

Abstract
Myxoma virus (MYXV) and vaccinia virus (VACV), two distinct members of the family Poxviridae, are both currently being developed as oncolytic virotherapeutic agents. Recent studies have demonstrated that ex vivo treatment with MYXV can selectively recognize and kill contaminating cancerous cells from autologous bone marrow transplants without perturbing the engraftment of normal CD34+ hematopoietic stem and progenitor cells. However, the mechanism(s) by which MYXV specifically recognizes and eliminates the cancer cells in the autografts is not understood. While little is known about the cellular attachment factor(s) exploited by MYXV for entry into any target cells, VACV has been shown to utilize cell surface glycosaminoglycans such as heparan sulfate (HS), the extracellular matrix protein laminin, and/or integrin β1. We have constructed MYXV and VACV virions tagged with the Venus fluorescent protein and compared their characteristics of binding to various human cancer cell lines as well as to primary human leukocytes. We report that the binding of MYXV or VACV to some adherent cell lines could be partially inhibited by heparin, but laminin blocked only VACV binding. In contrast to cultured fibroblasts, the binding of MYXV and VACV to a wide spectrum of primary human leukocytes could not be competed by either HS or laminin. Additionally, MYXV and VACV exhibited very different binding characteristics against certain select human leukocytes, suggesting that the two poxviruses utilize different cell surface determinants for the attachment to these cells. These results indicate that VACV and MYXV can exhibit very different oncolytic tropisms against some cancerous human leukocytes.
INTRODUCTION
Poxviruses are enveloped viruses with a large double-stranded DNA genome of about 200 kbp that encodes at least 150 to 200 functional open reading frames. Unlike most DNA viruses that replicate in the nucleus of infected cells, poxvirus replication takes place entirely in the cytoplasm of infected cells in a defined virus-induced organelle known as the viral factory (1). Vaccinia virus (VACV) belongs to the genus Orthopoxvirus and is the prototypical member of the Poxviridae family (1). VACV, which was used as a live-attenuated vaccine for the eradication of smallpox, has been extensively studied as the prototypic representative of the poxvirus family. VACV has also been developed as an oncolytic agent and is currently being tested in various clinical trials as an oncolytic virotherapeutic for the treatment of end-stage cancers, such as liver cancer or cancer that has metastasized to the liver (2–7). A second poxvirus with demonstrated oncolytic potential is myxoma virus (MYXV), which belongs to the genus Leporipoxvirus (8–10). Sequencing of the MYXV Lausanne strain genome has revealed that the genome is 161.8 kbp in size and encodes about 171 genes (11). The central region of the MYXV and VACV genomes includes viral genes that are highly conserved among all poxviruses. However, the terminal regions of both genomes are much less conserved and encode more unique genes that are involved in subverting the host immune system and circumventing various other antiviral responses of the infected host (8, 12, 13). Unlike VACV, which can infect a wide variety of vertebrate hosts, MYXV productively infects only lagomorphs and causes a lethal disease called myxomatosis in European rabbits (1, 9, 14, 15).
Despite its narrow host range in nature, MYXV has been shown to be able to productively infect various human cancer cells, and studies conducted in numerous nonrabbit animal models have revealed that this virus can selectively infect and kill a wide variety of cancer cells in both immunocompetent and immunodeficient hosts (8, 10, 16, 17). The host range determinants that mediate this cancer-specific tropism of MYXV outside the rabbit host are still being investigated, but at least two different intracellular pathways have been implicated in this cellular discrimination to date: (i) the failure of many cancer cells to induce an effective antiviral response, such as the synergistic interferon and tumor necrosis factor pathway that effectively aborts MYXV replication in primary nontransformed human cells (18, 19), and (ii) the constitutive activation of Akt in many cancer cells that favors permissive virus replication (20, 21). We have also recently shown that MYXV can selectively infect and kill primary human leukemic stem and progenitor cells while sparing normal human stem and progenitor cells derived from bone marrow in terms of differentiation potential in vitro and the ability to engraft recipient NOD/scid/IL2 receptor gamma-chain knockout (NSG) mice in vivo (22). Additionally, we recently showed that MYXV specifically binds and kills contaminating human CD138+ myeloma cells from primary patient bone marrow samples ex vivo, whereas MYXV does not bind and therefore does not infect normal CD34+ stem cells (23).
