

Abstract
Since 2005, a large poliomyelitis outbreak associated with type 2 circulating vaccine-derived poliovirus (cVDPV2) has occurred in northern Nigeria, where immunization coverage with trivalent oral poliovirus vaccine (tOPV) has been low. Phylogenetic analysis of P1/capsid region sequences of isolates from each of the 403 cases reported in 2005 to 2011 resolved the outbreak into 23 independent type 2 vaccine-derived poliovirus (VDPV2) emergences, at least 7 of which established circulating lineage groups. Virus from one emergence (lineage group 2005-8; 361 isolates) was estimated to have circulated for over 6 years. The population of the major cVDPV2 lineage group expanded rapidly in early 2009, fell sharply after two tOPV rounds in mid-2009, and gradually expanded again through 2011. The two major determinants of attenuation of the Sabin 2 oral poliovirus vaccine strain (A481 in the 5′-untranslated region [5′-UTR] and VP1-Ile143) had been replaced in all VDPV2 isolates; most A481 5′-UTR replacements occurred by recombination with other enteroviruses. cVDPV2 isolates representing different lineage groups had biological properties indistinguishable from those of wild polioviruses, including efficient growth in neuron-derived HEK293 cells, the capacity to cause paralytic disease in both humans and PVR-Tg21 transgenic mice, loss of the temperature-sensitive phenotype, and the capacity for sustained person-to-person transmission. We estimate from the poliomyelitis case count and the paralytic case-to-infection ratio for type 2 wild poliovirus infections that ∼700,000 cVDPV2 infections have occurred during the outbreak. The detection of multiple concurrent cVDPV2 outbreaks in northern Nigeria highlights the risks of cVDPV emergence accompanying tOPV use at low rates of coverage in developing countries.
INTRODUCTION
Amajor milestone for the World Health Organization (WHO) Global Polio Eradication Initiative has been the apparent eradication of indigenous wild poliovirus type 2 (WPV2), last detected in West Africa in the mid-1990s (F. Adu and C. Akoua-Koffi, unpublished data) and last detected worldwide in Uttar Pradesh, India, in 1999 (1). Key factors contributing to this achievement were the widespread use of supplemental immunization activities (SIAs) in the form of mass vaccination campaigns with oral poliovirus vaccine (OPV) (2), the high immunogenicity of the Sabin 2 OPV strain in trivalent OPV (tOPV) formulations (3), and the marked tendency of the Sabin 2 strain to spread to secondary contacts (4, 5), especially in settings with poor sanitation and high population densities (6).
Another important milestone has been the decline in WPV incidence in Nigeria, from 796 polio cases (719 WPV1 and 77 WPV3) reported in 2008 to 388 polio cases (75 WPV1 and 313 WPV3) reported in 2009 and 21 cases (8 WPV1 and 13 WPV3) reported in 2010 (7, 8). However, the reported number of WPV cases rose to 62 (47 WPV1 and 15 WPV3) in 2011 (for updates, see the Global Polio Eradication Initiative website [http://www.polioeradication.org/]), as immunization activities weakened in the northern states where polio is endemic (9). Until 2010, northern Nigeria had been an especially intense focus of WPV endemicity and the main exporter of WPV to countries in Africa and Asia (10–13). Key factors contributing to the decline in polio incidence have been the rising rates of OPV coverage in the SIAs following improved community engagement (8), the intensive use of monovalent OPV type 1 (mOPV1) starting in March 2006 and mOPV3 starting in July 2007 (14, 15), and continued sensitive acute flaccid paralysis (AFP) case surveillance linked to virologic surveillance to identify WPV reservoir communities (7, 8, 16). However, the emphasis on the use of mOPV1 and mOPV3 (and bivalent OPV [types 1 and 3] introduced in January 2010) (8, 17, 18) in SIAs to stop WPV transmission coupled with the persistently low rates of routine immunization coverage with tOPV have allowed a population immunity gap for poliovirus type 2 (PV2) to develop in northern Nigeria, leading to the emergence and spread of neurovirulent type 2 vaccine-derived poliovirus (VDPV2) (14, 15, 19–22). By 2005, all three poliovirus serotypes were again cocirculating in Nigeria, a condition not observed elsewhere during the preceding 6 years (1).
The principal biological mechanism underlying the emergence of VDPVs is the intrinsic genetic instability of the OPV strains, such that revertants with increased neurovirulence are selected for during replication of OPV in the human intestine (23). One clinical consequence of the genetic lability of OPV, the occurrence of cases of vaccine-associated paralytic poliomyelitis (VAPP) among OPV recipients and their close contacts (3), has been recognized for nearly 5 decades (24). The current worldwide incidence of VAPP is estimated to be 250 to 500 sporadically distributed cases per year (3). The serotype distribution of VAPP cases among immunologically healthy individuals is uneven: Sabin 1 is rarely implicated, Sabin 2 is most frequently associated with VAPP in unimmunized contacts of OPV recipients, and Sabin 3 is most frequently associated with VAPP in OPV recipients (25–27). Vaccine-related isolates from immunologically healthy VAPP patients typically show limited genetic divergence (<1% of nucleotide positions) from the parental OPV strains (apart from mosaic genomes arising from vaccine/vaccine recombination) (28, 29), consistent with durations of infections of only 1 to 2 months (30). Individuals with primary immunodeficiencies have an ∼3,000-fold-higher risk of VAPP (31) and, in rare instances, the further risk of prolonged infection (some lasting >10 years) with excretion of highly divergent immunodeficiency-associated VDPVs (iVDPVs) (3, 19, 21, 22, 32–36).
Quite distinct from iVDPVs are the circulating VDPVs (cVDPVs), which can emerge and spread in settings of low type-specific population immunity (32). cVDPV outbreaks have occurred and have been controlled in Egypt (37), Hispaniola (Haiti and the Dominican Republic) (38), the Philippines (39), Madagascar (40, 41), Indonesia (42), Cambodia (20), Myanmar (20), and China (43, 44). Within the past 3 years, cVDPV outbreaks have emerged in the Democratic Republic of Congo (type 2, 2008 to 2012), Ethiopia (type 2, 2008 to 2009, and type 3, 2009 to 2010), Somalia (type 2, 2008 to 2011), India (type 2, 2009 to 2010), Afghanistan (type 2, 2010 to 2012), Mozambique (type 1, 2011), Yemen (type 2, 2011), Chad (type 2, 2012), and Pakistan (type 2, 2012) (19, 21, 22, 45) (for updates, see the Global Polio Eradication Initiative website [http://www.polioeradication.org/]). Most cVDPV outbreaks involve Sabin types 2 and 1, rarely type 3. In addition to the well-defined cVDPV and iVDPV categories, VDPV isolates may be assigned to a third category, ambiguous VDPVs (aVDPVs), when there is no clear evidence of community circulation or immunodeficiency (19, 22, 46).
VDPVs have been defined for the purposes of global poliovirus surveillance as having >1% nucleotide sequence divergence (i.e., ≥10 nucleotide [nt] substitutions) from their parental Sabin strains in the ∼900-nt region encoding the major capsid protein, VP1 (16, 21, 32). This definition follows from the high rate of nucleotide sequence evolution in poliovirus (∼1% per year) (47) and the normal period of poliovirus excretion of less than 3 months (30, 32). The demarcation for VDPV2s has recently been lowered to >0.6% nucleotide sequence divergence (i.e., ≥6 nt substitutions) in view of the findings presented in this report (22, 48) and similar findings in the Democratic Republic of Congo (22, 48).
Nigeria has recorded the largest VDPV outbreak in terms of the number of reported cases, with a total of 403 cases associated with VDPV2 infection reported during 2005 to 2011 (14, 15, 19–22, 49). Here we describe the basic genetic properties of and genetic relationships among Nigerian VDPV2 outbreak isolates obtained from July 2005 to December 2011 and present evidence that the outbreak was associated with 23 independent VDPV2 emergences, 7 of which expanded into well-defined cVDPV2 lineage groups and several others of which may have signaled more limited cVDPV2 transmission. We further describe the population dynamics of the major cVDPV2 lineage group during the outbreak and the key biological properties of selected VDPV2 isolates. Finally, we discuss the implications of multiple, concurrent cVDPV outbreaks for current and future strategies to secure the cessation of all poliovirus transmission.
MATERIALS AND METHODS
Virus isolation and identification.
Viruses in stool specimens obtained from patients with AFP (see Table S1 in the supplemental material) were isolated in L20B (mouse L cells expressing the human CD155 poliovirus receptor [PVR]) (50, 51) and RD (human rhabdomyosarcoma cells; ATCC CCL-136) cell lines according to standard protocols (52). Isolates were characterized by reverse transcriptase PCR (RT-PCR) assays using enterovirus-specific and poliovirus group-, serotype-, and Sabin strain-specific primer sets (52–55) and enzyme-linked immunosorbent assays using highly specific cross-absorbed hyperimmune rabbit sera (52, 56). Since August 2006, vaccine-related poliovirus isolates have been screened for VDPVs by using a real-time RT-PCR assay (16, 48). VDPVs were identified by VP1 sequencing (37, 57).
Nucleic acid sequencing.
RNA extraction and RT-PCR amplification and cycle sequencing were performed as previously described (37, 57). Terminal sequences of 17 VDPV2 isolate genomes (all but one from lineage group 2005-8) were determined by using 5′ rapid amplification of the cDNA ends (RACE) kits (Life Technologies) according to the manufacturer's instructions. The first 19 nt at the 5′ ends of the remaining genomes were derived from the primer, which matched the 17 identical terminal sequences determined by RACE. Numbering of nucleotide positions followed that described previously for Sabin 2 (58, 59).
Phylogenetic analysis.
Sequence relationships (all nucleotide substitutions [KT]) in the P1/capsid region (nt 748 to 3384) among all VDPV2 isolates were summarized in phylogenetic trees constructed by Bayesian Markov chain Monte Carlo (MCMC) analysis implemented in BEAST v. 1.7 (60). Two independent MCMC chains were run for each BEAST analysis (200 million generations each for lineage group 2005-8 and 50 million generations each for the other lineage groups). High-performance computing was achieved by integrating the BEAGLE library (61) into BEAST runs. Sampling efficiency was examined by measuring the integrated autocorrelation time and effective sample size, as implemented in TRACER (MCMC Trace Analysis Tool, v1.5 [http://tree.bio.ed.ac.uk/software/tracer/]). Substitution parameters were estimated from the data set. Codon deletions at VP1-Thr24 were treated as single-nucleotide substitutions. Hypothesis testing within nested models of evolution was performed by using MODELTEST (62). Maximum clade credibility (MCC) trees including all VDPV2 isolates and representatives of all VDPV2 emergences were rooted to the P1/capsid region sequences of Sabin 2 and scaled to time by assuming a strict molecular clock with a KT substitution rate of 1.1 × 10−2 nt substitutions/site/year. Trees of individual lineage groups were constructed similarly, except that the root for lineage group 2005-8 was a simulated P1/capsid region sequence of a nonmutated S3/S2 recombinant homologue of the observed VDPV recombinants. Phylogenetic trees were displayed and edited by using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
A P1/capsid subtree including representative isolates from all lineages was inferred after two independent MCMC chains were run for 1 million generations each in Mr. Bayes (63), as implemented in the plug-in version for the Geneious v5.6 software package.
Phylogenetic resolution of separate emergences.
P1/capsid region sequences of isolates across and within emergences were compared in difference tables constructed by using MEGA5 software (64), with pairwise P1/capsid region nucleotide differences shown below the diagonal and the proportion of shared P1/capsid region nucleotide differences from Sabin 2 (excluding the codon for amino acid VP1143) shown above the diagonal.
Estimation of dates of the initiating OPV doses.
Dates of the tOPV doses that initiated each of the 23 observed emergences were estimated from KT values for P1/capsid region sequences by using the Bayesian inference method reported previously by Shapiro et al. (65), implemented in BEAST under the assumption of a uniform KT substitution rate of 1.1 × 10−2 nt substitutions/site/year. Mean estimated values and 95% highest posterior density (HPD) intervals for tOPV dose dates and inferred mean estimated times were calculated according to BEAST procedures, as described above.
Estimation of the number of lineages transiting the low-transmission season for poliovirus.
The number of lineages that continued through the seasonal lows for poliovirus transmission in northern Nigeria from December to January was estimated from the topology of the Bayesian MCC trees of the 7 lineage groups and the estimated duration of replication of virus of the 16 other separate emergences. KT substitution rates for lineage groups 2005-6, 2005-8, 2005-10, and 2007-4 were estimated from the data sets; otherwise, the rate was assumed to be 1.1 × 10−2 nt substitutions/site/year. A bifurcation of a branch in December was counted as two lineages, and one in January was counted as one lineage. Lineages whose last isolate was detected in January were not counted as transiting the low-transmission season. Long branches that spanned multiple years were counted across each seasonal low.
