were generated with the same plus primer, 5′-CTACGAAGCTTGTTATGGCACAGTCGGGTCATTATAAC-3′, and different minus primers, 5′-CTACGCTCGAGTTAAGCAGCTAAATCAATTGCAG-3′, 5′-CTACGCTCGAGTTACCAGGACTCATCAAACCCATTATG-3′, 5′-CTACGCTCGAGTTAGTCGACCTCGCCTAACAGTAA-3′, 5′-CTACGCTCGAGTTATTGTAACAATAAAACGGCCCCC-3′, 5′-CTACGCTCGAGTTAGTAAAGAGGACTTGCTGCCAAC-3′, 5′-CTACGCTCGAGTTACAATCCCACAAAACGGAAGTTC-3′, 5′-CTACGCTCGAGTTATAGGTCCACGGAGGGAATGG-3′, and 5′-CTACGCTCGAGTTACAAACCCAGAATTTCATTCCTG-3′. The HindIII (AAGCTT) and XhoI (CTCGAG) restriction sites are underlined, and the His sequence is shown in italics. PCR fragments were digested with HindIII and XhoI enzymes and then inserted into HindIII/XhoI-cut pcDNA3.1 vector. The full-length ORF11 protein expressing vector pcDNA3.1ORF11 was reported previously (22).
IP and analysis.
Melanoma cells were infected with ORF11 mutants or pOka, and protein lysates were collected with extraction buffer containing 0.1 M Tris base (pH 8.8), 0.1 M NaCl, 5 mM KCl, 1 mM CaCl2, 0.5 mM MgCl2, 1% SDS, 1% NP-40, and an EDTA-free protease inhibitor cocktail (Roche, CA). Protein lysates from pOkaΔ11-infected or uninfected melanoma cells were used as negative controls. Protein lysates were treated with RNase A (100 μg/ml) and DNase I (40 U/ml) (Invitrogen, Carlsbad, CA) for 30 min at 37°C before immunoprecipitation (IP) to assess possible RNA or DNA bridging in ORF11 protein binding to ORF9 protein. A rabbit antibody against the ORF11 protein N-terminal amino acids 1 to 245 (Ab11) was cross-linked to immobilized protein A (Pierce, Rockford, IL), and then antibody-bound beads were washed and incubated with lysates overnight at 4°C. IP samples were separated by SDS-polyacrylamide electrophoresis and either processed with GelCode blue staining (Thermo Fisher Scientific, Rockford, IL) or transferred to a nitrocellulose membrane. Transferred proteins were analyzed by immunoblotting using a rabbit antibody against full-length ORF9 protein (17); the blot was then stripped and reprobed with Ab11 antibody specific to ORF11 protein. For in vitro IP assays, HEK 293 cells were transfected with plasmids expressing ORF11 or the ORF11 truncations, along with the ORF9-expressing plasmid. Lysates were collected in radioimmunoprecipitation assay (RIPA) buffer containing the protease inhibitor cocktail (Roche Inc., Indianapolis, IN), and IP was performed with Ab11 and a rabbit anti-His antibody (His Ab) (Santa Cruz Biotech, Inc., Santa Cruz, CA). Normal mouse and rabbit sera (Vector Laboratories, Inc., Burlingame, CA) were used as negative controls. The antibodies were incubated with 30 μl of protein A+G beads for 2 h at 4°C (Santa Cruz Biotech, Inc.); the beads were washed in lysis buffer and then incubated with lysates overnight at 4°C. The samples were separated on a 7.5% polyacrylamide gel and transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore, Bedford, MA). For Western blotting, anti-ORF11 protein antibody Ab11 was used at a dilution of 1:20,000 and His Ab or mouse anti-His monoclonal antibody (His MAb) (Abcam Inc., San Francisco, CA) was used at a dilution of 1:1,000. Primary antibodies were detected with horseradish peroxidase-conjugated sheep anti-mouse and sheep anti-rabbit secondary antibodies (Amersham) at a dilution of 1:10,000 to 1:20,000 and visualized by ECL Plus (Amersham) and X-ray film (Kodak). The nuclear and cytoplasmic extracts from cells were prepared by using a nuclear extraction kit (Active Motif, Carlsbad, CA).
Mass spectrometry.
Melanoma cells (5 log10 cells/cm2) were inoculated with pOka (3 log10 PFU/cm2) and harvested 48 h postinfection (hpi), and the ORF9/ORF11 complex was immunoprecipitated as described above. Samples were separated by SDS-PAGE and proteins visualized using GelCode blue. Bands were excised and digested with trypsin, and the proteins were identified using an LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific, West Palm Beach, FL) at the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University.
Cryo-immuno-EM and confocal microscopy.
For cryo-immunoelectron microscopy (cryo-immuno-EM) experiments, HELF were seeded in a 75-mm2 flask at a density of 7.5 × 106 cells and infected with 4.9 log10 PFU of pOka, pOkaΔ11, and pOka11Δ549-793 (here referred to as pOka11Δ549) viruses. Samples for cryo-immuno-EM were collected at 2 days after infection and prepared as described previously (7). For immunogold staining, ultrathin sections (60 nm) were incubated in digoxigenin-blocking solution (Roche Diagnostics) for 30 min and then for 1 h with Ab11 or rabbit polyclonal anti-ORF9 protein antibodies at a dilution of 1:200. Samples were washed in phosphate-buffered saline (PBS) and incubated with protein A conjugated to 10-nm colloidal gold (PAG10 nm) from CMC (Utrecht, The Netherlands) at a dilution of 1:60. Samples were stained with 3.5% aqueous uranyl acetate for 10 min and with 0.2% lead citrate for 3 min and examined on a Joel 1230 transmission electron microscope.
For confocal microscopy, HELF were fixed with 4% formaldehyde at 48 hpi. After blocking with 1% fish gelatin for 1 h at room temperature (RT), cells were incubated with a rabbit anti-ORF9 protein antibody and an affinity-purified murine monoclonal anti-ORF11 protein antibody, clone 11N_12, which was raised against the N-terminal 400 amino acids of ORF11 protein. Cells were washed and incubated for 1 h at RT with fluorescein isothiocyanate-labeled anti-rabbit and Texas red-labeled anti-mouse secondary antibodies (Jackson ImmunoResearch, Inc.). Cell nuclei were counterstained with Hoechst 22358 (Invitrogen, Carlsbad, CA). Analysis was performed with a Leica TCSSP2 confocal laser scanning microscope (Heidelberg, Germany).
