

Abstract
Preparations of parainfluenza virus 5 (PIV5) that are potent activators of the interferon (IFN) induction cascade were generated by high-multiplicity passage in order to accumulate defective interfering virus genomes (DIs). Nucleocapsid RNA from these virus preparations was extracted and subjected to deep sequencing. Sequencing data were analyzed using methods designed to detect internal deletion and “copyback” DIs in order to identify and characterize the different DIs present and to approximately quantify the ratio of defective to nondefective genomes. Trailer copybacks dominated the DI populations in IFN-inducing preparations of both the PIV5 wild type (wt) and PIV5-VΔC (a recombinant virus that does not encode a functional V protein). Although the PIV5 V protein is an efficient inhibitor of the IFN induction cascade, we show that nondefective PIV5 wt is unable to prevent activation of the IFN response by coinfecting copyback DIs due to the interfering effects of copyback DIs on nondefective virus protein expression. As a result, copyback DIs are able to very rapidly activate the IFN induction cascade prior to the expression of detectable levels of V protein by coinfecting nondefective virus.
INTRODUCTION
The interferon (IFN) response is extremely potent at restricting virus replication and spread prior to activation of the adaptive immune system. IFN-α and -β are synthesized and secreted from cells in response to virus infection, and this leads to the establishment of an antiviral state in the infected cell and neighboring uninfected cells through the upregulation of hundreds of IFN-stimulated genes (ISGs) that together function to make the cell a hostile environment for virus replication. Triggering of the IFN-β promoter during infection with paramyxoviruses and other negative-sense viruses occurs through activation of cytosolic pattern recognition receptors (PRRs). The best characterized of these are RIG-I and mda-5, which become activated upon binding of viral pathogen-associated molecular patterns (PAMPs). The viral PAMPs that activate RIG-I and mda-5 are viral RNA molecules; RIG-I is thought to recognize primarily short double-stranded RNAs (dsRNAs) with an uncapped triphosphate moiety, while mda-5 is activated by longer dsRNAs (1–6). Following their activation by PAMP binding, RIG-I and mda-5 elicit a downstream signaling cascade that culminates in the nuclear translocation of the IFN regulatory factor 3 (IRF3) and NF-κB transcription factors and subsequent transcription from the IFN-β gene. In order to circumvent the powerful IFN response, most viruses have evolved mechanisms to evade it, by encoding viral factors that inhibit IFN induction, the ability of IFN to upregulate ISGs, or the function of certain ISG products (reviewed in reference 7).
Parainfluenza virus 5 (PIV5; formerly simian virus 5 [SV5]) is a prototype member of the genus Rubulavirus in the Paramyxoviridae family. The ∼15-kb negative-sense genomic RNA encodes eight gene products from its seven genes and carries noncoding leader (Le) and trailer (Tr) sequences at its 3′ and 5′ ends, respectively, that are essential for the control of transcription and replication. Early in infection, the 3′ genomic promoter directs synthesis of both viral mRNAs and antigenomes (the full-length complement of the genome) using genomic RNA as a template. Antigenomes serve as a template for the generation of progeny genomic RNA from the antigenomic (Tr′) promoter later in infection. The antigenomic promoter is thought to be stronger than the genomic promoter, reflecting the requirement of the virus to generate greater numbers of genomes than antigenomes (8).
Paramyxoviruses are known to spontaneously generate defective interfering virus genomes (DIs) due to errors during replication. These DIs are subgenomic and contain deletions (often extensive) that render the virus unable to complete a full replication cycle in the absence of a coinfecting, nondefective “helper” virus. Paramyxovirus DIs may be internal deletion or “copyback” in nature, and these two types of DIs differ considerably in their genome structures. Internal deletion DIs retain the Le and Tr sequences of the genome and therefore possess transcription and replication signals and have been shown to generate viral translation products (9, 10). In contrast, the 3′ genomic promoter in trailer copyback DI [DI(TrCB)] genomes has been replaced by a sequence complementary to the 5′ antigenomic promoter due to template switching from the antigenome to the nascent strand during synthesis of genomic RNA; the termini of DI(TrCB)s are thus complementary and form a dsRNA stem-loop structure when SDS treatment is used to dissociate the RNA genomes from encapsidating NP protein (11). This structure is thought to be responsible for the ability of DI(TrCB)s to act as potent inducers of IFN (12–15). DIs also efficiently inhibit the replication of nondefective genomes due to the replicative advantage conferred by their smaller size or through successful competition for viral or host factors that are required for genome replication (reviewed in reference 16). The substitution of the weak genomic promoter for the stronger antigenomic promoter in DI(TrCB) genomes additionally confers a significant replicative advantage over nondefective virus genomes, and this leads to their accumulation in virus stocks that are generated at high multiplicity.
To enable examination of IFN-β promoter activation in individual infected cells, we have generated an A549 cell line, A549/pr.IFN-β.GFP, which expresses green fluorescent protein (GFP) under the control of the IFN-β promoter and have shown that these cells faithfully report activation of the IFN induction cascade (14, 17). Following infection of these cells with a range of negative-sense RNA viruses (including PIV5, mumps virus, and influenza A virus), only a small minority of infected cells ever expressed GFP, indicating “heterocellular” activation of the IFN-β promoter (17). PIV5 encodes a potent IFN antagonist, the V protein, which targets the IFN induction cascade at multiple levels through inhibition of mda-5 and RIG-I activity and by acting as an alternative substrate for the IRF3 kinases TBK1 and IκB kinase ε (IKKε) (18–21). However, PIV5-VΔC, a recombinant PIV5 that does not express a functional IFN antagonist due to an extensive deletion in the V gene (22), also failed to activate the IFN-β promoter in the majority of infected cells (14). This led us to suggest that the viral PAMPs capable of activating the IFN induction cascade are not usually generated during the normal replication cycle of PIV5. Instead, activation of the IFN induction cascade appears to be associated primarily with the presence of DIs, since infection with DI-rich preparations of mumps virus or PIV5-VΔC rich in DIs could efficiently activate the IFN-β promoter in the majority of cells (14, 17). In this study, we generated preparations of PIV5-VΔC and the PIV5 wild type (wt) that are either poor or efficient inducers of IFN and subsequently used extensive deep sequencing analyses to identify and characterize the DIs present in these preparations.
MATERIALS AND METHODS
Cells, viruses, and IFN.