VDPV population dynamics during the outbreak.
The demographic history of all Nigerian VDPV2s was estimated under the Bayesian skyline plot model (66), as implemented in BEAST. The distribution of mean and 95% HPD effective population size values (Ne) for 2005 to 2011 was analyzed in TRACER.
Phenotypic characterization of VDPV isolates.
Phenotypic properties of representative isolates of each VDPV2 emergence were compared with those of Sabin 2 and MEF-1 (a neurovirulent WPV2 isolate whose robust growth favored its selection as the type 2 component of the inactivated poliovirus vaccine [IPV]) (67).
(i) Virus replication in HEK293 cells.
Monolayers of HEK293 cells (human neuron-derived cells; ATCC CRL-1573) (68) were infected with virus at a multiplicity of infection (MOI) of 10 PFU/cell and incubated for 24 h at 37.0°C and 39.5°C, as described previously by Campbell et al. (69). Virus was liberated by two freeze-thaw cycles, and infectivity titers were measured by plaque assays on HeLa cell monolayers incubated at 37.0°C, as described previously (70).
(ii) Neurovirulence testing in PVR-Tg21 mice.
Neurovirulence tests on VDPV2 isolates were carried out by using PVR-Tg21 (71) mice as previously described (39). The mice were purchased from the Central Laboratories for Experimental Animals (Kanagawa, Japan). The type 2 reference strains were Sabin 2 and MEF-1. Eight mice (equal numbers of males and females) were inoculated (30 μl/mouse) intracerebrally for each virus dilution (in 10-fold increments; range, 1.5 to 6.5 log 50% tissue culture infective doses [TCID50]/mouse and up to 7.0 log TCID50/mouse for Sabin 2). Mice were examined daily for 14 days postinoculation, and the times of paralysis or death were recorded. The virus titer that induced paralysis or death in 50% of inoculated mice (PD50) was calculated according to the method of Kärber (72).
(iii) Virus infectivity yields at supraoptimal temperatures.
Virus was grown to high titers in RD cell monolayers at 37.0°C, and infectivity yields were measured by plaque assays (70) on L20B cells incubated at 37.0°C and 39.5°C (73).
Nucleotide sequence accession numbers.
P1/capsid region sequences of all Nigerian VDPV2 isolates described here, including complete genomic sequences of the first isolates of each emergence, were deposited in the GenBank database under accession numbers JX274980 to JX275382 (see Table S1 in the supplemental material).
RESULTS
Identification of VDPV2 isolates.
The standard method for VDPV screening by the Global Polio Laboratory Network (GPLN) (52) has been to identify vaccine-related isolates by their genetic properties by using RT-PCR (55) (or other molecular methods) followed by antigenic characterization (74). Antigenic variants of the vaccine strains were flagged as candidate VDPVs (32) to be further characterized by sequencing of the VP1 capsid region (19). However, the early (2005-2006) VDPV2 isolates from Nigeria did not appear to be antigenic variants, such that the routine screening algorithm did not initially signal any VDPV emergence. An outbreak was first suspected in 2006 by the frequent isolation from AFP patients of vaccine-related PV2 (15), the serotype most frequently associated with cVDPV outbreaks (19, 21, 22, 32, 45), with temporal and geographic clustering in the northern states (Fig. 1). The higher isolation rate for PV2 appeared to be anomalous because WPV2 was believed to have been eradicated, many patients had no known exposure to tOPV, routine immunization coverage with tOPV was low, and only two of the six SIAs in 2006 used tOPV, as the remainder used mOPV1 (14, 15). Sequencing of the VP1 region identified most of the recent PV2 isolates from the north as VDPV2s (15). In recognition of the limitations of screening for VDPVs by antigenic methods, a new real-time RT-PCR assay was developed for VDPV screening (16, 19, 48) and applied retrospectively and prospectively to Nigerian PV2 isolates.
Fig 1.

Map of Nigeria showing state boundaries, population density by LandScan (http://www.ornl.gov/landscan/), and location (circles color-coded by estimated year of emergence) of first acute flaccid paralysis (AFP) cases found to be associated with each independent type 2 vaccine-derived poliovirus (VDPV2) emergence. The numbers 1 to 23 correspond to emergences listed in Table 1 according to the order of the estimated date of the initiating tOPV dose. The bold red line indicates the boundary between the northern and southern states. Abbreviations for Nigerian states (S) with VDPV2 cases (14, 15) are as follows: ADS, Adamawa; ANS, Anambra; BAS, Bauchi; BNS, Benue; BOS, Borno; JIS, Jigawa; KBS, Kebbi; KDS, Kaduna; KNS, Kano; KTS, Katsina; KGS, Kogi; LAS, Lagos; NAS, Nasarawa; NIS, Niger; PLS, Plateau; SOS, Sokoto; YBS, Yobe; ZAS, Zamfara.
Range of VP1 sequence divergence among Sabin 2-related case isolates.
During 2005 to 2011, 1,201 PV2s were isolated from individual AFP patients in Nigeria; all were related to Sabin 2. Of these, 695 (612 from the north and 83 from the south) were sequenced in the VP1 region. Most had been sequenced as candidate VDPV2s after screening by real-time RT-PCR, but isolates with <6 nt substitutions in VP1 had been sequenced independently of the screening assay results. The sequenced isolates diverged from Sabin 2 at 0 to 59 VP1 nucleotide positions (0% to 6.5%) (Fig. 2). Sabin 2-related isolates with ≥6 nt substitutions in the VP1 region are unusual and, on the basis of the findings reported here, have been redefined as VDPV2s (22, 48). Nearly all (401/403; 99.5%) of the VDPV2s detected were from cases in 15 northern states (Fig. 1 and 2A) (7, 15) known to have widening gaps of immunity to PV2 because of low routine immunization coverage with tOPV and a reduced frequency of SIAs with tOPV after 2005 (14, 15). Only two VDPV2 case isolates, of limited divergence (6 VP1 nucleotide substitutions), were found in the southern states (Fig. 1 and 2B). Among isolates with 0 to 5 VP1 nucleotide substitutions, in both the north and the south, the distribution of isolates showed a steep decline with increasing divergence from Sabin 2. However, in the north, the number of isolates with ≥6 nt substitutions increased (Fig. 2A). None of the Sabin 2-related isolates with ≤5 VP1 nucleotide substitutions appeared to be ancestral to any subsequent isolates. All isolates with a substitution in the VP1 region had a missense substitution within the codon for VP1143, a result of strong selection against the Sabin 2 allele (see below). Isolates with no VP1 substitutions are underrepresented in Fig. 2 because they were not flagged for sequencing by the real-time RT-PCR screening assay, which targets substitutions in VP1.
Fig 2.

Distribution of nucleotide substitutions in the VP1 regions of Sabin 2-related polioviruses isolated from patients with AFP from 2005 to 2011 in the northern (n = 612) (A) and southern (n = 83) (B) states of Nigeria. Each isolate is from an individual patient. The northern and southern states are identified in Fig. 1. Type 2 vaccine-derived polioviruses (VDPV2s) (black bars) are more divergent, having ≥6 nt substitutions in the VP1 region (48).
The 403 VDPV2s isolated from AFP patients (1 isolate per patient; includes 1 isolate from a healthy contact of an AFP patient) with onset during 2005 to 2011 (2005, 4; 2006, 39; 2007, 76; 2008, 66; 2009, 156; 2010, 27; 2011, 35) include 11 from patients who were coinfected with WPV1 or WPV3. Patients with such isolates were previously excluded from our VDPV2 case counts (15) because the lower paralytic attack rates of PV2 infections than of WPV1 and WPV3 infections (45) suggested that the etiologic agents of the observed AFP cases were most likely the coinfecting WPVs. Also excluded from our analyses were cVDPV2s exported from Nigeria into Niger (n = 6) and Chad (n = 1).
Independent emergence of multiple cVDPV2 lineages.
Although comparisons of VP1 region sequences (903 nt) provided a good overview of the outbreak, many of the VDPV2s were isolated soon after emergence, affording little time for divergence. Therefore, we sought to increase phylogenetic resolution among the closely related VDPV2 isolates by extending our sequence comparisons to the complete P1/capsid regions (2,637 nt) of all 2005-2011 Nigerian VDPV2 isolates (Fig. 3; see also Fig. S1 to S4 in the supplemental material). However, the relationships among time-ordered P1/capsid region sequences of all isolates were inconsistent with the poliovirus molecular clock (47), as some isolates from more recent cases had far fewer nucleotide substitutions than isolates from earlier cases. When the outbreak was resolved into 23 separate emergences, numbered by year and within years, ordered by the estimated dates of the initiating OPV doses (see below), compatibility with a molecular clock was restored (Fig. 4 and Table 1).
Fig 3.

Maximum clade credibility subtree of P1/capsid region sequences (2,637 nt) of 62 2005-2011 isolates representing the seven well-defined cVDPV2 lineage groups (in boldface type) and 16 other isolates that signaled additional independent VDPV2 emergences (lightface type). Lineage groups and emergences are numbered by year and ordered within years by the estimated dates of the initiating OPV doses. *, VDPV2/WPV1 coinfections. Color-coding of isolate identifiers corresponds to that used in Fig. 1.
Fig 4.

Timeline of independent VDPV2 emergences and spread in Nigeria from 2004 to 2011 based on the dates of VDPV2 specimen collection (specimens were collected 10 ± 6 days after onset of AFP). cVDPV2 lineage groups are indicated in boldface type. Dates of the initiating tOPV doses for the 14 emergences associated with single isolates are point estimates with indeterminate confidence intervals. Arrows indicate months of tOPV supplemental immunization activities (SIAs) (mass vaccination campaigns) in the northern states. The blue point symbolizes two superimposed points. The blue wedge in the lower left corner symbolizes the duration of type 2 vaccine-related poliovirus excretion in immunologically healthy primary-dose OPV recipients (30).
Table 1.
Independent VDPV2 emergences, Nigeria, 2004 to 2011g
| Estimated order of emergencea | Emergenceb | Earliest isolatec | Earliest isolate specimen date (day, mo, yr) | No. of nt substitutions of earliest isolate |
Estimated date of initiating tOPV dose (day, mo, yr) (95% HPD)d | Latest isolate specimen date (day, mo, yr)e | Total no. of isolates | Estimated duration of replication (mo) (95% HPD)f | |
|---|---|---|---|---|---|---|---|---|---|
| VP1 | P1 | ||||||||
| 1 | 2004-1 | BAS05-01 | 05 July 05 | 10 | 21 | 22 Sep 04 (03 Apr 04–20 Apr 05) | 08 June 06 | 3 | 21 (14–26) |
| 2 | 2005-1 | SOS05-01 | 05 Oct 05 | 7 | 19 | [07 Feb 05] | — | 1 | [8] |
| 3 | 2005-2 | LAS05-01 | 22 July 05 | 6 | 12 | [21 Feb 05] | — | 1 | [5] |
| 4 | 2005-3 | KDS05-01 | 03 Nov 05 | 7 | 16 | [16 Apr 05] | — | 1 | [7] |
| 5 | 2005-4 | KBS06-01 | 14 Jan 06 | 8 | 16 | [06 July 05] | — | 1 | [7] |
| 6 | 2005-5 | JIS06-06 | 17 July 06 | 15 | 28 | 13 Aug 05 (10 Apr–06 Dec 05) | 17 Oct 06 | 2 | 14 (10–20) |
| 7 | 2005-6 | BOS06-03 | 05 July 06 | 9 | 19 | 14 Sep 05 (15 Apr 05–01 Feb 06) | 07 Feb 08 | 6 | 29 (24–34) |
| 8 | 2005-7 | BOS06-01 | 31 May 06 | 9 | 18 | [16 Oct 05] | — | 1 | [7] |
| 9 | 2005-8 | JIS06-01 | 19 May 06 | 6 | 16h | 26 Oct 05 (22 July 05–12 Jan 06) | 17 Dec 11 | 361 | 74 (70–77) |
| 10 | 2005-9 | JIS06-04 | 15 June 06 | 9 | 18 | [31 Oct 05] | — | 1 | [7] |
| 11 | 2005-10 | SOS07-01 | 26 Feb 07 | 13 | 27 | 03 Dec 05 (11 May 05–16 July 06) | 05 Mar 09 | 6 | 40 (33–47) |
| 12 | 2005-11 | BAS06-01 | 07 Nov 06 | 11 | 25 | [27 Dec 05] | — | 1 | [10] |
| 13 | 2006-1 | BOS06-02 | 15 June 06 | 6 | 7 | [19 Mar 06] | — | 1 | [3] |
| 14 | 2006-2 | SOS08-09 | 02 Sep 08 | 20 | 50 | [11 Dec 06] | — | 1 | [21] |
| 15 | 2007-1 | NIS07-01 | 30 July 07 | 9 | 13 | [16 Feb 07] | — | 1 | [5] |
| 16 | 2007-2 | BAS08-06 | 07 Aug 08 | 10 | 40 | [22 Mar 07] | — | 1 | [17] |
| 17 | 2007-3 | ANS07-01 | 27 Oct 07 | 6 | 8 | [18 July 07] | — | 1 | [3] |
| 18 | 2007-4 | KBS08-03 | 01 Dec 08 | 12 | 30 | 05 Sep 07 (04 Apr 07–01 Feb 08) | 25 May 10 | 7 | 33 (28–38) |
| 19 | 2007-5 | BOS09-06 | 20 Apr 09 | 17 | 44 | 21 Oct 07 (14 Mar 07–07 June 08) | 02 June 09 | 2 | 19 (12–27) |
| 20 | 2007-6 | KTS09-05 | 27 Feb 09 | 16 | 37 | [18 Nov 07] | — | 1 | [15] |
| 21 | 2008-1 | KDS09-08 | 05 June 09 | 6 | 15 | [28 Nov 08] | — | 1 | [6] |
| 22 | 2010-1 | KBS11-01 | 22 Feb 11 | 6 | 13 | [10 Sep 10] | — | 1 | [5] |
| 23 | 2010-2 | NIS11-01 | 22 Nov 11 | 11 | 18 | [05 Apr 11] | — | 1 | [7] |
Numbered in estimated order of emergence (Fig. 1).
