

Abstract
Simian retrovirus type 4 (SRV-4), a simian type D retrovirus, naturally infects cynomolgus monkeys, usually without apparent symptoms. However, some infected monkeys presented with an immunosuppressive syndrome resembling that induced by simian immunodeficiency virus infection. Antiretrovirals with inhibitory activity against SRV-4 are considered to be promising agents to combat SRV-4 infection. However, although some antiretrovirals have been reported to have inhibitory activity against SRV-1 and SRV-2, inhibitors with anti-SRV-4 activity have not yet been studied. In this study, we identified antiretroviral agents with anti-SRV-4 activity from a panel of anti-human immunodeficiency virus (HIV) drugs using a robust in vitro luciferase reporter assay. Among these, two HIV reverse transcriptase inhibitors, zidovudine (AZT) and tenofovir disoproxil fumarate (TDF), potently inhibited SRV-4 infection within a submicromolar to nanomolar range, which was similar to or higher than the activities against HIV-1, Moloney murine leukemia virus, and feline immunodeficiency virus. In contrast, nonnucleoside reverse transcriptase inhibitors and protease inhibitors did not exhibit any activities against SRV-4. Although both AZT and TDF effectively inhibited cell-free SRV-4 transmission, they exhibited only partial inhibitory activities against cell-to-cell transmission. Importantly, one HIV integrase strand transfer inhibitor, raltegravir (RAL), potently inhibited single-round infection as well as cell-free and cell-to-cell SRV-4 transmission. These findings indicate that viral expansion routes impact the inhibitory activity of antiretrovirals against SRV-4, while only RAL is effective in suppressing both the initial SRV-4 infection and subsequent SRV-4 replication.
INTRODUCTION
Simian type D retroviruses (SRV/Ds) are prevalent among wild and colony-born macaque monkeys, including Macaca fascicularis (cynomolgus) and M. mulatta (rhesus) (1–3). Although SRV/D infection is asymptomatic in most of these monkeys, mild immunosuppression accompanied by anemia, diarrhea, and splenomegaly has been observed in infected cynomolgus monkeys (3, 4). Recently, Japanese macaques (M. fuscata) housed in the Primate Research Institute (PRI) of Kyoto University, Japan, died of a hemorrhagic syndrome with symptoms such as anorexia, pallor, and nasal hemorrhage (5). Extensive investigations revealed that this illness was caused by an infection with an SRV/D known as simian retrovirus type 4 (SRV-4) (5; M. Okamoto et al., unpublished data). SRV-4 has been reported to be distantly related to other SRV/Ds, including SRV-1, -2, -3, -5, -6, and -7; e.g., the previously isolated SRV-4 showed genome sequence similarities of 78, 76, and 74% to SRV-1, -2, and -3, respectively (6). Although there is more than 80% amino acid sequence identity between Gag, Prt, and Pol of SRV-4 and SRV-1, -2, or -3, the Env sequence of SRV-4 is relatively diverse (67 to 74%) compared to other SRV/Ds (6). Although SRV-4 asymptomatically infects cynomolgus monkeys (7), SRV-4 infection of Japanese macaques has not been reported to date. Because the cause of the high mortality observed only for SRV-4-infected Japanese monkeys at PRI remains unclear, it is important to study SRV-4 pathogenesis in Japanese monkeys and to develop a prevention/treatment strategy for controlling SRV-4 infection.
Human immunodeficiency virus (HIV) infection remains a significant threat to humans. Over 20 antiviral drugs have been approved for the treatment of HIV-1-infected individuals. Antiretroviral therapy (ART) can efficiently suppress viral load and enable the recovery of immune function in HIV-1-infected individuals. Some of these drugs suppress infections caused by other retroviruses, including murine leukemia virus (MLV) (8, 9), xenotropic murine leukemia-related retrovirus (XMRV) (10, 11), feline immunodeficiency virus (FIV) (12, 13), and human T-cell leukemia virus type 1 (HTLV-1) (14, 15), indicating that some anti-HIV drugs are widely active against several other retroviruses. There have been some reports on the anti-SRV/D activity of anti-HIV drugs. Tsai et al. reported that three nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs), zidovudine (AZT), zalcitabine (ddC), and 2′,3′-deoxyadenosine (ddA), exhibited inhibitory activity against SRV-2 infection in vitro (16). Moreover, although ddC treatment induced no major change in viral titers in pigtailed monkeys (M. nemestrina) naturally infected with SRV-2, the prophylactic use of ddC blocked de novo SRV-2 infection in this species (17). Rosenblum et al. reported that anti-SRV-1 and anti-SRV-2 activities of several NRTIs were relatively comparable with anti-HIV-1 activity (18). Furthermore, elvitegravir (EVG) and raltegravir (RAL), which are HIV-1 integrase strand transfer inhibitors (INSTIs), efficiently block SRV-3 (also known as Mason-Pfizer monkey virus) infection at nanomolar concentrations (19). Thus, some NRTIs and INSTIs exhibit anti-SRV/D activity; however, whether these drugs are active against SRV-4 infection remains unclear.
In this study, we extensively evaluated the anti-SRV-4 activity of a series of anti-HIV inhibitors, including NRTIs, nonnucleoside reverse transcriptase inhibitors (NNRTIs), an INSTI, and protease inhibitors (PIs), in vitro using single-round infection and multiround viral spread by cell-free and cell-to-cell transmission. Among the NRTIs tested, AZT and tenofovir disoproxil fumarate (TDF) efficiently blocked single-round infection and cell-free transmission of SRV-4, although they were less effective against cell-to-cell transmission. RAL, an INSTI, blocked single-round infection and cell-free transmission of SRV-4 within the nanomolar range, and notably, it was also effective against cell-to-cell SRV-4 transmission. These results indicate that AZT, TDF, and RAL are effective in blocking the initial SRV-4 infection, and particularly, RAL is the most promising drug for the control of SRV-4 replication.
MATERIALS AND METHODS
Antiviral agents.
Didanosine (ddI) (NRTI), lamivudine (3TC) (NRTI), stavudine (d4T) (NRTI), ddC (NRTI), AZT (NRTI), and nelfinavir (NFV) (PI) were purchased from Sigma (St. Louis, MO). Efavirenz (EFV) (NNRTI), nevirapine (NVP) (NNRTI), and saquinavir (SQV) (PI) were purchased from Toronto Research Chemicals (Ontario, Canada). Emtricitabine (FTC) (NRTI), TDF (NRTI), darunavir (DRV) (PI), and RAL (INSTI) were obtained through the AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH).
Cells and viruses.
TE671 (human rhabdomyosarcoma), 293T (human embryonic kidney), and 293T/SRV-4 (a persistently SRV-4-infected 293T cell line) cells, which have been established by the transfection of an SRV-4 infectious clone into 293T cells (Okamoto et al., unpublished), were grown in Dulbecco's modified Eagle's medium (DMEM). MT-2 cells (human T lymphocytes) were grown in RPMI 1640 medium. These media were supplemented with 10% fetal calf serum (FCS), 2 mM l-glutamine, 100 U/ml penicillin, and 50 μg/ml streptomycin. 293FT cells (Invitrogen, Carlsbad, CA) were cultured in DMEM supplemented with 0.5 mg/ml G418. Platinum-GP cells (Plat-GP; Cell Biolabs, San Diego, CA) were maintained in DMEM supplemented with 10 μg/ml blasticidin.
