

Abstract
Influenza virus is well recognized to modulate host tropism and pathogenesis based on mutations in the proteolytic cleavage site of the viral hemagglutinin (HA), which activates HA and exposes the fusion peptide for membrane fusion. Instead of the conventional trypsin-mediated cleavage event, modification of the cleavage site allows extended use of host cell proteases and enhanced spread in vivo. For H1N1 influenza viruses, the mouse-adapted A/WSN/33 strain is known to replicate in the brain based on recruitment of plasminogen by the viral neuraminidase (NA), as well as a Ser-Tyr substitution at the P2 position of the HA cleavage site. Here, we show that an equivalent Ser-Tyr substitution has occurred in the HA of naturally occurring human H1N1 influenza viruses. We characterize one of these viruses (A/Beijing/718/2009), as well as the prototype A/California/04/2009 with a Ser-Tyr substitution in the cleavage site, and show that these HAs are preferentially cleaved by plasmin. Importantly, cleavage activation by plasmin/plasminogen was independent of the viral NA, suggesting a novel mechanism for HA cleavage activation. We show that the viral HA itself can recruit plasminogen for HA cleavage. We further show that cellular factors, as well as streptokinase from bacteria commonly coinfecting the respiratory tract of influenza patients, can be a source of activated plasminogen for plasmin-mediated cleavage of influenza virus HAs that contain a Ser-Tyr substitution in the cleavage site.
INTRODUCTION
Influenza virus remains a major cause of morbidity and mortality in the human population (1), with the ever-present risk of new pandemics arising—especially as infections by emerging viruses from zoonotic transmission events. The influenza virus hemagglutinin (HA) mediates both receptor binding and membrane fusion during virus entry (2). A key step in activation of HA is its cleavage by host cell proteases (3–5), where proteolytic cleavage activation is directly related to exposure of the fusion peptide (6). Low-pathogenicity influenza virus strains contain a HA cleavage site with a single arginine residue and are thus described as having monobasic cleavage sites. These viruses can utilize trypsin (or other trypsin-like serine proteases) for activation, with the tissue distribution of the activating protease typically restricting infection to the respiratory and/or intestinal organs (7). In the case of highly pathogenic avian influenza (HPAI) viruses such as H5N1 and H7N7, it is well established that mutations in the region of the HA cleavage site lead to an insertion of several arginine or lysine residues in addition to the cleavage site arginine. Such HAs can be recognized by furin or related intracellular serine proteases found in many cell types, allowing a widening of the cell tropism of the virus (8). The presence of a polybasic cleavage site is critical for the systemic spread and increased virulence associated with HPAI.
Influenza A viruses exist as many different subtypes (H1 to H17), with H1 and H3 viruses currently infecting humans and H1N1 viruses responsible for the 2009 influenza pandemic (1, 9). With the exception of the 1918 influenza virus, H1 viruses are considered to have low pathogenicity and as such have a monobasic cleavage site. However, two H1 isolates, A/WSN/33 (WSN) and A/NWS/33 (NWS), have been selected to propagate in mouse brain and are thus considered to be highly pathogenic, neurovirulent viruses in mice (10, 11). The A/WSN/33 virus in particular has been used extensively for studies on influenza virus replication and pathogenesis, in part because this virus forms plaques in the absence of trypsin and serves as a model of highly pathogenic influenza virus. The HA of the A/WSN/33 virus was originally shown to be cleaved by plasmin, following activation of serum plasminogen in MDBK cells (12). The virulence properties of A/WSN/33 were subsequently linked to the neuraminidase (NA) gene (13) and the absence of a glycosylation site at position 130 of NA (14). In addition, the presence of a C-terminal lysine on NA was shown to be critical for the virulence properties of A/WSN/33, with the viral NA binding and sequestering plasminogen on the cell surface, leading to increased cleavage of HA (15, 16). Hiti and colleagues originally suggested that plasmin-mediated cleavage of A/WSN/33 HA may be influenced by residues neighboring the HA cleavage site arginine (17), and we have recently determined that a mutation in the HA cleavage site of A/WSN/33 is important for plasmin-mediated cleavage and neurovirulence (18). This is a Ser-Tyr substitution at the P2 cleavage site position, adjacent to the P1 cleavage site Arg. HA cleavage data are consistent with biochemical data showing that bulky hydrophobic residues in the P2 position of substrates strongly promote their plasmin-mediated cleavage (19–21).
While A/WSN/33 represents a specific and highly laboratory-adapted form of influenza virus, ongoing influenza surveillance studies have shown that an equivalent Ser-Tyr substitution in the P2 cleavage site position of HA has also occurred in certain naturally occurring influenza viruses. Here, we studied the cleavage activation of the HA of one of these naturally occurring influenza viruses (A/Beijing/718/09), as well as the prototype A/California/04/09 H1N1 virus with a Ser-Tyr substitution in the P2 cleavage site position of HA. We show that the presence of a P2 Tyr markedly enhances plasmin-mediated cleavage of naturally occurring influenza viruses. Significantly, plasmin-mediated cleavage activation occurs independently of the viral neuraminidase and so represents a distinct mechanism for enhanced cleavage activation of HA in vivo.
MATERIALS AND METHODS
Cells, viruses, plasmids, and reagents.