However, we still do not understand why MYXV selectively binds/infects so many types of human cancer cells but fails to bind or infect normal CD34+ stem and progenitor cells. Our working hypothesis is that CD34+ stem and progenitor cells simply do not express key cell surface molecules that allow MYXV to bind to the cell surface. To date, a specific cellular protein receptor(s) for the attachment of poxviruses to the surface of mammalian cells has not yet been identified for MYXV, but studies have shown that VACV recognizes and binds to target mammalian cells via both cell surface glycosaminoglycan (GAG)-dependent and -independent mechanisms (24–30). A few cellular attachment vaccinia virion proteins have also been identified to date, including A26, A27, D8, H3, and L1 (24–28, 30). The A27 and H3 viral proteins on the VACV intracellular mature virus (IMV) particle (also called mature virion [MV]) bind directly to cell surface heparan sulfate (HS) (25, 30). The VACV D8 and A26 IMV proteins bind chondroitin sulfate and the cell surface extracellular matrix (ECM) protein laminin, respectively (24, 28). It has been shown that pretreatment of vaccinia virus IMV with soluble heparin or laminin had an inhibitory effect on the virion attachment to target HeLa, BSC-40, and BSC-1 cells, indicating that VACV can utilize cell surface HS and laminin as attachment factors for these cells (24, 25, 31). A mutant vaccinia virus construct lacking A26 was shown to still be capable of binding to GAG-deficient cells (Sog9), indicating that VACV uses another additional, still unknown cellular receptor(s) or attachment factor(s), in addition to GAGs and cell surface ECM protein laminin (24). Foo et al. have reported that soluble vaccinia virus protein L1 binds to GAG-deficient cells and blocks virus entry, suggesting that virion L1 can also act as a cell receptor-binding protein (26). However, the specific cellular receptor(s) that L1 recognizes at the cell surface remains to be identified. Recently, Izmailyan et al. reported that VACV binding to HeLa cells was reduced when cell surface integrin β1 expression was knocked down using a small interfering RNA (siRNA), suggesting that integrin β1 can function as an attachment factor, at least for HeLa cells (29).
MYXV is structurally similar to VACV, and therefore, it was reasonable to propose that MYXV might attach to target mammalian cells in a similar manner as VACV. However, the tropism of these two poxviruses can be quite different for many human cancer cells (32), and detailed studies comparing the attachment of MYXV and VACV virions to target cells have not yet been conducted. To directly test the binding of MYXV and VACV to different cell types and under different conditions, we have constructed fluorescently tagged versions of MYXV and VACV and then compared the binding and infection of these viruses to various human normal and cancerous cells. To this end, we constructed a recombinant MYXV in which the M093L gene (the ortholog of the VAC A4L IMV structural protein [11, 33]) was replaced with the M093L gene fused with the coding sequence of fluorescent protein Venus at the amino terminus. This new recombinant virus (vMyx-Venus/M093) expresses Venus-fused M093 as a tagged virion component. Using vMyx-Venus/M093, we investigated the binding/infection of MYXV with various mammalian cells in parallel with a recombinant VACV, which expresses Venus-fused A4 in the virion (vVac-Venus/A4). In addition, we determined if the binding of MYXV and VACV to normal and cancerous mammalian cells was dependent on HS, the cell surface ECM protein laminin, or integrin β1. Our results demonstrate that the cell surface binding determinants are significantly different between these two oncolytic poxviruses, particularly for certain classes of human cancerous leukocytes such as T cell lymphoid cancers and multiple myeloma cells, and reflect the nonidentical potencies in their oncolytic potentials.
MATERIALS AND METHODS
Cell lines.
BSC-40 cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, and 100 U/ml of penicillin/streptomycin. U266, HuNS1, Jurkat, and CCRF-CEM cells were obtained from the American Type Culture Collection and maintained in RPMI 1640 (Lonza) supplemented with 10% FBS, 2 mM l-glutamine, and 100 U/ml of penicillin-streptomycin.
Construction of plasmids and recombinant viruses.
Construction of vMyx-GFP, which expresses enhanced green fluorescent protein (GFP) under the control of the vaccinia virus synthetic early/late promoter, has been described previously (34). To generate the amino (N)-terminal fluorescent protein Venus-fused M093, overlapping PCR was used to insert the coding sequence of Venus after the start codon of M093L. Primers were designed to amplify the partial coding sequence of M094L and the region containing the coding sequence of M093L and M092L using the genome of the MYXV Lausanne strain as the template (Table 1). The coding sequence of Venus was amplified by PCR using pDEST40-Venus as the template (Table 1). The resulting PCR products were adjoined into contiguity by overlapping PCR. The final PCR product was inserted into pCR2.1-TOPO (Invitrogen) to generate pVenus-M093L full-TOPO. To generate Venus-fused A4, the Venus-coding sequence was inserted immediately after the A4L start codon. A flexible glycine-serine linker encoded by the sequence GGTGGAGGCGGTTCA was introduced between Venus and A4L (Table 1). Cells infected with MYXV (Lausanne) or VACV (Western Reserve) were transfected with the plasmid encoding Venus-fused M093 or A4 at the N terminus, respectively. On the next day, cells were harvested and cell lysates were plated out on fresh BSC-40 cell monolayers. Fluorescent foci/plaques were picked and further purified until pure recombinants expressing Venus/M093 (vMyx-Venus/M093) or Venus/A4 (vVac-Venus/A4) were obtained.
Table 1.