Vero cells, A549 cells, and their derivatives were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37°C. PIV5 (strain W3A [23]) and PIV5-VΔC (22) were grown and titrated under appropriate conditions in Vero cells. Virus infections were carried out in DMEM supplemented with 2% fetal bovine serum. The construction and properties of the A549/pr(IFN-β).GFP cell line have been described extensively (14, 17). IFN-α (Roferon A; Roche) was used at 1,000 IU/ml.
Generation of DI-rich virus preparations.
DI virus-rich stocks of PIV5-VΔC and PIV5 wt were generated essentially as described previously (14). Briefly, Vero cells grown in 75-cm2 flasks were infected at a multiplicity of infection (MOI) of 5 PFU/cell with working stocks (vM0). The culture medium was harvested every 2 to 3 days: half was frozen at −70°C for subsequent analysis, while the other half was used to infect another 75-cm2 flask. Sequential preparations of these stocks are referred to as vM1, vM2, etc.
Immunofluorescence, immunoblotting, immunoprecipitation, and FACS analysis.
The procedures for immunoblotting, immunofluorescence, and immunoprecipitation have been described previously (18, 24). Antibodies used in these procedures included monoclonal antibodies against the PIV5 NP, P, M, HN, and L proteins (25), PIV5 V (raised against the C terminus; a kind gift from R. Lamb), phospho-IRF3 (Cell Signaling Technology), GFP (Roche), and β-actin (Sigma). The polyclonal antibodies used included those raised against ISG56 (Santa Cruz) and MxA (Santa Cruz). Immunofluorescence was examined using a Nikon Microphot-FXA immunofluorescence microscope or a Zeiss LSM 5 Exciter confocal microscope. For fluorescence-activated cell sorter (FACS) analysis, A549/pr(IFN-β).GFP cells were trypsinized to obtain a single-cell suspension and fixed in phosphate-buffered saline (PBS)–5% formaldehyde. Cells were then permeabilized and stained with anti-NP antibody and phycoerythrin-conjugated secondary antibody to determine PIV5 protein expression. Expression of GFP and NP was examined using a BD FACScan flow cytometer.
Interferon assays.
The amount of IFN secreted by cells was estimated by a cytopathic effect (CPE) reduction bioassay. Briefly, culture supernatants were harvested from infected cell monolayers, centrifuged at 1,500 × g for 10 min to pellet cellular debris, UV treated to inactivate residual virus, and then serially diluted 2-fold and added to monolayers of cells of the bovine diarrhea virus NPro-expressing cell line A549/BVDV-NPro (26) for 18 h prior to infection with encephalomyocarditis virus (EMCV) (0.05 PFU/cell). Monolayers were fixed 2 to 3 days postinfection (p.i.) (with PBS–5% formaldehyde), and CPE was monitored by staining with 0.1% crystal violet.
Isolation of RNA from purified RNPs and deep sequencing.
Viral RNPs were purified using an adaptation of a published protocol adapted from reference 27. Following high-multiplicity passage of virus in 300-cm2 flasks, culture supernatants were harvested from infected cell monolayers and used in assays to test activation of the IFN response. Infected cells were harvested from culture flasks using glass beads, washed with PBS, and pelleted at 700 × g for 10 min. The pellet was resuspended in ice-cold lysis buffer (150 mM NaCl, 50 mM Tris-HCl [pH 7.5], 0.6% NP-40 supplemented with protease inhibitor tablets [Roche]), incubated for 5 min, and then centrifuged at 4,200 × g for 5 min at 4°C. The supernatant was collected and held on ice, and the EDTA concentration was adjusted to 6 mM EDTA. Linear 30 to 35% cesium chloride gradients were prepared, overlaid with the cell extracts, and centrifuged overnight at 175,000 × g at 12°C. Nucleocapsid bands were harvested, and the associated RNA was isolated using TRIzol (Invitrogen) according to the manufacturer's instructions. Viral RNA isolated from PIV5 RNPs was sequenced in the Glasgow Polyomics Facility, University of Glasgow, using an Illumina GA2x platform. Reads were aligned to PIV5-VΔC and wild-type PIV5 (as appropriate to the source of the sample) using BWA (28) and visualized using Tablet (29).
Analysis of PIV5 DI genomes.
Illumina sequence reads containing DI breakpoints were detected and processed using a variant of the method used previously to detect splice junctions (30). The following steps were performed entirely using custom-made Perl scripts.
Detection of candidate reads spanning breakpoints.
Each FASTQ-formatted read was converted to FASTA format. The read was then aligned with the reference genome sequence using a regular-expression-based (regexp) method. Candidates for reads spanning a breakpoint were required to match two parts of the reference genome on either strand, with precise matching extending in each part for at least 21 nucleotides (nt). Four kinds of breakpoint were identified: (i) left copyback (DI), in which the left part of the read maps to the top strand of the reference genome and the right end maps to the complementary strand; (ii) right copyback [DI(TrCB)], which is the converse of left copyback; (iii) internal deletions, in which the left and right parts map to the same strand and are separated by at least 1 nt; and (iv) duplications, in which the left and right parts map to the same strand but the former is further downstream than the right. On the identification of a candidate breakpoint, the 17 nt on either side of the breakpoint were selected from the reference genome to make a 34-mer representing that particular break. This value was chosen on the basis that 17 is the shortest uniquely occurring sequence in the PIV5 genome (defined using the application Repeat from the GCG v.11.1-UNIX suite [31]). At the same time, a 112-mer was constructed by selecting 56 nt on either side of the break. The process outlined above was performed three times: on the original FASTQ-formatted reads and on the same reads trimmed at the 3′ end by 10 nt and then 15 nt. Since read-calling confidence decreased toward the 3′ end of each read, use of these trimmed sets found extra breakpoints that were not found by using the full-length reads.
Collection of candidate breakpoints.
After all candidate breakpoints had been collected, these were trimmed for redundancy, leaving one unique 34-mer and its corresponding 112-mer for each. The original FASTQ read file was then subsampled to retrieve all reads containing each of the 34-mers, outputting a separate file for each candidate breakpoint. This step was performed on 206-node CentOS compute cluster. Each subsampled FASTQ file was aligned to its corresponding 112-mer sequence using Maq (32). This step enabled quantification of the number of high-quality read matches to each candidate breakpoint, and the choice of a 112-mer sequence for matching ensured that the sequences flanking the breakpoint were unique in the reference genome. Note that any breakpoint occurring within 56 nt of the genome ends would not have been identified.