Emergences that expanded into well-defined cVDPV2 lineage groups (shown in boldface type) were previously described and numbered according to the date of detection of the earliest isolate (2004-1, 1; 2005-8, 2; 2005-6, 3; 2005-5, 4; 2005-10, 5; 2007-4, 6; 2007-5, 7) (15). All other emergences were previously described as “undefined.”
Abbreviations for state names are given in the legend to Fig. 1.
P1/capsid KT evolution rate was assumed to be constant at 1.1%/year (29 nt substitutions/year). HPD, highest posterior density.
Eight additional cases (three from Kano, three from Sokoto, and two from Kebbi) associated with cVDPV2, all from lineage group 2005-8, were reported in 2012. The most recent case had an onset date of 24 November 2012, and the isolate had 65 nt substitutions in VP1. One new VDPV2 emergence was associated with a 2012 case in the southern state of Edo (onset on 22 May 2012; the isolate had 6 nt substitutions in VP1). (Data as of 31 December 2012; for updates, see the Global Polio Eradication Initiative website [http://www.polioeradication.org/].) —, single isolate.
Calculated from the estimated date of the initiating tOPV dose to the date of the most recently isolated specimen (through 2011).
Values in brackets are point estimates with indeterminate confidence intervals.
Compared against a simulated unmutated Sabin 3/Sabin 2 recombinant with the recombination junction at the same location as in the natural isolates.
Of the 23 emergences, 7 expanded into observed cVDPV2 lineage groups containing 2 to 361 isolates. The two VDPV2 isolates from the southern states of Anambra and Lagos with only moderate levels of divergence from Sabin 2 were unlike most cVDPVs (19, 32, 75) by having either nonrecombinant genomes (ANS07-01) or vaccine/vaccine (Sabin 2/Sabin 3) (LAS05-01) recombinant genomes (data not shown). Because the corresponding cases occurred in states with relatively high rates of routine tOPV coverage and signaled independent emergences with no known secondary spread, they were not considered to be part of the outbreak (15).
Assignment of isolates to independent VDPV2 emergences and lineage groups was based primarily on pairwise P1/capsid region sequence differences across emergences (Table 2) and within lineage groups (see Tables S2 to S8 in the supplemental material). Substitutions within the codon for VP1143 were excluded from the pairwise comparisons because all VDPV2 isolates had a substitution within that codon. Other P1/capsid region substitutions were primarily synonymous changes, nearly randomly distributed, with a different pattern of substitution for each emergence. Among the P1/capsid region nucleotide differences from Sabin 2 outside the codon for VP1143, the proportion shared by the first isolates of each independent emergence averaged 2.4% (range, 0% to 13%), whereas the proportion shared by isolates within each lineage group averaged ∼42% (range, 11% to 95%; only a subset of isolates from lineage group 2005-8 were compared) (Table 2). The common node for sequences of the two isolates of lineage group 2005-5 was so deep as to suggest that they had emerged independently. However, the two isolates shared common recombination junctions near nt 485 and 4959 and close nucleotide identities (99.2% and 98.4%) in the flanking nonvaccine sequences (see below), thus confirming their emergence from a common tOPV dose. The structure of the recombinant genomes further supported the assignment of isolates to specific emergences and lineage groups (data not shown).
Table 2.
Nucleotide differences in the P1/capsid region across independent VDPV2 emergencesa
| Estimated order of emergence | Emergence | Earliest isolate | Value for emergence |
|||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9b | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | 23 | |||
| Sabin 2 | ||||||||||||||||||||||||||
| 1 | 2004-1 | BAS05-01 | 20 | 0.00 | 0.06 | 0.00 | 0.00 | 0.04 | 0.08 | 0.05 | 0.09 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.03 | 0.04 | 0.04 | 0.13 | 0.04 | 0.00 | 0.00 | 0.05 | |
| 2 | 2005-1 | SOS05-01 | 18 | 38 | 0.00 | 0.00 | 0.06 | 0.00 | 0.06 | 0.00 | 0.00 | 0.06 | 0.05 | 0.00 | 0.00 | 0.03 | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 | 0.02 | 0.00 | 0.00 | 0.00 | |
| 3 | 2005-2 | LAS05-01 | 12 | 30 | 30 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.03 | 0.00 | 0.00 | 0.08 | 0.04 | 0.11 | 0.05 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 4 | 2005-3 | KDS05-01 | 15 | 35 | 33 | 27 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.02 | 0.05 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.05 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 5 | 2005-4 | KBS06-01 | 15 | 35 | 31 | 27 | 30 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.05 | 0.00 | 0.00 | 0.00 | 0.07 | 0.00 | 0.00 | 0.05 | 0.00 | 0.00 | 0.07 | 0.00 | 0.00 | |
| 6 | 2005-5 | JIS06-06 | 27 | 45 | 45 | 39 | 42 | 42 | 0.00 | 0.05 | 0.02 | 0.00 | 0.08 | 0.00 | 0.06 | 0.03 | 0.05 | 0.00 | 0.00 | 0.00 | 0.06 | 0.10 | 0.05 | 0.10 | 0.02 | |
| 7 | 2005-6 | BOS06-03 | 18 | 35 | 34 | 30 | 33 | 33 | 45 | 0.00 | 0.00 | 0.06 | 0.00 | 0.00 | 0.00 | 0.06 | 0.03 | 0.00 | 0.00 | 0.04 | 0.05 | 0.04 | 0.00 | 0.00 | 0.00 | |
| 8 | 2005-7 | BOS06-01 | 17 | 35 | 35 | 29 | 32 | 32 | 42 | 35 | 0.00 | 0.00 | 0.05 | 0.00 | 0.09 | 0.03 | 0.00 | 0.05 | 0.00 | 0.04 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 9 | 2005-8b | JIS06-01 | 14 | 31 | 32 | 26 | 29 | 29 | 40 | 32 | 31 | 0.00 | 0.10 | 0.00 | 0.00 | 0.03 | 0.00 | 0.00 | 0.10 | 0.05 | 0.04 | 0.00 | 0.00 | 0.00 | 0.06 | |
| 10 | 2005-9 | JIS06-04 | 17 | 37 | 33 | 29 | 32 | 32 | 44 | 33 | 34 | 31 | 0.07 | 0.00 | 0.00 | 0.00 | 0.07 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 11 | 2005-10 | SOS07-01 | 26 | 46 | 42 | 38 | 40 | 39 | 49 | 44 | 41 | 36 | 40 | 0.00 | 0.00 | 0.11 | 0.00 | 0.00 | 0.00 | 0.04 | 0.00 | 0.03 | 0.00 | 0.00 | 0.00 | |
| 12 | 2005-11 | BAS06-01 | 24 | 44 | 42 | 35 | 37 | 39 | 51 | 42 | 41 | 38 | 41 | 50 | 0.00 | 0.03 | 0.00 | 0.06 | 0.06 | 0.00 | 0.05 | 0.08 | 0.00 | 0.00 | 0.00 | |
| 13 | 2006-1 | BOS06-02 | 6 | 26 | 24 | 18 | 21 | 21 | 31 | 24 | 21 | 20 | 23 | 32 | 30 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 14 | 2006-2 | SOS08-09 | 49 | 68 | 65 | 61 | 64 | 64 | 74 | 63 | 64 | 61 | 66 | 67 | 71 | 55 | 0.03 | 0.07 | 0.04 | 0.08 | 0.12 | 0.08 | 0.00 | 0.00 | 0.06 | |
| 15 | 2007-1 | NIS07-01 | 12 | 32 | 30 | 22 | 27 | 25 | 37 | 29 | 29 | 26 | 27 | 38 | 36 | 18 | 59 | 0.04 | 0.00 | 0.10 | 0.04 | 0.08 | 0.08 | 0.00 | 0.00 | |
| 16 | 2007-2 | BAS08-06 | 39 | 57 | 57 | 49 | 54 | 54 | 66 | 57 | 53 | 53 | 56 | 65 | 59 | 45 | 82 | 49 | 0.07 | 0.10 | 0.05 | 0.05 | 0.00 | 0.02 | 0.11 | |
| 17 | 2007-3 | ANS07-01 | 7 | 26 | 25 | 17 | 22 | 22 | 34 | 25 | 24 | 19 | 24 | 33 | 29 | 13 | 54 | 19 | 43 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.00 | |
| 18 | 2007-4 | KBS08-03 | 29 | 47 | 47 | 39 | 42 | 42 | 56 | 45 | 44 | 41 | 46 | 53 | 53 | 35 | 72 | 37 | 61 | 36 | 0.06 | 0.06 | 0.00 | 0.00 | 0.04 | |
| 19 | 2007-5 | BOS09-06 | 42 | 54 | 58 | 52 | 55 | 57 | 65 | 57 | 59 | 54 | 59 | 68 | 63 | 48 | 80 | 52 | 77 | 48 | 67 | 0.05 | 0.04 | 0.07 | 0.03 | |
| 20 | 2007-6 | KTS09-05 | 36 | 54 | 53 | 48 | 51 | 51 | 57 | 52 | 53 | 50 | 53 | 60 | 55 | 42 | 78 | 44 | 71 | 43 | 61 | 74 | 0.00 | 0.00 | 0.04 | |
| 21 | 2008-1 | KDS09-08 | 14 | 34 | 32 | 26 | 29 | 27 | 39 | 32 | 31 | 28 | 31 | 40 | 38 | 20 | 63 | 24 | 53 | 21 | 43 | 54 | 50 | 0.00 | 0.00 | |
| 22 | 2010-1 | KBS11-01 | 12 | 32 | 30 | 24 | 27 | 27 | 35 | 30 | 29 | 26 | 29 | 38 | 36 | 18 | 61 | 24 | 50 | 19 | 41 | 50 | 48 | 26 | 0.00 | |
| 23 | 2010-2 | NIS11-01 | 17 | 35 | 35 | 29 | 32 | 32 | 43 | 35 | 34 | 29 | 34 | 43 | 41 | 23 | 62 | 29 | 50 | 24 | 44 | 57 | 51 | 31 | 29 | |
Values below the diagonal are numbers of pairwise P1/capsid region nucleotide differences; values above the diagonal are fractions of P1/capsid region nucleotide differences from Sabin 2 that were shared among isolates. The codon for amino acid VP1143 was excluded. Well-defined cVDPV2 lineage groups are shown in boldface type.
Values exclude the first 93 nt in the P1/capsid region of JIS06-01, which were derived from Sabin 3.
When individual lineage groups were analyzed by Bayesian MCMC inference (see Fig. S1 to S4 in the supplemental material), the mean of the estimated substitution rates at all sites (KT) for the major lineage group, 2005-8, was 1.12 (1.05 to 1.20) × 10−2 nt substitutions/site/year, with an average rate of 1.14 × 10−2 nt substitutions/site/year for the four largest lineage groups (Table 3). The values obtained for this robust data set were very close to VP1 KT substitution rates described previously (36, 47, 57, 76, 77). Also, as previously described for WPV1 (47), synonymous transitions (A′S) were ∼10-fold more frequent than synonymous transversions (B′S) (Table 3 and Fig. 5).
Table 3.
