

Abstract
Herpes simplex virus type 1 particles are multilayered structures with a DNA genome surrounded by a capsid, tegument, and envelope. While the protein content of mature virions is known, the sequence of addition of the tegument and the intracellular compartments where this occurs are intensely debated. To probe this process during the initial stages of egress, we used two approaches: an in vitro nuclear egress assay, which reconstitutes the exit of nuclear capsids to the cytoplasm, and a classical nuclear capsid sedimentation assay. As anticipated, in vitro cytoplasmic capsids did not harbor UL34, UL31, or viral glycoproteins but contained US3. In agreement with previous findings, both nuclear and in vitro capsids were positive for ICP0 and ICP4. Unexpectedly, nuclear C capsids and cytoplasmic capsids produced in vitro without any cytosolic viral proteins also scored positive for UL36 and UL37. Immunoelectron microscopy confirmed that these tegument proteins were closely associated with nuclear capsids. When cytosolic viral proteins were present in the in vitro assay, no additional tegument proteins were detected on the capsids. As previously reported, the tegument was sensitive to high-salt extraction but, surprisingly, was stabilized by exogenous proteins. Finally, some tegument proteins seemed partially lost during egress, while others possibly were added at multiple steps or modified along the way. Overall, an emerging picture hints at the early coating of capsids with up to 5 tegument proteins at the nuclear stage, the shedding of some viral proteins during nuclear egress, and the acquisition of others tegument proteins during reenvelopment.
INTRODUCTION
The herpes simplex virus type 1 (HSV-1) particles are composed of four layers, namely, an inner core containing the viral genome, a protein shell called the capsid, a multiprotein layer, termed the tegument, and an envelope derived from cellular membranes that contains viral glycoproteins. The most commonly accepted model for HSV-1 egress proposes that capsids are assembled in the nucleus, incorporate viral DNA, bud through the inner nuclear membrane, lose that first envelope by fusion with the outer nuclear membrane, and are released naked into the cytoplasm (1–3). The capsids are then reenveloped later on at an intracellular organelle, where they acquire their mature envelope (4).
While the site of capsid assembly and genome packaging is well established, the addition of the tegument onto the capsid has not been elucidated in detail. It is believed to occur sequentially during virus egress in distinct cellular compartments, including the nucleus, the cytoplasm, and the site of secondary envelopment (2, 3, 5–7). This complexity is likely due to three factors. First, at least 23 different viral tegument proteins may be present in mature extracellular virions (8). Second, the tegument is involved in many facets of the viral life cycle, including the migration of capsids on microtubules (9–14), the anchorage of the capsids to nuclear pores (15–20), the transactivation of viral genes (21), the modulation of host protein expression (22, 23), viral latency (24), and the regulation of the immune response (13, 25–27). Finally, many tegument proteins also interact with each other and/or with viral glycoproteins and accumulate at the trans-Golgi network (TGN), where they altogether delineate the likely final envelopment site (4, 5, 28).
The first interactions of newly assembled capsids with other viral proteins take place in the nucleus. There, UL31 interacts with the membrane-anchored UL34 protein, binds to the capsids, and facilitates capsid budding through the inner nuclear membrane (3, 29–38). Both subunits of the UL31/UL34 complex are substrates for the US3 viral kinase (36, 38–43). UL31, UL34, and US3 are all believed to interact with the so-called nuclear C capsids at the inner nuclear membrane to promote primary envelopment (3, 41). Interestingly, deletion mutants for UL31, UL34, and US3 behave differently, with the first two arresting capsids in the nucleus (44–48), while US3 deletion causes the accumulation of perinuclear virions (40, 41, 46). Moreover, all three proteins are present on perinuclear virions, but only US3 is found on cytosolic capsids and in mature extracellular virions (8, 32, 33, 41, 49, 50). US3 thus is one of the early tegument proteins recruited onto capsids, while the UL31 and UL34 viral proteins are quickly shed from the capsids. Similarly, both the UL41 and UL49 tegument proteins were identified in mature virions and perinuclear virions (8, 50, 51) and qualify as early tegument proteins.
Other tegument proteins also may interact with nuclear capsids, but this is controversial. On one hand, both the UL36 and UL37 tegument proteins partially localize to the nucleus (19, 52–55). In the case of pseudorabies virus (PRV), a truncated form of UL36 was reported to preferentially localize to the nucleus, while full-length UL36 mainly localizes into the cytoplasm (56). Immunoblot analyses of enriched nuclear capsids detected both tegument proteins in some studies but not others (11, 14, 57, 58). In addition, immunogold labeling failed to see UL36 on perinuclear PRV capsids (59, 60). Finally, various reports debate the nuclear or cytoplasmic accumulation of capsids upon deletion of UL36, raising questions about the role UL36 plays during nuclear egress (61–67). On the other hand, an increasing volume of evidence suggests ICP0 and ICP4 are present on mature virions, but the intracellular compartment where this interaction occurs is open for debate (8, 14, 68–78). Finally, whether any other tegument proteins also interact with nuclear capsids presently is unknown.
At present, it is unclear which components of the tegument are added to capsids prior to or during nuclear egress (here defined as the primary tegument for the purpose of this study). It is equally unclear which viral proteins are added or removed later on in the cytoplasm or during the final envelopment of the capsids (defined here as the secondary tegument). To shed light on this complex viral maturation process, we used two complementary approaches. The first one relies on an in vitro nuclear egress assay we previously reported and which reproduces in the test tube the exit of HSV-1 capsids from infected nuclei into the cytoplasm (79). Critical to the current study, naked cytosolic capsids are produced in the presence of exogenous cytosol derived from uninfected cells. Since this exogenous cytosol is devoid of any cytosolic viral proteins, any tegument protein found on the cytoplasmic capsids is either already present on the nuclear capsids or is added as a result of capsid release from the nucleus. This represents a unique system to study nuclear egress itself as well as the tegumentation process at the early stages of egress. The second and classical assay relies on the analysis of nuclear capsids directly isolated from infected nuclei and enriched on a linear sucrose gradient (44, 57, 80–82).
We assessed primary tegumentation by immunoblotting the cytosolic capsids produced in our in vitro assay as well as the purified nuclear C capsids using a battery of antibodies. In addition, we took advantage of immunoelectron microscopy (immuno-EM) analysis of nuclear capsids to confirm these interactions and ruled out the possibility that tegument proteins identified by Western blotting were copurifying aggregates. As anticipated, the UL31 viral protein as well as the UL34 and gD integral membrane proteins were absent from these capsids. Interestingly, five tegument proteins were detected both on in vitro cytosolic capsids and in nuclear C capsids isolated on sucrose gradients (US3, ICP0, ICP4, UL36, and UL37). Not surprisingly, the bulk of these tegument proteins was removed by high salts. The main surprise was the stabilization of the tegument by irrelevant exogenous proteins. Interestingly, the comparison of mature extracellular virions and capsids produced in vitro suggests multiple rounds of addition for some of the tegument proteins, hinting at a dynamic multistep interaction between the capsids and the tegument proteins. Finally, an analysis of capsids produced in vitro in the presence of cytosol containing viral tegument proteins suggests that the bulk of remaining tegument components are recruited to the capsids during their final envelopment stage. Altogether, these results probe the initial stages of sequential addition of the tegument and may ultimately provide some insights into the mechanism of HSV-1 nuclear egress.
MATERIALS AND METHODS
Cells and viruses.
HeLa S3 cells, a HeLa strain adapted to culture in suspension, were grown in Joklik's modified Eagle's medium (JMEM; Sigma-Aldrich) supplemented with 5% fetal bovine serum (Medicorp) and 0.1 mM nonessential amino acids (Invitrogen). 143B, Vero, U2OS, HS30 (64), BD45 (61), and n-33 cells (83) were grown in Dulbecco's modified Eagle's medium (Sigma-Aldrich) supplemented with 10% fetal calf serum (FCS; HyClone) and 2 mM l-glutamine (Invitrogen) in 5% CO2. 143B cells were also supplemented with 15 μg/ml 5-bromo-2 deoxyuridine (BrdU; Sigma) except prior to infection. Wild-type (wt) HSV-1 17+ virus was provided by Beate Sodeik. Virus stocks were expanded on BHK cells and titrated on Vero cells as described previously (84). KΔUL36 and KΔUL37 were provided by Prashant Desai and were expanded and titrated on HS30- or BD45-complementing cells, respectively (61, 64). Null viruses lacking ICP0 (n212) (85) or ICP4 (n12) (83) were provided by Priscilla Schaffer and were expanded and titrated on U2OS or complementing n-33 cells, respectively. In the case of the deleted KΔUL36, KΔUL37, and n12 (ΔICP4) strains, nuclear capsids were analyzed by immuno-EM from noncomplementing Vero cells, while the n212 (ΔICP0) strains were produced on U2OS cells.
Antibodies.