293T, MDCK.1, MDCK.2, and MDCK cells (American Type Culture Collection [ATCC]) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco), 100 units/ml of penicillin (Pen), 10 μg/ml of streptomycin (Strep; Cellgro), and 25 mM HEPES (Cellgro) at 37°C in a 5% CO2 incubator. The CA0409 reverse genetic system was a generous gift from Toru Takimoto (University of Rochester). Recombinant CA0409 (rCA0409) wild-type (WT) and rCA0409 HA-328Y influenza virus strains were produced as described previously (22). Briefly, 12 plasmids were cotransfected in 293T/MDCK cells for 48 h using Lipofectamine 2000 (Invitrogen). The viruses were then plaque purified and amplified on MDCK cells grown in DMEM–0.2% bovine serum albumin (BSA)–HEPES (25 mM)–Pen/Strep (10 μg/ml) in the presence of 3 μg/ml of tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin. The HA and NA genes were sequenced and confirmed to be free of unwanted mutations. The A/Beijing/718/2009 (Bei718) HA gene was synthesized by GeneArt/Life Technologies based on the sequence in NBCI GenBank (accession no. ACZ98546). All the hemagglutinin genes were subcloned into the pEF4 vector (Invitrogen), while neuraminidase genes were subcloned into the pCAGGS vector unless otherwise specified. Point mutations were introduced by the use of a QuikChange site-directed mutagenesis kit (Aglient) following the user manual. TPCK-trypsin was obtained from Thermo Scientific, human plasmin (h-Pm) and plasminogen (h-Plg) were obtained from CalBiochem, streptokinase (group C) was obtained from Sigma, and 6-aminohexanoic acid was obtained from Alfa Aesar.
Surface biotinylation and Western blot analysis.
293T cells were grown on poly-d-lysine-treated 24-well plates to 60% to 70% confluence and transfected with 500 ng of plasmid using Lipofectamine 2000 for 18 h. Transfected cells were washed with phosphate-buffered saline (PBS) and underwent different treatments in DMEM–0.2% BSA as specified in each experiment. After the treatment, the plates were kept at 4°C on ice and were washed with PBS and incubated with sulfo-NHS-SS-biotin (Sigma) (250 μg/ml) for 30 min. Excess biotin was quenched with glycine (50 mM) for 10 min. Cells were washed with PBS and then lysed by the use of 1× radioimmunoprecipitation assay (RIPA) buffer (Millipore) with Complete protease inhibitor cocktail tablets (Roche) for 10 min. Lysed cells were centrifuged at 18,000 × g for 20 min at 4°C, and supernatant was collected and added to 30 μl of a 50% suspension of streptavidin agarose beads (Thermo) and incubated at 4°C on a rotating shaker for 18 h to pull down all the biotinylated proteins. The beads were washed 3 times with RIPA buffer and resuspended in 30 μl of 2× Laemmli sample buffer–10% beta-mercaptoethanol for Western blotting. HA bands were detected by primary goat α-PR8 H0 antibody (BEI Resources; catalog no. NR-3148) and secondary rabbit α-goat conjugated to horseradish peroxidase (Thermo Scientific).
Quantification of HA cleavage.
Western blot images were taken using a FujiFilm LAS-3000 imaging system. The pixel intensity of individual bands was measured by Image J, and relative cleavage efficiencies were calculated by the following equation: (HA1/HA0 + HA1) × 100%.
Immunoplaque assay.
MDCK cells were grown on 12-well plates to 90% to 100% confluence. Viruses were serially diluted in RPMI medium and incubated on MDCK cells for 1 h at 37°C. MDCK cells were washed 2 times with PBS before and after inoculation. Plaque assay medium (DMEM–0.2% BSA–HEPES–Pen/Strep–1% SeaPlaque agarose [Lonza]) was added along with proteases and/or inhibitors as specified in each experiment. Plates were incubated at room temperature for 10 min for the agarose to solidify and inversely incubated at 37°C in 5% CO2 in an incubator for 3 days. Cells were fixed in 10% formalin for 1 h and permeabilized with 0.5% Triton X-100 for 3 min. Virally infected cells were detected using primary mouse α-nucleoprotein (NP) and secondary goat α-mouse antibody conjugated to alkaline phosphatase (Jackson ImmunoResearch). Individual plaques were visualized by adding 5-bromo-4-chloro-3-indolylphosphate/Nitro Blue Tetrazolium (BCIP/NBT) substrate (Vector Labs). Plaques were counted, and viral titers were calculated.
Live-cell plasminogen binding assay and flow cytometry.
293T cells were grown on 12-well plates to 60% to 70% confluence and transfected with 1 μg of plasmid DNA using Lipofectamine 2000 for 18 h. Cells were gently washed with PBS and incubated in DMEM–0.2% BSA–HEPES for 30 min at 37°C to remove any trace plasminogen in FBS. The plates were chilled on ice, and cells were detached in 10 mM EDTA for 10 min. Cell pellets were collected by low-speed centrifugation at 4°C and resuspended in 100 μl of 2 μM human plasminogen–DMEM–0.2% BSA–HEPES for 30 min at 4°C on a rotating shaker. Nonbound plasminogen was removed by washing 3 times with Dulbecco's PBS (DPBS). H-Plg was labeled using primary mouse α-plasminogen (GeneTex) and secondary chicken α-mouse Alexa 647 (Invitrogen). Labeled cells were chilled on ice and immediately analyzed by fluorescence-activated cell sorter (FACS) analysis on an LSR II system (BD Biosciences, San Diego, CA). FACS data were further analyzed using FlowJo software (Tree Star, Ashland, OR).
Viral pathogenesis in mouse model.
