

Abstract
We previously reported that a recombinant pantothenate auxotroph of Mycobacterium bovis BCG expressing human immunodeficiency virus type 1 (HIV-1) subtype C Gag (rBCGpan-Gag) efficiently primes the mouse immune system for a boost with a recombinant modified vaccinia virus Ankara (rMVA) vaccine. In this study, we further evaluated the immunogenicity of rBCGpan-Gag in a nonhuman primate model. Two groups of chacma baboons were primed or mock primed twice with either rBCGpan-Gag or a control BCG. Both groups were boosted with HIV-1 Pr55gag virus-like particles (Gag VLPs). The magnitude and breadth of HIV-specific cellular responses were measured using a gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) assay, and the cytokine profiles and memory phenotypes of T cells were evaluated by polychromatic flow cytometry. Gag-specific responses were detected in all animals after the second inoculation with rBCGpan-Gag. Boosting with Gag VLPs significantly increased the magnitude and breadth of the responses in the baboons that were primed with rBCGpan-Gag. These responses targeted an average of 12 Gag peptides per animal, compared to an average of 3 peptides per animal for the mock-primed controls. Robust responses of Gag-specific polyfunctional T cells capable of simultaneously producing IFN-γ, tumor necrosis alpha (TNF-α), and interleukin-2 (IL-2) were detected in the rBCGpan-Gag-primed animals. Gag-specific memory T cells were skewed toward a central memory phenotype in both CD4+ and CD8+ T cell populations. These data show that the rBCGpan-Gag prime and Gag VLP boost vaccine regimen is highly immunogenic, inducing a broad and polyfunctional central memory T cell response. This report further indicates the feasibility of developing a BCG-based HIV vaccine that is safe for childhood HIV immunization.
INTRODUCTION
Development of safe, effective, and inexpensive prophylactic human immunodeficiency virus type 1 (HIV-1) vaccines remains a major health priority despite reports of a declining global incidence of HIV infection (1) and good progress in developing other preventive strategies (2, 3). A successful HIV vaccine is expected to induce both humoral and cell-mediated immune responses. Despite much interest following the discovery of newer technologies in generating broadly neutralizing antibodies (4, 5), it is widely acknowledged that T cell responses will be a critical component of an effective HIV vaccine (6, 7). While the precise correlates of HIV-1 immune protection have not been clearly elucidated (8, 9), studies of immune responses in HIV-1-infected long-term nonprogressors and nonhuman primate models of HIV/AIDS strongly suggest that induction of cellular HIV-specific immune responses is necessary for an effective vaccine strategy (10, 11). A number of studies have shown that immune responses directed to HIV-1 Gag (12–15) and an increased breadth of the Gag-specific CD4 T cell responses (15) correlate with lower viral loads, which illustrate the importance of the inclusion of Gag protein in candidate HIV vaccines. In the best-case scenario, T cell responses could destroy HIV-infected cells before the virus spreads to other cells and so prevent the infection from establishing in the infected individuals. However, it is unlikely that T cell responses will prevent all HIV infection, but they could control viral replication and lower the viral set point, resulting in lower transmission rates and extended time before progression to AIDS. Since the potential of heterologous prime-boost vaccination strategies in HIV-1 vaccine development was first demonstrated (16), a large body of research has shown that the use of live viral and bacterial vectors in prime-boost vaccinations is effective in evoking potent vaccine-specific immune responses to a wide variety of infectious diseases, as reviewed by Lu (17). Such immune responses ideally need to broadly target a range of antigens, be mediated by T cells with polyfunctional capacity, including those that are effective in killing virally infected cells, and possess both long-lived central memory T cell phenotypes as well as effector memory capable of trafficking to effector sites (18, 19).
Mycobacterium bovis BCG has been investigated as a live vaccine vector for a variety of human infections (20–25), including HIV-1 infections (26–30). An important and attractive feature of using BCG is its long safety record as a tuberculosis (TB) vaccine, having being injected into over 3 billion people worldwide. However, it has been reported to be unsafe in HIV-infected children (31–33) and to cause disseminated TB-like disease in simian immunodeficiency virus (SIV)-infected macaques (34, 35). Efforts to improve on the safety feature of BCG have led to generation of auxotrophic mutants of mycobacteria by deletion of genes that are necessary for growth such as panCD, leuD, and lysA (36–40). The ΔpanCD BCG auxotroph is unable to synthesize its own pantothenic acid, which is a key precursor of coenzyme A and is thus essential for several mycobacterial intracellular processes (41). Moreover, the ΔpanCD auxotrophic strains of Mycobacterium tuberculosis and M. bovis BCG have been shown to lack virulence in guinea pigs and mice, including SCID mice (36, 38), without losing their immunogenicity (37, 40). This safety feature makes auxotrophic mutants of BCG desirable HIV vaccine vectors, considering that immunocompromised and HIV-infected individuals might, inadvertently, be included in future campaigns of mass vaccination with a BCG-vectored HIV vaccine.
The use of the baboon as an animal model for evaluating the immunogenicity of candidate human vaccines, including HIV vaccines, has been reported (27, 42–47). Chacma baboons have been previously utilized to evaluate candidate HIV-1 vaccines (27, 42, 44), two of which are currently in clinical trial (48). Also, we have previously reported that a recombinant BCG (rBCG) expressing HIV-1 subtype C Gag is able to prime the immune system of chacma baboons for a boost with HIV-1 Pr55gag virus-like particles (Gag VLPs), resulting in induction of Gag-specific T cell responses (27). By modifying the mycobacterial shuttle vector, we were able to generate a more stable rBCG vaccine expressing HIV-1 subtype C Gag (rBCGpan-Gag) using a pantothenate auxotroph of Pasteur BCG (BCG ΔpanCD). The rBCG with improved stability elicited robust HIV-specific CD8+ T cell responses in BALB/c mice when used in a prime-boost combination with a recombinant modified vaccinia virus Ankara (rMVA) expressing a matching Gag antigen (49). In the current study, we investigated the immune response of baboons to a primary vaccination with rBCGpan-Gag and a boost with Gag VLPs. We show that this rBCG efficiently primed for a Gag VLP boost, resulting in enhancement of the magnitude and broadening of the breadth of immune responses. We further show that the prime-boost regimen generated high-magnitude, polyfunctional CD4+ and CD8+ T cell responses and memory T cells that were biased toward a central memory phenotype. These data support further development of HIV-1 vaccines based on BCG ΔpanCD, which can be used safely in children and immunocompromised individuals.
MATERIALS AND METHODS
Preparation of recombinant BCG vaccine stocks.
A recombinant BCG vaccine expressing the full-length HIV-1 subtype C gag gene (rBCGpan-Gag) was prepared as previously described by Chapman et al. (49), using M. bovis BCG mc26000 (BCG ΔpanCD), a pantothenic auxotroph strain derived from M. bovis BCG Pasteur 1172 P2 (Statens Serum Institute, Denmark). A control rBCG (rBCGpan-control) was prepared in an identical manner using BCG ΔpanCD that was transformed using an empty shuttle vector (not containing gag) designated pCONEPI (GenBank accession no. DQ191755). The genetic stability of vaccine stocks was confirmed in vitro and shown to be maintained after in vitro passage for 30 generations and in vivo by restriction enzyme mapping of DNA plasmids isolated from rBCG recovered from mice splenocytes (49). The vaccine and rBCG control stocks were stored at −80°C until further use. Prior to animal vaccinations, the vaccine and rBCG control were thawed on ice and passed through a 25-gauge syringe needle 10 times to disperse clumps.
Production and preparation of HIV-1 Pr55gag VLPs.
Gag VLPs were produced in Spodoptera frugiperda (Sf9)-derived insect cell suspension cultures as previously described (50). In brief, human codon-optimized HIV-1 gag with amino acid sequences derived from HIV-1 subtype C isolate Du422 (GenBank accession no. AF544010 [51]) was cloned into pFastBac as described previously (52). Sf9 cells were then infected with the recombinant baculovirus and the Gag VLPs harvested from the culture supernatant as described previously (50). Purified Gag VLPs were tested and shown to be negative for microbial contamination, and the endotoxin level was below 1.0 endotoxin units (EU)/ml. The Gag VLP vaccine stock was formulated by adding sterile trehalose solution to reach a final concentration of 15% and stored at 4°C. The Gag content and integrity of the vaccine stocks were evaluated as described previously (50) before animal vaccinations.