Primary antibodies for Western blotting and/or immuno-EM included the following: αUL6 and αUL31, provided by J. D. Baines (45, 86); αUL7 and αUL14, provided by Y. Nishiyama (87, 88); NC5 αVP23 and DL6 αgD, provided by R. J. Eisenberg and G. H. Cohen (89, 90); αUL20 and αUS3, provided by B. Roizman (91, 92); αUL34, provided by R. Roller (45); 147 αUL36, provided by B. Sodeik and A. Helenius (11); 780 αUL37, provided by F. J. Jenkins (54); ab5283 αVP13-14 LP1 and AGV031 αUL49, provided by G. Elliot (93, 94); αVP16, provided by H. Browne (95); and 3114 αgE, provided by D. Johnson (96). The remaining antibodies are commercially available, including α-ICP0 and α-ICP4 (Abcam), α-VP5 (East Coast Bio), α-PCNA (Chemicon Europe), α-calnexin (Stressgen), α-syntaxin 18 (Synaptic Systems), and anti-α-tubulin (Santa Cruz). Horseradish peroxidase-coupled or gold-labeled secondary antibodies (goat anti-mouse, goat anti-rabbit, and donkey anti-chicken) were purchased from Jackson ImmunoResearch or Cedarlane.
Isolation of nuclei.
Nuclei were isolated from HeLa S3 suspension cells as previously described (79, 97). Briefly, HeLa S3 cells grown in suspension were infected with HSV-1 17+ at a multiplicity of infection (MOI) of 3. For radiolabeled preparations, a protocol adapted from the work of Church and Wilson (98) was used as previously reported (79). Eight hours postinfection (hpi), cells were pelleted and resuspended in RSB (10 mM NaCl, 10 mM Tris-Cl, pH 8.4, 5 mM MgCl2) for less than 10 min. Cells were then broken mechanically. Nuclei were collected and enriched on a discontinuous iodixanol gradient at 10,000 × g for 30 min. The nuclear fraction was collected, adjusted to 50% glycerol and 1 mM dithiothreitol, and stored at −80°C.
Preparation of cytosol.
Cytosol from either uninfected or HSV-1-infected HeLa S3 cells were prepared as described before (79, 97). For this, mock-infected cells or cells infected with HSV-1 17+ at an MOI of 3 for 8 hpi were pelleted and resuspended in KEHM (50 mM KCl, 10 mM EGTA, 50 mM HEPES, pH 7.4, 2 mM MgCl2) supplemented with 1 mM dithiothreitol and a mixture of protease inhibitors (8.25 μM chymostatin, 1.05 μM leupeptin, 0.38 μM aprotinin, and 0.73 μM pepstatin A; Sigma-Aldrich). Cells were then broken and the cell lysates centrifuged at 267,000 × g for 30 min at 4°C. At that speed, virions, unenveloped capsids, and intracellular organelles and vesicles are quantitatively removed (79 and data not shown). The protein concentrations of these cytosol preparations were determined with a Bradford assay and the cytosol stored at −80°C.
In vitro assay.
Nuclei (described above) were incubated with nuclear buffer (20 mM Tris-Cl, pH 7.4, 5 mM MgCl2, 100 mM KCl, and 1 mM dithiothreitol) for 6 h at 37°C. Mock-treated or infected cytosol, usually 1 mg/ml, and an energy-regenerating system (17.3 mM creatine phosphate, 87 μg/ml creatine kinase, 2.17 mM ATP; Roche) were added as described before (79, 97, 99).
Capsid and virion purification.
Cytosolic capsids released in vitro were separated from nuclei with a spin column mounted with a 0.45-μm cellulose acetate filter (Costar) and centrifuged at 825 × g for 10 min at 4°C. The capsids were recovered from the permeate, diluted in TNE buffer (20 mM Tris, pH 7.5, 500 mM NaCl, 1 mM EDTA) containing 0.1% Triton X-100, and gently sonicated 10 times for 1 s each. The capsids were then concentrated using an Amicon centrifugal filter device (Millipore) and resuspended in TNE prior to their being loaded onto a 4-ml Sephacryl S-500 HR gel filtration column (GE Healthcare). Multiple 200-μl fractions were collected. Fractions specifically containing capsids were determined by liquid scintillation (to detect the radiolabeled viral DNA) and Western blotting (against capsid proteins VP23, VP5, and the cytoplasmic component α-tubulin). Fractions enriched in viral capsids were finally pooled and concentrated on a Microcon centrifugal filter device (Millipore) with a molecular weight cutoff (MWCO) of 100,000.
Extracellular virions were purified as followed. HeLa S3 cells were grown in suspension and infected with HSV-1 17+ wt at an MOI of 5. The extracellular milieu was collected at 24 hpi, centrifuged at 300 × g for 10 min at 4°C to remove intact cells, and treated with 50 μg/ml DNase I (Roche) for 30 min at 4°C. The supernatant was filtered on a 0.45-μm filter and centrifuged at 39,000 × g for 60 min at 4°C in a Beckman SW28 rotor. The pellet containing extracellular virus was resuspended in MNT (30 mM morpholineethanesulfonic acid [MES], 100 mM NaCl, and 20 mM Tris-HCl, pH 7.4) and stored at −80°C.
Nuclear B and C capsids were isolated from HSV-1-infected Vero cells at 18 hpi and resolved on a sucrose gradient as previously published (44, 57, 80–82), with some modifications. Briefly, infected cells were collected, washed with phosphate-buffered saline (PBS), and resuspended in NP-40 lysis buffer (150 mM NaCl, 10 mM Tris-HCl, pH 7.5, 2 mM MgCl2, 1% Igepal, 5 mM dithiothreitol) at 1 × 107 cells/ml for 30 min on ice. The cell lysate was then spun at 225 × g for 10 min and the nuclei resuspended in low-salt TNE (20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA) or high-salt TNE (500 mM NaCl), cracked by 3 cycles of freeze-thaw, DNase treated, and sonicated. These nuclear lysates were cleared at 11,000 × g for 30 min, and nuclear capsids were recovered using a 35% sucrose cushion. B and C capsids were finally isolated on a 20 to 50% linear sucrose gradient in low- or high-salt TNE at 100,000 × g for 1 h. The middle B capsids and lower C capsids were collected, pelleted at 100,000 × g, and stored at −80°C. To test the effect of salts and exogenous proteins on the stability of the tegument, C capsids initially isolated from low-salt gradients were incubated for 15 min at 37°C with high-salt TNE (500 mM NaCl) with or without 1 mg/ml of BSA or 1 mg/ml cytosol derived from noninfected cells, pelleted at 100,000 × g for 1 h, and stored at −80°C.
Electron microscopy.
The purity of the in vitro-produced capsids was analyzed by negative staining. Succinctly, purified capsids were adsorbed on hexagonal 200-mesh copper grids coated with Formvar and carbon (Canemco and Marivac). The samples were contrasted with 2% uranyl acetate (Canemco and Marivac), washed in distilled water, and dried on filter paper. For immuno-EM, nuclear C capsids were adsorbed on a nickel grid for 30 min. The grids were then incubated for 1 h in blocking buffer (PBS, 2% BSA, 0.2% gelatin) and then for 1 h with primary antibodies diluted in blocking buffer. Following extensive washes with PBS under magnetic agitation, the grids were incubated for 30 min with gold-coupled secondary antibodies, washed again in PBS, and contrasted as described above. All samples were examined on a Philips CM100 electronic microscope. Only beads directly in contact or no further than one bead from the capsid edge (i.e., 10 nm) were considered positive. Furthermore, we arbitrarily separated the results in two groups to discriminate strong labeling (≥5 beads/capsids) from weaker labeling (≤4 beads/capsids) as previously reported (100). Quantification was done from several independent fields and experiments (n = 200).
SDS-PAGE electrophoresis, Western blotting, and gel staining.
After boiling the samples for 10 min, they were loaded on 10 or 15% acrylamide SDS-PAGE gels in protein sample buffer (50 mM Tris-HCl, pH 6.8, 2% SDS, 0.1% bromophenol blue, 10% glycerol, and 2% β-mercaptoethanol). Proteins were then transferred from the gels to polyvinylidene difluoride (PVDF) membranes. The membranes were immersed for 1 h in blocking buffer (5% nonfat dry milk, 13.7 mM NaCl, 0.27 mM KCl, 0.2 mM KH2PO4, 1 mM Na2HPO4, and 0.1% Tween 20) and subsequently incubated for 2 to 4 h with primary antibodies diluted in blocking buffer. The blots were then washed and probed with secondary antibodies conjugated to horseradish peroxidase. The detection was done with the Super Signal West Pico chemiluminescent substrate (Pierce) on Kodak BioMax Light film or using a ChemiDoc MP system (Bio-Rad). When indicated, gels were instead stained with a 0.25% Coomassie 250 solution (Sigma) for 10 min and washed in destaining buffer (5% acetic acid–50% methanol) 3 times for 15 min. Alternatively, gels were stained with 0.1% silver nitrate (Sigma) for 30 min, developed in sodium carbonate buffer (3% sodium carbonate–0.04% formalin), washed in 5% acetic acid, and scanned.
Treatment of nuclei with trypsin.