Infection of human skin xenografts in SCIDhu mice.
Skin xenografts were made in homozygous CB-17scid/scid mice, using human fetal tissue supplied by Advanced Bioscience Resources (Alameda, CA) according to federal and state regulations (25). Animal use was in accordance with the Animal Welfare Act and was approved by the Stanford University Administrative Panel on Laboratory Animal Care. pOka and ORF11 mutant viruses were passaged three times in primary HELF before inoculation of the xenografts. The infectious virus titer was determined for each inoculum at the time of inoculation. Skin xenografts were harvested at 10 and 20 days postinoculation and analyzed by infectious-focus assay (24). Virus recovered from the tissues was tested by PCR and sequencing to confirm the expected deletions.
RESULTS
ORF11 protein interacts with ORF9 protein in VZV-infected cells.
Immunoprecipitation (IP) experiments to identify potential ORF11 protein binding partners were done using Ab11, a rabbit polyclonal antibody that recognizes the ORF11 protein N terminus (22), and lysates of VZV-infected and uninfected melanoma cells. A rabbit polyclonal antibody to VZV glycoprotein B (gB) was included in IP experiments and served as an unrelated antibody control. IP samples were separated by SDS-PAGE and proteins visualized using GelCode blue (Fig. 1A). A 110-kDa protein, which is the expected size of the ORF11 protein, was recovered from infected but not from uninfected cell lysates and gB IP extracts. Several polypeptides exhibiting apparent masses between 35 and 40 kDa were also detected in Ab11 infected IP extracts only. When these bands were excised from the SDS-PAGE gel and tested by mass spectroscopy, ORF9 polypeptides were detected, suggesting that ORF11 protein forms a complex with the ORF9 tegument protein in VZV replication. In addition, the VZV protein IE63 and cellular proteins annexin-2 and rRNA 2′-O-methyltransferase fibrillarin were identified in IPs with ORF11 protein.
Fig 1.

Identification of the interaction between ORF11 protein and ORF9 protein. (A) GelCode blue-stained gel of proteins immunoprecipitated from infected (I) or uninfected (U) cells using Ab11 and gB antibodies. The bands present only in infected Ab11 IP extracts (dashed-line box) were cut from the gel and subjected to mass spectroscopy. The arrow indicates a species of around 110 kDa, the predicted size of ORF11 protein. (B) Proteins immunoprecipitated by Ab11 and gB antibody from infected and uninfected cells were analyzed by Western blotting (WB) with Ab11 antibody. The ORF11 gene product (arrow) was immunoprecipitated by Ab11 from infected cells. (C) Proteins immunoprecipitated by Ab11 and gB antibody from infected and uninfected cells were analyzed by Western blotting with anti-ORF9 protein antibody; ORF9 protein was coprecipitated with ORF11 protein. Numbers on the left side of the blots indicate molecular mass, in kDa.
To verify the mass spectroscopy results, we next performed Western blot analyses of protein complexes that were coprecipitated from infected and uninfected cell lysates using Ab11 and gB antibody. The viral protein IE63 was not detected by Western blotting (data not shown), and therefore, the interaction between ORF11 and IE63 proteins was not confirmed. However, ORF11 protein was immunoprecipitated from VZV-infected cell lysates (Fig. 1B) and ORF9 protein was pulled down by Ab11, as shown using an antibody that recognizes the ORF9 gene product (Fig. 1C). In this experiment, ORF9 protein antibody also reacted with several polypeptides with sizes between 35 and 40 kDa, which was consistent with the mass spectroscopy analysis and a previous investigation of ORF9 expression in VZV-infected cells (7). No signal was detected by Western blotting using Ab11 or anti-ORF9 protein antibody in IPs done with uninfected cell lysates (Fig. 1B and C). IP with a rabbit polyclonal antibody to VZV gB did not yield proteins of the molecular masses that were recovered with Ab11 and anti-ORF9 protein antibodies (Fig. 1B and C). Thus, ORF9 protein binds to ORF11 protein in VZV-infected cells.
The interaction between ORF11 protein and ORF9 protein is independent of RNA, DNA, and other viral proteins.
Since ORF11 protein was previously identified as an RNA binding protein, immunoprecipitations using Ab11 were done with pOka- and pOkaΔ11-infected cell lysates prepared in the presence or absence of RNase A and DNase I. Multiple forms of ORF9 protein (35 to 40 kDa) was detected in pOka lysates with or without RNase A and DNase I but not in the pOkaΔ11 lysates (Fig. 2A), indicating that the interaction between ORF11 and ORF9 proteins is not dependent on the presence of RNA or DNA.
Fig 2.

Assessment of the interaction of ORF11 protein with ORF9 protein. (A) Immunoprecipitation was performed with Ab11 and pOka- or pOkaΔ11-infected cell lysates, with or without RNase A and DNase I treatment. Western blotting was done with anti-ORF9 protein antibody. Multiple forms of ORF9 protein were detected from pOka-infected cell lysates with or without RNase A and DNase I treatment and are indicated by arrowheads. (B) Expression of ORF11 protein and His-tagged ORF9 protein. HEK 293 cells were cotransfected with an ORF11-expressing plasmid and a plasmid encoding His-tagged ORF9 protein, harvested at 48 h posttransfection, and analyzed by Western blotting with rabbit anti-ORF11 antibody, Ab11 (left panel), or rabbit anti-His antibody (His Ab) (right panel). HEK 293 cells transfected with vector pcDNA3.1 alone served as a negative control. Molecular mass is indicated on the sides of the blots. (C) The left blot shows proteins immunoprecipitated with Ab11 or normal rabbit serum (control) from cotransfected HEK 293 cells, analyzed by Western blotting with mouse anti-His antibody (His MAb). ORF9 His-tagged protein was coprecipitated with ORF11 protein. The right blot shows proteins immunoprecipitated with rabbit anti-His Ab or normal rabbit serum (control) from cotransfected HEK 293 cells, analyzed by Western blotting with anti-ORF11 antibody Ab11. ORF11 protein was coprecipitated with His-tagged ORF9 protein.