For intranasal inoculation, 20 8-week-old male BALB/c mice, 8 for each experimental group and 4 for the PBS control, were inoculated intranasally with 105 PFU of virus diluted in 50 μl of PBS. Mouse body weights were measured daily for 6 days. Half of the mice were euthanized on day 3 and the other half on day 6. The indicated tissues were harvested, weighed, and stored at −80°C. An immunoplaque assay was performed on 20% tissue homogenate–PBS. For intracranial inoculation, 13 5-week-old male BALB/c mice were used, 4 for each experimental group, 3 for the WSN control, and 2 for an artificial cerebral spinal fluid (ACSF; Tocris) control. Each mouse was intracranially inoculated with 103 PFU of virus diluted in 5 μl of ACSF, using a Hamilton syringe. Mouse body weight was measured every 12 h for 3 days. All mice were euthanized on day 3. Brain tissues were collected, and viral titer determinations were performed as described for the intranasal inoculation. All work with animals was carried out according to the Cornell University Animal Care and Use program under Animal Welfare Assurance A3347-01 and complied with the Public Health Service Policy on Humane Care and Use of Laboratory Animals.
RESULTS
Bioinformatic analysis of the influenza virus H1 cleavage site.
A multiple-sequence alignment of the HA genes of representative H1 influenza viruses revealed an invariant Arg at position 329 (numbering based on H3 subtype virus A/Aichi/2/68), which comprises the P1 position of the proteolytic cleavage site (Fig. 1A). In addition to the invariant Arg at the cleavage site, H1 influenza viruses usually contain a highly conserved Ser at position 328 (the P2 cleavage position). A/WSN/33 is an exception to the consensus cleavage site, containing Tyr at position 328. The HAs of A/Beijing/718/2009 (NCBI accession no. ACZ98546) and A/Beijing/720/2009 (NCBI accession no. ACZ98560) also represent exceptions to the consensus cleavage site, as they also contain a Tyr at position 328. A/Beijing/718/2009 and A/Beijing/720/2009 were sequenced from human nasal swabs as part of an analysis of an outbreak of pandemic influenza in Beijing, China, in November 2009. No other information on these viruses is currently available. Additional viruses also contain an equivalent substitution (see Discussion) (Fig. 1B).
Fig 1.

Multiple-sequence alignment of the influenza virus H1 HA cleavage site and fusion peptide. (A) A total of 4,325 H1N1 HA amino acid (aa) sequences of H1N1 from the NCBI Influenza Virus Resource (http://www.ncbi.nlm.nih.gov/genomes/FLU/FLU.html) were aligned using muscle v3.8.31, and the aligned sequences of representative viruses at the HA cleavage site are shown along with the consensus sequence. Dots represent identical residues, P2 to P1′ represent positions of the cleavage site, and the location of the fusion peptide is shown. The representative strains selected are the following: A/South Carolina/1/1918 (accession number AAD17229), A/Puerto Rico/8/1934 (ABO21709), A/Wilson-Smith/1933 (ABD77796), A/New Caledonia/20/1999 (ABF21272), A/California/04/2009 (ACP41105), A/WSN/33 (AAA43209), A/Beijing/718/2009 (ACZ98546), and A/Beijing/720/2009 (ACZ98560). (B) List of H1 viruses with serine (S)-to-tyrosine (Y) mutation at the P2 position of the cleavage site, along with the information on subtype, accession number, and species of isolation.
Cleavage activation of A/Beijing/718/2009 HA by plasmin.
Based on the prediction that the presence of a Tyr at the P2 cleavage site position would increase HA cleavability by plasmin, we examined HA cleavage for the A/Beijing/718/2009 HA. An influenza virus A/Beijing/718/2009 HA gene was synthesized based on the database sequence. We compared this HA to a consensus A/California/04/09 HA as well as the A/Beijing/718/2009 HA with a mutation of Tyr-Ser to match the H1N1 consensus. HAs were expressed in 293T cells, and surface biotinylation was performed. Cells were then treated with human plasmin and HA detected by Western blotting to assess the efficiency of plasmin-mediated cleavage. Around 32% of the wild-type A/Beijing/718/2009 HA was cleaved by plasmin after 60 min (Fig. 2). In contrast, A/Beijing/718/2009 (Y328S) HA showed approximately four times less cleavage by plasmin (Fig. 2).
Fig 2.
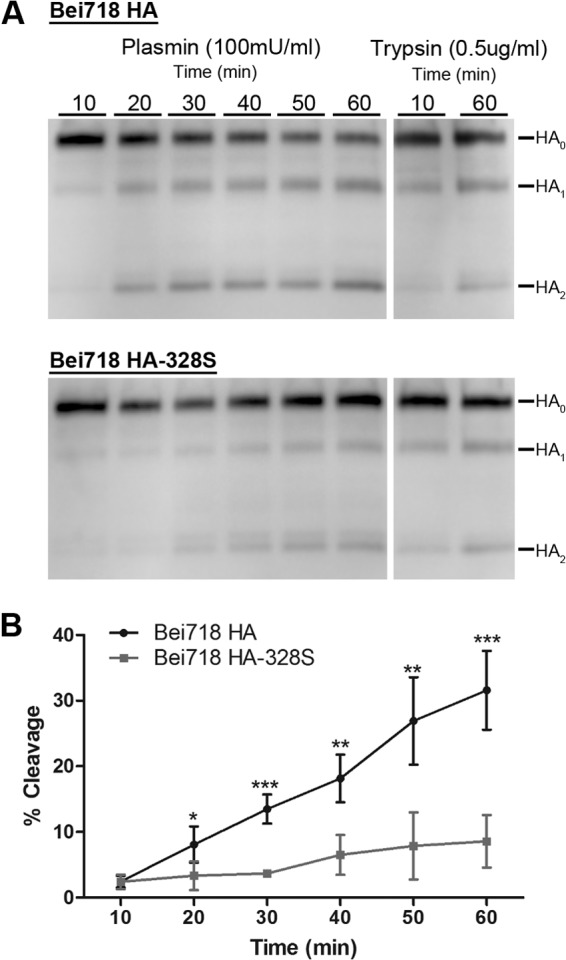
Cleavage of A/Beijing/718/2009 influenza virus HAs by human plasmin. (A) Human plasmin-dependent HA cleavage of A/Beijing/718/2009 (Bei718) HA and Bei718 HA-328S. Surface biotinylation was performed on 293T cells expressing each HA. Transfected cells were treated with human plasmin (100 mU/ml) at the indicated times, and the cleavage product(s) was detected by Western blot analysis using anti-HA antibody. (B) Quantification of the time-dependent human plasmin-mediated HA cleavage efficiency of Bei718 HA and Bei718 HA-328S. The error bars represent the standard deviations of the results of three independent experiments. Statistical analyses were performed using Student's t test (unpaired, one tail) by GraphPad Prism, comparing the individual treatment times (*, P < 0.05; **, P < 0.005; ***, P < 0.0001).