One hundred micrograms of uninfected or infected nuclei or cytosol was treated with or without 500 μl (11,500 BAEE units) of immobilized tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin (Pierce). Trypsin beads were thoroughly washed 3 times with digestion buffer (0.1 M NH4HCO3, pH 8.0) before being added to the samples. Every sample (treated and mock treated) was incubated in the digestion buffer for 2 h at 37°C with robust shaking. The trypsin beads were then removed from the sample by a 1-min centrifugation at 300 × g. The supernatant was collected and treated for SDS-PAGE and further processed for Coomassie blue staining or Western blotting.
RESULTS
Adaptation of the in vitro nuclear egress assay.
To identify the tegument present on HSV-1 capsids during the early stages of viral egress, we took advantage of our published in vitro nuclear egress assay (79, 97). The original assay relied on nuclei purified at 8 hpi from [3H]thymidine-labeled infected HeLa cells and quantification of viral egress 6 h later by liquid scintillation. We previously demonstrated that this assay produces naked cytoplasmic capsids in the presence of cytosol derived from infected cells or from cells that never encountered the virus (79, 97). We thus first asked which tegument proteins are present on these capsids. To ensure that no viral protein could coat the capsids after their release into the cytoplasm, the assays were initially performed with cytosol derived from uninfected cells. Following the in vitro egress reaction, the capsids were recovered according to the purification protocol described in Fig. 1. Briefly, they were passed over a 0.45-μm filter to remove the nuclei, which largely preserve their integrity in this assay (79, 97). Potential cytoplasmic capsid aggregates were then broken up with mild nonionic detergent and gentle sonication. The capsids were recovered from the sample by gel filtration over a high-porosity column with a broad resolution spectrum (20 kDa to 100 MDa). Unlike classical density centrifugation, which can concentrate nuclear capsids but also contaminants with similar densities, gel filtration allows the separation of small to very large entities and hence should provide a better purity at the expense of concentration (typical dilution of samples by a few folds). This is of course readily solved by concentrating the cleaner capsids afterwards, in our case with a Microcon centrifugal filter device that further serves to remove smaller particles below its cutoff. Gel filtration is particularly relevant here, as the expected capsid mass is around 200 MDa (101, 102), implying that the capsids should end up in the void volume of the column. In contrast, individual proteins and complexes typically can range from a few kDa to a few MDa and should be delayed and fractionated. Consequently, fractions from the gel filtration column were analyzed by liquid scintillation and Western blotting. Figure 1C indicates that cytoplasmic proteins, exemplified by the α-tubulin marker, eluted from the column in the 2- to 5-ml fractions. As expected, [3H]thymidine-labeled capsids released in vitro were found in the void volume of the column (i.e., between 1 to 2 ml), as confirmed by Western blotting against the VP23 and VP5 capsid proteins (Fig. 1C and data not shown). Capsid-enriched fractions were then pooled and concentrated using a Microcon centrifugal filter device. Similar results were obtained with capsids produced in the presence of cytosol prepared from infected cells (data not shown).
Fig 1.

Purification protocol. (A) Schematic illustration of the in vitro assay and expected results. In this scheme, infected nuclei were incubated in vitro in the presence of energy, nuclear buffer, and one of three conditions: no cytosol, cytosol derived from uninfected cells (mock-infected cytosol), or cytosol prepared from HSV-1-infected cells (infected cytosol). Note that in the absence of cytosol, very few capsids should be produced. (B) Depiction of the protocol used to purify HSV-1 cytoplasmic capsids produced in the in vitro assay. See Materials and Methods for details. (C) Enrichment of the in vitro capsids by gel filtration. The viral DNA was radiolabeled by the addition of [3H]thymidine to infected cells, and the nuclei were isolated as detailed in Materials and Methods. After their incubation in vitro with uninfected cytosol and preliminary steps of purification (as described for panel B), the cytoplasmic capsids were purified by gel filtration and fractions collected. An equivalent volume from each fraction was analyzed for its radioactivity level to detect viral DNA and probed by Western blotting against the capsid protein VP23 and against α-tubulin, a host protein. The box denotes fractions enriched in viral DNA and capsid proteins, which were subsequently pooled and concentrated before proceeding to further analysis.
To visualize the efficacy of the purification protocol, each purification step was analyzed by Western blotting. Figure 2A indicates the high abundance of VP5, VP23, gD, and α-tubulin in the starting in vitro assay and the complete removal of gD and α-tubulin in the final samples, indicating that the enrichment protocol worked well. To confirm these findings, we also examined the purity of the samples by EM negative staining for capsids released in the presence of mock-infected or infected cytosol and noted enriched capsids with some rare small debris (Fig. 2B). As before (79), all capsids were naked and therefore were well suited to having their tegument content analyzed.
Fig 2.
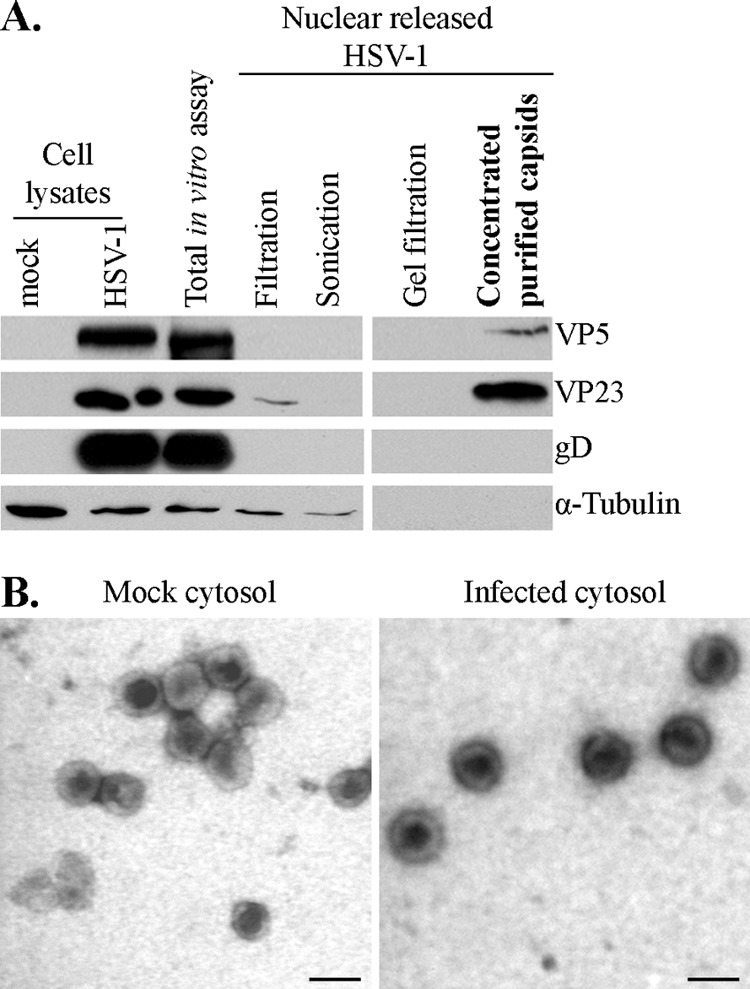
Purity of in vitro-released HSV-1 capsids. (A) Two-microgram aliquots from each step of the purification method were analyzed by SDS-PAGE and Western blotting to evaluate the purity of the samples following the in vitro assay done in the presence of mock-infected cytosol. The final step (concentrated purified capsids) was enriched in capsid proteins (VP5 and VP23) and devoid of gD, a glycoprotein associated with the viral envelope. Alpha-tubulin, a cellular component, was also absent from the final enriched material. (B) Capsids purified as described for panel A were subsequently visualized by negative staining and EM to visually inspect their purity. (Left) Capsids released from nuclei incubated in mock-infected cytosol. (Right) Capsids released from nuclei incubated in infected cytosol. Scale bar, 100 nm.
UL36, UL37, ICP0, and ICP4 are primary tegument proteins.
Given the controversy surrounding the putative UL36, UL37, ICP0, and ICP4 viral proteins as primary tegument proteins (see Introduction), we first evaluated their presence by Western blotting. To our surprise, full-length ICP0, UL36, and UL37 were strongly enriched in the capsid fraction produced in the absence of any cytoplasmic viral proteins. ICP4 was also readily and reproducibly detected in these samples but was not particularly enriched compared to its total nuclear pool (Fig. 3). This differential behavior for ICP4 may reflect its exclusive localization to the nucleus (103). Indeed, if only a small portion of the nuclear ICP4 coats the capsids, then little ICP4 enrichment would be anticipated. In contrast, only 15% of all ICP0 is nuclear at 8 hpi (72, 104), when the nuclei were harvested. Thus, only a small portion of the protein is present in the total in vitro assay, consequently ICP0 can be detected only after capsid enrichment. To examine if other tegument proteins are present, we probed several of them by Western blotting using an array of previously characterized antibodies (see Materials and Methods). VP22 and VP16 were not detected, and trace amounts of UL7, UL14, and VP13-14 were found, but due to their very low abundance, it was not clear if they were true components of the primary tegument (data not shown).
Fig 3.