In order to determine whether ORF11 and ORF9 proteins interact directly or through an intermediate viral protein, the proteins were expressed from plasmids in transient-transfection experiments. ORF11 protein and a His-tagged ORF9 protein were coexpressed in HEK 293 cells, and the expression of both proteins was confirmed by Western blot analysis of cell lysates. No signal was observed from vector alone-transfected HEK 293 cells (Fig. 2B). IP was then performed with Ab11, followed by Western blot analysis using a mouse anti-His monoclonal antibody (His MAb). ORF11 protein was coprecipitated with His-tagged ORF9 protein (Fig. 2C). The reciprocal IP was also performed with His Ab followed by Western blotting using Ab11. His-tagged ORF9 protein was coprecipitated with ORF11 protein (Fig. 2C). No ORF11 protein or His-tagged ORF9 protein was observed using control rabbit serum for IP (Fig. 2C). Therefore, ORF11 protein binds to ORF9 protein in the absence of other viral proteins.
Mapping of the region of ORF11 protein required for interaction with ORF9 protein.
To investigate the functional significance of the interaction between ORF11 and ORF9, we sought to identify the amino acids of ORF11 protein that are required for their binding. We first constructed two truncation mutants with deletions beginning from amino acid 254 or 549 of ORF11 protein (Fig. 3A). Expression of the ORF11 protein and its truncations together with His-tagged ORF9 protein was confirmed by Western blots of lysates from cotransfected cells (Fig. 3B). When these two ORF11 truncations were tested for their ability to bind ORF9 protein by IP, both abolished the interaction (Fig. 3C), suggesting that ORF11 protein C-terminal amino acids 549 to 793 are necessary for the binding. To more precisely define the ORF11 protein region that binds ORF9 protein, we generated a panel of ORF11 truncation mutants by stepwise deletions of the C terminus of ORF11 protein (Fig. 3A). These mutants were then cotransfected together with the plasmid expressing His-tagged ORF9 protein. All of the cotransfected ORF11 mutants and ORF9 protein were expressed (Fig. 3D). When the capacity of these ORF11 mutants to bind ORF9 protein was analyzed by IP, none showed an interaction with ORF9 protein except for the ORF11 mutant that included residues from amino acids 1 to 780. ORF9 protein pulldown by this ORF11 small truncation was similar to that of full-length ORF11 protein (1 to 793 amino acids) (Fig. 3E). These results suggested that the region including amino acid 767 to 780 within the C terminus of ORF11 protein was necessary for the interaction with ORF9 protein.
Fig 3.

Mapping of the ORF11 region required for interaction with ORF9 protein. (A) Schematic diagram of full-length ORF11 protein (wild type [wt]) and its C-terminal truncation mutants used for this study. The amino acids (aa) of the ORF11 protein RNA binding domain and their location are indicated. Plasmids expressing ORF11 or the mutants were separately transfected into HEK 293 cells, along with a plasmid expressing His-tagged ORF9 protein. Cell lysates were collected at 48 h posttransfection and tested by WB and IP. (B) WB with Ab11 antibody (left) for expression of ORF11, ORF11 548, and ORF11 253 in cotransfected HEK 293 cells. A WB with His Ab (right) shows the expression of His-tagged ORF9 protein in the three cotransfection assays. (C) Proteins immunoprecipitated with His Ab or control rabbit serum (control) from cotransfected HEK 293 cells were analyzed by WB with Ab11. (D) Western blots for expression of ORF11 and ORF11 mutant proteins (top) and ORF9 protein (bottom) in cotransfection experiments. (E) IP was performed with His Ab (+) and normal rabbit serum (−), followed by Western blotting using Ab11.
Effects of the ORF11 mutations on binding to the ORF9 protein during VZV replication.
Based on the in vitro mapping result, three VZV recombinants, pOka11Δ254-793 (hereinafter referred to as pOka11Δ254), pOka11Δ549-793 (hereinafter referred to as pOka11Δ549), and pOka11Δ767/780, were generated. The region of the ORF11 gene encoding the C-terminal amino acids 254 to 793 was deleted to make pOka11Δ254; the ORF11 sequences encoding amino acids 549 to 793 and amino acids 767 to 780 located within the ORF11 protein C terminus were deleted to make pOka11Δ549 and pOka11Δ767/780. The capacity of the mutant forms of ORF11 protein to interact with ORF9 protein was assessed in VZV-infected cells by IP using Ab11 antibody and Western blotting using antibody against the ORF9 gene product. pOka11Δ254, pOka11Δ549, and pOka11Δ767/780 proteins were expressed as ∼40-kDa, ∼75-kDa, and ∼100-kDa products, respectively (Fig. 4A and B). Full-length and mutant ORF11 proteins were immunoprecipitated by Ab11, and ORF9 protein was only coprecipitated with ORF11 from pOka-infected cells, but binding of ORF9 protein to ORF11 protein was not detected in lysates of pOka11Δ254-, pOka11Δ549-, or pOkaΔ11-infected cells or an uninfected control (Fig. 4A). This result is consistent with our in vitro binding assay and indicates that the interaction requires residues present within amino acids 549 to 793 of the ORF11 protein C terminus. When the interaction with ORF9 protein was assessed in pOka11Δ767/780- and wild-type pOka-infected cells, little ORF9 protein was detected from the IP sample of pOka11Δ767/780 (Fig. 4B), suggesting that the absence of 767 to 780 residues from the ORF11 protein C-terminal region significantly affected the ability of ORF11 protein to bind to ORF9 protein during VZV infection.
Fig 4.