As a control, we also mutated the A/California/04/09 HA to replace the P2 Ser with Tyr. Similar to Beijing/718/2009 HA, the Ser-Tyr mutation at the P2 position of A/California/04/09 HA significantly increased HA cleavage by plasmin (Fig. 3). The higher level of HA cleavage in (codon-optimized) A/Beijing/718/2009 than in (non-codon-optimized) A/California/04/09 likely correlates to the differences in expression level.
Fig 3.
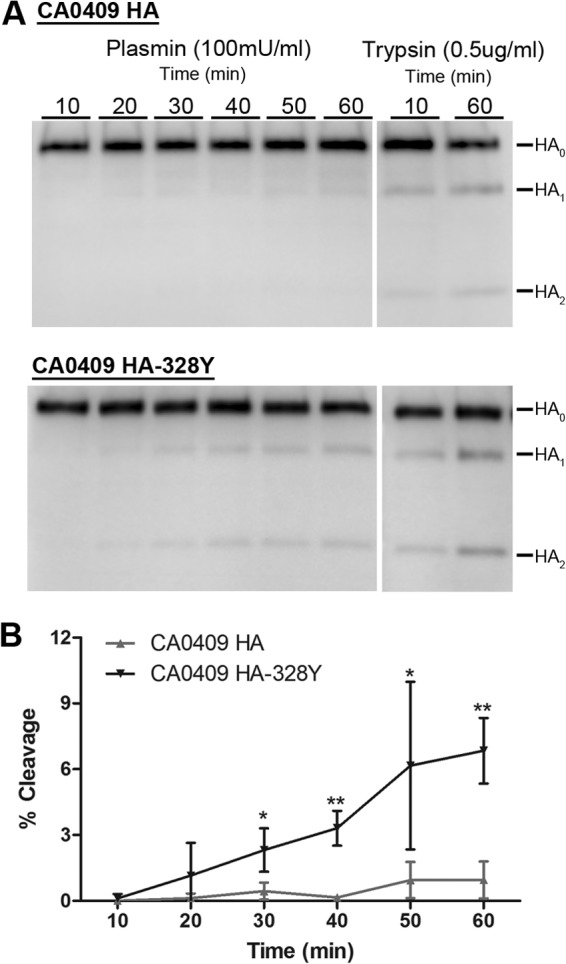
Cleavage of the A/California/04/2009 influenza virus HAs by human plasmin. (A) Human plasmin-dependent HA cleavage of A/California/04/09 (CA0409) HA and CA0409 HA-328Y. Surface biotinylation was performed on 293T cells expressing each HA. Transfected cells were treated with human plasmin (100 mU/ml) at the indicated times, and the cleavage product(s) was detected by Western blot analysis using anti-HA antibody. (B) Quantification of the time-dependent human plasmin-mediated HA cleavage efficiency of CA0409 HA and CA0409 HA-328Y. The error bars represent the standard deviations of the results of three independent experiments. Statistical analyses were performed using Student's t test (unpaired, one tail) by GraphPad Prism, comparing the individual treatment times (*, P < 0.05; **, P < 0.005).
Rescue of plasmin-activation H1N1 infection by introduction of Tyr at the P2 cleavage site position.
To determine whether the presence of Tyr at the P2 cleavage site position can rescue plasmin-mediated infection of pandemic H1N1 influenza virus, we introduced a S328Y mutation into the A/California/04/09 HA and rescued both wild-type and mutant recombinant viruses by reverse genetics and growth in embryonated chicken eggs. Despite our efforts, rescue of the intact A/Beijing/718/2009 HA on a A/California/04/09 background was unsuccessful. The introduction of the S328Y mutation into the A/California/04/09 HA resulted in increasing levels of plasmin-mediated cleavage such as were seen with A/Beijing/718/2009 (not shown). Whereas the wild-type A/California/04/09 formed plaques on MDCK cells after addition of exogenous trypsin, addition of plasmin failed to rescue plaque formation (Fig. 4A). In contrast, the A/California/04/09 HA S328Y mutant readily formed plaques in the presence of plasmin, as well as with trypsin (Fig. 4A). Plasmin-dependent plaque formation, but not trypsin-dependent plaque formation, was inhibited by the plasmin inhibitor 6-aminohexanoic acid (6-AHA). Interestingly, addition of serum alone did not rescue plaque formation for A/California/04/09 HA-S328Y, in contrast to the situation with A/WSN/33 (15, 16) (not shown). In addition to rescuing efficient plaque formation, the majority of plaques formed in the presence of plasmin were significantly bigger for the Y328 mutant compared to wild-type A/California/04/09 (Fig. 4B).