Concentrated SRV-4 was prepared as follows: 293T/SRV-4 cells (106 cells) were cultured in a T-75 flask. After 3 days, culture supernatants were recovered and filtered through a 0.45-μm membrane, followed by the addition of a 30% polyethylene glycol (PEG) solution and 1.2 M sodium chloride. The mixture was then incubated overnight at 4°C, followed by centrifugation at 3,000 rpm for 45 min at 4°C. The resultant pellet was resuspended in DMEM and used for assays immediately after titration.
Quantification of the proviral copy number.
Viral RNA and genomic DNA were prepared from concentrated SRV-4 by using the QIAamp viral RNA minikit (Qiagen, Hilden, Germany) and from SRV-4-infected 293T cells by using DNAzol (Invitrogen), respectively. The viral copy number was quantified by using the One Step PrimeScript reverse transcriptase PCR (RT-PCR) kit (TaKaRa, Otsu, Japan) and the StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA) with a known copy number control. The primer sets and a probe used for SRV-4 amplification were described previously (20). PCR conditions were 5 min at 42°C, 10 s at 95°C, and 55 cycles of 5 s at 95°C and 34 s at 62°C.
VSV-G-pseudotyped luciferase expression vectors.
An envelope-deleted SRV-4-based firefly luciferase expression vector, Δenv-SRV-4-luc (R. Yoshikawa et al., unpublished data), and a plasmid, pcDNA-VSV-G, encoding the vesicular stomatitis virus envelope glycoprotein (VSV-G) (provided by H. Miyoshi, Riken Bioresource Center, Tsukuba, Japan) were used to generate VSV-G-pseudotyped luciferase-expressing SRV-4. These plasmids were cotransfected into 293FT cells. After 48 h of transfection, culture supernatants were recovered, filtered through a 0.45-μm membrane, and stored at −80°C until use.
The VSV-G-pseudotyped luciferase-expressing HIV-1-based lentiviral vector was generated as reported previously (9). The Moloney MLV (MoMLV)-based retroviral vector was produced by cotransfection of the pDON-AI-2-luc plasmid, a firefly luciferase gene-containing pDON-AI-2 retroviral vector (TaKaRa) (provided by Y. Sakurai, Institute for Virus Research, Kyoto University, Kyoto, Japan), and pcDNA-VSV-G into a MoMLV-based packaging cell line, Plat-GP. The FIV-based lentiviral vector was prepared by cotransfection of a luciferase-encoding transfer vector, pCDF-luc-EF1-puro; a 34TF10-derived packaging vector, pFIV-34N (SBI System Biosciences, Mountain View, CA); and pcDNA-VSV-G into 293FT cells. All the recombinant viruses were collected and stored as mentioned above.
Evaluation of the anti-SRV-4 activities of NRTIs, NNRTIs, and an INSTI in single-round infection.
To evaluate the inhibitory activities of anti-HIV drugs against VSV-G-pseudotyped luciferase-expressing SRV-4, HIV-1, MoMLV, and FIV, TE671 cells (104 cells/well) were plated onto white 96-well flat plates. After 24 h of incubation, the cells were infected with each virus in the presence of various concentrations of inhibitors. Similarly, 3 × 105 MT-2 cells were infected separately. Luciferase activity was determined by using the Bright-Glo luciferase assay system (Promega, Madison, WI) and a TriStar LB 941 multimode microplate reader (Berthold, Bad Wildbad, Germany) at 48 h postinfection. Cytotoxicity of the inhibitors was measured by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay, as described previously (9). Antiviral activity and cytotoxicity of the inhibitors are presented as the concentration that blocks viral infection by 50% (50% effective concentration [EC50]) and the concentration that inhibits cell viability by 50% (50% cytotoxic concentration [CC50]), respectively.
Evaluation of the inhibitory activity of PI against SRV-4 production.
293T/SRV-4 cells were washed three times with phosphate-buffered saline (PBS) and plated at a density of 2 × 105 cells/well onto a six-well culture plate in the presence of various concentrations of PIs. After 72 h of incubation, culture supernatants were collected and concentrated as described above. The resultant pellet was solubilized with lysis buffer supplied in the Reverse Transcriptase Assay, colorimetric (Roche, Mannheim, Germany), and RT activity was quantified to evaluate viral production.
Effects of AZT, TDF, and RAL on SRV-4 replication.
To test cell-free SRV-4 infection, 293T cells were plated at a density of 2 × 105 cells/well onto a six-well plate and pretreated with inhibitors of approximately 10× EC50s determined by the single-round luciferase assay (AZT [400 nM], TDF [10 nM], and RAL [150 nM]) or dimethyl sulfoxide (DMSO) as a control for 4 h. Following this, culture media were replaced with fresh medium containing identical concentrations of each inhibitor, and the cells were infected with concentrated (37.5-fold) replication-competent SRV-4 at a multiplicity of infection (MOI) of 2.0 × 106 copies/cell.
For cell-to-cell SRV-4 infection, SRV-4-free 293T cells (2 × 105 cells) were pretreated with inhibitors as described above for the cell-free infection assay. Following this, 293T/SRV-4 cells (4 × 103 cells; proviral copy number, 5 × 101.3 copies/cell) were cocultured in the presence of identical concentrations of inhibitors.
In both the experimental approaches, culture supernatants were collected and replenished with an equal volume of fresh medium containing the corresponding inhibitors on days 1, 3, and 5 postinfection/postcoculture. SRV-4 in each collected supernatant was concentrated, and RT activity was quantified to monitor viral replication.
Statistical analysis.
Dunnett's test and the Bonferroni test were used to determine the statistical significance of anti-SRV-4 activity of inhibitors in single-round assays (Table 1) and of SRV-4 transmission in cell-free and cell-to-cell infection assays (Fig. 1), respectively.
Table 1.