Substitution rates for P1/capsid region sequences (2,637 nt) of cVDPV2 lineage groups
| Substitution parametera | Bayesian rate (nt substitutions/site/yr [10−2]) (95% HPD) for cVDPV2 lineage group |
|||
|---|---|---|---|---|
| 2005-6 | 2005-8 | 2005-10 | 2007-4 | |
| KT | 1.21 (0.91–1.50) | 1.12 (1.05–1.20) | 1.16 (0.81–1.47) | 1.09 (0.71–1.46) |
| K′S | 1.14 (0.86–1.43) | 0.88 (0.84–0.93) | 1.20 (0.85–1.54) | 1.05 (0.66–1.41) |
| K′A | 0.08 (0.006–0.16) | 0.12 (0.10–0.15) | 0.10 (0.004–0.20) | 0.09 (0.01–0.19) |
| A′S | 1.04 (0.78–1.31) | 0.80 (0.75–0.84) | 1.08 (0.75–1.38) | 0.98 (0.64–1.33) |
| B′S | 0.06 (0.003–0.14) | 0.08 (0.07–0.09) | 0.06 (0.006–0.12) | 0.11 (0.01–0.22) |
Substitution parameters (KT, total substitutions; K′S, synonymous substitutions; K′A, nonsynonymous substitutions; A′S, synonymous transitions; B′S, synonymous transversions) were normalized to total sites by using the following relationships: K′S = KS × LS/LT, K′A = KA × LN/LT, A′S = AS × LS/LT, and B′S = BS × LS/LT (LS, number of synonymous sites; LN, number of nonsynonymous sites; LT, total number of sites) (47). Estimates of the number of each category of site were calculated by implementation of the program MEGA5 (64) (LT sites = 2,637; LS sites ≈ 865; LN sites ≈ 1,708).
Fig 5.

Maximum likelihood estimates of relative frequencies of specific base changes (10−2) (black arrows, transitions; gray arrows, transversions) at all codon positions within the P1/capsid regions of the 361 2005-2011 cVDPV2 isolates from the major 2005-8 lineage group during divergence from the Sabin 2 root sequence.
Estimated dates of VDPV2 emergences.
The dates of the tOPV doses that initiated each of the 23 observed emergences were estimated from the total number of nucleotide substitutions in the P1/capsid region by assuming a constant P1/capsid KT substitution rate of 1.1 × 10−2 nt substitutions/site/year throughout the period of divergence (Table 1 and Fig. 4). The estimated time interval between the initiating tOPV doses (the corresponding vaccine recipients are unknown) and the first VDPV2 isolate averaged ∼9 months (range, 3 to 21 months) (Table 1 and Fig. 4), with confidence intervals (95% HPD intervals) of 7 to 15 months for the seven lineage groups. Uncertainties in the estimates were greatest for the least divergent isolates, such as BOS06-02 and ANS07-01, for which the random nature of nucleotide substitution is evident by the strong localization of substitutions to the VP1 region (Table 1), leading to nearly 3-fold-higher substitution rate estimates for the VP1 region than for the P1/capsid region. Although early selection against the Sabin 2 allele at VP1143 (and the possible effects of any hitchhiker substitutions) may violate our assumption of a constant rate of substitution into the P1/capsid region (77, 78), the primary source of uncertainty appears to derive from the stochastic nature of the clock.
The P1/capsid region sequence tree resolving the lineage groups generally had deep nodes (Fig. 3 and Table 4; see also Fig. S1 to S4 in the supplemental material), consistent with cVDPV2 transmission soon after administration of the initiating tOPV doses. Estimates of the mean time between the initiating tOPV dose and the dates of the first divergence within each lineage group ranged between 2.0 and 6.5 months (Fig. 4 and Table 4).
Table 4.
Estimated time to first divergence of cVDPV2 lineage groups
| Lineage group | Estimated date of initiating tOPV dose (day, mo, yr) (95% HPD) | Estimated date of first diverging node (day, mo, yr) (95% HPD) | Estimated mean time to first divergence (mo)a |
|---|---|---|---|
| 2004-1 | 22 Sep 04 (03 Apr 04–20 Apr 05) | 18 Mar 05 (12 Jan–17 May 05) | 5.9 |
| 2005-5 | 13 Aug 05 (10 Apr–06 Dec 05) | 24 Oct 05 (29 July 05–16 Jan 06) | 2.6 |
| 2005-6 | 10 Sep 05 (21 Apr 05–08 Feb 06) | 13 Jan 06 (09 Nov 05–13 Mar 06) | 4.1 |
| 2005-8 | 26 Oct 05 (22 July 05–12 Jan 06) | 27 Dec 05 (21 Nov 05–27 Jan 06) | 2.0 |
| 2005-10 | 03 Dec 05 (11 May 05–16 July 06) | 15 June 06 (18 Mar–16 Sep 06) | 6.4 |
| 2007-4 | 05 Sep 07 (04 Apr 07–01 Feb 08) | 07 Jan 08 (30 Oct 07–17 Mar 08) | 4.0 |
| 2007-5 | 21 Oct 07 (14 Mar 07–07 June 08) | 06 May 08 (01 Feb–09 Aug 08) | 6.5 |
Calculated from the estimated date of the initiating tOPV dose to the estimated date of the first diverging node. The P1/capsid KT evolution rate was assumed to be constant at 1.1%/year (29 nt substitutions/year).
The first VDPV2 isolate, BAS05-01, was isolated from a patient whose onset of AFP was ∼10 months after the estimated time (September 2004) of the initiating tOPV dose (Table 1). Independent emergences continued for nearly 4 years, into late 2008 (Fig. 1 to 4 and Table 1). Two additional VDPV2 isolates from cases in February and November 2011 signaled more recent emergences. The initiating tOPV dose for the major cVDPV2 lineage group, 2005-8, was estimated to have been given in October 2005, more than a year after the estimated date of the initiating dose for the first observed cVDPV2 lineage group, 2004-1. The last observed cVDPV2 lineage group, 2007-5, was estimated to have emerged from a tOPV dose given in late 2007. Thus, despite widespread circulation of cVDPV2, local gaps in immunity to PV2 persisted and permitted several new VDPV2 emergences to appear throughout the outbreak.
The most closely related isolates tended to cluster geographically (see Fig. S1 to S4 in the supplemental material). However, virus of the major lineage group, 2005-8, was disseminated widely across the northern states during its >6 years of transmission.
VDPV2 lineages transiting the low-transmission season for poliovirus.
The occurrence of VDPV2 infections (represented in Fig. 4 by dates of collection of VDPV2-positive specimens) showed a distinct seasonality (45), with a peak during March to June and a low during December to January (Fig. 4 and 6). Seasonal curves based on the appearance of polio cases would be shifted to the left because specimens were collected 10 ± 6 days after onset of AFP. Seasonal curves based on the estimated times of exposure would be shifted further to the left (by a total of ∼25 days) because paralysis usually occurs within 15 days of exposure to neurovirulent poliovirus (79). Seasonal variation is low in humid tropical climates and higher in dryer, more temperate zones (45) and may vary in its details from year to year. Superimposed on these natural cycles is the impact of immunization (including choice of vaccines, vaccine coverage rates, and population immunity profiles) on the virus population.
Fig 6.

Cumulative seasonality of specimens containing VDPV2 in Nigeria, 2004 to 2011. Note that AFP cases appeared ∼10 days earlier than specimen collection and that VDPV2 exposure likely occurred 3 to 4 weeks earlier than specimen collection.
The natural seasonality of polio reflects the fluctuation in the number of chains of poliovirus transmission during the year (80). To estimate the number of lineages (representing observed chains of transmission) that survived the seasonal bottlenecks in December to January, we counted the number of branches that transited across year boundaries (Fig. 4; see also Fig. S1 to S4 in the supplemental material), excluding branches that appeared to have terminated in January. The number of observed lineages estimated to have survived the seasonal bottlenecks rose sharply in 2006 to 2007 (∼63 lineages), declined slightly in 2007 to 2008 (∼48), peaked in 2008 to 2009 (∼144), declined sharply in 2009 to 2010 (∼22), and stabilized in 2010 to 2011 (∼25) (Fig. 7). Lineage group 2005-8 was predominant by 2006 to 2007, peaked at ∼135 lineages transiting the 2008-2009 seasonal low, and declined in 2009 to 2010 (∼21) and 2010 to 2011 (∼23) (Fig. 7).
Fig 7.

Estimated number of observed lineages that survived the 2004-2005 to 2010-2011 December-to-January seasonal bottlenecks. Color-coding of individual emergences and lineage groups (boldface type) does not correspond to that used in Fig. 1.
VDPV2 population dynamics during the outbreak.
The dynamics of virus populations throughout the year can be modeled by coalescent methods in which multiple lineages of a population are traced back in time through successive joining (coalescent) events to the most recent common ancestor (81). Virus population size and rates of growth and decline can be inferred from the topologies of phylogenetic trees (representing genealogies) constructed from temporally ordered sequences (66, 81). In rapidly evolving viruses, variation in the number of lineages can be used to estimate changes over time in the effective virus population size (Ne), a parameter used to model the fixation of “neutral” mutations in actual populations. Virus population dynamics can be visualized through Bayesian skyline plots (66) of the time dependence of Ne (approximately proportional to nucleotide diversity). We used the program BEAST (60) to analyze the population dynamics of all Nigerian VDPV2s from 2005 through mid-2011, comparing the Bayesian skyline plot with the total monthly case counts (Fig. 4 and 8). Ne increased ∼20-fold from mid-2005 to late 2006 (as new emergences were appearing), after which it fluctuated over a narrow range up to late 2008. In 2009, Ne increased by ∼10-fold for January to March, stabilized, and then declined ∼10-fold after two rounds of mass tOPV campaigns on 30 May and 1 August (Fig. 8). The sharp fluctuations in virus population dynamics are reflected in the monthly case counts in 2009 (Fig. 4). Ne stabilized near 2007-2008 levels from late 2009 to the end of 2010 and gradually increased in 2011 (Fig. 8), as two new emergences appeared and as virus of lineage group 2005-8 continued to circulate in eight northern states via at least 23 separate chains of transmission (see Fig. S2 in the supplemental material). As expected from the combined epidemic curves (Fig. 4), the skyline plot for lineage group 2005-8 (not shown) was nearly congruent with that for all Nigerian VDPV2s.
Fig 8.

Bayesian skyline plot of population dynamics of all Nigerian VDPV2s, 2005 to 2011. Shaded areas represent the 95% highest posterior density (HPD) interval around the mean of the effective virus population size (Ne) estimates. Arrows indicate months of tOPV SIAs in the northern states.
Relative substitution rates in VP1 and complete P1/capsid regions.
All WPV and candidate VDPV isolates are routinely characterized by VP1 sequencing by the GPLN (16, 19, 21). To assess the sensitivity of VP1 sequencing as a screening tool for divergent Sabin strain derivatives, we plotted the number of nucleotide substitutions in the VP1 region as a function of the number of nucleotide substitutions in the P1/capsid region for all 2005-2011 Nigerian VDPV2 isolates (Fig. 9). A strong linear correlation (slope = 0.93; R2 = 0.90) between the relative frequencies of VP1 and P1/capsid region nucleotide substitutions was observed. Although some early VDPV isolates had a higher density of nucleotide substitutions in the VP1 region than elsewhere in the P1/capsid region (Table 1), possibly reflecting early fixation of selected substitutions (see below), the differences tended to diminish over time (Fig. 9). Therefore, we conclude that VP1 sequencing has sufficient sensitivity to identify VDPVs, even at levels of divergence as low as 0.7% (i.e., 6 nt substitutions in VP1).
Fig 9.
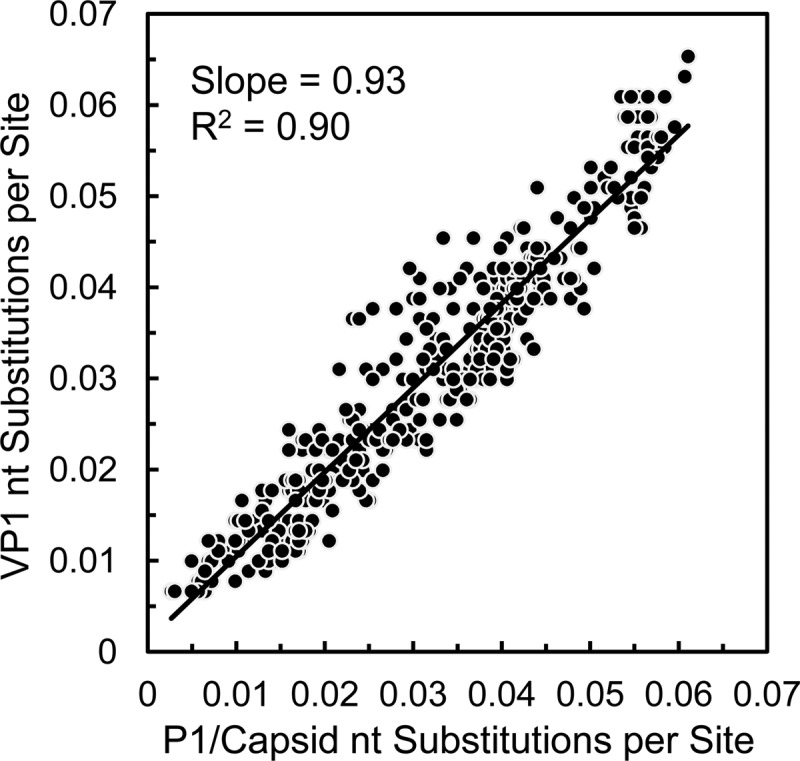
Frequency of nucleotide substitution into the VP1 region relative to the P1/capsid region among the 2005-2011 Nigerian VDPV2 isolates.