Animal vaccinations.
Twelve chacma baboons weighing between 6 and 15 kg were used in the study. Animals were selected for inclusion in the study based on low background reactivity in preimmunization peripheral blood mononuclear cells (PBMC) to peptide and protein reagents used in immunological assays. The animals were housed in the Animal Research Centre of the South African Medical Research Council (MRC) in Cape Town. Before the study, these baboons were shown to be healthy and negative for tuberculin skin test and SIV antibodies.
Animals were randomly divided into two groups and immunized as shown in Fig. 1. All animals were monitored for local reactions on the inoculation sites. Blood samples for PBMC isolation were obtained at preinoculation and various time points postinoculation. Blood samples at the termination of the study were unavailable from animals 737 and 743, which died from unrelated causes before the experimental endpoint. A mixture of ketamine hydrochloride (10 mg/kg body weight) and xylazine hydrochloride (0.6 mg/kg body weight) was used to anesthetize animals for all procedures. These experiments were reviewed and approved by the Animal Ethics Committee of the University of Cape Town (HSFAEC reference no. 08/033).
Fig 1.
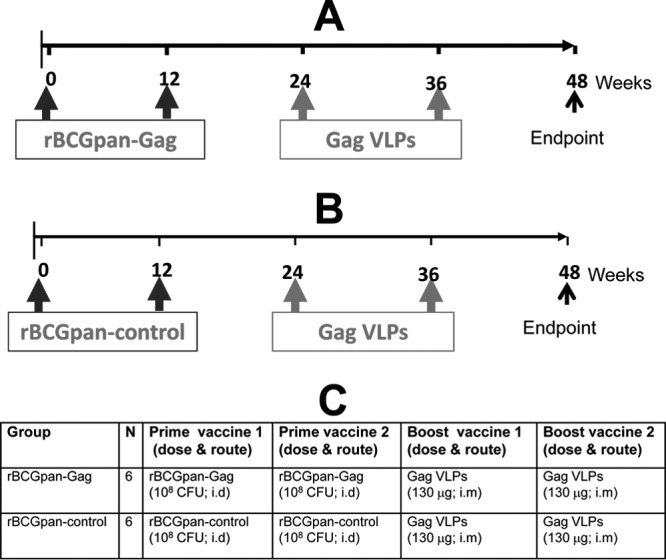
Experimental design and immunization schedule. (A and B) Two groups of chacma baboons were vaccinated twice (with the second vaccination administered 12 weeks after the first) with either rBCGpan-Gag (A) (n = 6) or rBCGpan-control (B) (n = 6) vaccine. All animals were subsequently boosted twice (with the second vaccination administered 12 weeks after the first) with Gag VLPs, and the experiment was terminated at week 48. (C) The dosage and route of the various vaccinations are shown in the table. i.d., intradermal route; i.m., intramuscular route.
Isolation of lymphocytes from blood.
PBMC were isolated from heparinized blood using a standard Ficoll gradient centrifugation method. Freshly isolated lymphocytes from blood were used in gamma interferon (IFN-γ) enzyme-linked immunosorbent spot (ELISPOT) assays, while the remainder were resuspended at 10 × 106 to 20 × 106 cells/ml in fetal bovine serum (FBS; Gibco) containing 10% dimethyl sulfoxide (DMSO; Sigma-Aldrich) and cryopreserved in liquid nitrogen until use in further immunological analyses.
IFN-γ ELISPOT assay.
Gag-specific T cells in the peripheral blood were quantified using an IFN-γ ELISPOT assay as previously described (44). The cells were incubated for 22 to 24 h in culture media containing synthetic Gag peptides (2 μg/ml) or culture medium alone. The Gag peptides, which spanned the entire HIV-1 Gag protein sequence, were based on the Du422 isolate (51) Gag sequences, were 15 to 18 amino acids in length, and overlapped by 10 amino acids. These peptides were prepared and pooled into three peptide pools corresponding to the amino acid sequences of the HIV-1 Gag p17, p24, and p15 regions for use in the IFN-γ ELISPOT assay. A response to any peptide pool that was equal to or greater than the peptide pool cutoff value of 30 spot-forming units (SFU)/106 PBMC after subtraction of the background response (cells and culture media alone) was considered positive. The cutoff value was determined by calculating the mean plus 3 standard deviations of the response of PBMC to the Gag peptide pools prior to animal immunizations. Values below this cutoff were set to zero when calculating the cumulative Gag response (sum of responses to the three individual peptide pools). Phytohematoglutinin-P (PHA-P; Sigma-Aldrich) was used as a positive control in all assays, and a cutoff value of 500 SFU/106 PBMC was used to validate the assay.
To evaluate the breadth of the Gag-specific responses, individual peptides to which PBMC responded at the peak immune response were identified using a pool-matrix IFN-γ ELISPOT method as previously described (27, 53).
Intracellular cytokine staining and polychromatic flow cytometry.
Cryopreserved PBMC obtained at the peak response time point (as measured by the ELISPOT assay) were used for detection of Gag-specific T cells by flow cytometry. Two monoclonal antibody staining panels were used in order to characterize the cytokine and memory profile of T cells. The following antibodies and fluorophores were used: CD3-fluorescein isothiocyanate (CD3-FITC), CD3-allophycocyanin-Cy7 (CD3-APC-Cy7), CD4-peridinin chlorophyll protein-Cy5.5 (CD4-PerCP-Cy5.5), CD8-Qdot605, CD28-FITC (clone CD28.2), CD95-APC, IFN-γ–Alexa 700, IFN-γ–phycoerythrin (IFN-γ-PE), interleukin-2–PE (IL-2-PE), tumor necrosis factor alpha-PE-Cy7 (TNF-α-PE-Cy7), TNF-α-PE (all from BD Biosciences), CD14-Pacific Blue, and CD19-Pacific Blue (Invitrogen). Titers of all antibodies were determined at optimal concentrations. Briefly, thawed and rested PBMC were washed and cultured in medium containing anti-CD28 (clone L293) and anti-CD49d (1 μg/ml) either alone or together with a single pool of Gag peptides at 1 μg/ml for 16 h in the presence of brefeldin A (Sigma) (10 μg/ml). Cells were then stained with violet amine reactive dye (“ViVid”; Molecular Probes) and then surface stained with CD4, CD8, CD14, and CD19 antibodies, with the inclusion of CD28 and CD95 for the memory panel, at room temperature. Cells were then fixed and permeabilized using Cytofix/Cytoperm buffer (BD) and stained intracellularly with CD3 and cytokine antibodies. For the memory panel, all cytokines were conjugated to PE and analyzed together. After washing, cells were resuspended in CellFix (BD). Approximately 500,000 total events were acquired per sample on an LSR II flow cytometer (BD), and analysis was performed using FlowJo (v8.5.3; Treestar). Dead cells (ViVid positive [ViVid+]), monocytes (CD14+), and B cells (CD19+) were excluded from the analysis. For characterizing cytokine profiles, cells were gated on singlets, lymphocytes, live CD3+, CD8+, or CD4+, and then IFN-γ, IL-2, or TNF-α or combinations of cytokine markers. For characterizing memory profiles, cells were gated on singlets, lymphocytes, live CD3+, CD8+, or CD4+, cytokine+ cells, and then memory T cells (CD28+ CD95+ and CD28− CD95+). Memory subsets were expressed as a percentage of total CD8+ or CD4+ memory cells (i.e., excluding naïve CD28+ CD95− cells). A positive cytokine response was defined as at least twice the level of the background (no antigen, only costimulatory antibodies), >0.05% after subtraction of the background, and at least 10 events. The latter criterion was introduced so as to minimize the possibility of error due to a low number of events that might arise from sequential subdivision of cell populations.
Anti-HIV-1 Gag antibody determinations by ELISA.
To monitor the development of humoral responses, the presence of Gag-specific antibodies in baboon sera was measured by an enzyme-linked immunosorbent assay (ELISA) method as previously described (44). ELISA plates were coated with HIV-1 recombinant Pr55Gag protein (Quality Biological) (0.05 μg per well). Sera obtained at prevaccination and 4 weeks after the second vaccination with rBCGpan and Gag VLPs were analyzed.
Statistical analysis.