RESULTS
Generation of DI-rich PIV5 preparations.
Our previous work demonstrated that the DI content and IFN-inducing capacity of a PIV5-VΔC preparation could be significantly increased by sequential high-multiplicity passage of the virus in Vero cells (which do not produce IFN [33] and can therefore be used to propagate both PIV5 wt and PIV5-VΔC). Thus, while a PIV5-VΔC preparation that had been generated by low-multiplicity passage so as to limit the propagation of DIs (termed vM0) activated the IFN-β promoter in only 8% of infected A549/prIFN-β.GFP reporter cells, infection with a stock that had been generated by two high-multiplicity passages (vM2) lead to the expression of GFP in over 80% of cells (Fig. 1A), consistent with an increase in other markers of activation of the IFN response, including IFN secretion, IRF3 phosphorylation, and expression of ISGs (14).
Fig 1.

Activation of the IFN response by PIV5 preparations generated by high-multiplicity passage. A549/pr(IFN-β).GFP reporter cells were infected with equivalent dilutions of PIV5-VΔC vM0 or vM2 (A) or (B) PIV5 wt vM0, vM3, vM6, vM9, or vM12 (B). Sixteen hours later, the culture media were harvested and the cells were fixed and immunostained for viral NP expression. Expression of GFP and virus proteins was analyzed by fluorescence microscopy (left panel). Multiplicity (PFU/cell) for each virus is indicated by the number shown in the NP panel. The amount of IFN present in the culture media was estimated by cell-based CPE reduction assay and is shown by the number in the GFP panel. Duplicate monolayers were trypsinized, fixed, and subjected to FACS analysis of GFP expression (right panel). n.d., not determined. (C) A549 cells were left uninfected (UI) or were infected with an equivalent multiplicity of PIV5 wt vM0, vM6, or vM12. Cell lysates were prepared 24 h later and subjected to SDS-PAGE and immunoblotting for phospho-IRF3 (p-IRF3), GFP, ISG56, MxA, viral NP, and actin.
Similarly, we generated preparations of PIV5 wt, which encodes a functional IFN antagonist, by high-multiplicity passages in Vero cells and examined these virus stocks for their ability to activate the IFN response. IFN secretion from infected cells and GFP expression following infection of A549/pr(IFN-β).GFP cells with PIV5 wt vM0, vM3, vM6, vM9, and vM12 preparations are shown in Fig. 1B. Both IFN secretion from infected cells and GFP expression steadily increased with the number of high-multiplicity passages until passages 9 to 12. Thus, monolayers infected with PIV5 wt vM6 and vM12 secreted 128 and 512 relative units of IFN, respectively, compared to 8 units for the vM0 preparation. The increase in GFP expression with the number of high-multiplicity passages could clearly be seen by both fluorescence microscopy and FACS, which demonstrated that 87.9% of cells infected with vM12 were positive for GFP compared to 2.81% for vM0. When analyzed by immunoblotting, PIV5 wt vM6- and vM12-infected cell lysates also exhibited considerably more IRF3 phosphorylation and GFP, ISG56, and MxA expression (the relevance of which is discussed further below) than their vM0 counterpart (Fig. 1C). Viruses that had been plaque purified from DI-rich PIV5 preparations and then amplified by low-MOI passage were comparable with vM0 stocks in their ability to activate the IFN response, indicating that it was the accumulation of DI viruses that led to an increase in IFN induction and not any genetic change in the properties of the nondefective virus that may be associated with high-MOI passage (data not shown). Thus, as we have shown previously for PIV5-VΔC (14), there is a clear correlation between activation of the IFN-β promoter and the presence of DIs in PIV5 wt preparations. Through the depletion or enrichment of DIs, it is therefore possible to generate stocks of PIV5 wt or PIV5-VΔC that are either poor or good inducers of the IFN-β promoter.
Sequence analysis of DI-rich PIV5 preparations.
We next performed deep sequencing analysis of selected virus preparations. Viral RNPs were isolated from infected cells, and the associated RNA was sequenced on an Illumina GA2x platform, giving sequencing reads 72 nt in length. Each read was mapped to the PIV5-VΔC or PIV5 wt genome as appropriate. The frequencies of reads at each nucleotide in the entire PIV5 genome for PIV5-VΔC (vM0 and vM2 preparations) and the PIV5 wt (vM0 and vM12 preparations) are shown in Fig. 2A and B, respectively. For the vM0 preparations of both viruses, approximately even coverage was obtained across the whole genome, with averages of 105,068 reads per nt for PIV5 wt and 134,118 reads per nt for PIV5-VΔC (equivalent to averages of 3,889 and 4,051 reads per base per million reads for PIV5 wt and PIV5-VΔC, respectively). For the IFN-inducing PIV5 preparations, PIV5-VΔC vM2 and PIV5 wt vM12, however, the overwhelming majority of sequence reads mapped to the 5′ end of the genome, indicating an abundance of DI(TrCB) genomes. A rough estimation of the number of DI(TrCB)s to nondefective genomes was obtained by dividing the average number of reads per nt in the appropriate genome region (from nt 14000 to the 5′ end of the genome for PIV5-VΔC vM2 and from nt 14500 to the 5′ end of the genome for PIV5 wt vM12) by the average reads per nucleotide in the genome prior to this point (e.g., nt 1 to 13999 for PIV5-VΔC and nt 1 to 14499 for PIV5 wt). Thus, there were approximately 19 DI(TrCB)s per nondefective genome for PIV5-VΔC vM2 and 59 for PIV5 wt vM12. These figures are likely to be an underestimate due to the low coverage at the extremities of the genome that is inherent in the sequencing process.
Fig 2.

Deep sequencing analyses of RNA isolated from PIV5-infected cells. Viral RNPs were extracted from Vero cells infected with PIV5-VΔC (A) or PIV5 wt (B). Associated RNA was subjected to deep sequencing using the Illumina GA2x platform, and sequencing reads were mapped to PIV5-VΔC (A) or PIV5 wt (B) reference genomes. The frequency of reads at each nucleotide is shown in red for vM0 virus preparations and in black for the DI-rich virus preparations PIV5-VΔC vM2 (A) and PIV5 wt vM12 (B). Coverage from nt 14000 to 15246 is shown as an inset in order to highlight the peaks at the 5′ end of the genome.
Detailed analysis of DI genomes in DI-rich PIV5 preparations.