Estimated number of VDPV2 infections.
The proportion of WPV infections that result in paralytic cases is lowest for PV2 (∼1:1,800) among the three poliovirus serotypes (45). Under the assumption that the case-to-infection ratio for VDPV2s is similar to that for WPV2, we estimate from the number of reported cases that ∼700,000 cVDPV2 infections have occurred in Nigeria since 2005. The actual number of infections is likely to be higher, as the relatively high frequency of isolates with no close relatives (representing “orphan” lineages, signaled by long branches on the trees) is an indication that cases had been missed in some areas.
Genetic properties of the VDPV2 isolates.
Two determinants are the main contributors to the attenuated phenotype of Sabin 2 in transgenic mice (82) and primates (58): A481 in the 5′-untranslated region (5′-UTR) and Ile143 in VP1. Both determinants were lost in all VDPV2 isolates (Table 5). For the 5′-UTR determinant, evidence of direct A481→G reversion was found for only a small proportion of isolates (20 of 403; 5.0%), representing 16 of the 23 emergences, usually those not associated with lineage groups. In the remainder, A481 had been exchanged out by recombination with other enteroviruses. Early lineage group 2005-8 isolates were Sabin 3/Sabin 2 5′-UTR recombinants with sequences containing the U472→C reversion of the major Sabin 3 attenuation determinant (83). A total of 12 distinct 5′-UTR recombinant classes were detected, some generated by serial recombination events (our unpublished results).
Table 5.
Genetic properties, growth yields, and temperature sensitivity in cultured cells and neurovirulence in PVR-Tg21 mice of selected Nigerian VDPV2 isolates and reference viruses
| Emergencea | Virus | 5′-UTR classb | VP1143 residue | No. of P1/capsid region nt substitutions | 3Dpol classc | Virus yield in HEK293 cells (log10 PFU/ml) at 37.0°Cd | Neurovirulence in PVR-Tg21 mice (PD50)e | Virus yield in RD cells (log10 PFU/ml) at 37.0°Cf | Efficiency of plating in L20B cells at 39.5°C/37.0°C |
|---|---|---|---|---|---|---|---|---|---|
| Sabin 2 | A (Sabin 2) | I | 3D:Sabin 2 | 8.43 | ≥7.5 | 8.72 | 1.4 × 10−4 | ||
| 2004-1 | BAS05-01 | A (Sabin 2*) | T | 21 | 3D:4-1/1 | 9.13 | ND | 8.63 | 0.42 |
| 2005-1 | SOS05-01 | A (Sabin 2*) | T | 19 | 3D:5-1/1 | 9.04 | ND | 9.12 | 0.57 |
| 2005-2 | LAS05-01 | A (Sabin 2*) | T | 12 | 3D:Sabin 3 | 8.97 | ND | 8.52 | 0.35 |
| 2005-3 | KDS05-01 | A (Sabin 2*) | N | 16 | 3D:5-3/1 | 8.50 | ND | 8.23 | 0.42 |
| 2005-4 | KBS06-01 | A (Sabin 2*) | T | 16 | 3D:5-4/1 | 8.96 | ND | 8.55 | 0.49 |
| 2005-5 | JIS06-06 | B | T | 28 | 3D:5-5/1 | 8.88 | ND | 9.18 | 0.45 |
| 2005-6 | BOS06-03 | C | T | 19g | 3D:5-6/1 | 8.93 | 2.4 | 9.42 | 0.47 |
| 2005-6 | BOS07-02 | C | T | 46g | 3D:5-6/1 | 9.04 | 2.1 | 9.21 | 0.60 |
| 2005-7 | BOS06-01 | A (Sabin 2*) | T | 18 | 3D:Sabin 1 | 8.95 | ND | 9.24 | 0.29 |
| 2005-8 | JIS06-01 | D1 (Sabin 3*) | T | 16 | 3D:5-8/1 | 8.82 | 3.3 | 9.22 | 0.66 |
| 2005-8 | KTS09-02 | D1 (Sabin 3*) | T | 70 | 3D:5-8/1 | 9.24 | 2.0 | 9.31 | 0.69 |
| 2005-9 | JIS06-04 | A (Sabin 2*) | T | 18 | 3D:5-9/1 | 8.83 | ND | 9.27 | 0.35 |
| 2005-10 | SOS07-01 | E | T | 27 | 3D:5-10/1 | 8.89 | 3.8 | 9.28 | 0.35 |
| 2005-11 | BAS06-01 | A (Sabin 2*) | T | 25 | 3D:5-11/1 | 8.75 | ND | 8.74 | 0.13 |
| 2006-1 | BOS06-02 | A (Sabin 2*) | T | 7 | 3D:Sabin 2 | 8.48 | 5.9 | 8.70 | 1.8 × 10−2 |
| 2006-2 | SOS08-09 | A (Sabin 2*) | T | 50 | 3D:6-2/1 | 8.80 | ND | 8.95 | 0.11 |
| 2007-1 | NIS07-01 | F | T | 13 | 3D:7-1/1 | 8.79 | ND | 8.97 | 0.15 |
| 2007-2 | BAS08-06 | G | T | 40 | 3D:7-2/1 | 8.89 | ND | 9.32 | 0.33 |
| 2007-3 | ANS07-01 | A (Sabin 2*) | V | 8 | 3D:Sabin 2 | 8.37 | ND | 8.55 | 6.4 × 10−4 |
| 2007-4 | KBS08-03 | H | T | 30 | 3D:7-4/1 | 8.81 | ND | 8.90 | 0.32 |
| 2007-5 | BOS09-06 | A (Sabin 2*) | T | 44 | 3D:7-5/1 | 9.02 | ND | 9.18 | 0.45 |
| 2007-6 | KTS09-05 | A (Sabin 2*) | T | 37 | 3D:7-6/1 | 8.83 | ND | 9.29 | 0.57 |
| 2008-1 | KDS09-08 | A (Sabin 2*) | T | 15 | 3D:Sabin 1 | 8.80 | ND | 8.96 | 2.5 × 10−4 |
| 2010-1 | KBS11-01 | A (Sabin 2*) | V | 13 | 3D:Sabin 1 | 9.01 | ND | 9.23 | 0.27 |
| 2010-2 | NIS11-01 | A (Sabin 2*) | T | 18 | 3D:Sabin 2 | 8.74 | ND | 9.08 | 0.24 |
| MEF-1 | MEF-1 | T | 3D:MEF-1 | 9.16 | 2.5 | 9.20 | 0.92 |
Well-defined cVDPV2 lineage groups are indicated in boldface type.
5′-UTR sequences not of Sabin 2 origin are labeled B to H. Asterisks indicate that all VDPV2 isolates with 5′-UTR sequences derived from Sabin 2 had a A481→G reversion and that all VDPV2 isolates with 5′-UTR sequences derived from Sabin 3 had a U472→C reversion.
3Dpol sequences of nonvaccine origin (see Table S1 in the supplemental material for GenBank accession numbers of complete or nearly complete genomic sequences) are numbered consecutively for each emergence (e.g., 3D:5-6/1 to 3D:5-6/3).
Virus was grown in HEK293 cell monolayers (37.0°C), and yields were determined by plaque assays on HeLa cells (37.0°C).
Neurovirulence was tested by intracerebral inoculation. ND, not done.
Virus was grown in RD cell monolayers, and yields were determined by plaque assays on L20B cells.
Compared against a simulated unmutated Sabin 3/Sabin 2 recombinant with the recombination junction at the same location as the in natural isolates.
The second critical determinant of the attenuated phenotype, VP1-Ile143, had reverted in all VDPV2 isolates. The majority of isolates (399 of 403 [99.0%]; 20 of 23 independent emergences) had the VP1-Ile143→Thr replacement to the WPV2 consensus (37), which remained fixed in the population, except for one lineage group 2005-8 isolate, KNS09-022, which had a superimposed (VP1-Thr143→Ala) replacement. Three other isolates, each representing independent emergences, had the alternative replacements VP1-Ile143→Asn (isolate KDS05-01) and VP1-Ile143→Val (isolates ANS07-01 and KBS11-01) (Table 5).
Apart from the codon for VP1143, nucleotide substitutions accumulated approximately uniformly across the P1/capsid region (Fig. 9), with evidence of purifying selection at most amino acid positions (Table 3). The detailed pattern of nucleotide substitution (Fig. 5), predominantly (∼80%) synonymous transitions (Table 3), closely matched that previously described for WPV1 lineages (47).
The codon for VP1-Thr24 was deleted in four isolates. Three of the isolates (BAS07-04, JIS07-03, and BAS08-01, of lineage group 2005-8) clustered closely on the tree (see Fig. S2 in the supplemental material), and their sequence relationships were consistent with localized circulation of the deletion variant for about a year. The fourth deletion variant, KTS09-22 (also of lineage group 2005-8), arose independently of the others, and there was no evidence of secondary spread (see Fig. S2 in the supplemental material). The 30 (PV3) or 32 (PV1 and PV2) amino-terminal residues of VP1 are disordered within the intact poliovirion (84); form a flexible, externalized amphipathic domain during virus uncoating (85); and can accommodate numerous amino acid substitutions within (47) and across (86) poliovirus serotypes. Variants with single-codon deletions in this domain occasionally emerge transiently within populations of circulating polioviruses of all three serotypes but constitute a very small proportion (<0.2%) of poliovirus isolates worldwide (our unpublished results).
All but seven isolates were vaccine/nonvaccine recombinants, with P2 and P3 noncapsid sequences derived from unidentified species C enteroviruses (87), a property typical of cVDPVs (19, 21, 32, 38, 40, 75) and associated with poliovirus circulation (88–90). Apart from isolates of the largest lineage group, 2005-8, in which the first 31 P1/capsid region codons were derived from Sabin 3, all other VDPV2 P1/capsid region sequences were derived exclusively from Sabin 2.
Phenotypic properties of VDPV2 isolates.
cVDPVs had recovered two key properties of WPVs: the capacity to cause paralytic poliomyelitis in humans and the capacity for sustained person-to-person transmission. Nearly all Nigerian VDPV2s were isolated from patients with AFP; >75% of those patients examined 60 days after onset of paralysis had residual asymmetric paralysis typical of poliomyelitis (14, 15). Moreover, the genetic relationships among the 403 cVDPV2 isolates document their capacity for widespread circulation for more than 6 years. Experimentally measurable virus properties that are possible surrogates for neuropathogenicity for humans and transmissibility are (i) virus yields in HEK293 cells, (ii) neurovirulence for transgenic mice expressing the human CD155 PVR, and (iii) virus yields during growth at supraoptimal temperatures.
(i) Virus yields in HEK293 cells.
HEK293 cells are derived from human neuronal cells (68), and neurovirulent polioviruses grow to higher yields in these cells than do attenuated strains (69). Sabin 2, MEF-1 (a neurovirulent WPV2 reference strain), and 25 Nigerian VDPV2 isolates (including the first isolates of each emergence) were grown in single-step growth experiments in HEK293 cells, and virus yields were measured by plaque assays on HeLa cells. Yields at 37.0°C were lowest for Sabin 2, ANS07-01, BOS06-02, and KDS05-01 and 3- to 5-fold higher for MEF-1, KTS09-02, BAS05-01, SOS05-01, BOS07-02, BOS09-06, and KBS11-01 (Table 5). Yields at 39.5°C were reduced by 1.5- to 15-fold for most isolates, but those of Sabin 2 dropped ∼700-fold, and isolates ANS07-01 and KDS09-08 did not grow at all (data not shown). The latter results reflect primarily the temperature-sensitive phenotype (see below), which in Sabin 2 is largely determined by A481 (91).
(ii) Neurovirulence for PVR-Tg21 transgenic mice.