Fig 4.

Plaque-forming properties of recombinant virus on MDCK cells. Representative pictures of immunoplaque assays of A/California/04/2009 (rCA0409) (A) and rCA0409 HA-328Y (B) virus on MDCK cells are shown. Plaque assays were performed by inoculating 30 to 40 PFU virus on MDCK cells in the presence of TPCK-trypsin (1 μg/ml), human plasmin (h-Pm) (3 mU/ml), or 5% FBS or were mock treated with or without addition of 6-AHA (5 mg/ml) for 3 days. Cells were fixed, and viral NP were stained with mouse α-NP antibody (Ab). Secondary α-mouse Ab conjugated with alkaline phosphatase (AP) was used to detect virally infected cell clumps visually. Images were taken using an Alpha DigiDoc AD-1200 imager. At least three individual experiments were performed for each condition.
Effects of introduction of Tyr at the P2 cleavage site position in a mouse model of H1N1 influenza virus.
To investigate the effects of introducing a Tyr at the P2 cleavage site position of HA, we challenged mice with A/California/04/09 and the A/California/04/09 HA-S328Y mutant via both intranasal and intracranial routes of inoculation. Mice were first challenged intranasally, and samples of lung, trachea, kidney, spleen, and brain were taken at 3 or 6 days postinfection. Tissue samples were then assayed for the presence of virus by plaque assay in MDCK cells. Both A/California/04/09 and A/California/04/09 HA-S328Y replicated well in the lung and, to a limited extent, in the trachea, with no discernible differences between the wild-type and mutant viruses (Fig. 5A). There was no evidence of spread to the kidney, spleen, and brain in either case (Fig. 5A). Mice inoculated with viruses lost about 15% of their weight at the end of experiment, with no mortality observed, indicated limited pathogenesis in mouse. However, there was no difference in weight loss between A/California/04/09 and A/California/04/09 HA-328Y (Fig. 5B).
Fig 5.

Mouse model of viral pathogenesis following intranasal and intracranial inoculation. (A) Summary of viral titer results at 3 days or 6 days postintranasal inoculation and 3 days postintracranial inoculation. Values inside the parentheses indicate the portion of positive samples for viral detection in each group. Average titers were calculated based on positive samples and were represented as log10 PFU/g of tissue. <, viral titer below the detection limit (i.e., 20 PFU/ml). (B and C) Percentage of initial weight loss of mice. Mouse body weights were measured every 24 h for intranasal inoculation (B) or every 12 h for intracranial inoculation (C). Body weights were normalized to the initial body weight prior to viral inoculation.
We also challenged mice intracranially, with brain samples taken at 3 days postinfection, and assayed for the presence of virus by a plaque assay in MDCK cells. A/WSN/33 was included as a control for these experiments. While A/WSN/33 replicated in the brain, there was no evidence of replication of either A/California/04/09 or A/California/04/09 HA-S328Y after intracranial inoculation (Fig. 5A). Only A/WSN/33-infected mice showed limited signs of infection, with 5% weight loss at the end of experiment. In contrast, A/California/04/09 and A/California/04/09 HA-328Y showed no sign of infection and no weight loss, as with the ASCF control (Fig. 5C).
Role of the viral neuraminidase in cleavage activation of A/Beijing/718/2009 HA.
It is well established for the neurotropic mouse-adapted influenza virus A/WSN/33 that the viral neuraminidase (NA) plays a major role in recruiting the plasminogen needed for plasmin-mediated HA activation. To assess the role of the NA in the context of A/Beijing/718/2009, we examined the effect of coexpression of A/California/04/2009 NA on HA cleavage. A/California/04/2009 NA was chosen because of its high sequence identity to A/Beijing/718/2009 NA (2 mutations, V106I and N248D, corresponding to the N2 subtype). The mutations were not predicted to have any effect on plasminogen recruitment. The NA from A/WSN/33 was used as a positive control. These experiments were performed in the presence of plasminogen, the inactive precursor of plasmin. As expected, A/WSN/33 NA coexpression resulted in a greatly enhanced cleavage of A/Beijing/718/2009 HA (Fig. 6). In contrast, coexpression of A/California/04/2009 NA resulted in very limited cleavage of its cognate HA (Fig. 6) and was at a level equivalent to that seen in the absence of any NA expression.
Fig 6.
HA cleavage by human plasminogen independent from its cognate neuraminidase. (A) Time-dependent human plasminogen-mediated cleavage of A/Beijing/718/2009 (Bei718) HA and Bei718 HA-328S. Surface biotinylation was performed as described for Fig. 2 by cotransfecting Bei718 HA with vector control, WSN NA, or CA0409 NA and treated with human plasminogen (6 μg/ml) for the indicated duration. (B) Quantification of three individual experiments. The error bars represent the standard deviations of the results of three independent experiments, and statistical analyses were performed as described for Fig. 2.
While short-term (1 h) exposure to plasminogen allowed only minimal or no HA cleavage in the presence of A/California/04/2009 NA or vector control, longer-term treatment (6 h) did result in some degree of HA cleavage. To investigate this further, we used conditioned medium from MDCK cells to determine if endogenous plasmin activators were present. Media recovered from MDCK cells after 24 h of cell growth allowed HA cleavage activation in the presence of plasminogen that was equivalent to the level of cleavage with activated plasmin (Fig. 7). It is likely that MDCK cells release cellular factors such as tissue plasminogen activator (tPA) or urokinase, which would account for the low level of plasminogen-mediated HA activation seen in Fig. 5.