Statistical analyses were performed using Prism version 5.0 (GraphPad Software, San Diego, CA). The t test for independent unpaired nonparametric comparisons was applied to assess the level of significance of comparisons between means. All tests were two tailed, and P values of 0.05 were considered significant.
RESULTS
Inoculation with rBCGpan induces self-limiting local reactions at the vaccination site.
Following vaccinations, animals were monitored for development of local reactions on the injection sites. All animals developed indurations, which culminated in skin ulcerations (5 to 15 mm in diameter) at inoculation sites within 2 weeks after receiving rBCGpan-Gag or rBCGpan-control. There were no differences in the sizes of the ulcerations. The second vaccination with rBCG induced ulcerations of comparable sizes. For both vaccinations, the ulcerations healed within 6 weeks postinoculation without any complication or need for medication. Vaccination with Gag VLPs did not result in any noticeable reaction at the inoculation site. With the exception of these local tissue reactions, the baboons remained healthy throughout the study period.
rBCGpan-Gag vaccine efficiently primes for a Gag VLP boost.
HIV-1 Gag-specific immune responses were measured in an IFN-γ ELISPOT assay using freshly isolated PBMC. A priming vaccination with the first dose of rBCGpan-Gag vaccine elicited low-magnitude cumulative Gag responses in 3 of 6 baboons (early responders), reaching a peak magnitude (mean ± standard deviation) of 313 ± 192 SFU/106 PBMC at 8 weeks postvaccination (Fig. 2A). These Gag-specific responses for the early responders waned with time but were still detectable at week 12 postvaccination. A second dose of rBCGpan-Gag which was given at week 12 boosted the responses in the 3 early responders and induced measurable responses in the other 3 late responders. The cumulative magnitude of responses reached the peak at 2 or 4 weeks after vaccination (378 ± 149 SFU/106 PBMC). These responses were directed predominantly to p15 (275 ± 261 SFU/106 PBMC in 6 of 6 baboons). The response to p17 was 100 ± 78 SFU/106 PBMC (for 4 of 6 baboons) and to p24 was 73 ± 41 SFU/106 PBMC (for 3 of 6 baboons). For the early responders, the Gag responses were maintained above detectable levels through to the time of booster vaccination, unlike 2 of 3 late responders, for which the Gag response was undetectable after 4 weeks. No Gag responses were detectable for the baboons that received the mock-prime inoculation with the rBCGpan-control (Fig. 2B).
Fig 2.

IFN-γ ELISPOT responses. PBMC obtained from various time points after vaccination with rBCGpan-Gag or rBCGpan-control and Gag VLPs were used in an IFN-γ ELISPOT assay using three Gag peptides spanning Gag p17, p24, and p15. Shown are the cumulative responses for rBCGpan-Gag-primed baboons at various time points (A) and week 26 (D) and the corresponding time points for rBCGpan-control baboons (B and E). Panel C shows a comparison of the mean cumulative responses of the two groups to Gag at week 26.
Boosting with Gag VLPs induced high-magnitude immune responses in animals receiving a rBCGpan-Gag prime that were 4- to 5-fold higher than those for the mock-prime group. These responses peaked at 2 weeks after boosting in both groups (Fig. 2A and B). At peak response, the mean cumulative Gag response was significantly higher (P < 0.0001) for the animals primed with rBCGpan-Gag (2,801 ± 293 SFU/106 PBMC) than for the mock-prime group (421 ± 116 SFU/106 PBMC; Fig. 2C). These responses were directed to all the three Gag peptide pools corresponding to p17, p24, and p15 for the rBCGpan-Gag group (Fig. 2D) and to only p24 and p15 for the mock-prime controls (Fig. 2E). Gag responses were consistently detected throughout the experimental period after the Gag VLP boost in all the animals in the rBCGpan-Gag group, in contrast to the results seen with the mock-primed group, where only 3 of 6 animals had consistent responses. At the experimental endpoint (week 48), the mean cumulative Gag response for the rBCGpan-Gag group (448 ± 108 SFU/106 PBMC) was significantly higher (P = 0.0273) than that for the mock-prime group (100 ± 47 SFU/106 PBMC).
The rBCGpan-Gag prime and Gag VLP boost regimen broadens the breadth of the Gag-specific response.
Next, we sought to evaluate the breadth of Gag responses by considering the number of responders to the individual Gag peptide pools and by identifying individual peptides to which the PBMC were responding for each animal in an IFN-γ ELISPOT assay. This was done at the peak IFN-γ ELISPOT response or 2 weeks after the first Gag VLP booster vaccination. All the animals in the rBCGpan-Gag group responded to all the three Gag peptide pools (p17, p24, and p15). In contrast, only a response to p15 was detectable in all the animals in the mock-prime control group, with only 3 of 6 and 5 of 6 animals responding to peptide pools corresponding to p17 and p24, respectively (data not shown). In addition, the mean cumulative responses to the three Gag peptide pools were significantly higher for the rBCGpan-Gag group than for the mock-prime controls (P = 0.012, P = 0.023, and P = 0.002 for the p17, p24, and p15 pools, respectively; data not shown). Also, in terms of magnitude, the responses to p15 were the most dominant of the responses to the 3 Gag domains for the rBCGpan-Gag group, while those to p24 were the most dominant for the mock-prime controls.
PBMC from animals primed with rBCGpan-Gag responded to an average of 12 peptides per baboon, with animal 738 responding to up to 19 peptides, whereas those from the mock-prime group responded to an average of 3 peptides per animal (Table 1). The distributions of Gag peptides being targeted by these PBMC also differed between the two groups. While PBMC from the rBCGpan-Gag group targeted more peptides corresponding to p15, p17, and p24, in that order, PBMC from the mock-prime group targeted peptides corresponding to p24 and only 1 or 2 or no peptides in the p17 and p15 domains.
Table 1.
Breadth of Gag IFN-γ ELISPOT responsesa
| Vaccine regimen | Animal ID | No. of Gag peptides targeted by PBMC at wk 26 (peak response) |
|||
|---|---|---|---|---|---|
| Gag p17 | Gag p24 | Gag p15 | Total | ||
| rBCGpan-Gag + Gag VLPs | 508 | 4 | 2 | 4 | 10 |
| 676 | 4 | 5 | 9 | 18 | |
| 710 | 4 | 3 | 4 | 11 | |
| 733 | 3 | 2 | 3 | 8 | |
| 734 | 2 | 1 | 4 | 7 | |
| 738 | 7 | 4 | 8 | 19 | |
| Total | 24 | 17 | 32 | 73 | |
| Avg | 4 | 3 | 5 | 12 | |
| rBCGpan-control + Gag VLPs | 675 | 0 | 0 | 2 | 2 |
| 687 | 0 | 2 | 0 | 2 | |
| 692 | 2 | 2 | 0 | 4 | |
| 732 | 0 | 3 | 1 | 4 | |
| 737 | 0 | 2 | 0 | 2 | |
| 743 | 0 | 4 | 1 | 5 | |
| Total | 2 | 13 | 4 | 19 | |
| Avg | <1 | 2 | <1 | 3 | |
PBMC obtained at week 26 (2 weeks after the first Gag VLP booster vaccination) were evaluated for the breadth of response using a Pool-Matrix ELISPOT mapping strategy. The table shows the number of Gag peptides to which the PBMC responded as identified using the Pool-Matrix IFN-γ ELISPOT method. ID, identification number.
The rBCGpan-Gag prime and Gag VLP boost regimen generates polyfunctional T-cell responses.
To further characterize the responses induced by the vaccine regimen, we investigated the T cell phenotype and functional profiles of the rBCGpan-Gag and Gag VLP prime-boost regimen using intracellular cytokine staining and polychromatic flow cytometry. Cellular responses were measured at the peak response time point, which coincided with 2 weeks after the first Gag VLP boost. As shown in Fig. 3A, Gag-specific T cell responses were both CD4+ and CD8 mediated, with 2-fold-higher median magnitudes of cytokine-producing CD4+ T cells in the prime-boost group. Only 2 of 6 animals in the mock-primed control group exhibited CD4+ or CD8+ responses (Fig. 3A). Gag-specific T cells produced all three cytokines in the order IFN-γ > TNF-α > IL-2 for both CD4+ and CD8+ T cells (Fig. 3B). When we examined the ability of these cells to simultaneously produce combinations of these cytokines (i.e., their polyfunctional nature), we detected these in both the CD4+ and CD8+ subsets (Fig. 3C and D). The proportion of CD4+ T cells simultaneously expressing all three cytokines was greater than the proportion of CD8+ T cells (approximately 55% versus 20%; Fig. 3D). In addition, >75% of responding CD4+ and CD8+ T cells simultaneously produced 2 or 3 cytokines (Fig. 3D). Thus, the combination of rBCGpan-Gag and Gag VLP candidate vaccines induced high frequencies of polyfunctional CD4+ and CD8+ T cell responses in the chacma baboons.