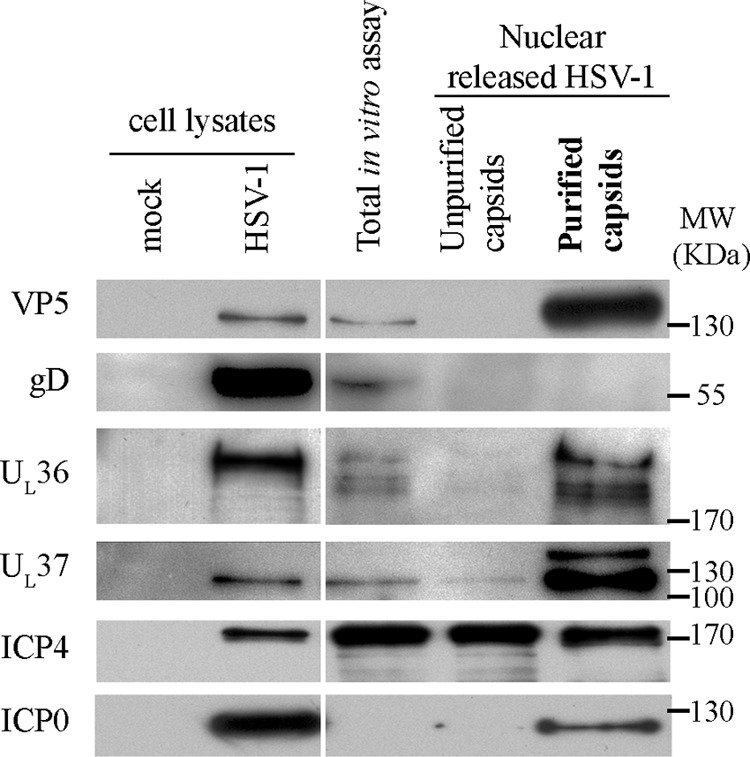
Western blot analysis of in vitro cytoplasmic capsids produced in the presence of mock-infected cytosol. HSV-1 17+ wt-infected nuclei were incubated for 6 h at 37°C with energy and uninfected cytosol. The capsids were then purified as described for Fig. 1. Five micrograms of mock- or HSV-1-infected cell lysates (antibody controls) and 10 μg of total in vitro assay, 0.45 μm filtrate (unpurified capsids), or concentrated purified capsids were loaded and analyzed by Western blotting, as indicated to the left of each panel. The results revealed that UL36, UL37, ICP0, and ICP4 were detected on the purified capsids.
Although the data suggested that, in addition to US3, full-length UL36, UL37, ICP0, and ICP4 were also primary tegument proteins, two alternative explanations could be invoked. The first one was the possible contamination of the nuclear preparations by cytosolic tegument proteins. The second possibility was that viral tegument proteins could be on the external face of the nuclei and were added to the capsids immediately following their release. To address the first issue, we examined whether the nucleus preparations used in the assay contained cytoplasmic viral proteins. To this end, stocks of nuclei were passed over a 0.45-μm column to remove the nuclei (79), and both control nuclei and filtrate were analyzed by SDS-PAGE and Western blotting. As seen in Fig. 4A, capsid and tegument proteins were found in the total nucleus preparation, as expected, but none were found in the filtrate. This not only ruled out the possible contamination of our nuclear preparations with cytoplasmic tegument proteins but also confirmed the integrity of the nuclei when filtered over the 0.45-μm column. It also excluded the presence of tegument aggregates in the preparations, which would end up in the filtrate over such large-pore filters, unless such aggregates were significantly larger than the capsids, which do pass through such filters (79). To examine the second scenario, the nuclei were subjected to trypsin treatment to digest the proteins from their exterior surfaces, reasoning that if a tegument protein was present outside the nuclei, it should be digested under these conditions. To prevent the passive diffusion of the small protease (23.2 kDa) through the nuclear pores, trypsin bound to 1-μm Sepharose beads was used. As shown in Fig. 4B, the enzyme efficiently digested all the proteins found in infected cytosol. In addition, treatment of nuclei efficiently removed calnexin and syntaxin 18, two integral membrane proteins found in the endoplasmic reticulum/outer nuclear envelope (Fig. 4C). This indicated that the trypsin was functional and could digest transmembrane proteins despite the large Sepharose beads. In contrast, the trypsin had no discernible effect on the tegument proteins when whole nuclei were treated with the enzyme (Fig. 4B), indicating that tegument proteins detected in our assay were exclusively inside the nuclei. Altogether, these results argued in favor of UL36, UL37, ICP0, and ICP4 as components of the primary tegument.
Fig 4.

Absence of cytosolic tegument proteins from nuclear preparations. (A) Five micrograms of nuclei used in the in vitro assay was incubated for 6 h at 37°C and filtered on a 0.45-μm column to gently remove the nuclei. All of the resulting filtrate was resolved by SDS-PAGE and analyzed by Western blotting. Controls included 5 μg of untreated nuclei. None of the viral proteins UL19, ICP0, ICP4, UL36, and UL37 was found in the nuclear filtrate. (B) To demonstrate that viral tegument proteins were not attached to the external face of the nuclei, 100 μg of nuclei was digested with Sepharose-bound trypsin. As a control for the enzyme activity, 100 μg of infected cytosol was also digested with the same amount of enzyme. The samples were analyzed by SDS-PAGE and Coomassie blue staining (upper panel). In the lower panels, 10-μg aliquots of proteins were used for Western blotting with various antibodies. (C) Fifteen micrograms of mock-infected or infected nuclei were digested with Sepharose-bound trypsin or were mock treated. Samples were analyzed by Western blotting with antibodies against the calnexin and syntaxin 18 integral membrane proteins. Mock-infected cell lysates were included as antibody controls.
Analysis of secondary tegument.
So far, our energies were focused on capsids produced in the presence of uninfected cytosol, hence defining the primary tegument. However, it is of upmost interest to determine whether additional tegument proteins are added to the capsids once they reach the cytoplasm. Fortunately, the in vitro assay theoretically can address this point by assaying the tegument content of capsids produced in the presence of infected cytosol, i.e., containing a full set of cytoplasmic tegument proteins but lacking intracellular organelles, such as the TGN and even small vesicles/tubules. It is thus possible to probe the sequential addition of the tegument in the nucleus and the cytoplasm and by deduction during reenvelopment. Infected nuclei thus were incubated in vitro in the presence of HSV-1-infected cytosol, and the released capsids were purified and analyzed by Western blotting as described above. Surprisingly, while the tegument proteins described above were present, none of the additional viral proteins tested were detected on the capsids under these conditions (data not shown). This observation argues that additional tegument proteins can be added primarily at the site of reenvelopment, although we cannot rule out that the in vitro assay inefficiently reproduces this tegumentation step due to, for example, a lower protein concentration in vitro (1 mg/ml) than in vivo (100 mg/ml) (105).
The tegumentation process seems to be dynamic.
Our next concern was the fate of the primary tegument proteins. To address this point, we compared by Western blotting the abundance of tegument proteins on cytosolic capsids produced in vitro to that of mature extracellular virions. Since equal amounts of virions and in vitro capsids were loaded (normalized for VP5) (Fig. 5), we could visually compare the relative abundance of each tegument protein. It was apparent that the abundances of ICP0, UL36, UL37, and US3 were all somewhat increased in mature virions, suggesting that these proteins are added in multiple rounds, i.e., at the nuclear stage and again later on. Once again, we could not rule out an inefficient in vitro tegumentation process or a loss of some tegument proteins during capsid purification, or that the extracellular virion preparations include L-particles with different tegument stoichiometries. Conversely, ICP4 seemed less abundant in mature virions. Finally, both UL37 and US3 qualitatively differed in the mature virions compared to the cytoplasmic capsids. In the first case, an upper 140-kDa band virtually disappeared in mature virions, while a 120-kDa immunoreactive protein was present in both samples (Fig. 5). For US3, while a 69-kDa band was stable in both samples, a 68-kDa band was apparent only on extracellular virions. If true, these findings argue for a dynamic interaction between the capsids and the tegument proteins and the possible processing of some tegument proteins along the egress route.
Fig 5.

Western blot analysis of primary tegument proteins (in vitro assay). HSV-1 17+ wt-infected nuclei were incubated for 6 h at 37°C with energy and mock-infected cytosol in the absence of any cytosolic viral proteins or organelles. Following their purification, the capsids released were analyzed by SDS-PAGE and Western blotting. As a control, extracellular virions were also loaded (normalized to have the same amount of VP5 as the in vitro capsids) to evaluate the relative quantities of the tegument proteins. Mock-treated or HSV-1-infected total cell lysates served as antibody controls. Controls +, viral capsid proteins expected in the purified capsids; controls −, viral proteins that should be absent from the purified capsids.
Nuclear C capsids are coated with the same tegument proteins as the in vitro cytosolic capsids.