Susceptibility of VSV-G-pseudotyped luciferase-expressing SRV-4 and related retroviruses/lentiviruses to NRTIs and an INSTI in single-round infectiona
| Target cell line and inhibitor | Mean EC50 ± SD (μM) (mean % inhibition ± SD) |
|||
|---|---|---|---|---|
| HIV-1 | SRV-4 | MoMLV | FIV | |
| TE671 | ||||
| NRTIs | ||||
| Thymidine analogs | ||||
| AZT | 0.018 ± 0.0074 | 0.042 ± 0.012* | 0.019 ± 0.0043 | 0.029 ± 0.0035 |
| d4T | 0.40 ± 0.10 | 0.17 ± 0.039 | 3.7 ± 0.82** | 0.52 ± 0.0055 |
| Inosine analog | ||||
| ddI | 10 ± 1.7 | 4.4 ± 1.3** | >10 (0) | 19 ± 1.7** |
| Cytidine analogs | ||||
| ddC | 5.6 ± 1.1 | 2.7 ± 0.13** | >10 (0)** | 3.2 ± 0.50** |
| 3TC | 4.4 ± 0.75 | 3.9 ± 1.2 | >10 (0)** | 2.2 ± 0.50* |
| FTC | 0.48 ± 0.15 | 0.50 ± 0.082 | >10 (0)** | 0.35 ± 0.030 |
| Adenosine analog | ||||
| TDF | 0.0043 ± 0.00058 | 0.00080 ± 0.00037** | 0.0035 ± 0.0012 | 0.0015 ± 0.00076** |
| INSTI | ||||
| RAL | 0.0031 ± 0.0015 | 0.015 ± 0.0065 | 0.0017 ± 0.00036 | 0.049 ± 0.00090* |
| MT-2 | ||||
| NRTIs | ||||
| Thymidine analogs | ||||
| AZT | 0.037 ± 0.014 | 0.11 ± 0.037 | 0.71 ± 0.36* | 1.4 ± 0.40** |
| d4T | 0.50 ± 0.13 | 2.3 ± 0.44 | 3.5 ± 0.61 | 21 ± 6.6** |
| Inosine analog | ||||
| ddI | 3.4 ± 0.70 | >10 (42 ± 4.4)* | >10 (0)* | 16 ± 4.7** |
| Cytidine analogs | ||||
| ddC | 7.2 ± 2.4 | 0.59 ± 0.45** | >10 (0) | 3.5 ± 1.1* |
| 3TC | 2.8 ± 1.0 | >10 (17 ± 3.3)** | >10 (0)** | 1.1 ± 0.10** |
| FTC | 0.52 ± 0.12 | 3.0 ± 0.40** | >10 (0)** | 0.22 ± 0.12 |
| Adenosine analog | ||||
| TDF | 0.0071 ± 0.0018 | 0.0016 ± 0.00025** | 0.0071 ± 0.00056 | 0.0039 ± 0.0021 |
| INSTI | ||||
| RAL | 0.0033 ± 0.0010 | 0.0024 ± 0.00068 | 0.00064 ± 0.00057 | 0.062 ± 0.032** |
Antiviral activities of NRTIs and an INSTI against VSV-G-pseudotyped SRV-4, HIV-1, MoMLV, and FIV were determined by using a luciferase assay. Data are shown as means and standard deviations obtained from three or more independent experiments, and statistical analyses were performed (*, P < 0.05; **, P < 0.01; not indicated, P ≥ 0.05 [determined by Dunnett's test against control HIV-1]). EC50s shown as >10 indicate that more than 10 μM drugs is required to block viral infection by 50%. In this case, percent inhibition of viral infection at 10 μM is shown in parentheses and is considered 10 μM for statistical analysis.
Fig 1.
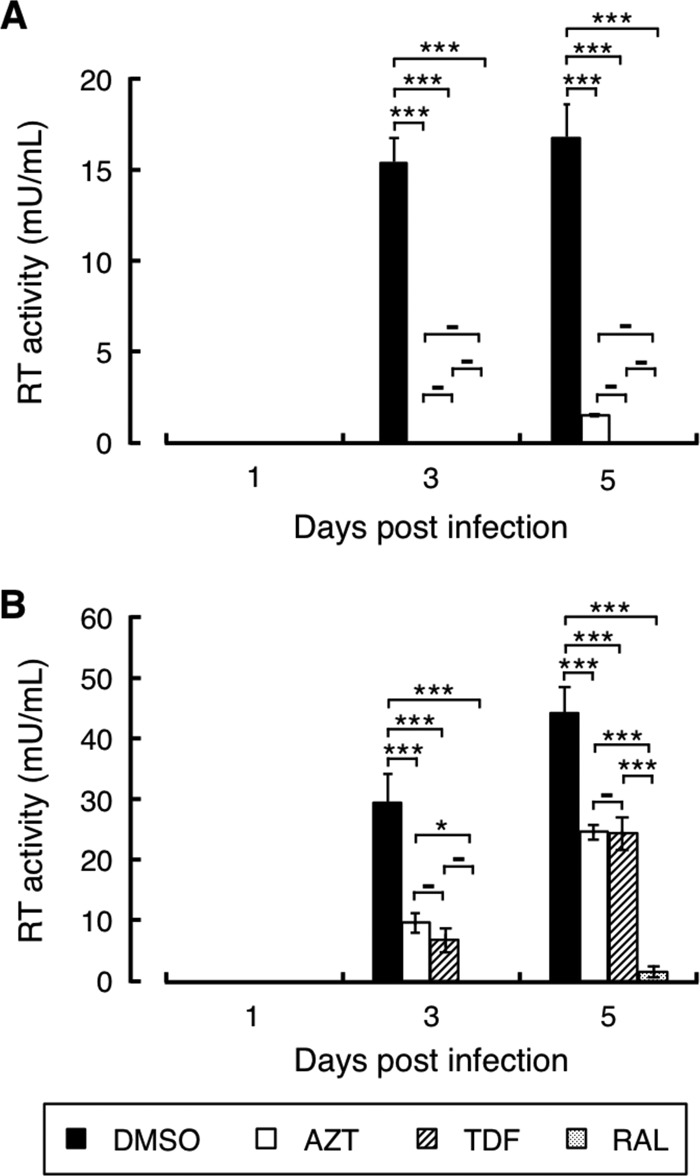
Effects of AZT, TDF, and RAL on SRV-4 replication. Anti-SRV-4 activities of AZT, TDF, and RAL were evaluated in cell-free transmission (A) and cell-to-cell transmission (B) models. SRV-4-free 293T cells were treated with AZT (400 nM), TDF (10 nM), RAL (150 nM), or vehicle (DMSO). After 4 h, culture media were replaced with fresh medium containing identical concentrations of each drug with replication-competent SRV-4 (A) or with SRV-4-infected 293T cells at a ratio of uninfected to infected cells of 50:1 (B). Culture supernatants were periodically collected, and SRV-4 was concentrated. The viral pellet was lysed, and reverse transcriptase activity derived from SRV-4 was quantified with a standard of known activity to monitor viral production. Data are shown as means and standard deviations obtained from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; −, P ≥ 0.05 (determined by the Bonferroni test).
Protein sequence alignment.
Standard amino acid sequences of SRV-4 (GenBank accession number NC_014474.1), HIV-1 (accession number NC_001802.1), MoMLV (accession number NC_001501.1), and FIV (accession number NC_001482.1) were aligned by using the program Clustal W (21), as described previously (9). Residues associated with drug resistance in HIV-1, reported in the Stanford University HIV Drug Resistance Database (22), are also shown.
RESULTS
Anti-SRV-4 activity of HIV NRTIs in single-round infection.