Primers used for construction of Venus fluorescent protein-tagged recombinant MYXV and VACV
| Recombinant virus | Amplifed fragment | Sequence |
|
|---|---|---|---|
| Forward | Reverse | ||
| vMyx-Venus/M093 | −500 bp to M093L start | CAGGATCCCCAGATAGTTGTTACGTACTTC | CCCTTGCTCACCATTTAGACGATTTAAAATTGAACGGAG |
| Venus | CGTCTAAATGGTGAGCAAGGGCGAGG | GAAGTCCATCTTGTACAGCTCGTCCATGC | |
| M093L start to +500 bp | GAGCTGTACAAGATGGACTTCATGGTGGAGTAC | GTCTCGAGCGCCAATAGCTCGAATAGTTC | |
| vVac-Venus/A4 | −500 bp to A4L Start | TAATATAGTCTAGATGGAATTTTAGACCATC | GCCCTTGCTCACCATTTAAGGCTTTAAAATTGAATTGCGATTATAAG |
| Venus | CAATTTTAAAGCCTTAAATGGTGAGCAAGGGCGAGGAG | GAAGTCTGAACCGCCTCCACCCTTGTACAGCTCGTCCATGCC | |
| A4L start to +500 bp | CTGTACAAGGGTGGAGGCGGTTCAGACTTCTTTAACAAGTTCTCACAGGGGCTG | GTTGGTAACGTCTGAGAAGGTTGG | |
Focus-forming assay.
Confluent BSC-40 cell monolayers were infected with vMyx (Lausanne), vMyx-GFP, or vMyx-Venus/M093. After virus adsorption at 37°C for 1 h, inoculum was removed and cells were overlaid with liquid medium. At 3 days after infection, foci were visualized using a Leica DMI6000 B inverted microscope. Fluorescence and phase-contrast images of foci were captured. Images were minimally processed and pseudocolored using Adobe Photoshop software (Adobe Systems).
Kinetics of synthesis of vMyx-Venus/M093.
Confluent BSC-40 cells grown in 6-well plates were mock infected or infected with vMyx-Venus/M093, which had been purified through a 36% sucrose cushion as described previously (35), at a multiplicity of infection (MOI) of 10.0 either in the presence or in the absence of 40 μg/ml of cytosine arabinoside (AraC; Sigma) at 37°C for 1 h. After adsorption, the inoculum was removed and cell monolayers were washed extensively with culture medium to remove unbound virions. Cells were maintained in medium with or without 40 μg/ml of AraC. At 2, 4, 6, 8, 12, and 24 h postinfection, cells were scraped into medium, pelleted, and stored at −80°C. To examine the synthesis of Venus/M093 protein, cell pellets were lysed in radioimmunoprecipitation assay buffer (0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% Nonidet P-40 [NP-40], 1% Triton X-100, 0.5× phosphate-buffered saline [PBS]) containing phenylmethylsulfonyl fluoride and protease inhibitor cocktail (Roche) on ice for 20 min. Clarified cell lysates were resolved on a 4 to 15% Tris-glycine gel (Bio-Rad), and proteins were transferred to a polyvinylidene difluoride membrane for Western blot analysis. The membrane was probed with an anti-GFP monoclonal antibody (MAb; Roche), followed by horseradish peroxidase (HRP)-conjugated goat anti-rat antibody (Jackson ImmunoResearch Laboratories). After detecting Venus/M093, the blot was sequentially reprobed with a rabbit anti-M071 (late protein) antisreum, a rabbit anti-M-T7 (early protein) antiserum, and an anti-β-actin MAb (Sigma), followed by an appropriate HRP-conjugated secondary antibody (Jackson ImmunoResearch Laboratories). The antibodies were stripped off from the blot prior to immunoblotting with a different antibody. Bound antibodies were detected using chemiluminescence reagent (Millipore) according to the manufacturer's instructions.
Virion component fractionation.
Virions purified through a 36% sucrose cushion were treated with 0.5% NP-40 with or without 50 mM dithiothreitol (DTT) at 37°C for 30 min. After treatment, insoluble fractions were separated from soluble fractions by centrifugation or left unfractionated. Total virion proteins and detergent-insoluble and soluble protein-containing fractions were dissolved in SDS sample buffer and subjected to SDS-PAGE. Western blot analyses were performed sequentially using an anti-GFP MAb and anti-M071 MAb, as described above.
Fluorescence microscopy.
For infection, BSC-40 cells grown on glass coverslips were infected with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 1.0. On the next day, cells were washed with PBS and fixed in 4% paraformaldehyde in PBS. For binding, prechilled BSC-40 cells were incubated with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 10.0 on ice for 1 h. After adsorption, unbound virions were removed by extensive washing with ice-cold culture medium, and cells were fixed as described above. Coverslips were placed on Vectashield mounting medium containing 4′,6-diamindino-2-phenylindole (DAPI; Vector Laboratories). Cells were visualized using a ×63 water-corrected immersion objective on a Leica laser scanning confocal microscope.
Virion binding and virus infection.
Virions purified through a 36% sucrose cushion were pretreated with 100 μg/ml of soluble heparin (HP; Sigma) or laminin (LN) from human placenta (Sigma) on ice for 1 h or mock treated. For cell monolayers, such as those of BSC-40 and HeLa cells, cells were detached from tissue culture dishes using 20 mM EDTA–PBS. For heparinase I (Hep I) treatment, cells were incubated with 2.5 U/ml of Hep I in suspension at 37°C for 30 min or mock treated. Cells were washed with ice-cold 10% FBS–PBS twice and chilled on ice. For binding, cells were incubated with virions at an MOI of 20.0 in suspension on ice for 1 h. Unbound virions were washed with 10% FBS–PBS after virion adsorption. Cells were fixed in 2% paraformaldehyde–PBS and analyzed using a BD FACSCalibur apparatus (BD Biosciences). For infection, human T lymphoblast cells, such as CCRF-CEM and Jurkat cells, and human multiple myeloma cells, such as U266 and HuNS1 cells, were infected with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 20.0 in suspension at 37°C for 1 h. After adsorption, cells were washed and incubated in a 37°C CO2 incubator for an additional 4 or 24 h. Cells were then fixed and analyzed by flow cytometry.