Fig 3.

Functional profile of vaccine-elicited Gag-specific T cells. PBMC obtained at week 26 (2 weeks after the first Gag VLP booster vaccination) were evaluated for CD4 and CD8 cytokine responses. (A) The total frequency of CD4+ and CD8+ T cells producing any cytokine (IFN-γ, TNF-α, or IL-2) in the CD4+ and CD8+ T cell subsets in group A (filled circles) and group B (open circles). (B) Individual cytokine responses in CD4+ and CD8+ T cells in group A (filled bars) and group B (open bars). (C) Polyfunctional profiles of Gag-specific CD4 and CD8+ T cells in group A. The frequencies of all possible combinations of the three cytokines (IFN-γ, TNF-α, and IL-2) in rBCGpan-Gag VLP prime-boost animals are shown for CD4+ (dark shading) and CD8+ (light shading) T cells. (D) Functional profiles are grouped and color coded according to number of functions and summarized as proportions of the total responses in the pie charts. Each slice of the pie corresponds to the median production of 3 cytokines (red), 2 cytokines (green), or 1 cytokine (blue).
The rBCGpan-Gag prime Gag VLP boost regimen induces central memory T cell responses.
We sought to determine the memory phenotype of vaccine-induced memory T cells in animals that received the combination of rBCGpan-Gag and Gag VLPs, using the phenotypic markers CD28 and CD95. PBMC from only three animals were available for the peak response time point after the first Gag VLP boost, while all six animals were tested 2 weeks after the second Gag VLP vaccination. Representative flow cytometry plots are shown in Fig. 4A, indicating the memory distribution of Gag-specific cytokine-producing T cells in the central and effector memory compartments. The majority of Gag-specific CD4+ T cells were found to be markedly skewed toward a central memory phenotype, with >95% of cytokine-producing CD4+ memory T cells expressing this phenotype 2 weeks after the first Gag VLP booster vaccination (Fig. 4B). Similarly, Gag-specific CD8+ T cells showed the same preference of skewing toward a central memory phenotype, with about 65% of total cytokine-positive cells expressing the central memory phenotype (Fig. 4B). The second Gag VLP vaccination did not change the distribution of memory phenotype in the CD8+ T cell subset but increased the proportion of CD4+ T cells expressing an effector memory phenotype to approximately 15%.
Fig 4.

Memory phenotype of Gag-specific T cells. Animals from group A (rBCGpan-Gag VLP prime-boost) were examined for Gag-specific memory T cells. Cytokine-producing CD4+ and CD8+ T cells were delineated into central (TCM) and effector (TEM) memory T cells based on CD28 and CD95 expression. (A) Representative flow cytometry plots of the memory profile of total CD4+ (upper panel) or CD8+ (lower panel) T cells are shown as a density plot and Gag-specific total cytokine+ T cells (blue dots) in one vaccinated animal (B734) at week 38. (B) Proportion of Gag-specific CD4+ (upper panel) or CD8+ (lower panel) T cells at weeks 26 and 38 (2 weeks after the first and second Gag VLP vaccinations, respectively). Data are shown as medians and summarized in pie charts, with the numbers of animals with a Gag response indicated below the pies.
Gag VLP vaccination elicits HIV-1 antibody responses.
We evaluated the induction of vaccine-specific humoral responses by determining the endpoint titers of anti-Gag antibodies in the sera obtained at three time points. As shown in Table 2, Gag-specific antibodies were detected only after booster vaccination with Gag VLPs. The titers for animals primed with rBCGpan-Gag were generally higher (median, >51,200; range, 6,400 to >51,200) than those for animals receiving a mock prime (median, 9,600; range, 200 to 51,200), suggesting a priming effect by rBCGpan-Gag vaccination.
Table 2.
Gag-specific antibody titersa
| Vaccine regimen | Animal ID | Gag-specific antibody titer |
||
|---|---|---|---|---|
| Pre-Vac (wk 0) | 4 wk post-rBCGpan-Vac (wk 16) | 4 wk post-Gag VLP-Vac (wk 40) | ||
| rBCGpan-Gag + Gag VLPs | 508 | <50 | <50 | >51,200 |
| 676 | <50 | <50 | >51,200 | |
| 710 | <50 | <50 | >51,200 | |
| 733 | <50 | <50 | >51,200 | |
| 734 | <50 | <50 | 6,400 | |
| 738 | <50 | <50 | 12,800 | |
| rBCGpan-control + Gag VLPs | 675 | <50 | <50 | 12,800 |
| 687 | <50 | <50 | 6,400 | |
| 692 | <50 | <50 | 1,600 | |
| 732 | <50 | <50 | 51,200 | |
| 737 | <50 | <50 | 200 | |
| 743 | <50 | <50 | 25,600 | |
Gag-specific antibodies were measured in the sera obtained at prevaccination (Pre-VAC) (week 0), 4 weeks after the second vaccination with rBCGpan (week 16), and 4 weeks after the second vaccination with Gag VLPs (week 40) using an ELISA method. The table shows the endpoint antibody titers of individual animals. ID, identification number.
DISCUSSION
In this study, a recombinant auxotrophic strain of BCG expressing HIV-1 subtype C Gag boosted with Gag VLPs was evaluated in a nonhuman primate model. We have previously shown that priming with this rBCG vaccine (rBCGpan-Gag), in a prime-boost combination with an antigen-matched recombinant MVA, elicits HIV-specific CD8+ T cells in mice (49). This study sought to further evaluate the immunogenicity of this rBCG vaccine prime/Gag VLP boost in nonhuman primates by characterizing the cellular immune response generated in immunized animals in terms of breadth, polyfunctionality, and phenotype of memory T cells.
Vaccinations with both rBCG-Gag and rBCG-control did not induce any systemic reactions in any animal, and the local cutaneous reactions were resolved without the need for medication, indicating that rBCG was well tolerated. This outcome was similar to our observations in previous studies where baboons were vaccinated with nonrecombinant BCG (54) and recombinant BCG (27). Intradermal inoculations of humans with wild-type BCG also result in development of similar local tissue reactions which are well tolerated (55, 56). However, future BCG-based vaccines which cause minimal or no reactogenicity will likely be more acceptable in clinical settings. This could possibly be achieved by the use of doses that are effective but lower than those used in the current study.
The ΔpanCD BCG (Pasteur) strain was used as a vector due to its improved safety, compared with that of wild-type BCG, as has been demonstrated with similar auxotrophs (36, 40). The safety of our ΔpanCD BCG vaccine was indicated by the development of fewer granulomas in the spleen and a reduced level of inflammation in the liver of vaccinated mice compared with the corresponding wild-type BCG recombinant results (49). The safety of BCG-vectored vaccines is a relevant issue, as vaccination of HIV-infected children with a clinical BCG (wild type) has been observed to be responsible for mycobacterial disease in 10% of these immunocompromised individuals (31, 33). In addition, BCG inoculation has been reported to enhance the pathogenicity of SIV infection in rhesus macaques, resulting in tuberculosis-like disease with disseminated granulomas (34, 35). However, no increase in viral load was seen in SIV-infected rhesus macaques vaccinated with a double panthothenate and leucine auxotroph of M. tuberculosis, and no mycobacteria could be detected in a variety of tissue samples or blood taken 54 weeks postvaccination (39).
Several studies have demonstrated superior immunogenicity from heterologous prime-boost vaccination strategies in HIV vaccine research (16) and as reviewed by Lu (17). rBCG vaccines have been shown to efficiently prime the immune response to the heterologous antigen in recombinant adenovirus, recombinant poxvirus, and protein prime-boost combinations (24, 26, 27, 30, 57, 58). We (27, 44, 52) and others (59) have previously shown that a Gag VLP boost, used in heterologous prime-boost modality, is a potent booster of Gag-specific responses. Further, we have shown that plant-produced Gag VLPs are immunogenic in mice (60), and others have shown that high yields of Gag VLPs can be made in tobacco chloroplasts (61), indicating the distinct prospect of producing affordable and cost-effective Gag-based HIV vaccines (62).