To date, there is no convenient assay system for evaluating the anti-SRV-4 activity of compounds; therefore, we first established a simple and quantitative assay system by employing VSV-G-pseudotyped luciferase-expressing SRV-4 as a model virus. The anti-SRV-4 activity of the test compounds was evaluated by using TE671 and MT-2 cells. TE671 cells, which are derived from human rhabdomyosarcoma, have frequently been used for infection experiments with several retroviruses, including SRVs (23). MT-2 cells, which are derived from human T lymphocytes, are also susceptible to some viruses, including HIV (24) and hepatitis C virus (25), and are routinely used for analysis of antiviral activity of inhibitors (9). Inhibitory activity against HIV-1 and FIV (lentiviruses) and MoMLV (gammaretrovirus) was also evaluated.
Some HIV NRTIs reportedly possess anti-SRV-1 and anti-SRV-2 activities (16, 18); therefore, we first evaluated the anti-SRV-4 activity of seven NRTIs which have been approved for the treatment of HIV-1-infected patients. When TE671 cells were used as targets, ddI, ddC, and 3TC exhibited weak anti-SRV-4 activities, with EC50s within the micromolar range (EC50, 2.7 to 4.4 μM), whereas d4T and FTC exhibited moderate anti-SRV-4 activities, with EC50s within the submicromolar range (EC50, 0.2 and 0.5 μM, respectively) (Table 1). Remarkably, AZT and TDF exhibited potent anti-SRV-4 activities, with EC50s of 42 and 0.8 nM, respectively. In contrast, almost all the NRTIs showed higher EC50s using MT-2 cells as targets than those using TE671 cells as targets (Table 1). However, AZT and TDF exerted potent anti-SRV-4 activities, with EC50s of 110 and 1.6 nM, respectively, even in the less sensitive MT-2 cells. Notably, all the NRTIs tested in this study exhibited no cytotoxicity against both cell types up to 100 μM, indicating that the observed anti-SRV-4 activity was not because of cell damage (data not shown).
One possible explanation for the difference in drug susceptibility between TE671 and MT-2 cells would be the different phosphorylation efficacies of NRTIs, which require sequential phosphorylations by cellular kinases to reach the active form (26, 27). To confirm this, we next evaluated anti-HIV-1, anti-FIV, and anti-MoMLV activities using the same assay system, in which TE671 or MT-2 cells were infected with VSV-G-pseudotyped luciferase-expressing HIV-1- or FIV-based lentiviral vectors or MoMLV-based retroviral vectors in the presence of various concentrations of inhibitors. HIV-1 infection was blocked by all the tested NRTIs to various extents (Table 1). Among these, AZT and TDF exhibited potent activities against SRV-4 and MoMLV infections, while d4T was less active than AZT (Table 1). In FIV infection, AZT and d4T were active within the submicromolar range only in TE671 cells; however, TDF exhibited potent anti-FIV activity in both cell types (Table 1). Importantly, variation of EC50s of NRTIs against HIV-1 was minimum between both target cell types (0.3- to 2.1-fold change in EC50s measured with TE671 and MT-2 cells), suggesting that cell-derived factors are not a major cause of target cell-based differences in anti-SRV-4 activity.
Taken together, these findings indicate that HIV NRTIs have inhibitory activity against SRV-4 infection to various extents. Among these, AZT showed preferential anti-SRV-4 activity, a trend different from that previously observed against SRV-1 and SRV-2 (18). In addition, TDF exhibited the most potent anti-SRV-4 activity in the single-round infection assay.
Inhibitory effect of HIV-1 NNRTIs on SRV-4 infection.
HIV-1 NNRTIs, including NVP and EFV, efficiently suppress HIV-1 infection by inhibiting HIV-1 RT activity by binding to a hydrophobic pocket near the RT polymerase active site (28, 29). In the present study, EFV showed slight cytotoxicity, with CC50 values of 57 and 48 μM in TE671 and MT-2 cells, respectively. However, both NVP and EFV potently inhibited HIV-1 infection, with EC50s within the nanomolar to subnanomolar range (0.57 to 82 nM) in both cell types. In contrast, NVP and EFV were completely inactive against SRV-4 infection as well as against MoMLV and FIV infections, even at 10 μM. These results correlate well with the impressive narrow spectrum of NNRTI activity; i.e., NNRTIs are active against HIV-1 but not against HIV-2 and other retroviruses (11, 30–32).
Inhibitory activity of an HIV INSTI against SRV-4 infection.
We next evaluated the inhibitory effect of RAL, the first INSTI approved for clinical use, on SRV-4 infection. RAL has potent anti-HIV-1 activity in addition to a broad antiviral spectrum including simian immunodeficiency virus (SIV) (33), MLV (34), XMRV (11), and SRV-3 (19). We also observed that RAL inhibited HIV-1 and MoMLV infections (Table 1). FIV was less susceptible to RAL than HIV-1 and MoMLV, although the RAL EC50 against FIV was at the nanomolar level. Most importantly, SRV-4 infection was potently inhibited by RAL within a nanomolar concentration (Table 1). We previously observed that EVG, a new INSTI contained within a recently approved anti-HIV drug, was active against not only HIV but also MoMLV and SIV (9), indicating that INSTIs are a preferential class of inhibitors for a wide range of retroviral infections. We report for the first time the potential blockage of SRV-4 infection by RAL without cytotoxicity.
Effect of HIV PIs on SRV-4 production.
We then evaluated the inhibitory activity of PIs against SRV-4 replication. It is impossible to evaluate the anti-SRV-4 activity of PIs with the replication-deficient SRV-4 used to evaluate the inhibitory activities of NRTIs, NNRTIs, and an INSTI. To overcome this limitation, we evaluated persistently SRV-4-infected cells, in which the production of progeny infectious virions from SRV-4-infected 293T cells was monitored in the presence of various concentrations of PIs. Viruses released into culture supernatants were quantified by virion-derived RT activity.
First, we measured the cytotoxicity of three PIs (NFV, SQV, and DRV) against 293T cells. Although DRV showed no cytotoxicity up to 100 μM, NFV and SQV decreased cell viability, with CC50 values of 22 and 28 μM, respectively. To exclude a cell toxicity-based reduction in viral production, we used 0.1 and 1 μM concentrations of PIs in this study, which are sufficiently high to exert anti-HIV-1 activity (11, 35, 36). However, none of the PIs inhibited late-phase SRV-4 replication steps even at 1 μM (data not shown), indicating that SRV-4 is intrinsically less susceptible to PIs.
Effects of AZT, TDF, and RAL on SRV-4 replication.
As observed in the early part of this study, two NRTIs (AZT and TDF) and one INSTI (RAL) efficiently inhibited replication-deficient SRV-4 infection in a single-cycle luciferase assay. To further elucidate the anti-SRV-4 property of these inhibitors, we assessed their effect on SRV-4 replication.
To precisely evaluate the inhibitory activity against SRV-4 replication, we distinguished the SRV-4 replication pattern into two viral expansion pathways: cell-free and cell-to-cell transmission. In the cell-free model, SRV-4-free 293T cells were infected with cell-free SRV-4 in the presence of inhibitors, and further viral expansion was monitored by virus-derived RT activity. In contrast, SRV-4-infected 293T cells were used as the source of infection for cell-to-cell transmission.