The mapping of sequence reads to a reference genome does not permit identification of the location of the breakpoints that gave rise to copyback or internal deletion DIs. To study the variety of DI species present in the genome population and their relative frequency in the DI population, the deep sequencing data were analyzed using methods specifically designed to identify the breakpoints that are associated with trailer copyback [DI(TrCB)], leader copyback [DI(LeCB)], and internal deletion DIs. While these analyses demonstrated an overwhelming enrichment of DI(TrCB)s in PIV5 wt vM12 and PIV5-VΔC vM2, consistent with the data presented in Fig. 2, we could also identify small numbers of potential internal deletion, duplication, and DI(LeCB) genomes. However, these were present at very low proportions in the DI population in the PIV5-VΔC vM2 and PIV5 wt vM12 preparations and, importantly, unlike DI(TrCB), their relative numbers did not increase upon high-multiplicity passage, indicating that they could not have contributed significantly to the IFN-inducing capacity of our DI-rich virus preparations. Furthermore, it is possible that at least some of the joins and deletions identified at low frequencies are an artifact of the sequencing techniques used, rather being derived from genuine DIs, and given their very low numbers, we have not yet attempted to validate their existence by other methods.
Analysis of the DI(TrCB) population indicated that, overall, reads mapping onto DI(TrCB)s were >22 and 87 times more abundant in PIV5-VΔC vM2 and PIV5 wt vM12, respectively, than in the original virus stocks. Several different DI(TrCB) species were detected in our virus preparations with various frequencies: the most abundant species for PIV5-VΔC and PIV5 wt are listed in Tables 1 and 2, respectively. For each unique DI(TrCB), the position of the breakpoint is listed along with the predicted total size of the DI, the length of the dsRNA stem-loop region, and the percentage of the total DI(TrCB) population that it comprises. These DI(TrCB)s varied considerably both in the total size of the genome (468 to 1,427 nt in length) and the length of the predicted dsRNA stem (89 to 412 nt). The points in the genome at which these DI(TrCB)s were generated did not fall into a particular region within the Tr, suggesting that no area was substantially more prone to template switching than others. The most prominent PIV5-VΔC DI(TrCB) species, which made up 62% of the total DI(TrCB) population in PIV5-VΔC vM2 (Table 1), was the one that we identified and cloned by reverse transcription (RT)-PCR previously (14). This DI(TrCB) was predicted to be 1,427 nt in length with a breakpoint such that either nt 14043 is transposed next to nt 15024 or nt 14044 is transposed to nt 15023 on the opposite strand, giving a predicted dsRNA stem of 223 bp. (A 2-nt redundancy makes it impossible to distinguish between these two possibilities.) However, the original cloned DI(TrCB) fragment we previously identified contained a single base insertion into the loop of the structure, thus generating a molecule of 1,428 nt which obeys the “rule of six” (34). Since we could not detect this inserted base in the bulk of sequences generated by deep sequencing, we repeated the DI cloning experiment and generated a further 14 clones. When these were sequenced, none of these had this base insert and only 2 clones had base inserts or deletions, but neither of these generated structures that obeyed the rule of six. The next most common DI(TrCB) species, comprising 15% of the population, was a much smaller DI, with a total length of 510 nt and a predicted dsRNA stem of 89 bp. RT-PCR and conventional sequencing also validated the presence of this particular DI(TrCB) in the preparation (data not shown). For PIV5 wt vM12, the most abundant DI(TrCB) [94% of the DI(TrCB) population] had nt 14496 transposed next to nt 15062 on the opposite strand, giving a total genome length of 936 nt and a predicted dsRNA stem of 184 bp (Table 2). The presence of both this DI(TrCB) and the second most abundant species was confirmed by RT-PCR, and the resulting PCR products were sequenced and found to be identical to those identified by deep sequencing (data not shown). An important observation from these analyses was that the most abundant DI(TrCB) species present in PIV5-VΔC vM2-infected cells could not be detected in PIV5 wt vM12-infected cells and vice versa.
Table 1.
Trailer copyback DIs in PIV5-VΔC vM2a
| Copyback junction (nt position) | DI genome size (nt) | Length of complementary region (bp) | Length of loop (nt) | Obeys rule of six? | % of DI(TrCB)s in vM2-infected cells |
|---|---|---|---|---|---|
| 14043/4-15023/4b | 1,427 | 223 | 981 | No (+5) | 62 |
| 14827-15157 | 510 | 89 | 332 | Yes | 15 |
| 14873-15153 | 468 | 93 | 282 | Yes | 5 |
| 14070-15153 | 1,271 | 93 | 1085 | No (+5) | 3 |
| 14338-15107 | 1,059 | 139 | 781 | No (+3) | 2 |
| Others | 13 |
DI(TrCB)s in PIV5-VΔC vM0 versus vM2, 1:22.
Our original description of this DI of nt 14043 transposed next to nt 15023 (14) was incorrect due to a data processing error.
Table 2.
Trailer copyback DIs in PIV5 wt vM12a
| Copyback junction (nt position) | DI genome size (nt) | Length of complementary region (bp) | Length of loop (nt) | Obeys rule of six? | % of DI(TrCB)s in vM12-infected cells |
|---|---|---|---|---|---|
| 14496-15062 | 936 | 184 | 568 | Yes | 94 |
| 14380/1-15147/8 | 966 | 99 | 768 | Yes | 5 |
| 14510-14834 | 1,150 | 412 | 326 | No (+4) | 0.5 |
| Others | <0.5 |
DI(TrCB)s in PIV5 wt vM0 versus vM12, 1:87.
The effect of DI(TrCB)s on the IFN antagonistic properties of nondefective PIV5.