Source leukocytes from healthy donors were obtained commercially from LifeSouth community blood centers (Gainesville, FL). Fresh primary bone marrow from patients with multiple myeloma was obtained from J. Moreb with the patients' consent (23). Mononuclear cells were enriched by centrifugation through a Ficoll gradient and incubated with mock-, heparin-, or laminin-treated vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 10.0 on ice for 1 h. Cells were washed after virus binding. For infection, cells were incubated at 37°C for 24 h after virion adsorption. After binding or infection, cells were stained with phycoerythrin-conjugated anti-CD45 and allophycocyanin (APC)-conjugated anti-CD14, anti-CD15, anti-CD19, or anti-CD3 (BD Biosciences). The percentage of virus-bound/infected cells within each cell population was determined by flow cytometry.
Cell surface integrin β1 expression.
HeLa cells were detached from tissue culture dishes as described above. HeLa, U266, and HuNS1 cells were stained with APC-conjugated anti-CD29 (BD Biosciences) in suspension at 4°C for 20 min. Unbound antibodies were removed by washing, and the level of integrin β1 expression on the cell surface was determined by flow cytometry.
RESULTS
vMyx-Venus/M093 replicates normally in cultured cells and produces fluorescent foci that are identical to those formed by the parental viruses.
We first constructed recombinant MYXV and VACV expressing a common Venus fluorescent fusion protein as a virion component to detect virus binding by flow cytometry. VACV that expresses the A4-GFP fusion or A4-yellow fluorescent protein (YFP) fusion at the N terminus has previously been used to study the binding of VACV to various mammalian cell lines (31, 36). Hence, we replaced M093L, the MYXV ortholog of VACV A4L, with M093L fused to the coding sequence of the fluorescent protein Venus at the N terminus and generated vMyx-Venus/M093 (Fig. 1A). Venus was chosen as the fluorescent tag because of its favorable signal-to-noise ratio compared to the background fluorescence signals encountered in some mammalian cells and tissues. To examine if this new recombinant vMyx-Venus/M093 behaves similarly to the wild-type MYXV, we compared the foci formed by infection with vMyx-Venus/M093 at a low MOI compared to those produced by the wild-type MYXV. As shown in Fig. 1B, vMyx-Venus/M093 produced similar-sized fluorescent foci on BSC-40 cell monolayers as the wild-type viruses vMyx (Lausanne) and vMyx-GFP, which has the coding sequence of enhanced green fluorescent protein under the control of the vaccinia virus synthetic early/late promoter inserted at an intergenic location between M135L and M136L. This indicates that vMyx-Venus/M093 has similar replication characteristics as the wild-type MYXV. Similarly, vVac-Venus/A4 forms plaques in the same cells that are indistinguishable from those of the parental VACV (data not shown).
Fig 1.

vMyx-Venus/M093 produces fluorescent foci that are similar in size to those produced by the parental viruses. (A) Construction of a recombinant vMyx-Venus/M093. M093 fused with Venus fluorescent protein at the amino terminus (Venus/M093) was generated by inserting the coding sequence of Venus in frame immediately following the start codon of the M093L gene. The arrow indicates the direction of transcription of M093L. aa, amino acids. (B) Characterization of fluorescent foci formed by vMyx-Venus/M093. BSC-40 cell monolayers were infected with the indicated viruses at 37°C for 1 h. At 1 h after infection, inoculum was removed and cell monolayers were overlaid with liquid medium. At 3 days postinfection, fluorescence and phase-contrast images of foci were captured.
M093, like VACV A4, is predominantly synthesized late during MYXV infection and is incorporated into the progeny intracellular mature virions.
We next examined the kinetics of the synthesis of Venus/M093 during MYXV infection for comparison with those of the published A4-fusion constructs of VACV (31). BSC-40 cells infected with vMyx-Venus/M093 were harvested at various time points, and the expression of Venus/M093 was examined by Western blotting. Our results showed that the expression of Venus/M093 could be detected as early as 2 h after infection and increased gradually over time (Fig. 2A). The expression of Venus/M093 protein was drastically reduced when infection was carried out in the presence of AraC (Fig. 2A; compare lanes 7 and 8), indicating that M093 is predominantly synthesized late during infection. Similar kinetics for the expression of Venus/A4 protein during vVac-Venus/A4 infection were observed (data not shown). We reprobed the blot against other MYXV proteins, M071 (late) and M-T7 (early). M071 has a similar kinetics profile as M093, since the production of M071 increased over time and was greatly reduced by AraC (Fig. 2A). M-T7, a secreted protein, is known to express early during MYXV infection (37). Consistent with the previous study (37), the maximal level of cell-associated M-T7 was detected at 2 h after infection (Fig. 2A), after which the protein was efficiently secreted from infected cells (data not shown).