Fig 7.
HA cleavage by the use of conditioned medium from MDCK cells. (A) Cleavage of A/Beijing/718/2009 HA. Plasmin (Plg)-MDCK-conditioned medium was prepared by incubating human plasmin (h-Plg) (5 μg/ml) on MDCK cells for 18 h. The conditioned medium was used to treat HA-transfected cells for 30 min. Human plasmin (100 mU/ml) was used as a positive control and MDCK medium alone as a negative control. (B) Quantification of plasminogen (Plg)-MDCK-conditioned medium-mediated HA cleavage. Relative cleavage efficiencies were calculated as described for Fig. 2. The error bars represent the standard deviations of the results of three independent experiments, and statistical analyses were performed as described for Fig. 2.
Recruitment of plasminogen by influenza virus hemagglutinin.
Unlike the situation with A/WSN/33, our data show that H1N1 viruses can undergo cleavage activation by the plasmin/plasminogen system in an NA-independent manner. A similar situation has been reported for the 1918 influenza virus, where the viral HA facilitated robust plasminogen binding (23). To determine whether an equivalent situation exists for other influenza H1N1 viruses, including A/Beijing/718/2009 HA, we tested plasminogen binding of HA using flow cytometry. As with A/South Carolina/1/18 HA, the A/Beijing/718/2009 HA bound plasminogen (Fig. 8). In fact, plasminogen binding was a common property all the HAs tested. A/PR/8/34 and A/California/04/09 HA showed the most robust binding (Fig. 8), with HAs such as A/NWS/33 and A/WS/33 showing binding that was less efficient. In agreement with the findings in Fig. 4, A/California/04/2009 NA-transfected cells failed to recruit any plasminogen, as shown by FACS analysis. In these studies, we also included A/WSN/33 NA, which served as a positive control for plasminogen binding, as well as a mutated version of A/WSN/33 NA (R146N, ΔK470 [corresponding to the N2 subtype]) as a negative control. Combined, these mutations were shown to abolish plasminogen binding (14, 15).
Fig 8.
Plasminogen binding on HA- and NA-transfected cells by flow cytometry. (A) Percentage of human plasminogen-binding cells. A/WSN/33 (WSN) NA was used as a positive control, pEF4 was used for background binding, and WSN NA R146N ΔK470 was used as a negative control. CA0409 NA, eight different H1 HAs (WSN, A/South Carolina/1/1918 [1918], CA0409, Bei178, A/NWS/1934 [NWS], A/Wilson-Smith/1933 [WS], A/Puerto Rico/8/1934 [PR8], and A/New Caledonia/20/1999 [New Caledonia]), and one H9 HA (not shown) were tested. The error bars represent the standard deviations of the results of three independent experiments. Statistical analyses were performed as described for Fig. 2 by comparing each sample to the negative control. (B) Representative FACS traces are shown. All plots were overlaid with the background control (pEF4 empty vector) to illustrate the peak shift on the x axis (allophycocyanin [APC] staining for plasminogen). The y axis data represent the percentage of cells normalized to the maximum (Max) cell number in a single channel within the plot.
Bacterial streptokinase can recruit plasminogen for cleavage activation of A/Beijing/718/2009 HA.
As our data clearly indicated that A/Beijing/718/2009 HA was preferentially activated by plasmin in an NA-independent manner, we also examined other ways that plasminogen could be recruited and activated for HA cleavage. One likely possibility is via bacterial virulence factors. Bacterial coinfections are common in more severe cases of influenza, with Staphylococcus aureus, S. pneumoniae, S. pyogenes, and H. influenzae commonly coinfecting the respiratory tract and contributing to the pneumonia often associated with influenza morbidity and mortality (24, 25). Many of these bacterial species express virulence factors that can recruit and activate plasminogen, including streptokinase and staphylokinase (26). As an example of a virulence factor, we tested the effects of streptokinase from S. pyogenes (27). A/Beijing/718/2009 HA, or the Y328S mutant, was expressed on the surface of cells and biotinylated. Cells were then exposed to inactive plasminogen, bacterial streptokinase, or a combination of both inactive plasminogen and bacterial streptokinase, and the cleavability of HA was determined. Cells were exposed to trypsin as a positive control and with media only as a negative control. Exposure to a combination of both inactive plasminogen and bacterial streptokinase led to a significant increase in the cleavage of HA for A/Beijing/718/2009 HA (Fig. 9). In contrast, treatment with plasminogen and streptokinase led to a much-reduced level of HA activation for the Y328S mutant (Fig. 9).
Fig 9.
HA cleavage by bacterial streptokinase-activated human plasminogen. (A) Surface biotinylation analysis was performed as mentioned above. 293T cells transfected with A/Beijing/718/2009 (Bei718) HA or HA-328S mutant were treated with different proteases as indicated for 1 h. Cleavage products were detected by Western blot analysis using anti-HA antibody. (B) Quantification of the streptokinase/plasminogen (Skc-Plg)-dependent HA cleavage efficiency. Cleavage efficiencies were calculated. The error bars represent the standard deviations of the results of three independent experiments, and statistical analyses were performed as described for Fig. 2.