The data presented above clearly demonstrate that high-multiplicity passage of PIV5 wt generates virus preparations that are efficient at activating the IFN response and that this ability correlates with an accumulation of DI(TrCB) genomes. These preparations activated the IFN-β promoter in the majority of cells, even in infections at a high multiplicity with regard to infectious particles (i.e., nondefective virus) (Fig. 1B). As such, every cell should have been infected with nondefective virus that encodes a functional V protein, yet there was no inhibition of IFN induction in these cells. It was also clear from these infections that, in addition to robust activation of the IFN response, the expression of viral NP was considerably lower in cells infected with PIV5 wt vM12 than that in cells infected with PIV5 wt vM0 (Fig. 1B and C). This indicates that DIs inhibited the synthesis of viral proteins expressed from nondefective genomes, which would likely impact the ability of the virus to antagonize the IFN response. Consistent with this finding, “nondefective” PIV5 wt vM0 is unable to inhibit IFN induction by coinfecting DI-rich PIV5 wt vM12 (Fig. 3A) and PIV5-VΔC vM2 (Fig. 3B). This was not due to an inherent inability of the V protein to prevent IFN induction by DI-related PAMPs generated during these infections, since preinfection of A549/pr(IFN-β).GFP cells with PIV5 wt was associated with a significant reduction in GFP expression when subsequently challenged with PIV5-VΔC vM2, compared to the level in cells that had not been preinfected (Fig. 3C). Furthermore, we have shown previously that the transient expression of PIV5 V from a plasmid inhibits IFN induction by a DI-rich IFN-inducing stock of PIV5-VΔC (35).
Fig 3.

PIV5 wt does not prevent activation of the IFN-β promoter by coinfecting copyback DIs, despite encoding an efficient IFN antagonist. (A) A549/pr(IFN-β).GFP cells were infected at an MOI of 10 PFU/cell with PIV5 wt vM0, PIV5 wt vM12, or a 50:50 mixture thereof. The cells were fixed at 20 h p.i., and GFP-positive cells and the distribution of NP (red), following immunostaining, were visualized by fluorescence microscopy. (B) A549/pr(IFN-β).GFP cells were treated as described above. At 18 h p.i., the cells were trypsinized, fixed, and immunostained for viral NP. Expression of GFP and NP was analyzed by FACS. Uninfected cells were included as a negative control. (C) Uninfected (UI) A549/pr(IFN-β).GFP cells or cells that had been preinfected with PIV5 wt vM0 (at 5 PFU/cell) for 24 h were infected with PIV5-VΔC vM2. The cells were fixed at 16 h p.i., and GFP was visualized by fluorescence microscopy.
The observation that cells infected with DI-rich preparations of PIV5 wt expressed high levels of the IFN-upregulated protein MxA (Fig. 1C and Fig. 4A) suggested that coinfecting DI(TrCB)s had impaired another function of the V protein: the ability to target STAT1 for proteasome-mediated degradation to prevent ISG expression in response to IFN (36). Consistent with this finding, A549/pr(IFN-β).GFP cells that had been coinfected with PIV5 wt vM0 and PIV5-VΔC vM2 and then subsequently treated with IFN-α, expressed considerable amounts of MxA, indicating that nondefective PIV5 wt was no longer able to interfere with STAT1 signaling (Fig. 4B). More specifically, cells that were GFP positive were also MxA positive and expressed very low levels of NP. This suggests that DI(TrCB)s had limited nondefective PIV5 replication and protein expression to the extent that the virus was unable to antagonize either the IFN induction or IFN signaling cascades.
Fig 4.

Copyback DIs impair the ability of PIV5 to limit IFN signaling. (A) Monolayers of A549/pr(IFN-β).GFP cells were infected with 1 PFU/cell of PIV5 wt vM0 or vM9. Cells were fixed at 18 h p.i. and immunostained for MxA and virus NP expression. GFP (green), MxA (blue), and NP (red) were visualized by confocal microscopy. (B) A549/pr(IFN-β).GFP cells were infected with 20 PFU/cell of either PIV5 wt vM0, PIV5-VΔC vM2, or a mixture of PIV5 wt vM0 and PIV5-VΔC vM2. At 8 h p.i., IFN-α (1,000 IU/ml) was added to the culture medium and the cells were fixed at 24 h p.i. Following immunostaining, GFP (green), MxA (blue), and NP (red) were visualized by confocal microscopy.
Given that STAT1 targeting occurs by a catalytic mechanism (37), whereas V-mediated inhibition of IFN induction occurs either through direct binding to mda-5 (19) or by indirectly targeting RIG-I through binding to LGP2 (21), the amount of V required to inhibit IFN signaling is much lower than that required to inhibit IFN induction. Consistent with this, cells stably expressing low levels of PIV5 V are completely STAT1 deficient and unable to respond to IFN (see below), yet are still able to produce IFN (data not shown). The inability of nondefective virus to inhibit STAT1 signaling in cells infected with DI(TrCB)s therefore suggests that the level of V in these cells is very low. Indeed, expression of V over an infection time course is severely impaired in cells coinfected with PIV5 wt vM0 and PIV5-VΔC vM2, compared to V expression during infection with PIV5 wt vM0 alone (Fig. 5). While V was detected from 6 h postinfection (p.i.) in cells infected with PIV5 wt vM0, during a coinfection full-length V was only detectable in very small amounts from 12 h p.i. In contrast, the IFN induction cascade was activated very rapidly in coinfected cells, since the active, phosphorylated form of IRF3 could be detected from as early as 2 h p.i. Thus, by the time detectable amounts of the V protein were expressed in these cells, IRF3 had already been strongly activated by the PIV5-VΔC DI(TrCB)s and GFP (and therefore IFN-β) had already been expressed.
Fig 5.
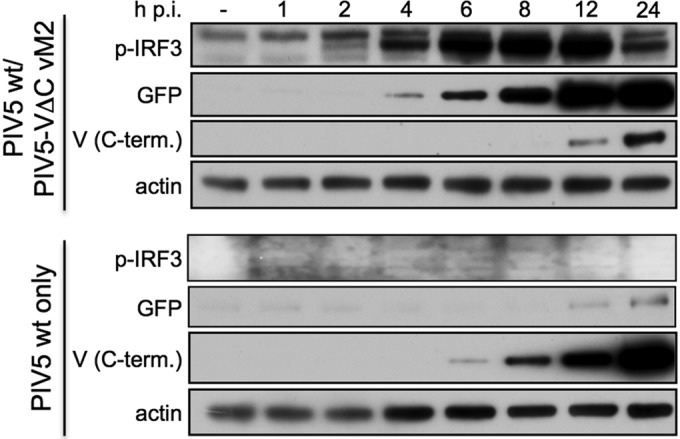
Coinfecting copyback DIs limit expression of the V protein by nondefective PIV5. Replicates of A549/pr(IFN-β).GFP cells were infected with PIV5 wt vM0 and PIV5-VΔC vM2 (5 PFU/cell of each virus; top panel) or PIV5 wt vM0 alone (5 PFU/cell; bottom panel). Cell lysates were prepared at various times p.i. (as indicated) and subjected to immunoblotting for phospho-IRF3 (p-IRF3), GFP, PIV5 V (C terminus [C-term.]), and actin.