Fig 2.

Venus/M093 fusion protein is incorporated into MYXV virions. (A) Temporal synthesis of Venus/M093 protein. BSC-40 cells were mock infected or infected with vMyx-Venus/M093 at an MOI of 10.0 in the absence or presence of cytosine arabinoside (AraC), indicated by +AraC, for 1 h at 37°C. After adsorption, the inoculum was removed and cells were washed. At the indicated time points, cells were harvested and lysed in radioimmunoprecipitation assay buffer. Clarified cell lysates were subjected to SDS-PAGE, and Western blotting was performed sequentially using the indicated antibodies, followed by an appropriate horseradish peroxidase-conjugated secondary antibody. (B) Venus/M093 fusion protein is incorporated into MYXV IMVs. IMVs purified through a 36% sucrose cushion were treated with NP-40 lysis buffer in the presence or absence of DTT. The detergent-insoluble fraction (I) was separated from the detergent-soluble fraction (S) or left unfractionated as total (T) input protein. The fractions were subjected to SDS-PAGE for Western blot analyses using the antibodies indicated in panel A.
Vaccinia virus A4 protein has been shown to be associated with the virion membrane fraction when the core proteins (detergent insoluble) of IMV are separated from the membrane proteins (detergent soluble) using NP-40 buffer containing DTT (38). We therefore examined if the Venus/M093 protein is also localized to the membrane fraction of intracellular MYXV virions. We treated purified virions with NP-40 buffer in the presence or absence of DTT to fractionate the core and membrane proteins. As shown in Fig. 2B, in the presence of DTT, the majority of Venus/M093 protein was detected in the membrane fraction, indicating that M093 is incorporated into progeny MYXV virions and is membrane associated (Fig. 2B; compare lanes 6 and 7). However, in the absence of the reducing agent, which disrupts the disulfide bonds in the virions, the majority of Venus/M093 protein was detected in the core fraction (Fig. 2B; compare lanes 9 and 10). This indicates that, similarly to VACV A4, M093 is incorporated into progeny MYXV virions and localized in proximity to the core (39).
Venus/M093- and Venus/A4-labeled virion-sized particles can be visualized within infected cells.
Our previous result indicates that the Venus/M093 protein is incorporated into progeny MYXV virions (Fig. 2B). We next examined if we could visualize Venus-labeled MYXV virions in infected cells and if the localization pattern of Venus/M093 was similar to that of Venus/A4 during vVac-Venus/A4 infection. To test this, we infected BSC-40 cells with either vMyx-Venus/M093 or vVac-Venus/A4 and visualized the localization of Venus/M093 or Venus/A4 by confocal microscopy. Our results showed that there was accumulation of Venus fluorescence over the viral factories for both virus infections. In addition, Venus-labeled virion-sized particles were present throughout the cytoplasm of infected cells (Fig. 3A), supporting our previous result. Similarly, Venus/A4 was localized to the viral factory and on the virion-sized particles in the cytoplasm, consistent with the localization pattern previously described for yellow fluorescent protein-A4 (Fig. 3A) (36).
Fig 3.

Venus-tagged MYXV and VACV virions visualized by fluorescence microscopy. (A) Fluorescence microscopy of cells infected with vMyx-Venus/M093 or vVac-Venus/A4. BSC-40 cells grown on glass coverslips were infected with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 0.1. At 24 h after infection, cells were fixed and glass coverslips were placed on mounting medium containing DAPI. Cells were visualized using a ×63 water-corrected immersion objective on a Leica TCS SP2 confocal microscope. (B) Binding of Venus-tagged MYXV or VACV to BSC-40 cells. BSC-40 cells grown on glass coverslips were washed with ice-cold medium and prechilled on ice. Cells were incubated with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 10.0 on ice for 1 h. After adsorption, unbound virions were removed by washing cell monolayers with ice-cold medium. Cells were fixed, mounted on DAPI containing mounting medium, and visualized as described for panel A. Yellow, Venus-labeled virions; blue, DNA in nuclei and viral factory. Bars, 10 μm (A) and 20 μm (B).
We next examined if the two Venus-tagged viruses would provide a useful tool to study comparative virus binding properties on various target cells. Therefore, purified vMyx-Venus/M093 or vVac-Venus/A4 virions were bound to BSC-40 cells on ice and washed, and virus binding was observed by confocal microscopy. As shown in Fig. 3B, the binding of vMyx-Venus/M093 or vVac-Venus/M093 to BSC-40 cell monolayers was detected, indicating that vMyx-Venus/M093 could be used to study the comparative levels of MYXV binding to various target cells in parallel with vVac-Venus/A4.
Venus-tagged MYXV binds two human multiple myeloma cell lines, U266 and HuNS1, much more efficiently than VACV.