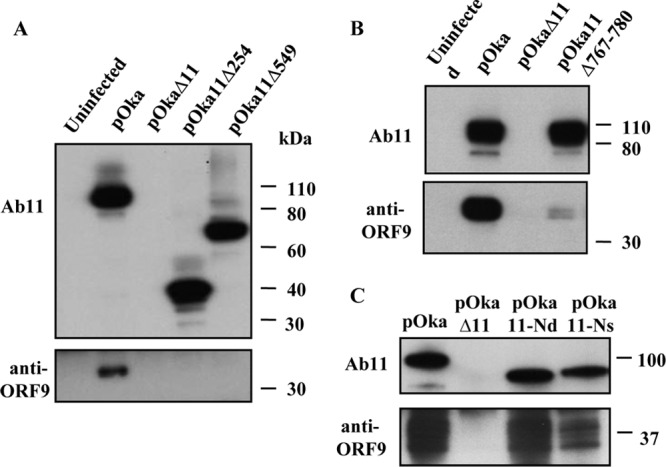
Immunoprecipitation of ORF11 and ORF9 proteins from melanoma cells infected with VZV and VZV ORF11 mutants. Melanoma cells were infected with VZV ORF11 mutants or pOka. The anti-ORF11 antibody Ab11 was used for immunoprecipitation, while Ab11 and the rabbit anti-ORF9 antibody were used for Western blotting. (A) The top blot is a Western blot with Ab11 showing that ORF11, ORF11Δ254, and ORF11Δ549 proteins were immunoprecipitated by Ab11 from cells infected with pOka, pOka11Δ254, and pOka11Δ549 but not from uninfected cells or cells infected with pOkaΔ11. The bottom blot is a Western blot with ORF9 antibody showing that ORF9 protein was immunoprecipitated only by Ab11 from the pOka-infected cells. (B) The top blot is a Western blot with Ab11 showing ORF11 and ORF11Δ767/780 protein detection from the input cell lysates used for IP. No signal was observed from uninfected or pOkaΔ11-infected cell lysates. The bottom blot is a Western blot with ORF9 antibody showing that ORF9 protein was immunoprecipitated by Ab11 from pOka-infected cells, and a trace of ORF9 protein was also observed from pOka11Δ767/780-infected cells. (C) The top blot is a Western blot with Ab11 showing that ORF11, ORF11-Nd, and ORF11-Ns proteins were immunoprecipitated by Ab11 from cells infected with pOka, pOka11-Nd, and pOka11-Ns. The bottom blot is a Western blot demonstrating that ORF9 proteins were immunoprecipitated by Ab11 from melanoma cells infected with pOka, pOka11-Nd, and pOka11-Ns but not from cells infected with pOkaΔ11. Molecular mass, in kilodaltons, is indicated on the right sides of the blots.
The interaction between the ORF11 and ORF9 proteins was also investigated in cells infected with two previously described ORF11 mutants, pOka11-Nd and pOka11-Ns (22). pOka11-Nd expresses ORF11 protein lacking amino acids 1 to 30, and pOka11-Ns has a mutation that disrupts the RNA binding domain that we mapped to amino acids 8 to 15 in the ORF11 protein N terminus. The ORF11-ORF9 protein interaction was preserved and similar to pOka in cells infected with pOka11-Nd and pOka11-Ns but was absent from pOkaΔ11-infected cells (Fig. 4C).
Characterization of VZV ORF11 mutant viruses in vitro.
The growth kinetics and plaque morphology of pOka11Δ254, pOka11Δ549, and pOka11Δ767/780 were investigated in infected melanoma cells. Comparisons of the growth kinetics of pOka with those of pOka11Δ254, pOka11Δ549, and pOkaΔ11 (Fig. 5A.I) and of pOka with pOka11Δ767/780 and pOkaΔ11 in melanoma cells showed no statistically significant differences over 6 days (Fig. 5A.III). Thus, as expected given our previous observations with pOkaΔ11 and confirmed in these experiments, none of the partial ORF11 deletions altered VZV replication as determined by plaque numbers in vitro. However, when plaque size was used to evaluate cell-cell spread of the pOka11Δ254, pOka11Δ549, pOka11Δ767/780, and pOkaΔ11 mutants compared to pOka in melanoma cells, all exhibited significantly reduced plaque sizes compared to those of pOka (Fig. 5A.II, A.IV, and B). Plaque sizes generated by pOka11Δ254, pOka11Δ549, and pOka11Δ767/780 were similar to those generated by pOkaΔ11. Thus, pOka11Δ767/780 showed the same phenotype for growth kinetics and plaque size as the VZV recombinants pOka11Δ254 and pOka11Δ549, with larger deletions.
Fig 5.

Growth kinetics of pOka, pOka11Δ549, pOka11Δ254, pOkaΔ11, and pOka11Δ767/780 in melanoma cells. Growth kinetics of pOka, pOkaΔ11, pOka11Δ549, and pOka11Δ254 (A.I) and pOka, pOkaΔ11, and pOka11Δ767/780 over 6 days (A.III) are shown. Bars indicate standard errors of the means. The plaque sizes of these viruses in melanoma cells at 4 days postinoculation are shown (A.II and A.IV). Bars indicate mean sizes of 50 plaques measured for each virus, with standard errors of the means. The asterisks indicate differences with a P value of <0.001 compared to the pOka plaque size. (B) Representative plaque morphologies of pOka, pOkaΔ11, pOka11Δ549, pOka11Δ254, and pOka11Δ767/780 in melanoma cells.
Effects of eliminating the ORF11-ORF9 protein interaction on the localization of ORF11 and ORF9 proteins in infected cells.
The cellular localization of ORF11 and ORF9 proteins was examined by confocal microscopy at 48 h postinfection (Fig. 6). ORF11 protein was present in both nuclei and cytoplasm, whereas ORF9 protein had a predominantly cytoplasmic localization in cells infected with pOka, pOka11Δ254, pOka11Δ549, and pOka11Δ767/780. This observation indicated that the ORF11 protein interaction with ORF9 protein occurs in the cytoplasm and that the ORF11 protein mutations had no detectable effect on the cellular localization of ORF11 or ORF9 as determined by confocal microscopy. Uninfected controls showed no reactivity with ORF11 or ORF9 protein antibodies (Fig. 6).
Fig 6.

Cellular localization of ORF11 and ORF9 proteins. The localization of ORF9 and ORF11 proteins in cells infected with pOka, pOka11Δ254, pOka11Δ549, and pOka11Δ767/780 was examined by confocal microscopy. Infected or uninfected HELF were fixed 48 hpi and stained with an anti-ORF9 protein (green) and a mouse monoclonal antibody to ORF11 protein (red). Cell nuclei were counterstained with Hoechst 22358 (blue). Size bars, 10 μm.