Our data illustrate that rBCGpan-Gag, on its own, is immunogenic and can achieve a 100% response rate after the second vaccination in a nonhuman primate model. This is a notable finding because, unlike the present baboon study results, no Gag-specific responses were detected in the murine model, in response to rBCGpan-Gag prime, before the MVA-Gag boost (49). In addition, the results of the present study are clearly better than those of our previous study in which a wild-type BCG recombinant was used (27). We further showed that a rBCGpan-Gag prime and Gag VLP boost regimen elicits a broad range of Gag epitope-containing peptides (range, 7 to 19) compared to the control animal results (range, 2 to 5). In rhesus macaque studies, a CD4 T cell response that targeted SIV Gag in vaccinated animals was associated with control of infection after SIV challenge (63) whereas both Gag- and non-Gag-antigen-specific CD8 T cells from vaccinated SIV controllers have been shown to suppress replication of SIV variants carrying cytotoxic T lymphocyte (CTL) escape mutations (64). Broad Gag-specific responses have also been associated with good control of HIV-1 replication in chronically infected HIV controllers (12–15, 65); in particular, the increased breadth of the Gag-specific CD4 T cell responses was significantly associated with effective immune control of viremia (15). Thus, our rBCGpan-Gag prime–Gag VLP boost regimen concurs with a potential correlate of AIDS-protective immunity.
Secretion of IFN-γ by CD4+ and CD8+ T cells has been associated with suppression of virus replication in HIV-1, SIV, and simian-human immunodeficiency virus (SHIV) infections (66–68), while cells secreting both IFN-γ and IL-2 have been shown to confer CD4-independent proliferation of HIV-1-specific CD8 T cells (69) and viral replication control in nonprogressive HIV-1 (70, 71) and SHIV89.6P (72) infection. Furthermore, polyfunctional CD4+ and CD8+ T cell responses that include the simultaneous release of IFN-γ, IL-2, and TNF-α have been associated with elite HIV-1 controllers (73–78). In the current report, we show that the rBCGpan-Gag and Gag VLP prime-boost vaccine regimen resulted in the induction of polyfunctional CD4+ (>50%) and CD8+ (∼25%) T cells, with the total frequency of cytokine-producing cells being greater in the CD4+ T cell subset. In addition, these T cells were significantly skewed toward a central memory phenotype, with >95% of the CD4+ and 65% of the CD8+ Gag-specific memory responses exhibiting a central memory phenotype. Effector memory cells are important as the first-line defense mechanism at the tissue sites where exposure to HIV occurs, and central memory cells may play a critical role, as they are long-lived and capable of replenishing the effector memory pool (79). HIV-specific memory T cells have been associated with better HIV-1 replication control, as indicated by preserved CD4+ T central and effector memory cells in HIV controllers (80) and higher proportions of HIV-specific CD8+ T central memory cells being associated with low viral set points in early HIV-1 infection (81). The role of effector memory T cells is further underscored by the findings of Hansen et al. (18, 19) that showed an association between vaccine-induced effector memory T cells and protection against mucosal SIV challenge.
Preexisting immunity to some environmental mycobacterial species has been reported to block in vivo multiplication of BCG (82), indicating potential reduction of vaccine efficacy of future BCG-based HIV vaccines. This is a crucial issue, particularly for sub-Saharan Africa, where vaccination against tuberculosis (TB) using BCG in infants is part of the WHO-recommended Extended Programme of Immunization (EPI) and where a cost-effective HIV vaccine such as that based on BCG would be expected to have a major impact due to the high HIV prevalence. Although this issue of preexisting anti-BCG immunity remains unresolved, a number of BCG studies in animals have shown enhanced immunologic effects attributable to prior BCG sensitization (83, 84) or coadministration with BCG (58). We observed an increase in the magnitude of BCG-specific IFN-γ ELISPOT responses after the second rBCGpan-Gag vaccination, suggesting that preexisting BCG immunity may not have a significant negative effect on the efficacy of future rBCG vaccines. Moreover, vaccination of infants with an efficacious BCG-vectored HIV vaccine has been proposed as a possible platform to prevent both TB and mother-to-child transmission of HIV (28).
In conclusion, this report demonstrates that a BCG-based HIV-1 vaccine boosted with Gag VLPs has the capacity to induce high-magnitude and broad Gag-specific responses which are characterized by generation of polyfunctional T cells and a memory phenotype skewed toward a central memory in nonhuman primates. These features are characteristic of an immune response that may be capable of controlling and containing the replication of HIV-1. Thus, our rBCGpan-Gag vaccine appears to be a promising candidate HIV vaccine when given in a prime-boost combination with a recombinant Gag-based HIV vaccine.
ACKNOWLEDGMENTS
We thank Tracey Muller and Evelyn Mojapelo for technical assistance with processing of samples and the staff at the MRC Animal Research Centre for animal care and assistance with animal vaccinations and sampling. We thank Andreia Soares, Cobus Olivier, and Marcel Tongo for critical reviews of the manuscript.
This study was supported by the South African AIDS Vaccine Initiative (SAAVI) and the U.S. National Institutes of Health (NIH) through funding from Phased Innovation Awards (PIA R21/R33) in AIDS Vaccine Research grant 5R33AI73182-4. This work is also based on research supported by NIH grant A121670, CFAR grant A151519, and the South African Research Chairs Initiative of the Department of Science and Technology and the National Research Foundation.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 28 February 2013
REFERENCES
- 1.UNAIDS. Global HIV/AIDS response: epidemic update and health sector progress towards universal access—progress report 2011. 2011. http://www.unaids.org/en/media/unaids/contentassets/documents/unaidspublication/2011/20111130_UA_Report_en.pdf.