We have previously shown that MYXV can efficiently infect two human multiple myeloma cell lines, U266 and HuNS1, and prevent the engraftment of these cancer cells into the bone marrow of immunodeficient mice (23). We therefore tested whether VACV could bind to those cell lines as efficiently as MYXV and therefore determined the levels of vVac-Venus/A4 binding to U266 and HuNS1 cells relative to those of vMyx-Venus/M093. As shown in Fig. 4A (left), vMyx-Venus/M093 bound essentially all U266 cells as efficiently as control BSC-40 and HeLa cells, whereas the binding of this virus to HuNS1 cells was detectable by flow cytometry on only approximately 50% of the cells. In the case of VACV, the binding of vVac-Venus/A4 to U266 and HuNS1 cells was detectable only to levels of 60% and 30%, respectively, compared to those to control BSC-40 cells (Fig. 4A, right). Therefore, our data indicate that VACV binds U266 and HuNS1 cells much less efficiently than MYXV, but at this point we could not distinguish whether VACV virions exhibit less affinity for the same binding determinants as MYXV or if they utilize different attachment receptors on myeloma cells.
Fig 4.

Venus-tagged MYXV binds to human myeloma cell lines more efficiently than VACV. (A) Binding of vMyx-Venus/M093 or vVac-Venus/A4 to control BSC-40 and HeLa cells compared to U266 and HuNS1 human multiple myeloma cells. Purified vMyx-Venus/M093 or vVac-Venus/A4 was added to prechilled cells at an MOI of 20.0 and incubated on ice for 1 h. After virion binding, cells were washed extensively, fixed with 2% paraformaldehyde, and analyzed by flow cytometry. The percentages of Venus-positive HeLa, U266, and HuNS1 cells detectable by flow cytometry were normalized to the percentage of BSC-40 cells detected. Statistical analyses between the two viruses for HeLa, U266, and HuNS1 cell lines were performed using the Student t test. (B) Venus-tagged MYXV binding to all tested cells, except HuNS1, is more sensitive to inhibition by soluble heparin than VACV binding. Cells were mock treated or treated with Hep I at 37°C for 30 min, washed, and chilled on ice. Purified vMyx-Venus/M093 or vVac-Venus/A4 was mock treated (−) or pretreated with soluble HP for 1 h. Virions were bound to prechilled cells at an MOI of 20.0 on ice for 1 h. After virion binding, cells were washed extensively and fixed with 2% paraformaldehyde. The relative levels of virion binding to cells were assessed by flow cytometry. (C) Soluble laminin blocks the binding of Venus-tagged VACV, but not MYXV, to HeLa cells. HeLa cells were incubated with mock-treated (−) or laminin-treated vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 20.0 on ice for 1 h. After binding, cells were processed as described for panel A and bound virions were detected by flow cytometry. The mean fluorescence intensity was determined and normalized to that for the untreated corresponding virus. Averages of two independent experiments are shown, and the error bars are plotted. Statistical analyses between the untreated and HP- or LN-treated groups for each virus were performed using the Student t test. * and **, P ≤ 0.05 and P ≤ 0.005, respectively, which are considered significant; a, P > 0.05, which is considered insignificant.
Venus-tagged MYXV and VACV virion binding to target cells is differentially inhibited by soluble heparin, but only VACV virion binding is inhibited by the ECM laminin.
Previous studies have shown that VACV binds to GAGs on the cell surface and that soluble heparin can competitively block the binding of VACV virions to cell surface HS, for example, on BSC-40 or HeLa cells (25, 31). To examine if MYXV comparably utilizes HS as a cell surface attachment factor, we performed binding assays on various mammalian cell lines. We mock treated or pretreated purified MYXV or VACV virions with soluble heparin, and the relative levels of virion binding to target cells were assessed by flow cytometry. Our data showed that binding of vVac-Venus/A4 to BSC-40 and HeLa cells, the cell lines used in previous binding studies, was reduced when virions were preincubated with soluble heparin (Fig. 4B, gray bars). Similarly, binding of vMyx-Venus/M093 to BSC-40 and HeLa cells was even more effectively competed by soluble heparin than it was for VACV, indicating that binding of MYXV to these cells is also mediated in large part by cell surface HS (Fig. 4B, dark bars). We next examined the GAG dependency of MYXV or VACV binding to two human multiple myeloma cell lines, U266 and HuNS1, that have previously been shown to be effectively infected and killed by ex vivo treatment with MYXV (23). As shown in Fig. 4B (bottom left), soluble heparin inhibited the binding of both vMyx-Venus/M093 and vVac-Venus/A4 to U266 cells, indicating that the binding of both MYXV and VACV to U266 cells is dependent on HS. In stark contrast, the binding of either virus to HuNS1 cells was not blocked by soluble heparin, indicating that attachment of MYXV or VACV to HuNS1 cells is completely independent of HS (Fig. 4B, bottom right).
To confirm and extend the results presented above, we treated BSC-40, HeLa, U266, and HuNS1 cells with heparinase I, which cleaves cell surface carbohydrate chains into smaller subunits. In agreement with our heparin treatment studies, the treatment of cells with heparinase I comparably reduced the binding of both MYXV and VACV to BSC-40, HeLa, and U266 cells but not to HuNS1 cells (Fig. 4B). This confirms that whereas the binding of both MYXV and VACV to BSC-40, HeLa, and U266 cells is GAG dependent for all cell lines, the binding of both viruses to HuNS1 cells is not mediated by HS.