To further investigate the intracellular distribution of ORF11 and ORF9 proteins, cells infected with pOka11Δ254, pOka11Δ549, pOka11Δ767/780, and pOka were fractionated into nuclear and cytoplasmic extracts (Fig. 7). As expected from the confocal microscopy analysis, ORF11 mutant proteins containing residues 1 to 253 and 1 to 548 or with deletions of residues 767 to 780 were present in both nuclear and cytoplasmic fractions, like intact ORF11 protein. In contrast, little or no ORF9 protein was detected in the nuclear fractions from cells infected with pOka and the VZV ORF11 mutants, including pOka11Δ254, pOka11Δ549, pOkaΔ11, and pOka11Δ767/780. α-Tubulin and RCC1 (regulator of chromosome condensation 1) were used to confirm the purity of the fractionations; α-tubulin was detected exclusively in the cytoplasmic fraction, while the nuclear protein RCC1 was only observed in the nuclear fraction (Fig. 7).
Fig 7.

Analysis of ORF9 and ORF11 proteins in nuclear and cytoplasmic fractions of VZV-infected cells. Cellular fractions were prepared from melanoma cells 48 h after infection with pOka, pOka11Δ254, pOka11Δ549, pOkaΔ11, or pOka11Δ767/780, separated on SDS-PAGE gels, and probed with either anti-ORF9 or anti-ORF11protein antibodies. The membranes were then stripped and reprobed for cellular protein α-tubulin and nuclear protein RCC1 to determine the purity of the fractions. Molecular mass is shown on the left. Nu, nuclear fractionation; Cyto, cytoplasmic fractionation.
Subcellular distribution of ORF11 protein relative to ORF9 protein in pOka11Δ549-infected cells.
It has been suggested that ORF9 protein accumulates in the trans-Golgi region of VZV-infected cells, where it might recruit other proteins for incorporation into the viral tegument (7), leading to the hypothesis that ORF11 protein binding to ORF9 protein may be required for its inclusion in the virus particle. The effects of blocking the interaction on the subcellular localization of these proteins were assessed by cryo-immuno-EM analysis of HELF at 48 h after infection with pOka11Δ549 or pOka. The distributions of ORF9 protein were similar in cells infected with these viruses; ORF9 was localized to Golgi cisternae and Golgi-derived membranes (Fig. 8A and B). In contrast, the subcellular localization of ORF11 protein was altered by deleting the ORF11 C terminus, which contains the interacting domain as shown in HELF infected with pOka11Δ549. ORF11 was detected primarily with the vesicular membranes present in the trans-Golgi region of cells infected with pOka (Fig. 8C, D, and E), whereas ORF11 was dispersed irregularly throughout the cytoplasm in cells infected with pOka11Δ549 (Fig. 8F and G). When HELF infected with pOkaΔ11 were examined, it was found that ORF9 retained the trans-Golgi region distribution (Fig. 8H), and ORF11 was not detected (Fig. 8I). Either no or very faint background labeling was detected in HELF infected with pOka and tested with the preimmune sera for the rabbit antibodies to ORF9 (Fig. 8J) and ORF11 (Fig. 8K) proteins. In addition, ORF11 protein incorporation into virions was significantly impaired in pOka11Δ549-infected cells, although the mutant ORF11 protein could be detected in some virions. pOka virions had a mean of 4.3 (standard deviation [SD], 1.6) gold particles/virion, compared to 1.8 (SD, 1.0) per pOka11Δ549 virion (P < 0.001), when the number of gold particles indicating ORF11 protein association with virions was counted in pOka- and pOka11Δ549-infected cells (53 virions each). Furthermore, compared with those infected with pOka, cells infected with pOka11Δ549 had fewer enveloped virions in the typical large cytoplasmic vacuoles observed in VZV-infected cells.
Fig 8.

Ultrastructural analysis of HELF infected with pOka, pOka11Δ549, and pOkaΔ11. Cells infected with pOka, pOka11Δ549, and pOkaΔ11 were labeled with antibodies against ORF9 (anti-ORF9) and ORF11 (Ab11) gene products or preimmune serum corresponding to the anti-ORF9 protein antibody or Ab11, followed by inoculation with gold-tagged secondary antibody, and evaluated by cryo-immuno-EM. (A and B) ORF9 protein cytoplasmic distribution in cells infected with pOka (A) and pOka11Δ549 (B). (C) Overview of ORF11 protein distribution in a pOka-infected cell. A representative extracellular VZV particle labeled with ORF11 was detected in pOka-infected cells and is shown in upper right corner. (D and E) Higher magnification to show the ORF11 cytoplasmic distribution in pOka-infected cells. (F and G) ORF11Δ549 cytoplasmic distribution in pOka11Δ549-infected cells. A representative extracellular VZV particle labeled with ORF11 protein was detected in pOka11Δ549-infected cells and is presented in upper right corner of panel F. (H) ORF9 cytoplasmic distribution in pOkaΔ11-infected cells. (I) Cryo-immuno-EM detection with Ab11 antibody in pOkaΔ11-infected cells. (J) Cryo-immuno-EM detection with preimmune serum to anti-ORF9 protein antibody in pOka-infected cells. (K) Cryo-immuno-EM detection with preimmune serum to Ab11 in pOka-infected cells. Scale bars, 0.2 μm (A, B, C, F, and G) and 0.1 μm (D, E, H, I, J, and K). Nuc, nucleus; Cyt, cytoplasm.
Effects of abrogating the interaction of ORF11 protein with ORF9 proteins on VZV virulence in human skin xenografts in vivo.