UL36, UL37, ICP0, ICP4, and US3 were detected on capsids produced in the absence of cytosolic viral proteins (Fig. 3). Given that these tegument proteins are unlikely to have coated the capsids from outside the nucleus (Fig. 4), presumably they were added to the capsids within the nucleus. Of these, only US3 is known to be on both enveloped perinuclear and mature extracellular virions (8, 41). To independently evaluate the composition of the primary tegument, we opted to isolate nuclear capsids by classical means to determine if our results are biologically relevant or simply an artifact of the in vitro assay. Past studies addressed this question by analyzing nuclear C capsid composition, sometimes with conflicting results (11, 14, 56, 57, 77, 78). For instance, both ICP0 and ICP4 are detectable on nuclear C capsids isolated in the presence of low salt (100 to 200 mM) (78) but not on any type of nuclear capsids in the presence of high salt (500 mM) (11, 14, 57). In addition, while some residual UL36 and UL37 are still discernible on nuclear C capsids using high salt (56, 57), others did not detect them using similar conditions (11, 14).
Considering the impact of salt concentrations on tegument stability, we decided to analyze in parallel our nuclear C capsids with both low and high concentrations of salt. Hence, nuclear C capsids and control B capsids were isolated at 18 hpi by classical sucrose sedimentation (44, 57, 80–82) in the presence of 150 or 500 mM NaCl (Fig. 6A). The capsids were then silver stained to visualize their protein pattern and visualized by EM negative staining for their relative purity (Fig. 6B and C). The EM negative staining revealed that B and C capsids were devoid of any aggregates and displayed the expected morphology and protein patterns (77, 78, 80, 86, 106). Western blot analysis subsequently showed that the C capsids were free of US3, UL36, UL37, ICP0, and ICP4 when incubated under high-salt conditions, whereas these full-length tegument proteins were all present on capsids incubated at a low salt concentration (Fig. 6C). This highlighted the sensitivity of the tegument to salts. Finally, we probed the presence of these tegument proteins on B capsids, which are expected to lack most tegument proteins (14, 78). Figure 6C indicates that this was indeed the case, confirming the specificity of the labeling and ruling out the presence of tegument aggregates that merely copurify with viral capsids along the sucrose gradient.
Fig 6.

Western blot analysis of nuclear C capsids. (A) Intranuclear capsids from infected Vero cells were separated on a 20 to 50% linear sucrose gradient in the presence of 150 or 500 mM NaCl. A, B, and C indicate the nuclear capsid types. (B) B and C capsids were visualized by negative staining and EM to visually inspect their purity. (C, top) Silver staining of B and C capsids isolated from sucrose gradient (top). (Bottom) B and C capsids from the sucrose gradients described for panel A were probed by Western blotting. (D) C capsids were initially isolated from the sucrose gradient in the presence of 150 mM NaCl and subsequently incubated in high salt (500 mM) in the presence or absence of exogenous proteins (either 1 mg/ml BSA or 1 mg/ml cytosol derived from noninfected cells), pelleted, and analyzed by SDS-PAGE and Western blotting. As antibody controls, mock-treated and infected cell lysates were also loaded in panels C and D (normalized to have the same amount of VP5 as the C capsids).
As detailed in Materials and Methods, the in vitro assay was performed in the presence of 1 mg/ml of cytosol, and the released capsids were enriched in the presence of 500 mM NaCl. Thus, the tegument proteins detected on the in vitro capsids seemed resistant to high salts, in apparent contradiction with the findings described above. One main difference between these in vitro samples and our nuclear capsid preparations is the presence of exogenous proteins in the form of cytosol. Since this cytosol was usually derived from uninfected cells, it could not be a source of exogenous tegument proteins. We thus investigated the impact of proteins on the stability of the tegument. Nuclear C capsids thus were isolated on 150 mM NaCl sucrose gradient in the absence of exogenous proteins, incubated with high salt (500 mM NaCl) in the presence of 1 mg/ml BSA or 1 mg/ml of mock-infected cytosol (derived from uninfected cells), and probed by Western blotting. Figure 6D shows that exogenous proteins during purification of C capsids did indeed stabilize tegument protein interactions with the capsids. This effect was not specific, as BSA could also stabilize the tegument to a large extent. Consequently, our results with the in vitro assay are fully consistent with those using purified nuclear C capsids despite the exposure of the in vitro capsids to high salts.
To confirm that the detection of tegument proteins in the capsid preparations was not due to contamination of unbound tegument proteins or aggregated tegument proteins that might cosediment with the capsids, we next quantified by immuno-EM the presence of ICP0, ICP4, UL36, or UL37 on nuclear C capsids. We also examined VP5, US3, the nuclear marker PCNA, the viral glycoproteins D and E, UL20, and UL31 as controls. Secondary antibodies alone were also used as control background signals. As further controls, the efficiency of antibodies against viral glycoprotein gD was tested on mature extracellular viruses. Not surprisingly, nearly all nuclear capsids (99%) were labeled with the major capsid protein VP5, with most of them (86%) harboring more than 5 beads/capsid (Fig. 7). Similarly, roughly 60% of the capsids scored positive for US3, albeit with a lower efficiency (14% with ≥5 beads/capsid), in agreement with their lower abundance on capsids (8, 107). As expected, none of the negative-control antibodies labeled the nuclear capsids (Fig. 7A), whereas nearly all extracellular virions were positive for the viral glycoprotein gD (98%) (data not shown). Interestingly, the nuclear capsids were also labeled for ICP0, ICP4, UL36, or UL37 with efficiencies similar to those of US3, ranging from 40% for ICP0 to 78% for UL36 (Fig. 7A). We did not notice any tegument aggregates by EM.
Fig 7.

Immunogold labeling of tegument proteins on nuclear C capsids. (A) Intranuclear wt C capsids were isolated from infected Vero cells on a 150 mM NaCl sucrose gradient and labeled by immunogold with antibodies specific for the following tegument proteins: US3, ICP0, ICP4, UL36, and UL37. Positive-control antibodies included VP5 (capsid control), while negative controls included the viral glycoproteins gD and gE, the viral proteins UL20 and UL31, and the nuclear host marker PCNA. Intranuclear capsids isolated from ICP0, UL36, or UL37 null mutants (ΔC capsids) were also isolated from noncomplementary cells (see Materials and Methods). (B) Intranuclear wt C capsids were isolated from infected Vero cells on a 500 mM NaCl sucrose gradient and labeled by immunogold with antibodies specific for the following tegument proteins: ICP0, ICP4, UL36, UL37, and US3. In panels B and C, quantification of labeled capsids was measured by counting 200 capsids for each sample. Only beads within 10 nm (i.e., one bead equivalent) were considered positive. Strong labeling (black) and weak labeling (white) were determined by the number of beads/capsid. Results are shown as percentages of labeled capsids.
To confirm the specificity of the labeling, we next purified nuclear C capsids on noncomplementing cells infected with viruses lacking ICP0 (n212; [85]), ICP4 (n12; [83]), UL36 (KΔUL36; [61]), or UL37 (KΔUL37; [64]). Unfortunately for n12 (ΔICP4), we were unable to produce nuclear capsids on noncomplementary cells, whereas for KΔUL36 and KΔUL37, de novo synthesis of UL36 or UL37 is not required for the assembly of nuclear capsids (61, 64). In the case of n212 (ΔICP0; [85]), it was produced on U2OS cells, which do not express any ICP0 but somehow complement the deletion (108). As expected, these null viruses were all negative for their respective deleted tegument proteins (Fig. 7A, ΔC capsids). Finally, to further confirm that the tegument is salt sensitive, we probed nuclear C capsids isolated from high-salt sucrose gradients (i.e., 500 mM NaCl). These immuno-EM analyses revealed that only residual amounts of these tegument proteins were detectable on the capsids, ranging between 10 and 25% (Fig. 7B), in agreement with previously reported values (11, 14). Our data thus are all consistent with at least five tegument proteins coating nuclear capsids and confirm past reports that these interactions are sensitive to high salt concentrations.
DISCUSSION
The tegument is among the least understood of the herpesvirus components and the subject of intense scrutiny. It is generally believed that in the initial stages of egress, the nuclear capsids interact with US3 in the nucleoplasm and with UL31 and UL34 viral proteins upon their interaction with the inner nuclear envelope (3, 31–33, 41, 49). UL31 and UL34 are present on perinuclear virions but are shed as the capsids enter the cytoplasm (8, 40, 41). In contrast, US3 remains capsid associated in mature virions and constitutes one of the first tegument proteins recruited onto the newly assembled capsids. A number of laboratories have addressed the issue by immuno-EM, deletion mutants, and immunofluorescence (see Introduction). In this study, we took advantage of a unique in vitro assay that reconstitutes HSV-1 nuclear egress in combination with a classical nuclear capsid sedimentation assay. These assays are most useful, since downstream viral intermediates are absent. The absence of UL31, UL34, gD, UL20, and α-tubulin (Fig. 3 and 5) and the enrichment of capsid proteins in the final viral preparations (Fig. 2 and 3) all were evidence that the in vitro assay performed as expected.
The present study revealed the presence of US3, ICP0, ICP4, UL36, and UL37 on cytosolic capsids produced in vitro (Fig. 3, 5, 6, and 7). Given the absence of cytosolic viral proteins and intracellular organelles (Fig. 3 and 5), these tegument proteins could only reasonably coat the capsids within the nucleus or at its external surface. The latter scenario is unlikely, since the tegument proteins were protected from digestion by trypsin outside the nuclei (Fig. 4). Consequently, the tegument proteins most likely were recruited in the nucleus prior to nuclear egress or during the budding process through the inner nuclear membrane. These findings are corroborated by the detection of the same proteins on nuclear C capsids purified in the presence of low salts (i.e., 150 mM NaCl). Moreover, immuno-EM analyses clearly demonstrated the specific coating of nuclear C capsids with US3, UL36, UL37, ICP0, and ICP4 (Fig. 7).