It has been shown that VACV protein A26 interacts with the extracellular matrix protein laminin on the surface and that pretreatment of VACV virions with laminin inhibits virus binding to HeLa and BSC-40 cells (24, 31). That previous study concluded that VACV utilizes cell surface laminin, in addition to GAGs, as a cell attachment factor (24). We next examined if MYXV binding to HeLa cells could be blocked by laminin. Therefore, we performed a binding assay in which we mock treated or pretreated purified vMyx-Venus/M093 and vVac-Venus/A4 virions with laminin prior to adsorption to HeLa cells and assessed relative virus binding levels by flow cytometry. Consistent with previous reports (24, 31), the binding of vVac-Venus/A4 to HeLa cells was partially blocked by the exogenous laminin (Fig. 4C). In contrast, however, the binding of vMyx-Venus/M093 was not affected by the treatment with laminin, indicating that MYXV does not require the cell surface ECM protein laminin for attachment to HeLa cells (Fig. 4C). Furthermore, laminin did not block the binding of either vMyx-Venus/M093 or vVac-Venus/A4 to U266 or HuNS1 cells, indicating that neither virus utilizes cell surface laminin as an attachment factor for binding to the multiple myeloma cells (data not shown).
Venus-tagged MYXV and VACV differentially bind and infect two human T lymphoblast-like cell lines, CCRF-CEM and Jurkat.
Our results indicate that both MYXV and VACV can bind comparably to human multiple myeloma cell lines in culture. To determine if this applied to other classes of human lymphoid cells, we next examined if the two viruses could bind to human T cell lines, specifically, CCRF-CEM and Jurkat. We determined the comparative levels of binding of vMyx-Venus/M093 and vVac-Venus/A4 at various MOIs to these two cell lines by measuring the mean fluorescence intensity. Our results showed that the mean fluorescence intensity of vMyx-Venus/M093 binding to Jurkat cells at an MOI of 100.0 was 2.5-fold higher than that of the binding to CCRF-CEM cells, indicating that MYXV does not bind to CCRF-CEM cells as avidly as Jurkat cells (Fig. 5A and B). In contrast, the mean fluorescence intensity of vVac-Venus/A4 binding to CCRF-CEM cells was 2-fold higher than that of the binding to Jurkat cells (Fig. 5A and B). This suggests that the binding of MYXV and VACV to CCRF-CEM and Jurkat cells utilizes nonidentical surface determinants.
Fig 5.
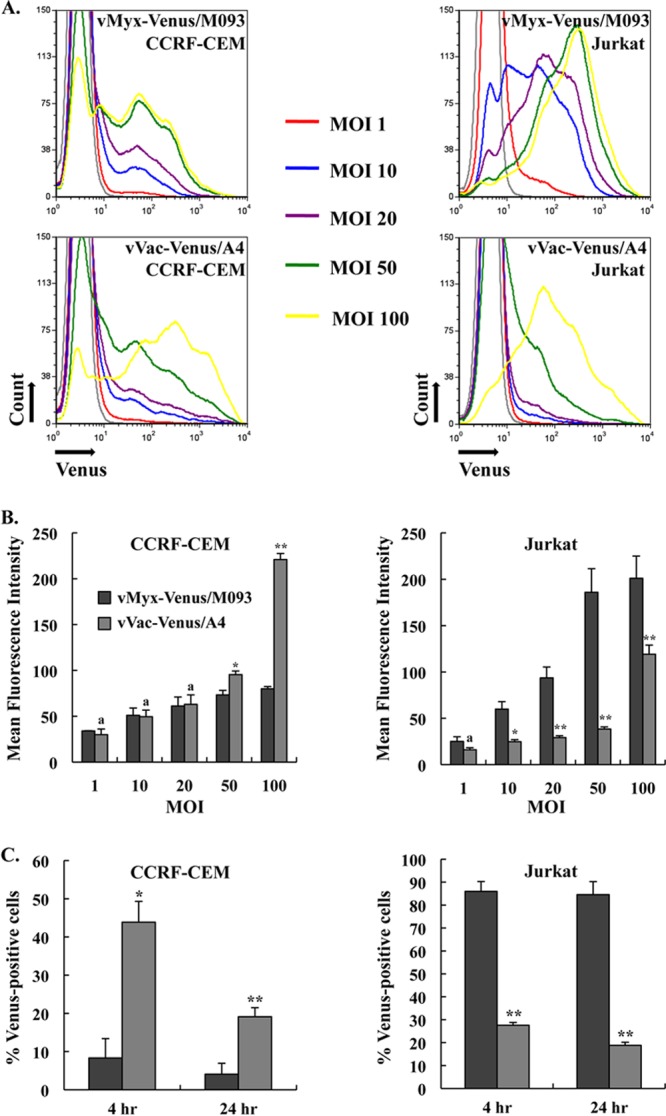
Venus-tagged MYXV and VACV bind and infect human CCRF-CEM and Jurkat T lymphoblastoid cells differently. (A) Histograms of the binding of Venus-tagged MYXV or VACV to CCRF-CEM and Jurkat cells at various MOIs. Prechilled CCRF-CEM or Jurkat cells were incubated with vMyx-Venus/M093 or vVac-Venus/A4 at the indicated MOIs for 1 h on ice. After binding, cells were washed and the amount of virions bound to the cells was determined by flow cytometry. Representative histograms of virus binding are shown. Mock infections are shown in filled gray histograms. (B) Differential binding of Venus-tagged MYXV versus VACV to Jurkat cells. Virion binding was performed as described for panel A. The mean fluorescence intensity was determined, and averages of two independent experiments are plotted. The error bars for each plot are shown. (C) Infection of CCRF-CEM and Jurkat cells with Venus-tagged MYXV or VACV. CCRF-CEM and Jurkat cells were infected with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 20.0 at 37°C for 1 h. After adsorption, unbound virions were removed by washing and cells were incubated at 37°C. At 4 and 24 h postinfection, cells were collected, fixed, and analyzed by flow cytometry. The percentage of Venus-positive cells was determined, and averages of two independent experiments are plotted. The error bars for each plot are shown. Statistical analyses between the two viruses for each cell line were performed using the Student t test. * and **, P ≤ 0.05 and P ≤ 0.005, respectively, which are considered significant; a, P > 0.05, which is considered insignificant.