Skin xenografts were inoculated with fibroblasts infected with pOka, pOka11Δ254, pOka11Δ549, or pOkaΔ11. pOka was the wild-type control and pOkaΔ11, which has shown to be severely attenuated in skin in vivo, was a negative control. The inoculum titers of the four viruses were similar (Fig. 9). Infected skin xenografts were harvested at 10 and 20 days after infection, and virus yields were determined by an infectious center assay. All skin xenografts infected with pOka yielded infectious virus. The mean titer of pOka was 2.8 log10 PFU/ml at day 10 and 3.6 log10 PFU/ml at day 20 postinoculation. In contrast to the in vitro studies, infectious virus was not recovered from any skin xenografts inoculated with pOka11Δ254, pOka11Δ549, or pOkaΔ11 at either day 10 or day 20 postinoculation (Fig. 9).
Fig 9.

Replication of pOka, pOkaΔ11, pOka11Δ254, and pOka11Δ549 in human skin xenografts in vivo. Titers of pOka, pOkaΔ11, pOka11Δ254, and pOka11Δ549 in human skin xenografts at 10 and 20 days after inoculation were determined by infectious focus assay; six xenografts were tested for each virus at each time point. “X” indicates that no virus was recovered from skin xenografts. Each bar represents the mean viral titer (log10), and error bars represent standard deviations. Inoculum titers of the viruses tested were not significantly different.
DISCUSSION
These experiments demonstrate that the ORF11 tegument protein of VZV interacts directly with ORF9 tegument protein, which is essential and constitutes a major component of the VZ virion tegument (7, 17). Eliminating the capacity of ORF11 protein to bind ORF9 protein was associated with a severe impairment of infection in human skin xenografts in the SCID mouse model. The deficiency was comparable to the effect of deleting the ORF11 gene on skin virulence (7), which was confirmed in this study. Thus, the ORF11-ORF9 protein interaction is likely to be important for VZV pathogenesis in skin, whereas interfering with the RNA binding capacity of ORF11 does not alter VZV replication in skin, as we showed previously (22).
The gene cluster that includes ORF9 to ORF12 is conserved in all of the alphaherpesviruses; these genes are UL49, UL48, UL47, and UL46 in HSV-1 and related viruses (1, 4). Analyses of HSV-1 virions show that the tegument proteins encoded by UL49, UL48, UL47, and UL46 are present at 1,000 to 2,000 copies, while the other known tegument proteins are present at fewer than 1,000 copies (26). These high concentrations suggest that the UL46 to UL49 proteins are likely to play a structural role in HSV-1 particles. Studies of PRV mutants that do not express these gene products also suggest their importance as structural components of the tegument (27–29). Quantitative analyses of VZV virions have been limited by the difficulty of recovering sufficient numbers of VZV virions from infected cells. However, the transcripts of ORF11 and ORF9 genes are among the most highly abundant viral mRNAs produced during lytic infection (30). Our previous report demonstrated that ORF11 and ORF9 proteins are readily detected in the VZV virion tegument and appear to be major components of the VZV virion tegument, like their HSV-1 and PRV homologues (7, 22).
In vitro binding assays indicated that the ORF11 protein C-terminal region of amino acids 549 to 793 is required for ORF11 protein interaction with ORF9 protein. Further mapping identified ORF11 protein amino acids 767 to 780 as the residues involved in ORF9 protein binding. Although plaque numbers were not reduced, pOka11Δ767/780, like pOka11Δ254, pOka11Δ549, and the full ORF11 deletion mutant, exhibited reduced capacity for cell-cell spread, based on the small-plaque phenotype. This capacity for cell-cell spread is important for VZV pathogenesis in skin.
We showed that ORF11 protein has both a nuclear and cytoplasmic distribution in pOka-, pOka11Δ254-, pOka11Δ549-, and pOka11Δ767/780-infected cells, whereas ORF9 is detected predominantly in the cytoplasm, implying that the interaction between these two proteins occurs in the cytoplasm. Like ORF11, the UL47 homologue is present in the nucleus and cytoplasm of HSV-1 infected cells (18). Precise information about the intracellular distribution of ORF11 and ORF9 proteins was provided by ultrastructure analysis and showed that both proteins were found predominantly with vesicle membranes in the trans-Golgi area of pOka-infected cells, which is the site of secondary envelopment. In contrast, ORF11 protein lacking the C terminus did not bind to ORF9 protein and was predominantly dispersed throughout the cytoplasm. Notably, this difference in the cytoplasmic distribution of ORF11 was not detected by confocal microscopy, which lacks the precision of cryo-immuno-EM. These observations suggest that ORF9 protein may be required to recruit ORF11 protein to the trans-Golgi region for virion assembly.
The tegument of alphaherpesvirus virions consists of numerous viral and several cellular proteins that appear to be recruited into the virus particle through a network of protein-protein interactions (31). Although the tegument has been described as amorphous, recent evidence suggests that this network of interactions causes an ordered addition of proteins that form the tegument during assembly. In HSV-1, PRV, and equine herpesvirus 1 (EHV-1)-infected cells, UL48 protein (VP16) interacts with capsid proteins, tegument proteins (including the other three encoded by the UL46-UL49 gene cluster), and viral glycoproteins (32–35). UL48 protein is a component of primary enveloped virions and is essential for addition of the outer tegument (36–39). Moreover, UL48 protein is critical for HSV-1 and PRV viral assembly (11). The sequence of events by which tegument proteins are incorporated into the VZV virion tegument is unknown. VZV ORF10 protein, the UL48 protein homologue, was associated with the tegument by analysis of purified virions (9). However, ORF10 protein is dispensable for viral replication in cultured cells, although it is a virulence factor for skin infection (40). Thus, ORF10 protein does not share the same properties as UL48 protein in other alphaherpesvirus orthologues.
ORF9 protein interacts with both tegument proteins and glycoproteins, suggesting that ORF9 protein is critical for facilitating tegument assembly and secondary envelopment of VZV virions. ORF9 protein binds to IE62 protein, the major VZV transactivator, indicating that ORF9 protein may recruit IE62 into the tegument and thereby incorporate the other known tegument proteins that bind to IE62 protein, including IE4, IE63, and ORF47 protein kinase (41–43). Moreover, the orthologues of HSV-1 and PRV UL49 proteins have been found to interact with glycoproteins (44–47). However, these studies did not determine whether UL49 protein was involved in recruiting viral proteins for assembly. ORF9 protein binds to gE, a major VZV envelope glycoprotein, suggesting that ORF9 protein is present in the outer tegument layer. ORF9 protein is an essential protein, which limits the generation of ORF9 mutants (21). ORF9 protein appears to be highly modified during infection, based on detection of multiple forms of ORF9 protein with anti-ORF9 antibody. Whether all or only some of these various-molecular-weight species of ORF9 protein bind to ORF11 protein was not determined.