- 2. Padian NS, McCoy SI, Karim SS, Hasen N, Kim J, Bartos M, Katabira E, Bertozzi SM, Schwartlander B, Cohen MS. 2011. HIV prevention transformed: the new prevention research agenda. Lancet 378:269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Quiñones-Mateu ME, Vanham G. 2012. HIV microbicides: where are we now? Curr. HIV Res. 10:1–2 [DOI] [PubMed] [Google Scholar]
- 4. Bonsignori M, Alam SM, Liao HX, Verkoczy L, Tomaras GD, Haynes BF, Moody MA. 2012. HIV-1 antibodies from infection and vaccination: insights for guiding vaccine design. Trends Microbiol. 20:532–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Verkoczy L, Kelsoe G, Moody MA, Haynes BF. 2011. Role of immune mechanisms in induction of HIV-1 broadly neutralizing antibodies. Curr. Opin. Immunol. 23:383–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barouch DH, Liu J, Li H, Maxfield LF, Abbink P, Lynch DM, Iampietro MJ, Sanmiguel A, Seaman MS, Ferrari G, Forthal DN, Ourmanov I, Hirsch VM, Carville A, Mansfield KG, Stablein D, Pau MG, Schuitemaker H, Sadoff JC, Billings EA, Rao M, Robb ML, Kim JH, Marovich MA, Goudsmit J, Michael NL. 2012. Vaccine protection against acquisition of neutralization-resistant SIV challenges in rhesus monkeys. Nature 482:89–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McDermott AB, Koup RA. 2012. CD8(+) T cells in preventing HIV infection and disease. AIDS 26:1281–1292 [DOI] [PubMed] [Google Scholar]
- 8. Burgers WA, Manrique A, Masopust D, McKinnon LR, Reynolds MR, Rolland M, Blish C, Chege GK, Curran R, Fischer W, Herrera C, Sather DN. 2012. Measurements of immune responses for establishing correlates of vaccine protection against HIV. AIDS Res. Hum. Retroviruses 28:641–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haynes BF, Gilbert PB, McElrath MJ, Zolla-Pazner S, Tomaras GD, Alam SM, Evans DT, Montefiori DC, Karnasuta C, Sutthent R, Liao HX, DeVico AL, Lewis GK, Williams C, Pinter A, Fong Y, Janes H, DeCamp A, Huang Y, Rao M, Billings E, Karasavvas N, Robb ML, Ngauy V, de Souza MS, Paris R, Ferrari G, Bailer RT, Soderberg KA, Andrews C, Berman PW, Frahm N, De Rosa SC, Alpert MD, Yates NL, Shen X, Koup RA, Pitisuttithum P, Kaewkungwal J, Nitayaphan S, Rerks-Ngarm S, Michael NL, Kim JH. 2012. Immune-correlates analysis of an HIV-1 vaccine efficacy trial. N. Engl. J. Med. 366:1275–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schmitz JE, Johnson RP, McClure HM, Manson KH, Wyand MS, Kuroda MJ, Lifton MA, Khunkhun RS, McEvers KJ, Gillis J, Piatak M, Lifson JD, Grosschupff G, Racz P, Tenner-Racz K, Rieber EP, Kuus-Reichel K, Gelman RS, Letvin NL, Montefiori DC, Ruprecht RM, Desrosiers RC, Reimann KA. 2005. Effect of CD8+ lymphocyte depletion on virus containment after simian immunodeficiency virus SIVmac251 challenge of live attenuated SIVmac239delta3-vaccinated rhesus macaques. J. Virol. 79:8131–8141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53 [DOI] [PubMed] [Google Scholar]
- 13. Pereyra F, Addo MM, Kaufmann DE, Liu Y, Miura T, Rathod A, Baker B, Trocha A, Rosenberg R, Mackey E, Ueda P, Lu Z, Cohen D, Wrin T, Petropoulos CJ, Rosenberg ES, Walker BD. 2008. Genetic and immunologic heterogeneity among persons who control HIV infection in the absence of therapy. J. Infect. Dis. 197:563–571 [DOI] [PubMed] [Google Scholar]
- 14. Prendergast A, Goodliffe H, Clapson M, Cross R, Tudor-Williams G, Riddell A, Daniels J, Williams A, Goulder P. 2011. Gag-specific CD4+ T-cell responses are associated with virological control of paediatric HIV-1 infection. AIDS 25:1329–1331 [DOI] [PubMed] [Google Scholar]
- 15. Ranasinghe S, Flanders M, Cutler S, Soghoian DZ, Ghebremichael M, Davis I, Lindqvist M, Pereyra F, Walker BD, Heckerman D, Streeck H. 2012. HIV-specific CD4 T cell responses to different viral proteins have discordant associations with viral load and clinical outcome. J. Virol. 86:277–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hu SL, Abrams K, Barber GN, Moran P, Zarling JM, Langlois AJ, Kuller L, Morton WR, Benveniste RE. 1992. Protection of macaques against SIV infection by subunit vaccines of SIV envelope glycoprotein gp160. Science 255:456–459 [DOI] [PubMed] [Google Scholar]
- 17. Lu S. 2009. Heterologous prime-boost vaccination. Curr. Opin. Immunol. 21:346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, Legasse AW, Chiuchiolo MJ, Parks CL, Axthelm MK, Nelson JA, Jarvis MA, Piatak M, Jr, Lifson JD, Picker LJ. 2011. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature 473:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hansen SG, Vieville C, Whizin N, Coyne-Johnson L, Siess DC, Drummond DD, Legasse AW, Axthelm MK, Oswald K, Trubey CM, Piatak M, Jr, Lifson JD, Nelson JA, Jarvis MA, Picker LJ. 2009. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 15:293–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arama C, Waseem S, Fernandez C, Assefaw-Redda Y, You L, Rodriguez A, Radosevic K, Goudsmit J, Kaufmann SH, Reece ST, Troye-Blomberg M. 2012. A recombinant Bacille Calmette-Guerin construct expressing the Plasmodium falciparum circumsporozoite protein enhances dendritic cell activation and primes for circumsporozoite-specific memory cells in BALB/c mice. Vaccine 30:5578–5584 [DOI] [PubMed] [Google Scholar]
- 21. Chapman R, Chege G, Shephard E, Stutz H, Williamson AL. 2010. Recombinant Mycobacterium bovis BCG as an HIV vaccine vector. Curr. HIV Res. 8:282–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dennehy M, Williamson AL. 2005. Factors influencing the immune response to foreign antigen expressed in recombinant BCG vaccines. Vaccine 23:1209–1224 [DOI] [PubMed] [Google Scholar]
- 23. Govan VA, Christensen ND, Berkower C, Jacobs WR, Jr, Williamson AL. 2006. Immunisation with recombinant BCG expressing the cottontail rabbit papillomavirus (CRPV) L1 gene provides protection from CRPV challenge. Vaccine 24:2087–2093 [DOI] [PubMed] [Google Scholar]
- 24. Hopkins R, Bridgeman A, Bourne C, Mbewe-Mvula A, Sadoff JC, Both GW, Joseph J, Fulkerson J, Hanke T. 2011. Optimizing HIV-1-specific CD8+ T-cell induction by recombinant BCG in prime-boost regimens with heterologous viral vectors. Eur. J. Immunol. 41:3542–3552 [DOI] [PubMed] [Google Scholar]
- 25. Shata MT, Stevceva L, Agwale S, Lewis GK, Hone DM. 2000. Recent advances with recombinant bacterial vaccine vectors. Mol. Med. Today 6:66–71 [DOI] [PubMed] [Google Scholar]
- 26. Cayabyab MJ, Korioth-Schmitz B, Sun Y, Carville A, Balachandran H, Miura A, Carlson KR, Buzby AP, Haynes BF, Jacobs WR, Letvin NL. 2009. Recombinant Mycobacterium bovis BCG prime-recombinant adenovirus boost vaccination in rhesus monkeys elicits robust polyfunctional simian immunodeficiency virus-specific T-cell responses. J. Virol. 83:5505–5513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chege GK, Thomas R, Shephard EG, Meyers A, Bourn W, Williamson C, Maclean J, Gray CM, Rybicki EP, Williamson AL. 2009. A prime-boost immunisation regimen using recombinant BCG and Pr55(gag) virus-like particle vaccines based on HIV type 1 subtype C successfully elicits Gag-specific responses in baboons. Vaccine 27:4857–4866 [DOI] [PubMed] [Google Scholar]
- 28. Im EJ, Saubi N, Virgili G, Sander C, Teoh D, Gatell JM, McShane H, Joseph J, Hanke T. 2007. Vaccine platform for prevention of tuberculosis and mother-to-child transmission of human immunodeficiency virus type 1 through breastfeeding. J. Virol. 81:9408–9418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosario M, Fulkerson J, Soneji S, Parker J, Im EJ, Borthwick N, Bridgeman A, Bourne C, Joseph J, Sadoff JC, Hanke T. 2010. Safety and immunogenicity of novel recombinant BCG and modified vaccinia virus Ankara vaccines in neonate rhesus macaques. J. Virol. 84:7815–7821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rosario M, Hopkins R, Fulkerson J, Borthwick N, Quigley MF, Joseph J, Douek DC, Greenaway HY, Venturi V, Gostick E, Price DA, Both GW, Sadoff JC, Hanke T. 2010. Novel recombinant Mycobacterium bovis BCG, ovine atadenovirus, and modified vaccinia virus Ankara vaccines combine to induce robust human immunodeficiency virus-specific CD4 and CD8 T-cell responses in rhesus macaques. J. Virol. 84:5898–5908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hesseling AC, Marais BJ, Gie RP, Schaaf HS, Fine PE, Godfrey-Faussett P, Beyers N. 2007. The risk of disseminated bacille Calmette-Guerin (BCG) disease in HIV-infected children. Vaccine 25:14–18 [DOI] [PubMed] [Google Scholar]