Others have shown that UL36 and UL37 have a tendency to smear across several fractions on density gradients, indicative of stickiness, oligomerization, and/or aggregation (57). Despite this, these potential tegument aggregates are an unlikely explanation for our findings for a number of reasons. First, if tegument aggregates were present in the in vitro assay, they would be present in the nuclear filtrate, since they would be anticipated to pass through the 0.45-μm filter (as do the capsids), but this was not the case (Fig. 4). The exception would be aggregates much larger than capsids, which should be visible by EM (see below). Second, we could not detect ICP0, ICP4 UL36, UL37, and US3 on B capsids, indicating that tegument aggregates do not randomly copurify with the capsids (Fig. 6). Third, two different approaches (classical nuclear C capsids and in vitro-generated cytosolic capsids) gave identical results (i.e., positive for US3, ICP0, ICP4, UL36, and UL37), and this occurred with two distinct techniques (Western blotting and immuno-EM). Fourth, these two capsid types were generated by radically different protocols involving very distinct biophysical properties, i.e., density (sedimentation) or shape and mass (gel filtration). It seems unlikely that putative tegument aggregates would, by chance, behave as capsids under both conditions. Fifth, the Sephacryl S-500 column used in this study separates macromolecules ranging from 20 to 100,000 kDa, with anything larger than 100,000 kDa ending up in the void volume. Published estimates evaluate the HSV-1 viral capsids at 200,000 kDa (101, 102), meaning it should end up in the void volume of the column, as we indeed found (Fig. 1). If present, only very large aggregates visible by EM could end up in the same fraction as the capsids, which was not the case. Moreover, the antibody-labeled gold beads did not interact with large structures other than capsids. Thus, neither smaller (filterable) nor larger (EM) aggregates were found. Finally, B capsids were devoid of detectable UL36, UL37, US3, ICP0, and ICP4 (Fig. 6), arguing that these tegument proteins were not detected on C capsids because they form aggregates that are largely dispersed along the gradient. These findings are strong evidence these tegument proteins are indeed components of the primary tegument coating nuclear C capsids. They also confirm the usefulness and relevance of the in vitro nuclear egress assay to study the complex tegumentation process.
The nuclear recruitment of most tegument proteins seen in this study is consistent with some past findings reporting their presence in the nucleus or their detection on nuclear or perinuclear capsids (53–57, 78, 109). This contrasts with other reports that did not see UL36, UL37, ICP0, or ICP4 in those locations (11, 14, 59, 60, 77). This apparent discrepancy may in part be explained by technical issues. First and foremost, the stability of the tegument is clearly salt dependent (this study and references 11, 14, 76, and 78), and past studies looking at nuclear capsids typically relied on high salts to get clean capsids, including those addressing tegument composition (11, 14, 32, 33, 56, 57, 110–113). Second, the tegument is stabilized by the addition of carrier proteins like BSA or even mock-infected cytosol (Fig. 6), which explains why in our in vitro assay, the tegument is still capsid bound despite treatment with high salts. Another element is a recent study reporting that while a minor population of UL36 is detectable in the nuclei of infected Vero cells, none could be seen in rabbit skin cells (19), arguing that UL36 is in very low abundance in the nucleus or that the presence of UL36 in the nucleus is cell type specific. Interestingly, Leelawong and colleagues (56) recently suggested that for PRV, this very minor nuclear population of UL36 is mainly composed of a C-terminally truncated form of the protein using an anti-UL36 peptide antibody targeting amino acids 2818 to 2836. However, our data are rather consistent with the full-length UL36 being on nuclear capsids (Fig. 3, 5, 6). It is not clear what justifies such a discrepancy.
Interestingly, Cardone and colleagues noted a penton-capping density on heated T36 capsids that is absent from nuclear capsids (111). T36 capsids are extracellular virions that are extracted with 0.5 M NaCl and Triton and heated to 37°C to additionally strip UL37. Under these conditions (114), many tegument proteins fall off the capsid, and the authors concluded that the penton-capping density can only be UL36, since other tegument proteins were not anticipated. They consequently concluded that nuclear capsids, which lack this density (58), must be devoid of UL36. Unfortunately, it is possible that a number of tegument proteins do remain capsid associated in this scenario. For example, ICP0 and ICP4, which were not examined in the above-described study, have been shown to resist high salts in some studies (14, 76–78). This may also be true of other uncharacterized tegument proteins. It thus remains to be confirmed if the penton densities truly are UL36. Another explanation is that tegument stability differs between extracellular viruses, cell-associated viruses, and nuclear capsids. Indeed, Newcomb and Brown demonstrated that upon Triton and salt treatment, while extracellular virus tegument remains stable, cell-associated virus tegument is lost (115). The same conclusion may explain why the penton-capping density is lost on C capsids and not on extracellular viruses after purification.
The presence of ICP0 and ICP4 on nuclear C capsids is not surprising, given that both are present in that compartment at some point. Similarly, UL36 as a primary tegument is intuitive, since it is an inner tegument protein, possesses a nuclear localization signal (NLS), may allow capsid transport along the cytoskeleton during egress, and presumably brings the capsids to the site of reenvelopment (9–12). Given that UL37 binds to UL36 (59, 116), it may simply come along. The absence of the UL49 tegument protein in our study, identified in mature virions (8) and in a recent proteomic analysis of perinuclear HSV-1 virions (50), is puzzling. In principle, it should have been detected on nuclear capsids, but we cannot explain this discrepancy. It may not have been detected here due to the purification conditions and tegument stability, since outer tegument proteins display weaker interactions with the capsids than inner tegument proteins. Despite this caveat, the present study suggests that some of the tegument proteins present in mature virions are acquired at the nuclear stage of viral egress.
The addition of the primary tegument proteins to the capsids in the early phase of egress raises an important question as to their roles. Several plausible options come to mind, though additional work is needed to explore these avenues. Given the lack of accumulation of capsids in the nucleus upon the deletion of UL36 in some studies (61, 62, 65, 67), it does not appear to modulate nuclear egress per se. Some of the primary tegument may be involved in the transport and/or targeting of the capsids to the inner nuclear membrane within the nucleus or in the actual passage through the two nuclear envelopes, serve as a scaffold for the addition of secondary tegument proteins, transport and/or dock the capsids to site of final envelopment, or simply be required for the next round of infection.
Despite the addition of infected cytosol during the in vitro assay, no additional proteins were detected on the cytosolic capsids produced in vitro. It may of course be that the process of tegumentation is less efficient in vitro than in infected cells. However, it is tempting to speculate that many of these additional tegument proteins are added to the capsids further downstream at the site of reenvelopment. The presence of several tegument proteins at the TGN (5) argues in favor of such a scenario.
It should be pointed out that addition of a tegument protein to the capsid in the nucleus does not need to be exclusive, since multiple rounds of additions are possible. This may be supported by the apparent greater abundance of ICP0, UL36, and UL37 in mature extracellular virions compared to the capsids released in vitro (Fig. 5). Qualitative differences in the US3 and UL37 protein patterns are also suggestive of such dynamism (Fig. 5). We also cannot rule out that these differences are due to the presence of L-particles in the extracellular virus preparations, which may change the relative abundance of some tegument proteins compared to VP5. However, the prospect of a dynamic tegument is a stimulating option that has been reported by others, including a recent report documenting the maturation of the tegument (51, 56, 115, 117, 118).
The current study attempts to probe the complex tegumentation process. Several viral proteins were identified as potential components of the primary tegument, i.e., those added to nuclear capsids. The results also suggest that several tegument proteins are acquired principally during the reenvelopment step. Finally, the total loss of UL31, partial loss of some tegument proteins during egress, possible repeated addition of other tegument proteins, and potential maturation of UL37, US3, and UL36 underline the likely dynamic nature of the interactions between the capsids and its tegument. Future research will undoubtedly better define the molecular mechanism underlying these interactions.
ACKNOWLEDGMENTS
We sincerely thank J. D. Baines, H. Browne, G. H. Cohen, R. Courtney, G. Elliot, R. J. Eisenberg, D. Johnson, A. Helenius, F. J. Jenkins, Y. Nishiyama, B. Roizman, R. J. Roller, and B. Sodeik for the generous gift of antibodies and P. Schaffer and P. Desai for mutant viruses and complementary cell lines. We are also indebted to all laboratory members and Kerstin Radtke and Sylvie Laboissière for critical reading of the manuscript. We thank Beate Sodeik for her excellent suggestion to use Sepharose-coupled trypsin.
This research was funded by the Canadian Institutes for Health Research (to R.L.; MOP 49547 and MOP 82921).