Ultrastructural analysis showed that the interaction with ORF9 protein is not absolutely required for bringing ORF11 protein to the site of secondary assembly in cultured cells in vitro, but incorporation of ORF11 into the virion tegument is markedly diminished and reduced ORF11 protein in virions is likely associated with an impaired production of enveloped VZV virions. No changes in the morphology of the mature virions could be detected, suggesting that other tegument or cellular proteins compensated for limited ORF11 protein in the tegument structure, as has been reported for HSV-1 and PRV (14, 48). Importantly, the capacity of ORF11 protein to bind to ORF9 protein appears to be critical for VZV infection of skin xenografts, and any mechanisms that may have compensated for the disruption of the ORF11-ORF9 protein interaction in vitro are not sufficient for replication in the human skin tissue microenvironment in vivo. This requirement for ORF11 protein was not detected in skin organ cultures infected in vitro (16) but was present for VZV skin pathogenesis in vivo under conditions where the viruses must overcome intrinsic antiviral responses. Since it is not yet known why ORF9 protein is an essential VZV protein, it is possible that ORF11 protein might affect its recruitment as a structural component, resulting in impaired skin infection. That the ORF11-ORF9 protein interaction is needed helps to account for the severe attenuation of the ORF11 deletion mutant in vivo. As a major tegument component, ORF11 protein may be involved in recruitment of other viral proteins for virion assembly. The deleted ORF11 protein region appears to have functions that contribute to the phenotype caused by ORF11 protein C-terminal truncation.
In summary, we have shown that the ORF11 and ORF9 proteins form a complex in VZV infection, which highly depends on the presence of amino acid residues 767 and 780 in the ORF11 C-terminal domain. The loss of the ORF11-ORF9 protein interaction markedly affected the deposition of ORF11 protein in the trans-Golgi region, the site for secondary assembly. While ORF11 protein may have other functions, its inclusion in the virion tegument at normal levels appears to be needed for skin infection in vivo. The proteins encoded by the conserved alphaherpesvirus genes in the cluster corresponding to the VZV ORF9 to ORF12 genes have some virus-specific differences in their individual contributions to tegument assembly. However, these observations about VZV ORF11 and ORF9 proteins and the reports of functions of related proteins in other alphaherpesviruses suggest that they play overlapping roles in tegument assembly. Finally, interactions of tegument proteins may not be required in vitro but may be critical for replication in differentiated host tissues, as shown for VZV infection of skin in vivo.
ACKNOWLEDGMENTS
We thank Leigh Zerboni, Michelle Lai, and Phillip Sung for their help with the animal experiments and Kevin Mo for his help with the viral replication assay.
This work was supported by NIH grant AI053846 to the Arvin laboratory and a grant from the Bayerisches Staatsministerium für Wissenschaft, Forschung und Kunst (BayGene) to the Haas laboratory.
Footnotes
Published ahead of print 20 February 2013
REFERENCES
- 1. Cohen I, Straus SE, Arvin AM. 2007. Herpes simplex viruses, p 2773–2818 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Kinchington PR, Cohen JI. 2000. Viral proteins, p 74–104 In Arvin AM, Gershon AA. (ed), Varicella zoster virus virology and clinical management. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 3. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 4. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5. Kelly JB, Fraefel C, Cunningham AL, Diefenbach RJ. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 145:173–265 [DOI] [PubMed] [Google Scholar]
- 6. Besser J, Sommer MH, Zerboni L, Bagowski CP, Ito H, Ku CC, Arvin AM. 2003. Differentiation of varicella-zoster virus ORF47 protein kinase and IE62 protein binding domains and their contributions to replication in human skin xenografts in the SCID-hu mouse. J. Virol. 77:5964–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Che X, Reichelt M, Sommer MH, Rajamani J, Zerboni L, Arvin AM. 2008. Functions of the ORF9-to-ORF12 gene cluster in varicella-zoster virus replication and in the pathogenesis of skin infection. J. Virol. 82:5825–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kinchington PR, Bookey D, Turse SE. 1995. The transcriptional regulatory proteins encoded by varicella-zoster virus open reading frames (ORFs) 4 and 63, but not ORF 61, are associated with purified virus particles. J. Virol. 69:4274–4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kinchington PR, Hougland JK, Arvin AM, Ruyechan WT, Hay J. 1992. The varicella-zoster virus immediate-early protein IE62 is a major component of virus particles. J. Virol. 66:359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Spengler M, Niesen N, Grose C, Ruyechan WT, Hay J. 2001. Interactions among structural proteins of varicella zoster virus. Arch. Virol. Suppl. 17:71–79 [DOI] [PubMed] [Google Scholar]
- 11. Roizman B, Campadelli-Fiume G. 2007. Alphaherpes viral genes and their functions, p 70–92 In Arvin AM, Campadelli-Fiume C, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 12. Dorange F, Tischer BK, Vautherot JF, Osterrieder N. 2002. Characterization of Marek's disease virus serotype 1 (MDV-1) deletion mutants that lack UL46 to UL49 genes: MDV-1 UL49, encoding VP22, is indispensable for virus growth. J. Virol. 76:1959–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kopp M, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. 2002. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 76:8820–8833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, McKnight JL. 1993. Herpes simplex virus type 1 UL46 and UL47 deletion mutants lack VP11 and VP12 or VP13 and VP14, respectively, and exhibit altered viral thymidine kinase expression. J. Virol. 67:1482–1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y, Sirko DA, McKnight JL. 1991. Role of herpes simplex virus type 1 UL46 and UL47 in alpha TIF-mediated transcriptional induction: characterization of three viral deletion mutants. J. Virol. 65:829–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang Z, Selariu A, Warden C, Huang G, Huang Y, Zaccheus O, Cheng T, Xia N, Zhu H. 2010. Genome-wide mutagenesis reveals that ORF7 is a novel VZV skin tropic factor. PLoS Pathog. 