What was unclear from the data presented in Fig. 5 was whether the rapid activation of antiviral responses by DI(TrCB)s was itself responsible for the inhibition of nondefective virus protein synthesis, by upregulating an ISG product that is inhibiting nondefective virus replication. In this regard, we have recently shown that ISG56 is the predominant ISG responsible for the inhibitory effect of IFN on PIV5 replication (38). To elucidate the role of the antiviral response in inhibition of nondefective virus replication by DI(TrCB)s, we examined the effects of DI(TrCB)s on virus replication in cells with impaired antiviral responses (Fig. 6). A549/V cells expressing the V protein of PIV5 are STAT1 deficient and thus unable to respond to IFN (36), while A549/NPro cells expressing NPro of BVDV, rendering them IRF3 deficient, are thus unable to mount IRF3-dependent antiviral responses, such as the production of IFN-β or ISG56 (26) (Fig. 6A). Consistent with our previous results, nondefective virus protein synthesis is markedly reduced in naïve A549 cells infected with the DI(TrCB)-rich PIV5-VΔC vM2 stock, with synthesis of L and M impaired to such an extent that they are undetectable even following immunoprecipitation (Fig. 6B and C). In A549/V cells, levels of viral NP and P proteins were marginally increased compared to A549 cells, while expression of NPro increased viral protein synthesis slightly more, as indicated by an increase in NP, P, L, and M expression; however, nondefective virus replication in these cell lines was still a fraction of the level of replication that occurs in the absence of DI(TrCB)s. Thus, while impairment of either IFN- or IRF3-dependent antiviral responses slightly relieved the inhibitory effect of DI(TrCB)s on nondefective viral protein synthesis, inhibition of nondefective virus replication by DI(TrCB)s was largely independent of innate antiviral responses. Taken together, these data are consistent with a scenario in which DI(TrCB)s simultaneously and independently both activate the IFN induction cascade and inhibit nondefective virus replication. The outcome of this is that the rapid activation of the IFN induction cascade by DI(TrCB)s occurs prior to the expression of detectable levels of V by coinfecting nondefective virus: i.e., insufficient V protein is expressed early enough during these infections to limit IFN induction by coinfecting DI(TrCB)s.
Fig 6.

Inhibition of nondefective virus replication by copyback DIs is independent of the antiviral response. (A) A549, A549/V, and A549/NPro cells were treated with IFN-α (1,000 IU/ml), infected with PIV5-VΔC vM2, or left untreated. Cell lysates were prepared after 16 h and subjected to immunoblotting for IRF3, ISG56, STAT1, PIV5 P, and actin. (B) A549 (-), A549/NPro (N), and A549/V (V) monolayers were infected at an MOI of 5 PFU/cell with PIV5 wt vM0, PIV5-VΔC vM0, or PIV5-VΔC vM2 or were uninfected. At 20 h p.i., the monolayers were radioactively labeled with [35S]methionine for 1 h. Labeled polypeptides in total cell extracts were immunoprecipitated using monoclonal antibodies to viral NP, P, M, HN, and L proteins and visualized by SDS-PAGE analysis and autoradiography. (C) Autoradiogram of total cell extracts used in panel B. (D) Coomassie staining of total cell extracts used in panel B. The positions of the viral proteins are indicated.
DISCUSSION
In this study, we have extended our previous work on the role of DIs in IFN induction by PIV5, utilizing powerful deep sequencing methods to examine DIs in virus preparations. To our knowledge, this study is the first to analyze deep sequencing data using programs designed specifically to identify the sites of DI(TrCB)s, DI(LeCB)s, internal deletions, and duplications. This allowed us to conduct an extensive analysis of the types of DIs present in virus stocks, the points in the genome at which these errors were generated, and the frequency with which they occurred. Furthermore, we used these techniques to compare the variation of virus genomes from virus stocks that are efficient activators of the IFN response with those that are not. We demonstrated that PIV5 preparations generated by high-multiplicity passage are very efficient at activating the IFN response, regardless of whether the parental virus encodes a functional IFN antagonist or not. Deep sequencing of viral genomes revealed a correlation between the IFN-inducing capacity of a virus stock and the presence of high numbers of DI(TrCB)s, consistent with previous studies indicating a role for paramyxovirus DI(TrCB)s in IFN induction (12–15, 39). We identified a range of distinct DI(TrCB)s, and these varied considerably in the site of the copyback error, the length of the predicted dsRNA stem, and the size of the DI genome. Furthermore, no major DI(TrCB) species were detected that were present in both PIV5 wt and PIV5-VΔC DI-rich preparations. The lack of conserved copyback points suggested that there is no particular part of the Tr in which a template switching error is substantially more likely to occur. This is consistent with a previous study in which a range of DI(TrCB)s generated during undiluted passage of PIV5 W3 were cloned and sequenced by conventional methods (34); these DIs also lacked conservation in the transposition points, the sizes of the genomes, and the lengths of the dsRNA stems. None of the DI(TrCB)s identified in the study by Murphy et al. (34) were found in our sequencing analysis, despite using the same virus strain and the same cell line for high-multiplicity passage. Several of the DI(TrCB)s identified in this previous study contained identical copyback error sites, yet differed in their total genome length, indicating additional internal deletion errors. The nature of deep sequencing does not permit identification of these types of DI(TrCB)s, although we have not yet observed any such DIs by cloning and conventional sequencing. Given the variation in the lengths of the dsRNA stem among DI(TrCB)s identified here and previously, it is highly likely that these DIs will differ in their ability to activate the IFN response since the ability of dsRNA to activate RIG-I and mda-5 is dependent on its length (6).
Our analyses also identified small populations of potential DI(LeCB), internal deletion, and duplication DIs in our virus preparations. It is possible that some of these are artifacts generated during the RT-PCR step of the deep sequencing analysis due to template switching by the reverse transcriptase (40, 41). We are therefore currently attempting to validate the existence of these DIs by other methods. However, it is clear that even when present in low numbers, these DIs could not have contributed significantly to the IFN-inducing capacity of the virus preparations since they were no more prevalent in virus stocks that were efficient IFN inducers than in the original vM0 stocks. Naturally occurring internal deletion mutants of Sendai virus have been identified previously (9, 42, 43), and consistent with our observations, Strahle and colleagues have shown that a virus stock composed predominantly of internal deletion species was a poorer inducer of IFN than a stock exclusively containing DI(TrCB)s (12).