We observed that in the cell-free model, SRV-4 efficiently infected 293T cells and reached the maximum level at 3 days postinfection (Fig. 1A). Similarly, viral expansion through de novo SRV-4 transmission was observed in the cell-to-cell model (Fig. 1B). However, SRV-4 expanded more efficiently through the cell-to-cell mechanism than through the cell-free mechanism, as judged by the 2- to 3-fold-higher RT activity observed in the cell-to-cell model at 5 days postinfection, indicating that cell-derived SRV-4 is a favorable source of SRV-4. Under these conditions, the 10-fold-higher EC50s of AZT, TDF, and RAL, previously measured in single-round infection assays, completely inhibited cell-free SRV-4 infection up to 3 days (Fig. 1A). However, on day 5, only 7% of the viral production was observed in the presence of AZT, whereas TDF and RAL still almost completely blocked SRV-4 expansion. This tendency was well correlated with the antiviral activity measured during the single-round SRV-4 infection (Table 1). In contrast, when cell-associated SRV-4 was used as the infectious source, inhibitory activities of AZT and TDF were only partial; therefore, de novo SRV-4 transmission was ongoing at 3 and 5 days postinfection (Fig. 1B). Notably, we sequenced the RT regions of the proviral DNA at the end of this study, and no changes from the original SRV-4 were observed (data not shown). Thus, drug resistance was not associated with insufficient activity. However, only 3% to 5% of viral replication was observed in the presence of RAL on day 5 (P < 0.001, compared to AZT and TDF), indicating that RAL potently inhibited SRV-4 replication; therefore, it should be highly effective in controlling SRV-4 infection and replication.
DISCUSSION
To date, several SRV serotypes have been identified, and their distributions in monkeys have been revealed (1–3, 37–41). For example, SRV-4 and SRV-5 infect cynomolgus and rhesus monkeys, respectively, while the Japanese monkey is not a natural host of these SRVs (7, 42). However, the recent outbreak of SRV-4 at PRI revealed that Japanese monkeys are susceptible to SRV-4 (5), since fatal disease could be induced in some of them (43) (Okamoto et al., unpublished). These epidemics reflect the necessity for effective drugs against SRV-4 infection. In addition, human SRV infection has been reported, although no associated diseases have been identified (44). This finding also suggests that the identification of anti-SRV drugs is important to prevent the entry of these viruses into the human population.
Among the identified SRVs, the inhibitory activity of anti-HIV drugs against SRV-1 and SRV-2 has been relatively well analyzed. In these studies, the evaluation of anti-SRV activity was performed by time-consuming, cost-intensive, and hazardous procedures, e.g., using wild-type SRVs and infected monkeys (17, 18). In the present study, we used a VSV-G-pseudotyped luciferase reporter SRV-4 to screen inhibitors with anti-SRV-4 activity from a panel of clinically approved anti-HIV drugs. In this system, the luciferase reporter gene enabled sensitive and rapid evaluation. Moreover, replacement of the intrinsic envelope with VSV-G avoids the restriction of target cell tropism, thereby enabling the direct comparison of antiviral activity with other viruses in the same cells. Using this assay system, we reported for the first time that two anti-HIV NRTIs (AZT and TDF) and one INSTI (RAL) efficiently inhibited SRV-4 infection. The tendency for drug susceptibility of SRV-4 is different from that of SRV-1 and SRV-2, as reported in a previous study in which SRV-1 and SRV-2 infections were more potently inhibited by ddC than by AZT, 3TC, and d4T (18). Reportedly, SRV-4 is genetically distinct from SRV-1 and SRV-2 (3), suggesting that this intrinsic diversity reflects drug susceptibility.
Among the NRTIs tested, AZT and TDF exhibited potent anti-SRV-4 activities in single-round infection and cell-free viral transmission and also inhibited HIV-1, MoMLV, and FIV infections to various extents. However, the inhibitory activities of some NRTIs, particularly the thymidine analogs AZT and d4T, against SRV-4, MoMLV, and FIV infections were markedly (>10-fold) varied between TE671 and MT-2 cells (Table 1). A similar variation was previously reported for several viruses (18, 45). Major factors accounting for the different sensitivities of viruses to NRTIs in different target cells include the endogenous levels of some kinases as well as the levels of the intracellular pool of nucleotides (45–47). Moreover, although HIV-1 preferentially infects lymphoid cells, SRV infects a wide variety of cells, including not only CD4+, CD8+, and B cells in vivo but also lung fibroblast and kidney cells of monkeys in vitro (48). It is likely that the nature of the virus and assay conditions affects the susceptibility of SRV-4 to NRTIs in different cells, although further analyses are required to completely elucidate this phenomenon. TDF preferentially inhibited all the tested retroviruses. All the nucleoside-type RT inhibitors required three sequential phosphorylations, whereas TDF requires only a two-step phosphorylation to be active (49, 50), suggesting that this kinetic advantage reflects potent antiviral properties.
To gain deeper insights into the drug susceptibility of SRV-4, amino acid sequences of regions corresponding to the RT-polymerase domain (residues 63 to 234 of HIV-1) and the integrase catalytic core domain (IN-CCD) (residues 50 to 212) were compared with those of HIV-1, MoMLV, and FIV (Fig. 2). Because genotypic studies to elucidate drug susceptibility based on amino acid changes have been extensively performed for HIV-1 (22, 51, 52), we applied those observations to genotypic analysis of SRV-4. Overall, we confirmed that some amino acid residues in SRV-4 are identical to reported mutations affecting drug susceptibility in HIV-1. For example, HIV-1 RT mutations at positions 41, 67, 70, 210, 215, and 219, known as thymidine analog mutations (TAMs), are frequently observed in AZT and D4T resistance (52–54). In SRV-4 RT, some residues corresponding to TAMs differ from those of wild-type HIV-1 (Fig. 2A), although they must not be involved in drug susceptibility of SRV-4, since AZT and d4T inhibited SRV-4 infection at a similar or superior level compared to HIV-1 infection. In addition, although mutations at Q151 in the LPQG motif and M184 in the YMDD motif are involved in higher-level resistance to some NRTIs (55–57), these motifs are completely conserved in SRV-4 RT. In contrast, MoMLV showed complete insensitivity to certain NRTIs, including 3TC, at 10 μM (Table 1), in agreement with previous reports (8, 18). Taken together, as apparent from genotypic analysis of SRV-4 RT, AZT and TDF are thought to be potent therapeutic agents for the inhibition and control of SRV-4.
Fig 2.

Protein sequence alignments of RT and IN. Reference amino acid sequences of SRV-4 (GenBank accession number NC_014474.1), HIV-1 (accession number NC_001802.1), MoMLV (accession number NC_001501.1), and FIV (accession number NC_001482.1) were aligned by using the program Clustal W, and the regions corresponding to the RT-polymerase domain (residues 63 to 234) (A) and the IN catalytic core domain (residues 50 to 212) (B) of HIV-1 are shown. The amino acid numbering of SRV-4 IN is based on that of SRV-3 (68). Absolutely conserved residues and conserved substitutions are shown in black and gray boxes, respectively. For symbols above the sequences, closed circles indicate residues associated with drug resistance for HIV-1, and asterisks indicate catalytic residues.