DISCUSSION
We show here that plasmin-plasminogen-mediated activation of influenza virus HA can be influenced by mutations in the HA cleavage site of circulating virus strains. While cleavage activation of influenza virus mediated by plasmin-plasminogen has been established for some time, its importance for virus infection has been restricted to the highly laboratory-adapted A/WSN/33 strain, which is known to specifically recruit plasminogen via the viral NA protein, with a Ser-Tyr mutation in the P2 position of the HA cleavage site contributing to efficient HA cleavage and enhanced viral pathogenesis. Here we show that an equivalent Ser-Tyr mutation in the HA of a circulating human H1N1 strain also contributes to enhanced cleavage by plasmin-plasminogen but without recruitment of plasminogen by NA.
Our study focused on one pandemic H1N1 virus, A/Beijing/718/2009; however, it should be noted that this is likely not an isolated example of a virus that is preferentially activated by plasmin. In addition to the A/Beijing/720/2009 virus isolated from the same surveillance site as A/Beijing/718/2009, a pandemic H1N1 virus with a cleavage P2 Ser-Tyr substitution, A/Taiwan/97196/2009, was also isolated in a separate surveillance study in humans (28), and several H1 viruses were also isolated from swine (see Fig. 1B). As with A/Beijing/718/2009, no clinical information is available for these viruses.
In addition to A/WSN/33, a role for plasmin-plasminogen has been reported for other influenza strains, with plasminogen activation mediated by the host cellular protein annexin II, which is incorporated into the virus particles (29). A role for plasmin-plasminogen has also been proposed for the 1918 influenza virus via direct binding to the viral HA (23). Our data show that sequestration of plasminogen by HA is a common property of distinct viral strains. However, in general, plasminogen binding by HA was less efficient than for the A/WSN/33 NA (not shown). HA-mediated binding of plasminogen likely works in concert with cellular factors such as annexin II, tPA, or urokinase and mutations in the HA cleavage site to promote HA cleavage activation.
One possible cell type where plasminogen is known to be highly active is endothelial cells, which are suggested to be important in the “cytokine storms” associated with highly pathogenic influenza viruses (30). Both A/California/04/09 and the Ser-Tyr mutant were able to infect blood microvascular endothelial cells (HMVEC-LBI; Lonza), as detected by immunofluorescence assays for viral HA and NP (not shown). However, experiments to address any increased virus replication in endothelial cells based on a Ser-Tyr cleavage site mutation or use of plasmin-plasminogen failed to show any productive rounds of virus infection compared to A/WSN/33, which appeared to replicate to a limited degree (not shown).
Although in vitro data show enhanced usage of plasmin for HA activation with Ser-Tyr substitutions at the P2 cleavage site position, data from the mouse model show no discernible differences between A/California/04/09 and A/California/04/09 HA-328Y. The situation in humans remains unresolved, with the lack of effect in the mouse possibly due to differences in the activating protease. This discrepancy could be addressed in cell culture models of the human respiratory epithelium. A lack of obvious in vivo effects may also be explained by the relatively poor cleavage of A/California/04/09 HA-328Y compared to A/Beijing/718/2009 HA. The data from intracranial inoculation showing that A/California/04/09 HA-328Y is unable to replicate in brain also indicate that additional mutations in HA are required for neurotropism. Given that bacterial coinfections are common with influenza and based on the data reported here on the role of bacterial streptokinase, one possible outcome is that P2 Ser-Tyr substitution is important in the context of a bacterial coinfection, e.g., with plasminogen-activating S. aureus or S. pyogenes. One possibility is that a Ser-Try mutant can be selected in the respiratory tract of a patient with ongoing influenza virus-bacterial coinfection, which would allow extended use of proteases such as plasmin. In the absence of clinical data from humans, it will be interesting to test this in mouse models of influenza virus-bacterial coinfections.
Despite extensive efforts regarding influenza surveillance and the large amount of sequence information in the database, molecular characterization of clinically relevant viruses identified in surveillance studies is still lacking. The combination of bioinformatic analysis and experimental virology plays an important role is translating the sequence information to a real understanding of the heterogeneous viral population. Here, we provide an example of a distinct variant human pandemic virus with discrete biological properties and describe its molecular properties both in vitro and in vivo. Such studies can provide important information for pandemic planning.
ACKNOWLEDGMENTS
We thank Yueting Zhang, Jean K. Millet, Alice M. Hamilton, Tamar Friling, and all the members in the Whittaker laboratory for helpful discussions. We also thank Lu Huang for helpful discussion on flow cytometry. We also thank Toru Takimoto for provision of the recombinant virus system.
These studies were funded by U.S. Department of Health and Human Services contract HHSN266200700008C (NIAID Centers of Excellence for Influenza Research and Surveillance). Work in the laboratory of G.R.W. is also supported by a research grant from the National Institutes of Health (R01 AI48678).