It took significantly more passages (between 9 and 12) to generate a stock that was efficient at inducing IFN for the PIV5 wt than it did for PIV5-VΔC (which required only 2 passages). The reasons for this are unclear but are under investigation. The V protein itself may play a role in regulating DI generation, leading to PIV5-VΔC accumulating DIs at a higher rate than PIV5 wt. In this regard, V is known to be able to interact with RNA (44) and the viral NP and P proteins (45). In addition, V has been reported to regulate PIV5 transcription and replication (46) and to limit the synthesis of aberrant RNAs that may otherwise activate antiviral responses (47, 48). V may also affect the properties of the DIs generated, since the major DI(TrCB) identified in PIV5-VΔC preparations failed to obey the rule of six (dictating that genome lengths divisible by 6 are more efficiently replicated than those that are not [34]), whereas the major DI(TrCB) in the PIV5 wt vM12 preparation did obey this rule.
Although the V protein is able to inhibit IFN-β induction by PIV5 DI-derived PAMPs, it is only able to do so if present in sufficiently large amounts before DI virus PAMPs are detected. During our PIV5 wt vM12 infections, and during coinfections of PIV5 wt vM0 and DI-rich virus preparations, interference from coinfecting DI(TrCB)s had impaired the replication of nondefective PIV5 wt (and therefore V protein expression) to the extent that it failed to block the induction of IFN or IFN signaling by DI(TrCB)-related PAMPs. In cells in which DI(TrCB) are initially generated, however, the levels of V synthesized by nondefective virus prior to copyback generation (and therefore any interference with nondefective virus replication) are likely to be sufficient to prevent activation of the IFN induction cascade. We have previously presented compelling evidence that neither viral genome replication nor viral protein synthesis is required for IFN induction by DI-rich preparations of several paramyxoviruses (49). Thus, in this system, we suggest that the IFN induction cascade would normally be activated only if a nondefective virus fails to generate sufficient V protein quickly enough to block activation of the IFN induction cascade by coinfecting DI(TrCB) or in cells infected with DI(TrCB)s in the absence of a coinfecting nondefective virus.
ACKNOWLEDGMENTS
This work was supported by The Wellcome Trust (087751/A/08/Z). J.A.L.S. is indebted to the BBSRC for a Ph.D. studentship.
We thank Julie Galbraith and Pawel Herzyk of the University of Glasgow for performing the deep sequencing.
The University of St. Andrews is a charity registered in Scotland (no. SC013532).
Footnotes
Published ahead of print 28 February 2013
REFERENCES
- 1. Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, Reis e Sousa C. 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314:997–1001 [DOI] [PubMed] [Google Scholar]
- 2. Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. 2009. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J. Virol. 83:10761–10769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann KK, Schlee M, Endres S, Hartmann G. 2006. 5′-triphosphate RNA is the ligand for RIG-I. Science 314:994–997 [DOI] [PubMed] [Google Scholar]
- 4. Schlee M, Roth A, Hornung V, Hagmann CA, Wimmenauer V, Barchet W, Coch C, Janke M, Mihailovic A, Wardle G, Juranek S, Kato H, Kawai T, Poeck H, Fitzgerald KA, Takeuchi O, Akira S, Tuschl T, Latz E, Ludwig J, Hartmann G. 2009. Recognition of 5′ triphosphate by RIG-I helicase requires short blunt double-stranded RNA as contained in panhandle of negative-strand virus. Immunity 31:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmidt A, Schwerd T, Hamm W, Hellmuth JC, Cui S, Wenzel M, Hoffmann FS, Michallet MC, Besch R, Hopfner KP, Endres S, Rothenfusser S. 2009. 5′-triphosphate RNA requires base-paired structures to activate antiviral signaling via RIG-I. Proc. Natl. Acad. Sci. U. S. A. 106:12067–12072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S. 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 205:1601–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Randall RE, Goodbourn S. 2008. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 89:1–47 [DOI] [PubMed] [Google Scholar]
- 8. Le Mercier P, Garcin D, Hausmann S, Kolakofsky D. 2002. Ambisense Sendai viruses are inherently unstable but are useful to study viral RNA synthesis. J. Virol. 76:5492–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hsu CH, Re GG, Gupta KC, Portner A, Kingsbury DW. 1985. Expression of Sendai virus defective-interfering genomes with internal deletions. Virology 146:38–49 [DOI] [PubMed] [Google Scholar]
- 10. Re GG, Morgan EM, Kingsbury DW. 1985. Nucleotide sequences responsible for generation of internally deleted Sendai virus defective interfering genomes. Virology 146:27–37 [DOI] [PubMed] [Google Scholar]
- 11. Kolakofsky D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555 [DOI] [PubMed] [Google Scholar]
- 12. Strahle L, Garcin D, Kolakofsky D. 2006. Sendai virus defective-interfering genomes and the activation of interferon-beta. Virology 351:101–111 [DOI] [PubMed] [Google Scholar]
- 13. Baum A, Sachidanandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303–16308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Killip MJ, Young DF, Ross CS, Chen S, Goodbourn S, Randall RE. 2011. Failure to activate the IFN-beta promoter by a paramyxovirus lacking an interferon antagonist. Virology 415:39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shingai M, Ebihara T, Begum NA, Kato A, Honma T, Matsumoto K, Saito H, Ogura H, Matsumoto M, Seya T. 2007. Differential type I IFN-inducing abilities of wild-type versus vaccine strains of measles virus. J. Immunol. 179:6123–6133 [DOI] [PubMed] [Google Scholar]
- 16. Re GG. 1991. Deletion mutants of paramyxoviruses, p 275–298 In Kingsbury DW. (ed), The paramyxoviruses. Plenum Press, New York, NY [Google Scholar]