Footnotes
Published ahead of print 13 February 2013
REFERENCES
- 1. Enquist LW, Husak PJ, Banfield BW, Smith GA. 1998. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 51:237–347 [DOI] [PubMed] [Google Scholar]
- 2. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 3. Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 9:382–394 [DOI] [PubMed] [Google Scholar]
- 4. Henaff D, Radtke K, Lippe R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449 [DOI] [PubMed] [Google Scholar]
- 5. Mettenleiter TC. 2006. Intriguing interplay between viral proteins during herpesvirus assembly or: the herpesvirus assembly puzzle. Vet. Microbiol. 113:163–169 [DOI] [PubMed] [Google Scholar]
- 6. Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, Mettenleiter TC. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 75:3675–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baines JD, Hsieh CE, Wills E, Mannella C, Marko M. 2007. Electron tomography of nascent herpes simplex virus virions. J. Virol. 81:2726–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J. Virol. 82:8605–8618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sodeik B, Ebersold MW, Helenius A. 1997. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 136:1007–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee GE, Murray JW, Wolkoff AW, Wilson DW. 2006. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 80:4264–4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wolfstein A, Nagel C-H, Radtke K, Döhner K, Allan VJ, Sodeik B. 2006. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 7:227–237 [DOI] [PubMed] [Google Scholar]
- 12. Shanda SK, Wilson DW. 2008. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 82:7388–7394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelly BJ, Fraefel C, Cunningham AL, Diefenbach RJ. 2009. Functional roles of the tegument proteins of herpes simplex virus type 1. Virus Res. 145:173–186 [DOI] [PubMed] [Google Scholar]
- 14. Radtke K, Kieneke D, Wolfstein A, Michael K, Steffen W, Scholz T, Karger A, Sodeik B. 2010. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 6:e1000991 doi:10.1371/journal.ppat.1000991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 20:4922–4931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Copeland AM, Newcomb WW, Brown JC. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 83:1660–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 83:6610–6623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liashkovich I, Hafezi W, Kuhn JM, Oberleithner H, Shahin V. 2011. Nuclear delivery mechanism of herpes simplex virus type 1 genome. J. Mol. Recognit. 24:414–421 [DOI] [PubMed] [Google Scholar]
- 19. Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. 2012. A nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J. Virol. 86:8998–9014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 86:3682–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rajčáni J, Andrea V, Ingeborg R. 2004. Peculiarities of herpes simplex virus (HSV) transcription: an overview. Virus Genes 28:293–310 [DOI] [PubMed] [Google Scholar]
- 22. Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mbong EF, Woodley L, Dunkerley E, Schrimpf JE, Morrison LA, Duffy C. 2012. Deletion of the herpes simplex virus 1 UL49 gene results in mRNA and protein translation defects that are complemented by secondary mutations in UL41. J. Virol. 86:12351–12361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Everett RD. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761–770 [DOI] [PubMed] [Google Scholar]
- 25. Mossman KL, Smiley JR. 2002. Herpes simplex virus ICP0 and ICP34.5 counteract distinct interferon-induced barriers to virus replication. J. Virol. 76:1995–1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jurak I, Silverstein LB, Sharma M, Coen DM. 2012. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J. Virol. 86:10093–10102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xing J, Wang S, Lin R, Mossman KL, Zheng C. 2012. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J. Virol. 86:3528–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Omar OS, Simmons AJ, Andre NM, Wilson DW, Gross ST. 2013. Pseudorabies virus and herpes simplex virus type 1 utilize different tegument-glycoprotein interactions to mediate the process of envelopment. Intervirology 56:50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klupp BG, Granzow H, Mettenleiter TC. 2000. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 74:10063–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yamauchi Y, Shiba C, Goshima F, Nawa A, Murata T, Nishiyama Y. 2001. Herpes simplex virus type 2 UL34 protein requires UL31 protein for its relocation to the internal nuclear membrane in transfected cells. J. Gen. Virol. 82:1423–1428 [DOI] [PubMed] [Google Scholar]
- 31. Fuchs W, Klupp BG, Granzow H, Osterrieder N, Mettenleiter TC. 2002. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 76:364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leelawong M, Guo D, Smith GA. 2011. A physical link between the pseudorabies virus capsid and the nuclear egress complex. J. Virol. 85:11675–11684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang K, Baines JD. 2011. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. U. S. A. 108:14276–14281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Roller RJ, Bjerke SL, Haugo AC, Hanson S. 2010. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J. Virol. 84:3921–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roller RJ, Haugo AC, Kopping NJ. 2011. Intragenic and extragenic suppression of a mutation in herpes simplex virus 1 UL34 that affects both nuclear envelope targeting and membrane budding. J. Virol. 85:11615–11625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 78:399–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J. Gen. Virol. 81:2397–2405 [DOI] [PubMed] [Google Scholar]
- 38. Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 83:3115–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 65:5757–5764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Klupp BG, Granzow H, Mettenleiter TC. 2001. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol. 82:2363–2371 [DOI] [PubMed] [Google Scholar]
- 41. Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 76:8939–8952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 79:9325–9331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mou F, Wills E, Baines JD. 2009. Phosphorylation of the UL31 protein of herpes simplex virus 1 by the US3 encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 83:5181–5191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 74:117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(l)31 and u(l)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 75:8803–8817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wagenaar F, Pol JM, Peeters B, Gielkens AL, de Wind N, Kimman TG. 1995. The US3-encoded protein kinase from pseudorabies virus affects egress of virions from the nucleus. J. Gen. Virol. 76:1851–1859 [DOI] [PubMed] [Google Scholar]
- 47. Chang YE, Van Sant C, Krug PW, Sears AE, Roizman B. 1997. The null mutant of the U(L)31 gene of herpes simplex virus 1: construction and phenotype in infected cells. J. Virol. 71:8307–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ott M, Tascher G, Hassdenteufel S, Zimmermann R, Haas J, Bailer SM. 2011. Functional characterization of the essential tail anchor of the herpes simplex virus type 1 nuclear egress protein pUL34. J. Gen. Virol. 92:2734–2745 [DOI] [PubMed] [Google Scholar]
- 49. Granzow H, Klupp BG, Mettenleiter TC. 2004. The pseudorabies virus US3 protein is a component of primary and of mature virions. J. Virol. 78:1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Padula ME, Sydnor ML, Wilson DW. 2009. Isolation and preliminary characterization of herpes simplex virus 1 primary enveloped virions from the perinuclear space. J. Virol. 83:4757–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Read GS, Patterson M. 2007. Packaging of the virion host shutoff (Vhs) protein of herpes simplex virus: two forms of the Vhs polypeptide are associated with intranuclear B and C capsids, but only one is associated with enveloped virions. J. Virol. 81:1148–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. McNabb DS, Courtney RJ. 1992. Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology 190:221–232 [DOI] [PubMed] [Google Scholar]
- 53. Abaitua F, O'Hare P. 2008. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 82:5234–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schmitz JB, Albright AG, Kinchington PR, Jenkins FJ. 1995. The UL37 protein of herpes simplex virus type 1 is associated with the tegument of purified virions. Virology 206:1055–1065 [DOI] [PubMed] [Google Scholar]
- 55. Lee JI, Luxton GW, Smith GA. 2006. Identification of an essential domain in the herpesvirus VP1/2 tegument protein: the carboxy terminus directs incorporation into capsid assemblons. J. Virol. 80:12086–12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J. Virol. 86:6303–6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bucks MA, O'Regan KJ, Murphy MA, Wills JW, Courtney RJ. 2007. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 361:316–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-filled HSV-1 capsids. Mol. Cell 26:479–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Klupp BG, Fuchs W, Granzow H, Nixdorf R, Mettenleiter TC. 2002. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 76:3065–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mohl BS, Bottcher S, Granzow H, Kuhn J, Klupp BG, Mettenleiter TC. 2009. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 83:9641–9651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 74:11608–11618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abaitua F, Souto RN, Browne H, Daikoku T, O'Hare P. 2009. Characterization of the herpes simplex virus (HSV)-1 tegument protein VP1-2 during infection with the HSV temperature-sensitive mutant tsB7. J. Gen. Virol. 90:2353–2363 [DOI] [PubMed] [Google Scholar]
- 63. Klupp BG, Granzow H, Mundt E, Mettenleiter TC. 2001. Pseudorabies virus ul37 gene product is involved in secondary envelopment. J. Virol. 75:8927–8936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Desai P, Sexton GL, McCaffery JM, Person S. 2001. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 75:10259–10271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Luxton GW, Lee JI, Haverlock-Moyns S, Schober JM, Smith GA. 2006. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 80:201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Leege T, Granzow H, Fuchs W, Klupp BG, Mettenleiter TC. 2009. Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J. Gen. Virol. 90:1560–1568 [DOI] [PubMed] [Google Scholar]