The above-described findings, together with clinical reports and the sequence properties of the Nigerian VDPV2 isolates, suggested that they would be neurovirulent when introduced into the central nervous systems of experimental animals (58, 82). We selected six isolates, representing four independent VDPV2 emergences (lineage groups 2005-6, 2005-8, and 2005-10, and 2006-1) with various degrees of genetic difference from the parental Sabin 2 strain, for neurovirulence testing by intracerebral inoculation of PVR-Tg21 transgenic mice (39) (Table 5). All but one (JIS06-01) of the six VDPV2s tested were isolated from AFP patients reported to have had residual paralysis at 60 days (15). Isolates BOS06-03, BOS07-02, and KTS09-02 were highly neurovirulent (PD50 = 2.0 to 2.4), similar to MEF-1 (PD50 = 2.5) and unlike Sabin 2 (PD50 ≥ 7.5) (Table 5). Isolates JIS06-01 and SOS07-01 were slightly less neurovirulent (PD50 = 3.3 to 3.8) and nonrecombinant isolate BOS06-02 was much less neurovirulent (PD50 = 5.9) than the other VDPV2 isolates but more neurovirulent than Sabin 2. Although the number of isolates compared was small, increased yields in HEK293 cells at 37.0°C correlated with neurovirulence in PVR-Tg21 mice (Table 5). Within lineage groups 2005-6 and 2005-8, neurovirulence appeared to increase with increased divergence from Sabin 2.
(iii) Virus yields at supraoptimal temperatures.
The Sabin OPV strains produce lower virus yields at supraoptimal temperatures than do WPVs (92). Higher virus yields at elevated temperatures may facilitate person-to-person transmission by increasing the amount of infectious virus excreted by febrile individuals. Sabin 2, all 25 VDPV2 isolates tested, and MEF-1 grew to high titers (8.23 to 9.42 log10 PFU/ml) in RD cells incubated at 37.0°C when measured by plaque assays on L20B cells also incubated at 37.0°C (Table 5) (73, 91). However, when cells were incubated at 39.5°C for plaque assays, the efficiency of plating of Sabin 2 on L20B cells dropped ≥6,000-fold, whereas that of MEF-1 was largely unaltered. Two VDPV2 isolates (ANS07-01 and KDS09-08) were nearly as temperature sensitive as Sabin 2, and one isolate (BOS06-02) had an intermediate temperature-sensitive phenotype (Table 5). These three isolates had retained some vaccine-like traits despite reversion of the two determinants thought to contribute most to the attenuated and temperature-sensitive phenotypes. However, these isolates also had among the fewest P1/capsid region nucleotide substitutions (7 to 15), were not vaccine/nonvaccine recombinants, and had no known progeny. The remaining 22 VDPV2 isolates were not temperature sensitive, having efficiencies of plating at 39.5°C that were only 1.3- to 8.4-fold lower than that of MEF-1. Loss of the temperature-sensitive phenotype in these viruses appeared to have occurred early during most VDPV2 emergences, did not always accompany reversion at A481 or VP1143, and did not strictly correlate with the extent of P1/capsid region divergence from Sabin 2 at substitution levels above ∼1%. All isolates with heterologous 5′-UTR and 3Dpol sequences had lost the temperature-sensitive phenotype, as had one nonrecombinant isolate (NIS11-01).
DISCUSSION
The large, prolonged, and widespread cVDPV2 outbreak in northern Nigeria since 2005 underscores the risks associated with the use of OPV at suboptimal rates of coverage (14, 15, 19, 21, 22, 32). Localization of the cVDPV2 outbreak almost entirely to the northern states follows the pattern of WPV1 and WPV3 transmission in Nigeria since 2005 (7, 8, 93), and the concurrent decline in the incidence of polio cases associated with all three serotypes is likely the result of improved OPV coverage in northern communities where polio is endemic. The Nigerian cVDPV outbreak is surpassed only by the ∼1983-1993 cVDPV2 outbreak in Egypt in duration and is by far the largest in total number of reported VDPV cases in a single outbreak (45). Its final magnitude and duration remain to be determined, as circulation, while apparently sharply reduced, continued into 2012 (7, 19, 22). Although the cVDPV2 outbreak in Egypt was probably of a comparable scale (49), its full extent is unknown because of the absence at that time of sensitive nationwide AFP surveillance (37); in contrast, surveillance in Nigeria was generally strong throughout the outbreak. In both countries, the cVDPVs had reestablished endemic PV2 transmission through successive low-transmission seasons.
Several key risk factors contributed to cVDPV2 emergence and spread in northern Nigeria: (i) the suspension of SIAs in the northern states in 2003 to 2004 (93), (ii) continued weakness in routine immunization with tOPV (7), (iii) insufficient tOPV coverage in previous SIAs (7), and (iv) the shift from tOPV to mOPV1, mOPV3, and bivalent OPV in SIAs conducted after March 2006 (14, 15). The particular emphasis on mOPV1 in SIAs addressed the serious threat posed by the continued circulation of WPV1, the most paralytogenic (45) and transmissible (10–12) of the three WPV serotypes. However, the background of weak routine tOPV immunization permitted a widening gap in immunity to PV2 to develop. These conditions favored concurrent independent cVDPV2 emergences at numerous locations across northern Nigeria, as the threshold of population immunity necessary to limit PV2 spread fell below critical levels (14, 15). In contrast, population immunity to PV2 in southern Nigeria remained sufficiently high to block most (if not all) local cVDPV2 emergences and limit transmission of any cVDPV2 introduced from the north to levels below detection by the AFP surveillance system. The likelihood that the immunity threshold required to block poliovirus transmission is higher in the more densely populated and humid tropical south than in the less populous and semiarid north (15, 45) offers encouraging prospects for the ultimate success of intensified SIAs in the northern states, provided that high rates of OPV coverage are attained (9) and a proper balance between vaccination with bivalent OPV (17, 18) and tOPV (14, 15) is maintained.
The occurrence of a large cVDPV2 outbreak in a setting of comparatively sensitive surveillance (7) offered an unprecedented view of early outbreak events, including detection of multiple independent emergences, first suggested by studies in Madagascar (40, 41). Similar dynamics may typify other cVDPV outbreaks, especially those associated with PV2 (19, 22). However, previous cVDPV outbreaks have usually occurred in settings of both low OPV coverage and weak surveillance, and the outbreaks were detected months or years after the first emergences (37, 38). For example, AFP surveillance was not yet implemented at the start of the cVDPV2 outbreak in Egypt, and the earliest available cVDPV2 outbreak isolates were from cases that occurred about 5 years after the first initiating tOPV dose (because the early outbreak isolates had been discarded). The Egypt cVDPV2 tree also had deep nodes (37), possibly signaling separate emergences, and the atypically low apparent rate of VP1 evolution observed during the Egypt cVDPV2 outbreak (37, 47) may be explained by the occurrence of separate emergences over time.
The cVDPV2 population showed exponential growth during the initial emergence and spread in 2006, stabilization, and then a second phase of exponential growth in early 2009. Four tOPV SIAs in late 2006 and in 2007, despite gaps in coverage (14, 15), may have limited further expansion of the 2005-8 lineage group. However, the absence of tOPV SIAs between 1 September 2007 and 30 May 2009 permitted the accumulation of a population of young children with no immunity to PV2, setting the stage for multiple chains of transmission transiting the 2008-2009 low-transmission season and the burst of cases in early 2009. Implementation of two tOPV SIAs in May and August 2009 was followed by a sharp decline in the number of polio cases, a decrease in the effective population size, and fewer observed lineages transiting the 2009-2010 low-transmission season. Despite the high immunogenicity of Sabin 2 in tOPV (3) and the implementation of seven SIAs using tOPV in 2010 to 2011, circulation of cVDPV2 continued (7, 19, 22) along several separate chains of transmission, indicating that critical numbers of unimmunized children continue to be missed in the tOPV SIAs in the remaining reservoirs of endemicity in northern Nigeria (9).
The observation that one lineage group predominated does not necessarily imply any fundamental difference in the potential transmissibilities among lineage groups. Random events, such as early VDPV introduction into dense populations of nonimmune people or transmission to highly mobile populations, may have shaped the observed patterns of spread. Indeed, viruses of the minor cVDPV2 lineage groups had sustained transmission for an estimated 15 to 35 months, had also diverged into separate chains of transmission within a few months of the initiating tOPV doses, and had recovered robust growth phenotypes when measured experimentally. Moreover, emergences represented by single isolates were estimated to have sustained replication for 3 to 21 months, considerably longer than the 1- to 2-month duration of vaccine virus infections in immunocompetent OPV recipients (30). Although isolates from these emergences would be classified as aVDPV2s, circulation is suspected from the frequency of their appearance in states with low levels of population immunity to PV2 (15) and known gaps in surveillance (7). Indeed, the current outbreak was foreshadowed in 2002 by the isolation of an aVDPV2 from an underimmunized child with AFP in the Plateau state (46). Like most aVDPVs described here, the Plateau isolate resembled cVDPVs, with 2.5% P1/capsid region divergence from Sabin 2 and a mosaic genome with 5′-UTR and 3Dpol region sequences derived from nonvaccine sources (46).
The genetic mechanisms for reversion of an attenuated OPV strain to a cVDPV phenotypically indistinguishable from WPV are incompletely understood and may differ in their specifics among the three Sabin strains. The Sabin 2 strain, derived from a WPV isolate with intrinsically low neurovirulence and subjected to the shortest selection pathway to attenuation (94), retained more of the biological properties of its WPV parent than did the other Sabin strains, especially the capacity for secondary spread (4–6, 25–27, 95). Because key determinants of the attenuated and temperature-sensitive phenotypes of Sabin 2 are strongly selected against during replication in the human intestine (58, 82), reversion to the WPV phenotype may occur well before the accumulation of 6 to 10 substitutions in the VP1 region. Thus, the definitions for VDPVs are not grounded in the underlying genetics of phenotypic reversion but on the poliovirus molecular clock and the need to balance sensitivity with specificity in global poliovirus surveillance. Accordingly, review of the epidemiologic settings (for cVDPVs) and clinical features (for iVDPVs) is crucial for the evaluation of surveillance data. Some early emergences, including three with noncapsid sequences derived from OPV strains, appeared not to have recovered the full biological properties of WPVs when characterized in the laboratory, suggesting that changes in noncapsid sequences may be necessary for full phenotypic reversion (75). Nonetheless, all but one (JIS06-01) of the eight VDPV2s tested in PVR-Tg21 mice were isolated from AFP patients reported to have had residual paralysis at 60 days postonset.
The high genetic lability of Sabin 2, coupled with the lower paralytic attack rate for PV2 infections (45), highlights the need to maintain sensitive AFP and poliovirus surveillance worldwide (16). Implementation of the recently developed real-time RT-PCR screening method by the GPLN (16, 19) has resulted in the recent detection of cVDPV2 emergences in the Democratic Republic of Congo, Ethiopia, Somalia, India, Afghanistan, Yemen, Chad, and Pakistan (19, 21, 22, 45). The observation that several of these recent cVDPV2 outbreaks were also associated with multiple independent emergences has prompted a sharp upward reassessment of the risks of cVDPV2 emergence and spread in settings of low population immunity to PV2 (19, 22, 32, 37, 40, 96, 97) and consideration by the WHO of a globally synchronized switch from tOPV to bivalent OPV following control of cVDPV2 outbreaks (19, 98).
Supplementary Material
ACKNOWLEDGMENTS
We thank Pamela Mitula (WHO-Nigeria), Michael Mwanza (WHO-Nigeria), Goitom Weldegebriel (WHO-Nigeria), Sue Gerber (CDC), and Julie Jenks (CDC) for their contributions to immunization and surveillance in Nigeria. We also thank the field surveillance teams who investigated AFP cases, laboratory staff of the WHO National Polio Laboratories in Maiduguri and Ibadan, Naomi Dybdahl-Sissoko (CDC), Ray Campagnoli (CDC), Karen Ching (CDC), Nhien Tran (CDC), and Ling Wei (CDC) for excellent technical assistance. We thank Matthias Gromeier for advice on the use of HEK293 cells for phenotypic characterization of poliovirus isolates.
This work was supported in part by the Centers for Disease Control and Prevention; a grant from the Polio Research Committee of the World Health Organization; a Grand Challenges Exploration grant from the Bill and Melinda Gates Foundation; grants-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; and grants-in-aid for research on emerging and reemerging infectious diseases from the Ministry of Health, Labor, and Welfare, Japan.
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the funding agencies.
Footnotes
Published ahead of print 13 February 2013
This paper is dedicated to Walter Dowdle in recognition of his distinguished career of service to global public health.
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02954-12.