- 17. Chen S, Short JA, Young DF, Killip MJ, Schneider M, Goodbourn S, Randall RE. 2010. Heterocellular induction of interferon by negative-sense RNA viruses. Virology 407:247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. U. S. A. 101:17264–17269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Childs KS, Andrejeva J, Randall RE, Goodbourn S. 2009. Mechanism of mda-5 inhibition by paramyxovirus V proteins. J. Virol. 83:1465–1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu LL, Puri M, Horvath CM, Sen GC. 2008. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J. Biol. Chem. 283:14269–14276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Childs K, Randall R, Goodbourn S. 2012. Paramyxovirus V proteins interact with the RNA helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 86:3411–3421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. He B, Paterson RG, Stock N, Durbin JE, Durbin RK, Goodbourn S, Randall RE, Lamb RA. 2002. Recovery of paramyxovirus simian virus 5 with a V protein lacking the conserved cysteine-rich domain: the multifunctional V protein blocks both interferon-beta induction and interferon signaling. Virology 303:15–32 [DOI] [PubMed] [Google Scholar]
- 23. Choppin PW. 1964. Multiplication of a myxovirus (Sv5) with minimal cytopathic effects and without interference. Virology 23:224–233 [DOI] [PubMed] [Google Scholar]
- 24. Carlos TS, Fearns R, Randall RE. 2005. Interferon-induced alterations in the pattern of parainfluenza virus 5 transcription and protein synthesis and the induction of virus inclusion bodies. J. Virol. 79:14112–14121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Randall RE, Young DF, Goswami KK, Russell WC. 1987. Isolation and characterization of monoclonal antibodies to simian virus 5 and their use in revealing antigenic differences between human, canine and simian isolates. J. Gen. Virol. 68:2769–2780 [DOI] [PubMed] [Google Scholar]
- 26. Hilton L, Moganeradj K, Zhang G, Chen YH, Randall RE, McCauley JW, Goodbourn S. 2006. The NPro product of bovine viral diarrhea virus inhibits DNA binding by interferon regulatory factor 3 and targets it for proteasomal degradation. J. Virol. 80:11723–11732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leppert M, Rittenhouse L, Perrault J, Summers DF, Kolakofsky D. 1979. Plus and minus strand leader RNAs in negative strand virus-infected cells. Cell 18:735–747 [DOI] [PubMed] [Google Scholar]
- 28. Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. 2010. Tablet—next generation sequence assembly visualization. Bioinformatics 26:401–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gatherer D, Seirafian S, Cunningham C, Holton M, Dargan DJ, Baluchova K, Hector RD, Galbraith J, Herzyk P, Wilkinson GW, Davison AJ. 2011. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. U. S. A. 108:19755–19760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Devereux J, Haeberli P, Smithies O. 1984. A comprehensive set of sequence analysis programs for the VAX. Nucleic Acids Res. 12:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li H, Ruan J, Durbin R. 2008. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 18:1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 2:955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murphy SK, Parks GD. 1997. Genome nucleotide lengths that are divisible by six are not essential but enhance replication of defective interfering RNAs of the paramyxovirus simian virus 5. Virology 232:145–157 [DOI] [PubMed] [Google Scholar]
- 35. Poole E, He B, Lamb RA, Randall RE, Goodbourn S. 2002. The V proteins of simian virus 5 and other paramyxoviruses inhibit induction of interferon-beta. Virology 303:33–46 [DOI] [PubMed] [Google Scholar]
- 36. Didcock L, Young DF, Goodbourn S, Randall RE. 1999. The V protein of simian virus 5 inhibits interferon signalling by targeting STAT1 for proteasome-mediated degradation. J. Virol. 73:9928–9933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Precious BL, Carlos TS, Goodbourn S, Randall RE. 2007. Catalytic turnover of STAT1 allows PIV5 to dismantle the interferon-induced anti-viral state of cells. Virology 368:114–121 [DOI] [PubMed] [Google Scholar]
- 38. Andrejeva J, Norsted H, Habjan M, Thiel V, Goodbourn S, Randall RE. 2013. ISG56/IFIT1 is primarily responsible for interferon-induced changes to patterns of parainfluenza virus type 5 transcription and protein synthesis. J. Gen. Virol. 94:59–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Strahle L, Marq JB, Brini A, Hausmann S, Kolakofsky D, Garcin D. 2007. Activation of the beta interferon promoter by unnatural Sendai virus infection requires RIG-I and is inhibited by viral C proteins. J. Virol. 81:12227–12237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mader RM, Schmidt WM, Sedivy R, Rizovski B, Braun J, Kalipciyan M, Exner M, Steger GG, Mueller MW. 2001. Reverse transcriptase template switching during reverse transcriptase-polymerase chain reaction: artificial generation of deletions in ribonucleotide reductase mRNA. J. Lab. Clin. Med. 137:422–428 [DOI] [PubMed] [Google Scholar]
- 41. Cocquet J, Chong A, Zhang G, Veitia RA. 2006. Reverse transcriptase template switching and false alternative transcripts. Genomics 88:127–131 [DOI] [PubMed] [Google Scholar]
- 42. Engelhorn M, Stricker R, Roux L. 1993. Molecular cloning and characterization of a Sendai virus internal deletion defective RNA. J. Gen. Virol. 74:137–141 [DOI] [PubMed] [Google Scholar]
- 43. Amesse LS, Pridgen CL, Kingsbury DW. 1982. Sendai virus DI RNA species with conserved virus genome termini and extensive internal deletions. Virology 118:17–27 [DOI] [PubMed] [Google Scholar]
- 44. Lin GY, Paterson RG, Lamb RA. 1997. The RNA binding region of the paramyxovirus SV5 V and P proteins. Virology 238:460–469 [DOI] [PubMed] [Google Scholar]
- 45. Randall RE, Bermingham A. 1996. NP:P and NP:V interactions of the paramyxovirus simian virus 5 examined using a novel protein:protein capture assay. Virology 224:121–129 [DOI] [PubMed] [Google Scholar]
- 46. Lin Y, Horvath F, Aligo JA, Wilson R, He B. 2005. The role of simian virus 5 V protein on viral RNA synthesis. Virology 338:270–280 [DOI] [PubMed] [Google Scholar]
- 47. Gainey MD, Dillon PJ, Clark KM, Manuse MJ, Parks GD. 2008. Paramyxovirus-induced shutoff of host and viral protein synthesis: role of the P and V proteins in limiting PKR activation. J. Virol. 82:828–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dillon PJ, Parks GD. 2007. Role for the phosphoprotein P subunit of the paramyxovirus polymerase in limiting induction of host cell antiviral responses. J. Virol. 81:11116–11127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Killip MJ, Young DF, Precious BL, Goodbourn S, Randall RE. 2012. Activation of the beta interferon promoter by paramyxoviruses in the absence of virus protein synthesis. J. Gen. Virol. 93:299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]