RAL, an HIV INSTI, showed potent inhibitory activity against SRV-4 infection as well as against HIV-1, MoMLV, and FIV infections. HIV-1 acquires high-level RAL resistance by mutations such as Q148H/R/K and N155H (52, 58). Although SRV-4 IN contains H166, which corresponds to N155 in HIV-1 (Fig. 2B), SRV-4 retained susceptibility within levels similar to those of wild-type HIV-1 (Table 1). Reportedly, SRV-3 also contains amino acids corresponding to N155H and F121Y, which are other INSTI resistance mutations; however, SRV-3 shows complete susceptibility to RAL (19). In contrast, bovine immunodeficiency virus (BIV) reportedly showed 23-fold resistance to RAL compared to wild-type HIV-1, although BIV contains a histidine (H) residue at the position corresponding to N155 (19), as seen in SRV-4, indicating that N155H is not a determinant of RAL susceptibility in retroviruses other than HIV. Although in the present study, FIV showed less susceptibility to RAL than the other retroviruses/lentiviruses tested (Table 1), FIV does not contain major INSTI resistance mutations. However, one distinct difference was observed: FIV IN carries G145, whereas it corresponds to Y143 in HIV-1 (Fig. 2B). The Y143G mutation has rarely been observed in RAL-treated patients (59); therefore, the precise effect of this mutation on RAL resistance remains unclear. However, it was speculated that that the Y143G mutation lacks the interaction with RAL (19), and interestingly, Y143G reportedly affects proviral formation (60), although this is apparent in nondividing cells (61), likely suggesting that FIV IN G145 affects susceptibility of FIV not only to INSTIs but also to NRTIs.
To expand viral infection in vitro and in vivo, viruses utilize two main pathways: cell-free and cell-to-cell transmission. However, the transmission pathway depends on the nature of the viruses. For example, cell-free HIV-1 efficiently infects CD4+ T cells and also spreads in a cell-to-cell manner, whereas HTLV-1 transmits exclusively by a cell-to-cell pathway (62–66). In the present study, we compared the inhibitory activities of some inhibitors against SRV-4 replication in both cell-free and cell-to-cell transmission. We observed that although AZT and TDF could almost completely block cell-free SRV-4 transmission, they showed only marginal effects on cell-to-cell SRV-4 transmission (Fig. 1). In contrast, RAL completely suppressed SRV-4 replication in both cell-free and cell-to-cell transmission. These results indicate that a favorable pathway is intrinsically present in anti-HIV-1 drugs; AZT and TDF preferentially block cell-free infection, whereas RAL is active in both the pathways. A similar observation was reported for HIV-1, in which tenofovir preferentially suppressed cell-free transmission compared with cell-to-cell transmission (67). These observations may highlight the importance of the kinetics of viral replication and drug activation because AZT and TDF require tri- and diphosphorylation, respectively, to become active metabolites, whereas RAL does not require any modification to exert its antiviral activity. In addition, it is likely that the kinetics of SRV-4 replication steps, including reverse transcription and integration, vary between cell-free and cell-to-cell transmission, as seen for HIV-1; this may be another determinant of viral transmission pathway-dependent anti-SRV-4 activities.
Taken together, the present study demonstrated that AZT, TDF, and RAL potently inhibited SRV-4 infection. These inhibitors suppressed single-round infection and cell-free virus transmission of SRV-4; however, cell-to-cell transmission was blocked only by RAL. To effectively control SRV-4 infection and maintain a minimum risk of the emergence of drug resistance, a combination therapy of drugs such as ART in HIV-1 infection is important.
ACKNOWLEDGMENTS
We thank H. Miyoshi and Y. Sakurai for providing lentiviral vectors and the pDON-AI-2-luc vector, respectively. The following reagents were obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: emtricitabine, tenofovir disoproxil fumarate, darunavir (from Tibotec, Inc.), and raltegravir (from Merck & Company, Inc.).
This work was supported in part by a JSPS KAKENHI grant-in-aid for young scientists (B) to K.S. (grant number 24791021) and a grant-in-aid for scientific research (B) to M.O. (grant number 24300153).
Footnotes
Published ahead of print 30 January 2013
REFERENCES
- 1. Daniel MD, King NW, Letvin NL, Hunt RD, Sehgal PK, Desrosiers RC. 1984. A new type D retrovirus isolated from macaques with an immunodeficiency syndrome. Science 223:602–605 [DOI] [PubMed] [Google Scholar]
- 2. Marx PA, Maul DH, Osborn KG, Lerche NW, Moody P, Lowenstine LJ, Henrickson RV, Arthur LO, Gilden RV, Gravell M. 1984. Simian AIDS: isolation of a type D retrovirus and transmission of the disease. Science 223:1083–1086 [DOI] [PubMed] [Google Scholar]
- 3. Montiel NA. 2010. An updated review of simian betaretrovirus (SRV) in macaque hosts. J. Med. Primatol. 39:303–314 [DOI] [PubMed] [Google Scholar]
- 4. Henrickson RV, Maul DH, Lerche NW, Osborn KG, Lowenstine LJ, Prahalada S, Sever JL, Madden DL, Gardner MB. 1984. Clinical features of simian acquired immunodeficiency syndrome (SAIDS) in rhesus monkeys. Lab. Anim. Sci. 34:140–145 [PubMed] [Google Scholar]
- 5. CDC-KUPRI 2010. Information of hemorrhagic syndrome of Japanese macaques (provisional designation). Primate Res. 26:69–71 [Google Scholar]
- 6. Zao CL, Armstrong K, Tomanek L, Cooke A, Berger R, Estep JS, Marx PA, Trask JS, Smith DG, Yee JL, Lerche NW. 2010. The complete genome and genetic characteristics of SRV-4 isolated from cynomolgus monkeys (Macaca fascicularis). Virology 405:390–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zao CL, Ward JA, Tomanek L, Cooke A, Berger R, Armstrong K. 2011. Virological and serological characterization of SRV-4 infection in cynomolgus macaques. Arch. Virol. 156:2053–2056 [DOI] [PubMed] [Google Scholar]