REFERENCES
- 1. Centers for Disease Control and Prevention 2001. Apparent global interruption of wild poliovirus type 2 transmission. MMWR Morb. Mortal. Wkly. Rep. 50:222–224 [PubMed] [Google Scholar]
- 2. World Health Organization 2012. Progress towards global interruption of wild poliovirus transmission, January 2011-March 2012. Wkly. Epidemiol. Rec. 87:195–200 [PubMed] [Google Scholar]
- 3. Sutter RW, Kew OM, Cochi SL, Aylward RB. 2013. Poliovirus vaccine—live, p 598–645 In Plotkin SA, Orenstein WA, Offit PA. (ed), Vaccines, 6th ed WB Saunders Company, Philadelphia, PA [Google Scholar]
- 4. Benyesh-Melnick M, Melnick JL, Rawls WE, Wimberley I, Barrera-Oro J, Ben-Porath E, Rennick V. 1967. Studies on the immunogenicity, communicability, and genetic stability of oral poliovaccine administered during the winter. Am. J. Epidemiol. 86:112–136 [DOI] [PubMed] [Google Scholar]
- 5. Chen RT, Hausinger S, Dajani AS, Hanfling M, Baughman AL, Pallansch MA, Patriarca PA. 1996. Seroprevalence of antibody against poliovirus in inner-city preschool children. Implications for vaccination policy in the United States. JAMA 275:1639–1645 [PubMed] [Google Scholar]
- 6. Aylward RB, Porta D, Fiore L, Ridolfi B, Chierchini P, Forastiere F. 1997. Unimmunized Gypsy populations and implications for the eradication of poliomyelitis in Europe. J. Infect. Dis. 175(Suppl 1):S86–S88 doi:10.1093/infdis/175.Supplement_1.S86 [DOI] [PubMed] [Google Scholar]
- 7. World Health Organization 2011. Progress towards eradicating poliomyelitis—Nigeria, January 2010-June 2011. Wkly. Epidemiol. Rec. 86:356–36321837845 [Google Scholar]
- 8. World Health Organization 2010. Progress towards eradicating poliomyelitis in Nigeria, January 2009-June 2010. Wkly. Epidemiol. Rec. 85:273–280 [PubMed] [Google Scholar]
- 9. Donaldson L, Toole M, El Sayed N, de Quadros C, Koplan J, Mogedal S, Nduati R, Singhal A. 2012. Report of the Independent Monitoring Board of the Global Polio Eradication Initiative, June 2012. World Health Organization, Geneva, Switzerland: http://www.polioeradication.org/imb.aspx [Google Scholar]
- 10. Centers for Disease Control and Prevention 2010. Outbreaks following wild poliovirus importations—Europe, Africa, and Asia, January 2009-September 2010. MMWR Morb. Mortal. Wkly. Rep. 59:1393–1399 [PubMed] [Google Scholar]
- 11. Centers for Disease Control and Prevention 2006. Resurgence of wild poliovirus type 1 transmission and consequences of importation—21 countries, 2002-2005. MMWR Morb. Mortal. Wkly. Rep. 55:145–150 [PubMed] [Google Scholar]
- 12. Centers for Disease Control and Prevention 2009. Wild poliovirus type 1 and type 3 importations—15 countries, Africa, 2008-2009. MMWR Morb. Mortal. Wkly. Rep. 58:357–362 [PubMed] [Google Scholar]
- 13. World Health Organization 2011. Progress in interrupting wild poliovirus circulation in countries with re-established transmission: Africa, 2009-2010. Wkly. Epidemiol. Rec. 86:104–112 [PubMed] [Google Scholar]
- 14. Jenkins HE, Aylward RB, Gasasira A, Donnelly CA, Mwanza M, Garnier S, Chauvin C, Abanida E, Pate MA, Adu F, Baba M, Grassly NC. 2010. Implications of a circulating vaccine-derived poliovirus in Nigeria. N. Engl. J. Med. 362:2360–2369 [DOI] [PubMed] [Google Scholar]
- 15. Wassilak S, Pate MA, Wannemuehler K, Jenks J, Burns C, Chenoweth P, Abanida EA, Adu F, Baba M, Gasasira A, Iber J, Mkanda P, Williams AJ, Shaw J, Pallansch M, Kew O. 2011. Outbreak of type 2 vaccine-derived poliovirus in Nigeria, 2005-2009: emergence and widespread circulation in an underimmunized population. J. Infect. Dis. 203:898–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Centers for Disease Control and Prevention 2009. Laboratory surveillance for wild and vaccine-derived polioviruses—worldwide, January 2008-June 2009. MMWR Morb. Mortal. Wkly. Rep. 58:950–954 [PubMed] [Google Scholar]
- 17. Sutter RW, John TJ, Jain H, Agarkhedkar S, Ramanan PV, Verma H, Deshpande J, Singh AP, Sreevatsava M, Malankar P, Burton A, Chatterjee A, Jafari H, Aylward RB. 2010. Immunogenicity of bivalent types 1 and 3 oral poliovirus vaccine: a randomised, double-blind, controlled trial. Lancet 376:1682–1688 [DOI] [PubMed] [Google Scholar]
- 18. World Health Organization 2010. Polio vaccines and polio immunization in the pre-eradication era: WHO position paper. Wkly. Epidemiol. Rec. 85:213–228 [PubMed] [Google Scholar]
- 19. Centers for Disease Control and Prevention 2012. Update on vaccine-derived polioviruses—worldwide, April 2011-June 2012. MMWR Morb. Mortal. Wkly. Rep. 61:741–746 [PubMed] [Google Scholar]
- 20. Centers for Disease Control and Prevention 2007. Update on vaccine-derived polioviruses—worldwide, January 2006-August 2007. MMWR Morb. Mortal. Wkly. Rep. 56:996–1001 [PubMed] [Google Scholar]
- 21. Centers for Disease Control and Prevention 2009. Update on vaccine-derived polioviruses—worldwide, January 2008-June 2009. MMWR Morb. Mortal. Wkly. Rep. 58:1002–1006 [PubMed] [Google Scholar]
- 22. Centers for Disease Control and Prevention 2011. Update on vaccine-derived polioviruses—worldwide, July 2009-June 2011. MMWR Morb. Mortal. Wkly. Rep. 60:846–850 [PubMed] [Google Scholar]
- 23. Minor PD, Almond JW. 2002. Poliovirus vaccines: molecular biology and immune response, p 381–390 In Semler BL, Wimmer E. (ed), Molecular biology of picornaviruses. ASM Press, Washington, DC [Google Scholar]
- 24. Henderson DA, Witte JJ, Morris L, Langmuir AD. 1964. Paralytic disease associated with oral polio vaccines. JAMA 190:41–48 [DOI] [PubMed] [Google Scholar]
- 25. Nkowane BM, Wassilak SGF, Orenstein WA, Bart KJ, Schonberger LB, Hinman AR, Kew OM. 1987. Vaccine-associated paralytic poliomyelitis. United States: 1973 through 1984. JAMA 257:1335–1340 [PubMed] [Google Scholar]
- 26. Schonberger LB, McGowan JE, Jr, Gregg MB. 1976. Vaccine-associated poliomyelitis in the United States, 1961-1972. Am. J. Epidemiol. 104:202–211 [DOI] [PubMed] [Google Scholar]
- 27. Strebel PM, Sutter RW, Cochi SL, Biellik RJ, Brink EW, Kew OM, Pallansch MA, Orenstein WA, Hinman AR. 1992. Epidemiology of poliomyelitis in the United States one decade after the last reported case of indigenous wild virus-associated disease. Clin. Infect. Dis. 14:568–579 [DOI] [PubMed] [Google Scholar]
- 28. Cammack N, Phillips A, Dunn G, Patel V, Minor PD. 1988. Intertypic genomic rearrangements of poliovirus strains in vaccinees. Virology 167:507–514 [PubMed] [Google Scholar]
- 29. Kilpatrick DR, Ching K, Iber J, Campagnoli R, Freeman CJ, Mishrik N, Pallansch MA, Kew OM. 2004. Multiplex PCR method for identifying recombinant vaccine-related polioviruses. J. Clin. Microbiol. 42:4313–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alexander JP, Jr, Gary HE, Jr, Pallansch MA. 1997. Duration of poliovirus excretion and its implications for acute flaccid paralysis surveillance: a review of the literature. J. Infect. Dis. 175:S176–S182 [DOI] [PubMed] [Google Scholar]
- 31. Sutter RW, Prevots R. 1994. Vaccine-associated paralytic poliomyelitis among immunodeficient persons. Infect. Med. 11:426–438 [DOI] [PubMed] [Google Scholar]
- 32. Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA. 2005. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu. Rev. Microbiol. 59:587–635 [DOI] [PubMed] [Google Scholar]
- 33. Kew OM, Sutter RW, Nottay B, McDonough M, Prevots DR, Quick L, Pallansch M. 1998. Prolonged replication of a type 1 vaccine-derived poliovirus in an immunodeficient patient. J. Clin. Microbiol. 36:2893–2899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Khetsuriani N, Prevots DR, Quick L, Elder ME, Pallansch M, Kew O, Sutter RW. 2003. Persistence of vaccine-derived polioviruses among immunodeficient persons with vaccine-associated paralytic poliomyelitis. J. Infect. Dis. 188:1845–1852 [DOI] [PubMed] [Google Scholar]
- 35. MacLennan C, Dunn G, Huissoon AP, Kumararatne DS, Martín J, O'Leary P, Thompson RA, Osman H, Wood P, Minor P, Wood DJ, Pillay D. 2004. Failure to clear persistent vaccine-derived neurovirulent poliovirus infection in an immunodeficient man. Lancet 363:1509–1513 [DOI] [PubMed] [Google Scholar]
- 36. Martín J, Dunn G, Hull R, Patel V, Minor PD. 2000. Evolution of the Sabin strain of type 3 poliovirus in an immunodeficient patient during the entire 637-day period of virus excretion. J. Virol. 74:3001–3010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang C-F, Naguib T, Yang S-J, Nasr E, Jorba J, Ahmed N, Campagnoli R, van der Avoort H, Shimizu H, Yoneyama T, Miyamura T, Pallansch MA, Kew O. 2003. Circulation of endemic type 2 vaccine-derived poliovirus in Egypt, 1983 to 1993. J. Virol. 77:8366–8377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kew OM, Morris-Glasgow V, Landaverde M, Burns C, Shaw J, Garib Z, André J, Blackman E, Freeman CJ, Jorba J, Sutter R, Tambini G, Venczel L, Pedreira C, Laender F, Shimizu H, Yoneyama T, Miyamura T, van der Avoort H, Oberste MS, Kilpatrick D, Cochi S, Pallansch M, de Quadros C. 2002. Outbreak of poliomyelitis in Hispaniola associated with circulating type 1 vaccine-derived poliovirus. Science 296:356–359 [DOI] [PubMed] [Google Scholar]
- 39. Shimizu H, Thorley B, Paladin FJ, Brussen KA, Stambos V, Yuen L, Utama A, Tano Y, Arita M, Yoshida H, Yoneyama T, Benegas A, Roesel S, Pallansch M, Kew O, Miyamura T. 2004. Circulation of type 1 vaccine-derived poliovirus in the Philippines in 2001. J. Virol. 78:13512–13521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rakoto-Andrianarivelo M, Gumede N, Jegouic S, Balanant J, Andriamamonjy SN, Rabemanantsoa S, Birmingham M, Randriamanalina B, Nkolomoni L, Venter M, Schoub BD, Delpeyroux F, Reynes J-M. 2008. Reemergence of recombinant vaccine-derived poliovirus outbreak in Madagascar. J. Infect. Dis. 197:1427–1435 [DOI] [PubMed] [Google Scholar]
- 41. Rousset D, Rakoto-Andrianarivelo M, Razafindratsimandresy R, Randriamanalina B, Guillot S, Balanant J, Mauclère P, Delpeyroux F. 2003. Recombinant vaccine-derived poliovirus in Madagascar. Emerg. Infect. Dis. 9:885–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Estívariz C, Watkins MA, Handoko D, Rusipah R, Deshpande JM, Rana BJ, Irawan E, Widhiastuti D, Pallansch MA, Thapa A, Imari S. 2008. A large vaccine-derived poliovirus outbreak on Madura Island—Indonesia, 2005. J. Infect. Dis. 197:347–354 [DOI] [PubMed] [Google Scholar]
- 43. Liang X, Zhang Y, Xu W, Wen N, Zou S, Lee LA, Yu J. 2006. An outbreak of poliomyelitis caused by type 1 vaccine-derived poliovirus in China. J. Infect. Dis. 194:545–551 [DOI] [PubMed] [Google Scholar]
- 44. Yan D, Li L, Zhu S, Zhang Y, An J, Wang D, Wen N, Jorba J, Liu W, Zhong G, Huang L, Kew O, Liang X, Xu W. 2010. Emergence and localized circulation of a vaccine-derived poliovirus in an isolated mountain community in Guangxi, China. J. Clin. Microbiol. 48:3274–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nathanson N, Kew OM. 2010. From emergence to eradication: the epidemiology of poliomyelitis deconstructed. Am. J. Epidemiol. 172:1213–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Adu FD, Iber J, Bukbuk D, Gumede N, Yang S-J, Jorba J, Campagnoli R, Sule WF, Yang C-F, Burns C, Pallansch M, Harry T, Kew O. 2007. Isolation of recombinant type 2 vaccine-derived poliovirus (VDPV) from a Nigerian child. Virus Res. 127:17–25 [DOI] [PubMed] [Google Scholar]
- 47. Jorba J, Campagnoli R, De L, Kew O. 2008. Calibration of multiple poliovirus molecular clocks covering an extended evolutionary range. J. Virol. 82:4429–4440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. World Health Organization 2010. Sixteenth Informal Consultation of the Global Polio Laboratory Network, 22 to 23 September 2010. World Health Organization, Geneva, Switzerland: http://www.polioeradication.org/Portals/0/Document/Resources/GPLN_publications/GPLN_Meeting_recommendations_2010.pdf [Google Scholar]