6:e1000971 doi:10.1371/journal.ppat.1000971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cilloniz C, Jackson W, Grose C, Czechowski D, Hay J, Ruyechan WT. 2007. The varicella-zoster virus (VZV) ORF9 protein interacts with the IE62 major VZV transactivator. J. Virol. 81:761–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donnelly M, Elliott G. 2001. Nuclear localization and shuttling of herpes simplex virus tegument protein VP13/14. J. Virol. 75:2566–2574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elliott G, Hafezi W, Whiteley A, Bernard E. 2005. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J. Virol. 79:9735–9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pomeranz LE, Blaho JA. 2000. Assembly of infectious herpes simplex virus type 1virions in the absence of full-length VP22. J. Virol. 74:10041–10054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tischer B, Kaufer B, Sommer M, Wussow F, Arvin AM, Osterrieder N. 2007. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J. Virol. 81:13200–13208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Che X, Oliver SL, Sommer MH, Rajamani J, Reichelt M, Arvin AM. 2011. Identification and functional characterizations of varicella-zoster virus ORF11 gene product. Virology 412:156–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Niizuma T, Zerboni L, Sommer MH, Ito H, Hinchliffe S, Arvin AM. 2003. Construction of varicella-zoster virus recombinants from parent Oka cosmids and demonstration that ORF65 protein is dispensable for infection of human skin and T cells in the SCID-hu mouse model. J. Virol. 77:6062–6065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moffat JF, Stein MD, Kaneshima H, Arvin AM. 1995. Tropism of varicella-zoster virus for human CD4+ and CD8+ T lymphocytes and epidermal cells in SCID-hu mice. J. Virol. 69:5236–5242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Moffat JF, Zerboni L, Kinchington PR, Grose C, Kaneshima H, Arvin AM. 1998. Attenuation of the vaccine Oka strain of varicella-zoster virus and role of glycoprotein C in alphaherpesvirus virulence demonstrated in the SCID-hu mouse. J. Virol. 72:965–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fuchs W, Klupp BG, Granzow H, Hengartner C, Brack A, Mundt A, Enquist LW, Mettenleiter TC. 2002. Physical interaction between envelope glycoproteins E and M of pseudorabies virus and the major tegument protein UL49. J. Virol. 76:8208–8217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fuchs W, Granzow H, Mettenleiter TC. 2003. A pseudorabies virus recombinant simultaneously lacking the major tegument proteins encoded by the UL46, UL47, UL48, and UL49 genes is viable in cultured cells. J. Virol. 77:12891–12900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fuchs W, Granzow H, Klupp BG, Kopp M, Mettenleiter TC. 2002. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 76:6729–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohrs RJ, Hurley MP, Gilden DH. 2003. Array analysis of viral gene transcription during lytic infection of cells in tissue culture with varicella-zoster virus. J. Virol. 77:11718–11732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mettenleiter TC. 2006. Intriguing interplay between viral proteins during herpesvirus assembly or: the herpesvirus assembly puzzle. Vet. Microbiol. 113:163–169 [DOI] [PubMed] [Google Scholar]
- 32. Elliott G, Mouzakitis G, O'Hare P. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hafezi W, Bernard E, Cook R, Elliott G. 2005. Herpes simplex virus tegument protein VP22 contains an internal VP16 interaction domain and a C-terminal domain that are both required for VP22 assembly into the virus particle. J. Virol. 79:13082–13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kato K, Daikoku T, Goshima F, Kume H, Yamaki K, Nishiyama Y. 2000. Synthesis, subcellular localization and VP16 interaction of the herpes simplex virus type 2 UL46 gene product. Arch. Virol. 145:2149–2162 [DOI] [PubMed] [Google Scholar]
- 35. Lee J, Vittone V, Diefenbach E, Cunningham A, Diefenbach R. 2008. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 378:347–354 [DOI] [PubMed] [Google Scholar]
- 36. Gross ST, Harley CA, Wilson DW. 2003. The cytoplasmic tail of herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 317:1–12 [DOI] [PubMed] [Google Scholar]
- 37. Kamen D, Gross S, Girvin M, Wilson D. 2005. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 79:6134–6141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Naldinho-Souto R, Browne H, Minson T. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 80:2582–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu Q, Courtney RJ. 1994. Chemical cross-linking of virion envelope and tegument proteins of herpes simplex virus type 1. Virology 204:590–599 [DOI] [PubMed] [Google Scholar]
- 40. Che X, Zerboni L, Sommer MH, Arvin AM. 2006. Varicella-zoster virus open reading frame 10 is a virulence determinant in skin cells but not in T cells in vivo. J. Virol. 80:3238–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Baiker A, Bagowski C, Ito H, Sommer M, Zerboni L, Fabel K, Hay J, Ruyechan W, Arvin AM. 2004. The immediate-early 63 protein of varicella-zoster virus: analysis of functional domains required for replication in vitro and for T-cell and skin tropism in the SCIDhu model in vivo. J. Virol. 78:1181–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lynch JM, Kenyon TK, Grose C, Hay J, Ruyechan WT. 2002. Physical and functional interaction between the varicella zoster virus IE63 and IE62 proteins. Virology 302:71–82 [DOI] [PubMed] [Google Scholar]
- 43. Spengler ML, Ruyechan WT, Hay J. 2000. Physical interaction between two varicella zoster virus gene regulatory proteins, IE4 and IE62. Virology 272:375–381 [DOI] [PubMed] [Google Scholar]
- 44. Chi J, Harley C, Mukhopadhyay A, Wilson D. 2005. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 86:253–261 [DOI] [PubMed] [Google Scholar]
- 45. Farnsworth A, Wisner TW, Johnson DC. 2007. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 81:319–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. O'Regan KJ, Bucks MA, Murphy MA, Wills JW, Courtney RJ. 2007. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 358:192–200 [DOI] [PubMed] [Google Scholar]
- 47. Stylianou J, Maringer K, Cook R, Bernard E, Elliott G. 2009. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. J. Virol. 83:5204–5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Michael K, Klupp BG, Mettenleiter TC, Karger A. 2006. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J. Virol. 80:1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]