- 8. Powell SK, Artlip M, Kaloss M, Brazinski S, Lyons R, McGarrity GJ, Otto E. 1999. Efficacy of antiretroviral agents against murine replication-competent retrovirus infection in human cells. J. Virol. 73:8813–8816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, Watanabe Y, Ohata Y, Doi S, Sato M, Kano M, Ikeda S, Matsuoka M. 2008. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J. Virol. 82:764–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sakuma R, Sakuma T, Ohmine S, Silverman RH, Ikeda Y. 2010. Xenotropic murine leukemia virus-related virus is susceptible to AZT. Virology 397:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Smith RA, Gottlieb GS, Miller AD. 2010. Susceptibility of the human retrovirus XMRV to antiretroviral inhibitors. Retrovirology 7:70 doi:10.1186/1742-4690-7-70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu YQ, Remington KM, North TW. 1996. Mutants of feline immunodeficiency virus resistant to 2′,3′-dideoxy-2′,3′-didehydrothymidine. Antimicrob. Agents Chemother. 40:1983–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. North TW, North GL, Pedersen NC. 1989. Feline immunodeficiency virus, a model for reverse transcriptase-targeted chemotherapy for acquired immune deficiency syndrome. Antimicrob. Agents Chemother. 33:915–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matsushita S, Mitsuya H, Reitz MS, Broder S. 1987. Pharmacological inhibition of in vitro infectivity of human T lymphotropic virus type I. J. Clin. Invest. 80:394–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Miyazato P, Yasunaga J, Taniguchi Y, Koyanagi Y, Mitsuya H, Matsuoka M. 2006. De novo human T-cell leukemia virus type 1 infection of human lymphocytes in NOD-SCID, common gamma-chain knockout mice. J. Virol. 80:10683–10691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tsai CC, Follis KE, Benveniste RE. 1988. Antiviral effects of 3′-azido-3′-deoxythymidine, 2′,3′-dideoxycytidine, and 2′,3′-dideoxyadenosine against simian acquired immunodeficiency syndrome-associated type D retrovirus in vitro. AIDS Res. Hum. Retroviruses 4:359–368 [DOI] [PubMed] [Google Scholar]
- 17. Tsai CC, Follis KE, Yarnall M, Blakley GA. 1989. Toxicity and efficacy of 2′,3′-dideoxycytidine in clinical trials of pigtailed macaques infected with simian retrovirus type 2. Antimicrob. Agents Chemother. 33:1908–1914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenblum LL, Patton G, Grigg AR, Frater AJ, Cain D, Erlwein O, Hill CL, Clarke JR, McClure MO. 2001. Differential susceptibility of retroviruses to nucleoside analogues. Antivir. Chem. Chemother. 12:91–97 [DOI] [PubMed] [Google Scholar]
- 19. Koh Y, Matreyek KA, Engelman A. 2011. Differential sensitivities of retroviruses to integrase strand transfer inhibitors. J. Virol. 85:3677–3682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. White JA, Todd PA, Rosenthal AN, Yee JL, Grant R, Lerche NW. 2009. Development of a generic real-time PCR assay for simultaneous detection of proviral DNA of simian betaretrovirus serotypes 1, 2, 3, 4 and 5 and secondary uniplex assays for specific serotype identification. J. Virol. Methods 162:148–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J, Shafer RW. 2003. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 31:298–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tailor CS, Nouri A, Zhao Y, Takeuchi Y, Kabat D. 1999. A sodium-dependent neutral-amino-acid transporter mediates infections of feline and baboon endogenous retroviruses and simian type D retroviruses. J. Virol. 73:4470–4474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Harada S, Koyanagi Y, Yamamoto N. 1985. Infection of HTLV-III/LAV in HTLV-I-carrying cells MT-2 and MT-4 and application in a plaque assay. Science 229:563–566 [DOI] [PubMed] [Google Scholar]
- 25. Kato N, Nakazawa T, Mizutani T, Shimotohno K. 1995. Susceptibility of human T-lymphotropic virus type I infected cell line MT-2 to hepatitis C virus infection. Biochem. Biophys. Res. Commun. 206:863–869 [DOI] [PubMed] [Google Scholar]
- 26. Cihlar T, Ray AS. 2010. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antiviral Res. 85:39–58 [DOI] [PubMed] [Google Scholar]
- 27. Piliero PJ. 2004. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J. Acquir. Immune Defic. Syndr. 37(Suppl 1):S2–S12 http://download.bioon.com.cn/upload/201101/17/101159hswyecdhh77cw6xv.attach.pdf [DOI] [PubMed] [Google Scholar]
- 28. Ren J, Stammers DK. 2008. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 134:157–170 [DOI] [PubMed] [Google Scholar]
- 29. Huang H, Chopra R, Verdine GL, Harrison SC. 1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282:1669–1675 [DOI] [PubMed] [Google Scholar]
- 30. Auwerx J, Esnouf R, De Clercq E, Balzarini J. 2004. Susceptibility of feline immunodeficiency virus/human immunodeficiency virus type 1 reverse transcriptase chimeras to non-nucleoside RT inhibitors. Mol. Pharmacol. 65:244–251 [DOI] [PubMed] [Google Scholar]
- 31. Ren J, Bird LE, Chamberlain PP, Stewart-Jones GB, Stuart DI, Stammers DK. 2002. Structure of HIV-2 reverse transcriptase at 2.35-A resolution and the mechanism of resistance to non-nucleoside inhibitors. Proc. Natl. Acad. Sci. U. S. A. 99:14410–14415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Witvrouw M, Pannecouque C, Switzer WM, Folks TM, De Clercq E, Heneine W. 2004. Susceptibility of HIV-2, SIV and SHIV to various anti-HIV-1 compounds: implications for treatment and postexposure prophylaxis. Antivir. Ther. 9:57–65 [PubMed] [Google Scholar]
- 33. Lewis MG, Norelli S, Collins M, Barreca ML, Iraci N, Chirullo B, Yalley-Ogunro J, Greenhouse J, Titti F, Garaci E, Savarino A. 2010. Response of a simian immunodeficiency virus (SIVmac251) to raltegravir: a basis for a new treatment for simian AIDS and an animal model for studying lentiviral persistence during antiretroviral therapy. Retrovirology 7:21 doi:10.1186/1742-4690-7-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Beck-Engeser GB, Eilat D, Harrer T, Jäck HM, Wabl M. 2009. Early onset of autoimmune disease by the retroviral integrase inhibitor raltegravir. Proc. Natl. Acad. Sci. U. S. A. 106:20865–20870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koh Y, Nakata H, Maeda K, Ogata H, Bilcer G, Devasamudram T, Kincaid JF, Boross P, Wang YF, Tie Y, Volarath P, Gaddis L, Harrison RW, Weber IT, Ghosh AK, Mitsuya H. 2003. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 47:3123–3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Patick AK, Mo H, Markowitz M, Appelt K, Wu B, Musick L, Kalish V, Kaldor S, Reich S, Ho D, Webber S. 1996. Antiviral and resistance studies of AG1343, an orally bioavailable inhibitor of human immunodeficiency virus protease. Antimicrob. Agents Chemother. 40:292–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hara M, Sata T, Kikuchi T, Nakajima N, Uda A, Fujimoto K, Baba T, Mukai R. 2005. Isolation and characterization of a new simian retrovirus type D subtype from monkeys at the Tsukuba Primate Center, Japan. Microbes Infect. 7:126–131 [DOI] [PubMed] [Google Scholar]
- 38. Marx PA, Bryant ML, Osborn KG, Maul DH, Lerche NW, Lowenstine LJ, Kluge JD, Zaiss CP, Henrickson RV, Shiigi SM. 1985. Isolation of a new serotype of simian acquired immune deficiency syndrome type D retrovirus from Celebes black macaques (Macaca nigra) with immune deficiency and retroperitoneal fibromatosis. J. Virol. 56:571–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nandi JS, Bhavalkar-Potdar V, Tikute S, Raut CG. 2000. A novel type D simian retrovirus naturally infecting the Indian Hanuman langur (Semnopithecus entellus). Virology 277:6–13 [DOI] [PubMed] [Google Scholar]