- 32. Hesseling AC, Rabie H, Marais BJ, Manders M, Lips M, Schaaf HS, Gie RP, Cotton MF, van Helden PD, Warren RM, Beyers N. 2006. Bacille Calmette-Guerin vaccine-induced disease in HIV-infected and HIV-uninfected children. Clin. Infect. Dis. 42:548–558 [DOI] [PubMed] [Google Scholar]
- 33. Hesseling AC, Schaaf HS, Hanekom WA, Beyers N, Cotton MF, Gie RP, Marais BJ, van Helden P, Warren RM. 2003. Danish bacille Calmette-Guérin vaccine-induced disease in human immunodeficiency virus-infected children. Clin. Infect. Dis. 37:1226–1233 [DOI] [PubMed] [Google Scholar]
- 34. Shen Y, Zhou D, Chalifoux L, Shen L, Simon M, Zeng X, Lai X, Li Y, Sehgal P, Letvin NL, Chen ZW. 2002Induction of an AIDS virus-related tuberculosis-like disease in macaques: a model of simian immunodeficiency virus-mycobacterium coinfection. Infect. Immun. 70:869–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou D, Shen Y, Chalifoux L, Lee-Parritz D, Simon M, Sehgal PK, Zheng L, Halloran M, Chen ZW. 1999. Mycobacterium bovis bacille Calmette-Guerin enhances pathogenicity of simian immunodeficiency virus infection and accelerates progression to AIDS in macaques: a role of persistent T cell activation in AIDS pathogenesis. J. Immunol. 162:2204–2216 [PubMed] [Google Scholar]
- 36. Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, Morris SL, Jacobs WR. 2006. Mycobacterium tuberculosis Delta RD1 Delta panCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24:6309–6320 [DOI] [PubMed] [Google Scholar]
- 37. Sambandamurthy VK, Derrick SC, Jalapathy KV, Chen B, Russell RG, Morris SL, Jacobs WR. 2005. Long-term protection against tuberculosis following vaccination with a severely attenuated double lysine and pantothenate auxotroph of Mycobacterium tuberculosis. Infect. Immun. 73:1196–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sampson SL, Dascher CC, Sambandamurthy VK, Russell RG, Jacobs WR, Bloom BR, Hondalus MK. 2004. Protection elicited by a double leucine and pantothenate auxotroph of Mycobacterium tuberculosis in guinea pigs. Infect. Immun. 72:3031–3037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sampson SL, Mansfield KG, Carville A, Magee DM, Quitugua T, Howerth EW, Bloom BR, Hondalus MK. 2011. Extended safety and efficacy studies of a live attenuated double leucine and pantothenate auxotroph of Mycobacterium tuberculosis as a vaccine candidate. Vaccine 29:4839–4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tullius MV, Harth G, Maslesa-Galic S, Dillon BJ, Horwitz MA. 2008. A replication-limited recombinant Mycobacterium bovis BCG vaccine against tuberculosis designed for human immunodeficiency virus-positive persons is safer and more efficacious than BCG. Infect. Immun. 76:5200–5214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sambandamurthy VK, Wang X, Chen B, Russell RG, Derrick S, Collins FM, Morris SL, Jacobs WR., Jr 2002. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 8:1171–1174 [DOI] [PubMed] [Google Scholar]
- 42. Burgers WA, Chege GK, Muller TL, van Harmelen JH, Khoury G, Shephard EG, Gray CM, Williamson C, Williamson AL. 2009. Broad, high-magnitude and multifunctional CD4+ and CD8+ T-cell responses elicited by a DNA and modified vaccinia Ankara vaccine containing human immunodeficiency virus type 1 subtype C genes in baboons. J. Gen. Virol. 90:468–480 [DOI] [PubMed] [Google Scholar]
- 43. Casimiro DR, Tang A, Chen L, Fu TM, Evans RK, Davies ME, Freed DC, Hurni W, Aste-Amezaga JM, Guan L, Long R, Huang L, Harris V, Nawrocki DK, Mach H, Troutman RD, Isopi LA, Murthy KK, Rice K, Wilson KA, Volkin DB, Emini EA, Shiver JW. 2003. Vaccine-induced immunity in baboons by using DNA and replication-incompetent adenovirus type 5 vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 77:7663–7668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chege GK, Shephard EG, Meyers A, van Harmelen J, Williamson C, Lynch A, Gray CM, Rybicki EP, Williamson AL. 2008. HIV-1 subtype C Pr55gag virus-like particle vaccine efficiently boosts baboons primed with a matched DNA vaccine. J. Gen. Virol. 89:2214–2227 [DOI] [PubMed] [Google Scholar]
- 45. Leung L, Srivastava IK, Kan E, Legg H, Sun Y, Greer C, Montefiori DC, Zur MJ, Barnett SW. 2004. Immunogenicity of HIV-1 Env and Gag in baboons using a DNA prime/protein boost regimen. AIDS 18:991–1001 [DOI] [PubMed] [Google Scholar]
- 46. Locher CP, Witt SA, Ashlock BM, Polacino P, Hu SL, Shiboski S, Schmidt AM, Agy MB, Anderson DM, Staprans SI, Zur MJ, Levy JA. 2004. Human immunodeficiency virus type 2 DNA vaccine provides partial protection from acute baboon infection. Vaccine 22:2261–2272 [DOI] [PubMed] [Google Scholar]
- 47. Murthy KK, Salas MT, Carey KD, Patterson JL. 2006. Baboon as a nonhuman primate model for vaccine studies. Vaccine 24:4622–4624 [DOI] [PubMed] [Google Scholar]
- 48. Williamson AL, Rybiki E, Shephard E, Gray G, Bekker LG, Downing K, Williamson C. 2012. South African HIV-1 vaccine candidates—the journey from the bench to clinical trials. S. Afr. Med. J. 102:452–455 [DOI] [PubMed] [Google Scholar]
- 49. Chapman R, Shephard E, Stutz H, Douglass N, Sambandamurthy V, Garcia I, Ryffel B, Jacobs W, Williamson AL. 2012. Priming with a recombinant pantothenate auxotroph of Mycobacterium bovis BCG and boosting with MVA elicits HIV-1 Gag specific CD8(+) T cells. PLoS One 7:e32769 doi:10.1371/journal.pone.0032769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lynch A, Meyers AE, Williamson AL, Rybicki EP. 2012. Stability studies of HIV-1 Pr55gag virus-like particles made in insect cells after storage in various formulation media. Virol. J. 9:210 doi:10.1186/1743-422X-9-210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Williamson C, Morris L, Maughan MF, Ping LH, Dryga SA, Thomas R, Reap EA, Cilliers T, van Harmelen J, Pascual A, Ramjee G, Gray G, Johnston R, Karim SA, Swanstrom R. 2003. Characterization and selection of HIV-1 subtype C isolates for use in vaccine development. AIDS Res. Hum. Retroviruses 19:133–144 [DOI] [PubMed] [Google Scholar]
- 52. Jaffray A, Shephard E, van Harmelen J, Williamson C, Williamson AL, Rybicki EP. 2004. Human immunodeficiency virus type 1 subtype C Gag virus-like particle boost substantially improves the immune response to a subtype C gag DNA vaccine in mice. J. Gen. Virol. 85:409–413 [DOI] [PubMed] [Google Scholar]
- 53. Anthony DD, Lehmann PV. 2003. T-cell epitope mapping using the ELISPOT approach. Methods 29:260–269 [DOI] [PubMed] [Google Scholar]
- 54. Chege GK, Williamson AL, Passmore JS, Bourn W, Ryffel B, Shephard EG. 2005. The immune response of the Chacma baboon to bacille Calmette Guerin: development of a primate model for BCG-based vaccine research. Vaccine 23:5783–5791 [DOI] [PubMed] [Google Scholar]
- 55. Hoft DF, Leonardi C, Milligan T, Nahass GT, Kemp B, Cook S, Tennant J, Carey M. 1999. Clinical reactogenicity of intradermal bacille Calmette-Guerin vaccination. Clin. Infect. Dis. 28:785–790 [DOI] [PubMed] [Google Scholar]
- 56. Kemp EB, Belshe RB, Hoft DF. 1996. Immune responses stimulated by percutaneous and intradermal bacille Calmette-Guerin. J. Infect. Dis. 174:113–119 [DOI] [PubMed] [Google Scholar]