- 49. Wringe A, Fine PEM, Sutter RW, Kew OM. 2008. Estimating the extent of vaccine-derived poliovirus infection. PLoS One 3:e3433 doi:10.1371/journal.pone.0003433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hovi T, Stenvik M. 1994. Selective isolation of poliovirus in recombinant murine cell line expressing the human poliovirus receptor gene. J. Clin. Microbiol. 32:1366–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pipkin PA, Wood DJ, Racaniello VR, Minor PD. 1993. Characterisation of L cells expressing the human poliovirus receptor for the specific detection of polioviruses in vitro. J. Virol. Methods 41:333–340 [DOI] [PubMed] [Google Scholar]
- 52. World Health Organization 2004. Polio laboratory manual, 4th ed World Health Organization, Geneva, Switzerland [Google Scholar]
- 53. Kilpatrick DR, Nottay B, Yang C-F, Yang S-J, da Silva E, Peñaranda S, Pallansch M, Kew O. 1998. Serotype-specific identification of polioviruses by PCR using primers containing mixed-base or deoxyinosine residues at positions of codon degeneracy. J. Clin. Microbiol. 36:352–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kilpatrick DR, Nottay B, Yang C-F, Yang S-J, Mulders MN, Holloway BP, Pallansch MA, Kew OM. 1996. Group-specific identification of polioviruses by PCR using primers containing mixed-base or deoxyinosine residues at positions of codon degeneracy. J. Clin. Microbiol. 34:2990–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kilpatrick DR, Yang C-F, Ching K, Vincent A, Iber J, Campagnoli R, Mandelbaum M, De L, Yang S-J, Nix A, Kew OM. 2009. Rapid group-, serotype-, and vaccine strain-specific identification of poliovirus isolates by real-time reverse transcription-PCR using degenerate primers and probes containing deoxyinosine residues. J. Clin. Microbiol. 47:1939–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van der Avoort HG, Reimerink JH, Ras A, Mulders MN, van Loon AM. 1995. Isolation of epidemic poliovirus from sewage during the 1992-3 type 3 outbreak in The Netherlands. Epidemiol. Infect. 114:481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liu H-M, Zheng D-P, Zhang L-B, Oberste MS, Pallansch MA, Kew OM. 2000. Molecular evolution of a type 1 wild-vaccine poliovirus recombinant during widespread circulation in China. J. Virol. 74:11153–11161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Macadam AJ, Pollard SR, Ferguson G, Skuce R, Wood D, Almond JW, Minor PD. 1993. Genetic basis of attenuation of the Sabin type 2 vaccine strain of poliovirus in primates. Virology 192:18–26 [DOI] [PubMed] [Google Scholar]
- 59. Rezapkin GV, Fan L, Asher DM, Fibi MR, Dragunsky EM, Chumakov KM. 1999. Mutations in Sabin 2 strain of poliovirus and stability of attenuated phenotype. Virology 258:152–160 [DOI] [PubMed] [Google Scholar]
- 60. Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29:1969–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, Huelsenbeck JP, Ronquist F, Swofford DL, Cummings MP, Rambaut A, Suchard MA. 2012. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst. Biol. 61:170–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Posada D, Crandall KA. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817–818 [DOI] [PubMed] [Google Scholar]
- 63. Ronquist F, Huelsenbeck JP. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574 [DOI] [PubMed] [Google Scholar]
- 64. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shapiro B, Ho SYW, Drummond AJ, Suchard MA, Pybus OG, Rambaut A. 2011. A Bayesian phylogenetic method to estimate unknown sequence ages. Mol. Biol. Evol. 28:879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Drummond AJ, Rambaut A, Shapiro B, Pybus OG. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22:1185–1192 [DOI] [PubMed] [Google Scholar]
- 67. Vidor E, Plotkin SA. 2013. Poliovirus vaccine—inactivated, p 573–597 In Plotkin SA, Orenstein WA, Offit PA. (ed), Vaccines, 6th ed WB Saunders Company, Philadelphia, PA [Google Scholar]
- 68. Shaw G, Morse S, Ararat M, Graham FL. 2002. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK 293 cells. FASEB J. 16:869–871 [DOI] [PubMed] [Google Scholar]
- 69. Campbell SA, Lin J, Dobrikova EY, Gromeier M. 2005. Genetic determinants of cell type-specific poliovirus propagation in HEK 293 cells. J. Virol. 79:6281–6290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Burns CC, Shaw J, Campagnoli R, Jorba J, Vincent A, Quay J, Kew OM. 2006. Modulation of poliovirus replicative fitness in HeLa cells by deoptimization of synonymous codon usage in the capsid region. J. Virol. 80:3259–3272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Koike S, Taya C, Kurata T, Abe S, Ise I, Yonekawa H, Nomoto A. 1991. Transgenic mice susceptible to poliovirus. Proc. Natl. Acad. Sci. U. S. A. 88:951–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kärber G. 1931. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Arch. Exp. Pathol. Pharmakol. 162:480–483 [Google Scholar]
- 73. Macadam AJ, Ferguson G, Stone DM, Meredith J, Knowlson S, Auda G, Almond JW, Minor PD. 2006. Rational design of genetically stable, live-attenuated poliovirus vaccines of all three serotypes: relevance to poliomyelitis eradication. J. Virol. 80:8653–8663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van der Avoort HG, Hull BP, Hovi T, Pallansch MA, Kew OM, Crainic R, Wood DJ, Mulders MN, van Loon AM. 1995. A comparative study of five methods of intratypic differentiation of polioviruses. J. Clin. Microbiol. 33:2562–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Jegouic S, Joffret M-L, Blanchard C, Riquet FB, Perret C, Pelletier I, Colbere-Garapin F, Rakoto-Andrianarivelo M, Delpeyroux F. 2009. Recombination between polioviruses and co-circulating coxsackie A viruses: role in the emergence of pathogenic vaccine-derived polioviruses. PLoS Pathog. 5:e1000412 doi:10.1371/journal.ppat.1000412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gavrilin GV, Cherkasova EA, Lipskaya GY, Kew OM, Agol VI. 2000. Evolution of circulating wild poliovirus and of vaccine-derived poliovirus in an immunodeficient patient: a unifying model. J. Virol. 74:7381–7390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Odoom JK, Yunus Z, Dunn G, Minor PD, Martín J. 2008. Changes in population dynamics during long-term evolution of Sabin type 1 poliovirus in an immunodeficient patient. J. Virol. 82:9179–9190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. van der Sanden S, Pallansch MA, van de Kassteele J, El-Sayed N, Sutter RW, Koopmans M, van der Avoort H. 2009. Shedding of vaccine viruses with increased antigenic and genetic divergence after vaccination of newborns with monovalent type 1 oral poliovirus vaccine. J. Virol. 83:8693–8704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Bodian D, Horstmann DM. 1965. Polioviruses, p 430–473 In Horsfall FL, Tamm I. (ed), Viral and rickettsial infections of man, 4th ed JB Lippincott, Philadelphia, PA [Google Scholar]
- 80. Shulman LM, Handsher R, Yang S-J, Yang C-F, Manor J, Vonsover A, Grossman Z, Pallansch M, Mendelson E, Kew OM. 2000. Resolution of the pathways of poliovirus type 1 transmission during an outbreak. J. Clin. Microbiol. 38:945–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Kingman JFC. 1982. On the genealogy of large populations. J. Appl. Probab. 19:27–43 [Google Scholar]
- 82. Ren R, Moss EG, Racaniello VR. 1991. Identification of two determinants that attenuate vaccine-related type 2 poliovirus. J. Virol. 65:1377–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Westrop GD, Wareham KA, Evans DM, Dunn G, Minor PD, Magrath DI, Taffs F, Marsden S, Skinner MA, Schild GC, Almond JW. 1989. Genetic basis of attenuation of the Sabin type 3 oral poliovirus vaccine. J. Virol. 63:1338–1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hogle JM, Chow M, Filman DJ. 1985. The three-dimensional structure of poliovirus at 2.9 Å resolution. Science 229:1358–1365 [DOI] [PubMed] [Google Scholar]
- 85. Lin J, Cheng N, Chow M, Filman DJ, Steven AC, Hogle JM, Belnap DM. 2011. An externalized polypeptide partitions between two distinct sites on genome-released poliovirus particles. J. Virol. 85:9974–9983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Toyoda H, Kohara MM, Kataoka Y, Suganuma T, Omata T, Imura N, Nomoto A. 1984. Complete nucleotide sequences of all three poliovirus serotype genomes: implication for genetic relationship, gene function and antigenic determinants. J. Mol. Biol. 174:561–585 [DOI] [PubMed] [Google Scholar]
- 87. Brown B, Oberste MS, Maher K, Pallansch M. 2003. Complete genomic sequencing shows that polioviruses and members of human enterovirus species C are closely related in the noncapsid coding region. J. Virol. 77:8973–8984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Dahourou G, Guillot S, Le Gall O, Crainic R. 2002. Genetic recombination in wild-type poliovirus. J. Gen. Virol. 83:3103–3110 [DOI] [PubMed] [Google Scholar]
- 89. Guillot S, Caro V, Cuervo N, Korotkova E, Combiescu M, Persu A, Aubert-Combiescu A, Delpeyroux F, Crainic R. 2000. Natural genetic exchanges between vaccine and wild poliovirus strains in humans. J. Virol. 74:8434–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Liu H-M, Zheng D-P, Zhang L-B, Oberste MS, Kew OM, Pallansch MA. 2003. Serial recombination during circulation of type 1 wild-vaccine recombinant polioviruses in China. J. Virol. 77:10994–11005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Macadam AJ, Pollard SR, Ferguson G, Dunn G, Skuce R, Almond JW, Minor PD. 1991. The 5′ noncoding region of the type 2 poliovirus vaccine strain contains determinants of attenuation and temperature sensitivity. Virology 181:451–458 [DOI] [PubMed] [Google Scholar]
- 92. Sabin AB. 1959. Recent studies and field tests with a live attenuated poliovirus vaccine, p 14–33 In Live poliovirus vaccines: papers presented and discussions held at the First International Conference on Live Poliovirus Vaccines Pan American Sanitary Bureau, Washington, DC [Google Scholar]
- 93. World Health Organization 2005. Progress towards poliomyelitis eradication in Nigeria, January 2004-July 2005. Wkly. Epidemiol. Rec. 80:305–311 [PubMed] [Google Scholar]
- 94. Sabin AB, Boulger LR. 1973. History of Sabin attenuated poliovirus oral live vaccine strains. J. Biol. Stand. 1:115–118 [Google Scholar]
- 95. Fox JP, LeBlanc DR, Gelfand HM, Clemmer DJ, Potash L. 1960. Spread of a vaccine strain of poliovirus in southern Louisiana communities, p 144–155 In Second International Conference on Live Poliovirus Vaccines Pan American Health Organization scientific publication no 60. Pan American Health Organization, Washington, DC [Google Scholar]
- 96. Tebbens RJD, Pallansch MA, Kew OM, Caceres VM, Jafari H, Cochi SL, Sutter RW, Aylward RB, Thompson KM. 2006. Risks of paralytic disease due to wild or vaccine-derived poliovirus after eradication. Risk Anal. 26:1471–1505 [DOI] [PubMed] [Google Scholar]
- 97. Korotkova EA, Park R, Cherkasova EA, Lipskaya GY, Chumakov KM, Feldman E, Kew OM, Agol VI. 2003. Retrospective analysis of a local cessation of vaccination against poliomyelitis: a possible scenario for the future. J. Virol. 77:12460–12465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. World Health Organization 2012. Meeting of the Strategic Advisory Group of Experts on Immunization, April 2012—conclusions and recommendations. Wkly. Epidemiol. Rec. 87:201–216 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.