Footnotes
Published ahead of print 28 February 2013
REFERENCES
- 1. Cox NJ, Subbarao K. 2000. Global epidemiology of influenza: past and present. Annu. Rev. Med. 51:407–421 [DOI] [PubMed] [Google Scholar]
- 2. Wiley DC, Skehel JJ. 1987. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 56:365–394 [DOI] [PubMed] [Google Scholar]
- 3. Klenk H-D, Garten W. 1994. Activation cleavage of viral spike proteins by host proteases, p 241–280 In Wimmer E. (ed), Cellular receptors for animal viruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 4. Klenk HD, Garten W. 1994. Host cell proteases controlling virus pathogenicity. Trends Microbiol. 2:39–43 [DOI] [PubMed] [Google Scholar]
- 5. Choi SY, Bertram S, Glowacka I, Park YW, Pohlmann S. 2009. Type II transmembrane serine proteases in cancer and viral infections. Trends Mol. Med. 15:303–312 [DOI] [PubMed] [Google Scholar]
- 6. Steinhauer DA. 1999. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 258:1–20 [DOI] [PubMed] [Google Scholar]
- 7. Perdue ML. 2008. Molecular determinants of pathogenesis for avian influenza viruses, p 23–41 In Swayne DE. (ed), Avian influenza. Blackwell, Ames, IA [Google Scholar]
- 8. Kawaoka Y, Webster RG. 1988. Sequence requirements for cleavage activation of influenza virus hemagglutinin expressed in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 85:324–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neumann G, Noda T, Kawaoka Y. 2009. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 459:931–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stuart-Harris CH. 1939. A neurotropic strain of human influenza virus. Lancet 233:497–499 [Google Scholar]
- 11. Stuart-Harris CH, Schild GC. 1976. Influenza: the viruses and the disease. Edward Arnold, London, England [Google Scholar]
- 12. Lazarowitz SG, Goldberg AR, Choppin PW. 1973. Proteolytic cleavage by plasmin of the HA polypeptide of influenza virus: host cell activation of serum plasminogen. Virology 56:172–180 [DOI] [PubMed] [Google Scholar]
- 13. Schulman JL, Palese P. 1977. Virulence factors of influenza A viruses: WSN virus neuraminidase required for plaque production in MDBK cells. J. Virol. 24:170–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li S, Schulman J, Itamura S, Palese P. 1993. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J. Virol. 67:6667–6673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Goto H, Kawaoka Y. 1998. A novel mechanism for the acquisition of virulence by a human influenza A virus. Proc. Natl. Acad. Sci. U. S. A. 95:10224–10228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goto H, Wells K, Takada A, Kawaoka Y. 2001. Plasminogen-binding activity of neuraminidase determines the pathogenicity of influenza A virus. J. Virol. 75:9297–9301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hiti AL, Davis AR, Nayak DP. 1981. Complete sequence analysis shows that the hemagglutinins of the H0 and H2 subtypes of human influenza virus are closely related. Virology 111:113–124 [DOI] [PubMed] [Google Scholar]
- 18. Sun X, Tse LV, Ferguson AD, Whittaker GR. 2010. Modifications to the hemagglutinin cleavage site control the virulence of a neurotropic H1N1 influenza virus. J. Virol. 84:8683–8690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Weinstein MJ, Doolittle RF. 1972. Differential specificities of the thrombin, plasmin and trypsin with regard to synthetic and natural substrates and inhibitors. Biochim. Biophys. Acta 258:577–590 [DOI] [PubMed] [Google Scholar]
- 20. Robbins KC, Summaria L. 1970. Human plasminogen and plasmin. Methods Enzymol. 19:184–199 [DOI] [PubMed] [Google Scholar]
- 21. Hervio LS, Coombs GS, Bergstrom RC, Trivedi K, Corey DR, Madison EL. 2000. Negative selectivity and the evolution of protease cascades: the specificity of plasmin for peptide and protein substrates. Chem. Biol. 7:443–453 [DOI] [PubMed] [Google Scholar]
- 22. Bussey KA, Bousse TL, Desmet EA, Kim B, Takimoto T. 2010. PB2 residue 271 plays a key role in enhanced polymerase activity of influenza A viruses in mammalian host cells. J. Virol. 84:4395–4406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chaipan C, Kobasa D, Bertram S, Glowacka I, Steffen I, Tsegaye TS, Takeda M, Bugge TH, Kim S, Park Y, Marzi A, Pohlmann S. 2009. Proteolytic activation of the 1918 influenza virus hemagglutinin. J. Virol. 83:3200–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. CDC 2009. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)—United States, May-August 2009. MMWR Morb. Mortal. Wkly. Rep. 58:1071–1074 [PubMed] [Google Scholar]
- 25. Wang XY, Kilgore PE, Lim KA, Wang SM, Lee J, Deng W, Mo MQ, Nyambat B, Ma JC, Favorov MO, Clemens JD. 2011. Influenza and bacterial pathogen coinfections in the 20th century. Interdiscip. Perspect. Infect. Dis. 2011:146376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scheiblauer H, Reinacher M, Tashiro M, Rott R. 1992. Interactions between bacteria and influenza A virus in the development of influenza pneumonia. J. Infect. Dis. 166:783–791 [DOI] [PubMed] [Google Scholar]
- 27. McArthur JD, Cook SM, Venturini C, Walker MJ. 2012. The role of streptokinase as a virulence determinant of Streptococcus pyogenes—potential for therapeutic targeting. Curr. Drug Targets 13:297–307 [DOI] [PubMed] [Google Scholar]
- 28. Chen GW, Tsao KC, Huang CG, Gong YN, Chang SC, Liu YC, Wu HH, Yang SL, Lin TY, Huang YC, Shih SR. 2012. Amino acids transitioning of 2009 H1N1pdm in Taiwan from 2009 to 2011. PLoS One 7:e45946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. LeBouder F, Morello E, Rimmelzwaan GF, Bosse F, Pechoux C, Delmas B, Riteau B. 2008. Annexin II incorporated into influenza virus particles supports virus replication by converting plasminogen into plasmin. J. Virol. 82:6820–6828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Teijaro JR, Walsh KB, Cahalan S, Fremgen DM, Roberts E, Scott F, Martinborough E, Peach R, Oldstone MB, Rosen H. 2011. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell 146:980–991 [DOI] [PMC free article] [PubMed] [Google Scholar]