- 40. Nandi JS, Tikute SA, Chhangani AK, Potdar VA, Tiwari-Mishra M, Ashtekar RA, Kumari J, Walimbe A, Mohnot SM. 2003. Natural infection by simian retrovirus-6 (SRV-6) in Hanuman langurs (Semnopithecus entellus) from two different geographical regions of India. Virology 311:192–201 [DOI] [PubMed] [Google Scholar]
- 41. Nandi JS, Van Dooren S, Chhangani AK, Mohnot SM. 2006. New simian beta retroviruses from rhesus monkeys (Macaca mulatta) and langurs (Semnopithecus entellus) from Rajasthan, India. Virus Genes 33:107–116 [DOI] [PubMed] [Google Scholar]
- 42. Li B, Axthelm MK, Machida CA. 2000. Simian retrovirus serogroup 5: partial gag-prt sequence and viral RNA distribution in an infected rhesus macaque. Virus Genes 21:241–248 [DOI] [PubMed] [Google Scholar]
- 43. Cyranoski D. 2010. Japanese monkey deaths puzzle. Nature 466:302–303 [DOI] [PubMed] [Google Scholar]
- 44. Lerche NW, Switzer WM, Yee JL, Shanmugam V, Rosenthal AN, Chapman LE, Folks TM, Heneine W. 2001. Evidence of infection with simian type D retrovirus in persons occupationally exposed to nonhuman primates. J. Virol. 75:1783–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dahlberg JE, Mitsuya H, Blam SB, Broder S, Aaronson SA. 1987. Broad spectrum antiretroviral activity of 2′,3′-dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 84:2469–2473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Balzarini J. 2000. Effect of antimetabolite drugs of nucleotide metabolism on the anti-human immunodeficiency virus activity of nucleoside reverse transcriptase inhibitors. Pharmacol. Ther. 87:175–187 [DOI] [PubMed] [Google Scholar]
- 47. Ray AS. 2005. Intracellular interactions between nucleos(t)ide inhibitors of HIV reverse transcriptase. AIDS Rev. 7:113–125 [PubMed] [Google Scholar]
- 48. Maul DH, Zaiss CP, MacKenzie MR, Shiigi SM, Marx PA, Gardner MB. 1988. Simian retrovirus D serogroup 1 has a broad cellular tropism for lymphoid and nonlymphoid cells. J. Virol. 62:1768–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. De Clercq E, Holý A. 2005. Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat. Rev. Drug Discov. 4:928–940 [DOI] [PubMed] [Google Scholar]
- 50. De Clercq E. 2009. The history of antiretrovirals: key discoveries over the past 25 years. Rev. Med. Virol. 19:287–299 [DOI] [PubMed] [Google Scholar]
- 51. Shafer RW. 2006. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 194(Suppl 1):S51–S58 doi:10.1086/505356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Johnson VA, Calvez V, Günthard HF, Paredes R, Pillay D, Shafer R, Wensing AM, Richman DD. 2011. 2011 update of the drug resistance mutations in HIV-1. Top. Antivir. Med. 19:156–164 [PMC free article] [PubMed] [Google Scholar]
- 53. Gao Q, Gu ZX, Parniak MA, Li XG, Wainberg MA. 1992. In vitro selection of variants of human immunodeficiency virus type 1 resistant to 3′-azido-3′-deoxythymidine and 2′,3′-dideoxyinosine. J. Virol. 66:12–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lin PF, Samanta H, Rose RE, Patick AK, Trimble J, Bechtold CM, Revie DR, Khan NC, Federici ME, Li H. 1994. Genotypic and phenotypic analysis of human immunodeficiency virus type 1 isolates from patients on prolonged stavudine therapy. J. Infect. Dis. 170:1157–1164 [DOI] [PubMed] [Google Scholar]
- 55. Quan Y, Gu Z, Li X, Liang C, Parniak MA, Wainberg MA. 1998. Endogenous reverse transcriptase assays reveal synergy between combinations of the M184V and other drug resistance-conferring mutations in interactions with nucleoside analog triphosphates. J. Mol. Biol. 277:237–247 [DOI] [PubMed] [Google Scholar]
- 56. Shirasaka T, Kavlick MF, Ueno T, Gao WY, Kojima E, Alcaide ML, Chokekijchai S, Roy BM, Arnold E, Yarchoan R. 1995. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. U. S. A. 92:2398–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tisdale M, Kemp SD, Parry NR, Larder BA. 1993. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3′-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A. 90:5653–5656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fransen S, Gupta S, Danovich R, Hazuda D, Miller M, Witmer M, Petropoulos CJ, Huang W. 2009. Loss of raltegravir susceptibility by human immunodeficiency virus type 1 is conferred via multiple nonoverlapping genetic pathways. J. Virol. 83:11440–11446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Canducci F, Sampaolo M, Marinozzi MC, Boeri E, Spagnuolo V, Galli A, Castagna A, Lazzarin A, Clementi M, Gianotti N. 2009. Dynamic patterns of human immunodeficiency virus type 1 integrase gene evolution in patients failing raltegravir-based salvage therapies. AIDS 23:455–460 [DOI] [PubMed] [Google Scholar]
- 60. Ikeda T, Nishitsuji H, Zhou X, Nara N, Ohashi T, Kannagi M, Masuda T. 2004. Evaluation of the functional involvement of human immunodeficiency virus type 1 integrase in nuclear import of viral cDNA during acute infection. J. Virol. 78:11563–11573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tsurutani N, Kubo M, Maeda Y, Ohashi T, Yamamoto N, Kannagi M, Masuda T. 2000. Identification of critical amino acid residues in human immunodeficiency virus type 1 IN required for efficient proviral DNA formation at steps prior to integration in dividing and nondividing cells. J. Virol. 74:4795–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, Tanaka Y, Osame M, Bangham CR. 2003. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science 299:1713–1716 [DOI] [PubMed] [Google Scholar]
- 63. Jolly C, Kashefi K, Hollinshead M, Sattentau QJ. 2004. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 199:283–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sattentau Q. 2008. Avoiding the void: cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 6:815–826 [DOI] [PubMed] [Google Scholar]
- 65. Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, Gout O, Alcover A, Thoulouze MI. 2010. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat. Med. 16:83–89 [DOI] [PubMed] [Google Scholar]
- 66. Matsuoka M, Jeang KT. 2007. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 7:270–280 [DOI] [PubMed] [Google Scholar]
- 67. Sigal A, Kim JT, Balazs AB, Dekel E, Mayo A, Milo R, Baltimore D. 2011. Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477:95–98 [DOI] [PubMed] [Google Scholar]
- 68. Snásel J, Krejcík Z, Jencová V, Rosenberg I, Ruml T, Alexandratos J, Gustchina A, Pichová I. 2005. Integrase of Mason-Pfizer monkey virus. FEBS J. 272:203–216 [DOI] [PubMed] [Google Scholar]