- 57. Ami Y, Izumi Y, Matsuo K, Someya K, Kanekiyo M, Horibata S, Yoshino N, Sakai K, Shinohara K, Matsumoto S, Yamada T, Yamazaki S, Yamamoto N, Honda M. 2005. Priming-boosting vaccination with recombinant Mycobacterium bovis bacillus Calmette-Guerin and a nonreplicating vaccinia virus recombinant leads to long-lasting and effective immunity. J. Virol. 79:12871–12879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yu JS, Peacock JW, Jacobs WR, Jr, Frothingham R, Letvin NL, Liao HX, Haynes BF. 2007. Recombinant Mycobacterium bovis bacillus Calmette-Guerin elicits human immunodeficiency virus type 1 envelope-specific T lymphocytes at mucosal sites. Clin. Vaccine Immunol. 14:886–893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Buonaguro L, Devito C, Tornesello ML, Schroder U, Wahren B, Hinkula J, Buonaguro FM. 2007. DNA-VLP prime-boost intra-nasal immunization induces cellular and humoral anti-HIV-1 systemic and mucosal immunity with cross-clade neutralizing activity. Vaccine 25:5968–5977 [DOI] [PubMed] [Google Scholar]
- 60. Meyers A, Chakauya E, Shephard E, Tanzer FL, Maclean J, Lynch A, Williamson AL, Rybicki EP. 2008. Expression of HIV-1 antigens in plants as potential subunit vaccines. BMC Biotechnol. 8:53 doi:10.1186/1472-6750-8-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Scotti N, Alagna F, Ferraiolo E, Formisano G, Sannino L, Buonaguro L, De Stradis A, Vitale A, Monti L, Grillo S, Buonaguro FM, Cardi T. 2009. High-level expression of the HIV-1 Pr55gag polyprotein in transgenic tobacco chloroplasts. Planta 229:1109–1122 [DOI] [PubMed] [Google Scholar]
- 62. Rybicki EP. 2009. Plant-produced vaccines: promise and reality. Drug Discov. Today 14:16–24 [DOI] [PubMed] [Google Scholar]
- 63. Giraldo-Vela JP, Bean AT, Rudersdorf R, Wallace LT, Loffredo JT, Erickson P, Wilson NA, Watkins DI. 2010. Simian immunodeficiency virus-specific CD4+ T cells from successful vaccinees target the SIV Gag capsid. Immunogenetics 62:701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Iwamoto N, Tsukamoto T, Kawada M, Takeda A, Yamamoto H, Takeuchi H, Matano T. 2010. Broadening of CD8+ cell responses in vaccine-based simian immunodeficiency virus controllers. AIDS 24:2777–2787 [DOI] [PubMed] [Google Scholar]
- 65. Honeyborne I, Prendergast A, Pereyra F, Leslie A, Crawford H, Payne R, Reddy S, Bishop K, Moodley E, Nair K, van der Stok M, McCarthy N, Rousseau CM, Addo M, Mullins JI, Brander C, Kiepiela P, Walker BD, Goulder PJ. 2007. Control of human immunodeficiency virus type 1 is associated with HLA-B*13 and targeting of multiple gag-specific CD8+ T-cell epitopes. J. Virol. 81:3667–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Boaz MJ, Waters A, Murad S, Easterbrook PJ, D'Sousa E, van Wheeley C, Vyakarnam A. 2003. CD4 responses to conserved HIV-1 T helper epitopes show both negative and positive associations with virus load in chronically infected subjects. Clin. Exp. Immunol. 134:454–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boaz MJ, Waters A, Murad S, Easterbrook PJ, Vyakarnam A. 2002. Presence of HIV-1 Gag-specific IFN-gamma+IL-2+ and CD28+IL-2+ CD4 T cell responses is associated with nonprogression in HIV-1 infection. J. Immunol. 169:6376–6385 [DOI] [PubMed] [Google Scholar]
- 68. Migueles SA, Sabbaghian MS, Shupert WL, Bettinotti MP, Marincola FM, Martino L, Hallahan CW, Selig SM, Schwartz D, Sullivan J, Connors M. 2000. HLA B*5701 is highly associated with restriction of virus replication in a subgroup of HIV-infected long term nonprogressors. Proc. Natl. Acad. Sci. U. S. A. 97:2709–2714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zimmerli SC, Harari A, Cellerai C, Vallelian F, Bart PA, Pantaleo G. 2005. HIV-1-specific IFN-gamma/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc. Natl. Acad. Sci. U. S. A. 102:7239–7244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Emu B, Sinclair E, Favre D, Moretto WJ, Hsue P, Hoh R, Martin JN, Nixon DF, Mccune JM, Deeks SG. 2005. Phenotypic, functional, and kinetic parameters associated with a apparent T-cell control of human immunodeficiency virus replication in individuals with and without antiretroviral treatment. J. Virol. 79:14169–14178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Harari A, Petitpierre S, Vallelian F, Pantaleo G. 2004. Skewed representation of functionally distinct populations of virus-specific CD4 T cells in HIV-1-infected subjects with progressive disease: changes after antiretroviral therapy. Blood 103:966–972 [DOI] [PubMed] [Google Scholar]
- 72. Mooij P, Nieuwenhuis IG, Knoop CJ, Doms RW, Bogers WMJM, ten Haaft PJF, Niphuis H, Koornstra W, Bieler K, Kostler J, Morein B, Cafaro A, Ensoli B, Wagner R, Heeney JL. 2004. Qualitative T-helper responses to multiple viral antigens correlate with vaccine-induced immunity to simian/human immunodeficiency virus infection. J. Virol. 78:3333–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Almeida JR, Price DA, Papagno L, Arkoub ZA, Sauce D, Bornstein E, Asher TE, Samri A, Schnuriger A, Theodorou I, Costagliola D, Rouzioux C, Agut H, Marcelin AG, Douek D, Autran B, Appay V. 2007. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 204:2473–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ferrando-Martínez S, Casazza JP, Leal M, Machmach K, Munoz-Fernandez MA, Viciana P, Koup RA, Ruiz-Mateos E. 2012. Differential Gag-specific polyfunctional T cell maturation patterns in HIV-1 elite controllers. J. Virol. 86:3667–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ferre AL, Hunt PW, Critchfield JW, Young DH, Morris MM, Garcia JC, Pollard RB, Yee HF, Jr, Martin JN, Deeks SG, Shacklett BL. 2009. Mucosal immune responses to HIV-1 in elite controllers: a potential correlate of immune control. Blood 113:3978–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ferre AL, Hunt PW, McConnell DH, Morris MM, Garcia JC, Pollard RB, Yee HF, Jr, Martin JN, Deeks SG, Shacklett BL. 2010. HIV controllers with HLA-DRB1*13 and HLA-DQB1*06 alleles have strong, polyfunctional mucosal CD4+ T-cell responses. J. Virol. 84:11020–11029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kannanganat S, Kapogiannis BG, Ibegbu C, Chennareddi L, Goepfert P, Robinson HL, Lennox J, Amara RR. 2007. Human immunodeficiency virus type 1 controllers but not noncontrollers maintain CD4 T cells coexpressing three cytokines. J. Virol. 81:12071–12076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lanzavecchia A, Sallusto F. 2005. Understanding the generation and function of memory T cell subsets. Curr. Opin. Immunol. 17:326–332 [DOI] [PubMed] [Google Scholar]
- 80. Potter SJ, Lacabaratz C, Lambotte O, Perez-Patrigeon S, Vingert B, Sinet M, Colle JH, Urrutia A, Scott-Algara D, Boufassa F, Delfraissy JF, Theze J, Venet A, Chakrabarti LA. 2007. Preserved central memory and activated effector memory CD4+ T-cell subsets in human immunodeficiency virus controllers: an ANRS EP36 study. J. Virol. 81:13904–13915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Burgers WA, Riou C, Mlotshwa M, Maenetje P, Rosa DD, Brenchley J, Mlisana K, Douek DC, Koup R, Roederer M, de Bruyn G, Karim SA, Williamson C, Gray CM. 2009. Association of HIV-specific and total CD8(+) T memory phenotypes in subtype C HIV-1 infection with viral set point. J. Immunol. 182:4751–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brandt L, Cunha JF, Olsen AW, Chilima B, Hirsch P, Appelberg R, Andersen P. 2002. Failure of the Mycobacterium bovis BCG vaccine: some species of environmental mycobacteria block multiplication of BCG and induction of protective immunity to tuberculosis. Infect. Immun. 70:672–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gheorghiu M, Lagranderie MR, Gicquel BM, Leclerc CD. 1994. Mycobacterium bovis BCG priming induces a strong potentiation of the antibody response induced by recombinant BCG expressing a foreign antigen. Infect. Immun. 62:4287–4295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Kameoka M, Nishino Y, Matsuo K, Ohara N, Kimura T, Yamazaki A, Yamada T, Ikuta K. 1994. Cytotoxic T lymphocyte response in mice induced by a recombinant BCG vaccination which produces an extracellular alpha antigen that fused with the human immunodeficiency virus type 1 envelope immunodominant domain in the V3 loop. Vaccine 12:153–158 [DOI] [PubMed] [Google Scholar]