- 67. Roberts AP, Abaitua F, O'Hare P, McNab D, Rixon FJ, Pasdeloup D. 2009. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 83:105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Elliott G, Hafezi W, Whiteley A, Bernard E. 2005. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J. Virol. 79:9735–9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Sedlackova L, Rice SA. 2008. Herpes simplex virus type 1 immediate-early protein ICP27 is required for efficient incorporation of ICP0 and ICP4 into virions. J. Virol. 82:268–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Yao F, Courtney RJ. 1989. A major transcriptional regulatory protein (ICP4) of herpes simplex virus type 1 is associated with purified virions. J. Virol. 63:3338–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Yao F, Courtney RJ. 1992. Association of ICP0 but not ICP27 with purified virions of herpes simplex virus type 1. J. Virol. 66:2709–2716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kalamvoki M, Qu J, Roizman B. 2008. Translocation and colocalization of ICP4 and ICP0 in cells infected with herpes simplex virus 1 mutants lacking glycoprotein E, glycoprotein I, or the virion host shutoff product of the UL41 gene. J. Virol. 82:1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Yang TY, Courtney RJ. 1995. Influence of the host cell on the association of ICP4 and ICP0 with herpes simplex virus type 1. Virology 211:209–217 [DOI] [PubMed] [Google Scholar]
- 74. McLauchlan J, Addison C, Craigie MC, Rixon FJ. 1992. Noninfectious L-particles supply functions which can facilitate infection by HSV-1. Virology 190:682–688 [DOI] [PubMed] [Google Scholar]
- 75. Szilagyi JF, Cunningham C. 1991. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 72:661–668 [DOI] [PubMed] [Google Scholar]
- 76. Delboy MG, Siekavizza-Robles CR, Nicola AV. 2010. Herpes simplex virus tegument ICP0 is capsid associated, and its E3 ubiquitin ligase domain is important for incorporation into virions. J. Virol. 84:1637–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maringer K, Elliott G. 2010. Recruitment of herpes simplex virus type 1 immediate-early protein ICP0 to the virus particle. J. Virol. 84:4682–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Loret S, Lippe R. 2012. Biochemical analysis of infected cell polypeptide (ICP)0, ICP4, UL7 and UL23 incorporated into extracellular herpes simplex virus type 1 virions. J. Gen. Virol. 93:624–634 [DOI] [PubMed] [Google Scholar]
- 79. Remillard-Labrosse G, Guay G, Lippe R. 2006. Reconstitution of herpes simplex virus type 1 nuclear capsid egress in vitro. J. Virol. 80:9741–9753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lamberti C, Weller SK. 1998. The herpes simplex virus type 1 cleavage/packaging protein, UL32, is involved in efficient localization of capsids to replication compartments. J. Virol. 72:2463–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Newcomb WW, Homa FL, Brown JC. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 80:6286–6294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Sheaffer AK, Newcomb WW, Brown JC, Gao M, Weller SK, Tenney DJ. 2000. Evidence for controlled incorporation of herpes simplex virus type 1 UL26 protease into capsids. J. Virol. 74:6838–6848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yeh L, Schaffer PA. 1993. A novel class of transcripts expressed with late kinetics in the absence of ICP4 spans the junction between the long and short segments of the herpes simplex virus type 1 genome. J. Virol. 67:7373–7382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 79:8847–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. DeLuca NA, Schaffer PA. 1987. Activities of herpes simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense peptides. Nucleic Acids Res. 15:4491–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Taus NS, Salmon B, Baines JD. 1998. The herpes simplex virus 1 UL 17 gene is required for localization of capsids and major and minor capsid proteins to intranuclear sites where viral DNA is cleaved and packaged. Virology 252:115–125 [DOI] [PubMed] [Google Scholar]
- 87. Nozawa N, Daikoku T, Yamauchi Y, Takakuwa H, Goshima F, Yoshikawa T, Nishiyama Y. 2002. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genes 24:257–266 [DOI] [PubMed] [Google Scholar]
- 88. Wada K, Goshima F, Takakuwa H, Yamada H, Daikoku T, Nishiyama Y. 1999. Identification and characterization of the UL14 gene product of herpes simplex virus type 2. J. Gen. Virol. 80:2423–2431 [DOI] [PubMed] [Google Scholar]
- 89. Cohen GH, Ponce de Leon M, Diggelmann H, Lawrence WC, Vernon SK, Eisenberg RJ. 1980. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J. Virol. 34:521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Isola VJ, Eisenberg RJ, Siebert GR, Heilman CJ, Wilcox WC, Cohen GH. 1989. Fine mapping of antigenic site II of herpes simplex virus glycoprotein D. J. Virol. 63:2325–2334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ward PL, Campadelli-Fiume G, Avitabile E, Roizman B. 1994. Localization and putative function of the UL20 membrane protein in cells infected with herpes simplex virus 1. J. Virol. 68:7406–7417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Munger J, Chee AV, Roizman B. 2001. The U(S)3 protein kinase blocks apoptosis induced by the d120 mutant of herpes simplex virus 1 at a premitochondrial stage. J. Virol. 75:5491–5497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Donnelly M, Verhagen J, Elliott G. 2007. RNA binding by the herpes simplex virus type 1 nucleocytoplasmic shuttling protein UL47 is mediated by an N-terminal arginine-rich domain that also functions as its nuclear localization signal. J. Virol. 81:2283–2296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Elliott G, O'Reilly D, O'Hare P. 1996. Phosphorylation of the herpes simplex virus type 1 tegument protein VP22. Virology 226:140–145 [DOI] [PubMed] [Google Scholar]
- 95. McLean C, Buckmaster A, Hancock D, Buchan A, Fuller A, Minson A. 1982. Monoclonal antibodies to three non-glycosylated antigens of herpes simplex virus type 2. J. Gen. Virol. 63:297–305 [DOI] [PubMed] [Google Scholar]
- 96. Cross AM, Hope RG, Marsden HS. 1987. Generation and properties of the glycoprotein E-related 32K/34K/35K and 55K/57K polypeptides encoded by herpes simplex virus type 1. J. Gen. Virol. 68:2093–2104 [DOI] [PubMed] [Google Scholar]
- 97. Remillard-Labrosse G, Lippe R. 2011. In vitro nuclear egress of herpes simplex virus type 1 capsids. Methods 55:153–159 [DOI] [PubMed] [Google Scholar]
- 98. Church GA, Wilson DW. 1997. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 71:3603–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Lippe R, Miaczynska M, Rybin V, Runge A, Zerial M. 2001. Functional synergy between Rab5 effector Rabaptin-5 and exchange factor Rabex-5 when physically associated in a complex. Mol. Biol. Cell 12:2219–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Aggarwal A, Miranda-Saksena M, Boadle RA, Kelly BJ, Diefenbach RJ, Alam W, Cunningham AL. 2012. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 86:6123–6137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Tang L, Johnson JE. 2002. Structural biology of viruses by the combination of electron cryomicroscopy and X-ray crystallography. Biochemistry 41:11517–11524 [DOI] [PubMed] [Google Scholar]
- 102. Zhou ZH, Dougherty M, Jakana J, He J, Rixon FJ, Chiu W. 2000. Seeing the herpesvirus capsid at 8.5 A. Science 288:877–880 [DOI] [PubMed] [Google Scholar]
- 103. Zhu Z, Schaffer PA. 1995. Intracellular localization of the herpes simplex virus type 1 major transcriptional regulatory protein, ICP4, is affected by ICP27. J. Virol. 69:49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Lopez P, Van Sant C, Roizman B. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 75:3832–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zeskind BJ, Jordan CD, Timp W, Trapani L, Waller G, Horodincu V, Ehrlich DJ, Matsudaira P. 2007. Nucleic acid and protein mass mapping by live-cell deep-ultraviolet microscopy. Nat. Methods 4:567–569 [DOI] [PubMed] [Google Scholar]
- 106. Ren Y, Bell S, Zenner HL, Lau SY, Crump CM. 2012. Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J. Gen. Virol. 93:319–329 [DOI] [PubMed] [Google Scholar]
- 107. Heine JW, Honess RW, Cassai E, Roizman B. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Yao F, Schaffer PA. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Morrison EE, Stevenson AJ, Wang YF, Meredith DM. 1998. Differences in the intracellular localization and fate of herpes simplex virus tegument proteins early in the infection of Vero cells. J. Gen. Virol. 79:2517–2528 [DOI] [PubMed] [Google Scholar]
- 110. Yang K, Wills EG, Baines JD. 2012. Release of the herpes simplex virus 1 protease by self cleavage is required for proper conformation of the portal vertex. Virology 429:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Cardone G, Newcomb WW, Cheng N, Wingfield PT, Trus BL, Brown JC, Steven AC. 2012. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J. Virol. 86:4058–4064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Cockrell SK, Sanchez ME, Erazo A, Homa FL. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J. Virol. 83:47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. 2011. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J. Virol. 85:4875–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Newcomb WW, Brown JC. 2010. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 84:9408–9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Newcomb WW, Brown JC. 2009. Time-dependent transformation of the herpesvirus tegument. J. Virol. 83:8082–8089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. del Rio T, DeCoste CJ, Enquist LW. 2005. Actin is a component of the compensation mechanism in pseudorabies virus virions lacking the major tegument protein VP22. J. Virol. 79:8614–8619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Michael K, Klupp BG, Mettenleiter TC, Karger A. 2006. Composition of pseudorabies virus particles lacking tegument protein US3, UL47, or UL49 or envelope glycoprotein E. J. Virol. 80:1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]