We next examined if virion binding was correlated to subsequent infection levels. Therefore, we infected CCRF-CEM and Jurkat cells with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 20.0 and determined the percentage of cells that were expressing Venus-fusion protein after 4 versus 24 h of infection at 37°C by flow cytometry. Consistent with the binding data at 4°C, our results showed that almost 90% of Jurkat cells were infected by vMyx-Venus/M093 both at 4 h and at 24 h postinfection, while less than 10% of CCRF-CEM cells were infected with this virus (Fig. 5C). In contrast, vVac-Venus/A4 infected CCRF-CEM cells more efficiently than Jurkat cells (50% versus 25%) at 4 h postinfection. It should be noted that the absolute number of CCRF-CEM cells infected with VACV significantly decreased at 24 h of infection, indicating that infection with VACV caused considerable cell death (Fig. 5C) (23). Taken together, our data indicate that MYXV and VACV differentially bind to and therefore differentially initiate infection of both CCRF-CEM and Jurkat cells (Fig. 5).
Venus-tagged MYXV and VACV utilize different cellular binding determinants to attach to CCRF-CEM T lymphoblastoid cells.
Our results indicated differential aspects of the binding of Venus-tagged MYXV and VACV to both T lymphoblast-like cell lines. We next examined if the binding of MYXV or VACV to CCRF-CEM and Jurkat cells was dependent on cell surface HS. We mock treated or pretreated purified vMyx-Venus/M093 or vVac-Venus/A4 virions with soluble heparin prior to adsorption to cells and assessed the relative levels of virion binding by flow cytometry. Interestingly, soluble heparin did not inhibit the binding of vMyx-Venus/M093 to CCRF-CEM cells, while it reduced the binding of vVac-Venus/A4, indicating that MYXV utilizes another still unknown cell surface determinant to attach to CCRF-CEM (Fig. 6A). In the case of Jurkat cells, treatment with soluble heparin reduced the binding of both viruses, albeit to different extents (Fig. 6A).
Fig 6.

Binding of Venus-tagged MYXV, but not VACV, to human CCRF-CEM T lymphoblastoid cells is heparan sulfate independent. (A) Soluble heparin does not block Venus-tagged MYXV binding to CCRF-CEM cells. Venus-tagged MYXV or VACV that was mock treated (−) or pretreated with soluble HP was bound to CCRF-CEM or Jurkat cells at an MOI of 100.0 on ice for 1 h. After binding, cells were washed and virion binding was determined by flow cytometry. The mean fluorescence intensity was determined, and averages of two independent experiments are plotted. The error bars for each plot are shown. Statistical analyses between the untreated and HP- or LN-treated groups for each virus were performed using the Student t test. (B) Competitive binding assay. BSC-40, CCRF-CEM, or Jurkat cells were mock adsorbed or incubated with untagged vMyx (Lausanne) or vVac (Western Reserve) at an MOI of 100.0 on ice for 1 h. After binding with primary viruses, cells were washed twice and incubated with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 10.0 for BSC-40 cells or 100.0 for CCRF-CEM and Jurkat cells on ice for 1 h. Unbound virions were removed by washing, and cells were fixed with 2% paraformaldehyde. The amount of vMyx-Venus/M093 or vVac-Venus/A4 bound to the cells was determined by flow cytometry. The mean fluorescence intensity was determined and normalized to that for the untreated corresponding virus. Averages of two independent experiments are shown, and the error bars are plotted. Statistical analyses between mock-adsorbed cells and cells incubated with untagged vMyx or vVac were performed for each cell line using the Student t test. * and **, P ≤ 0.05 and P ≤ 0.005, respectively, which are considered significant; a, P > 0.05, which is considered insignificant.
Our results indicated that binding of MYXV to CCRF-CEM human T lymphoblastoid cells was independent of HS, while the binding of VACV showed some HS dependence. This led us to hypothesize that MYXV and VACV utilize a different cellular determinant(s) to attach to CCRF-CEM cells. Therefore, we performed a competitive binding assay in which we prebound nonfluorescently tagged viruses to CCRF-CEM and Jurkat cells prior to binding with Venus-tagged virus. We then quantitated the relative amount of Venus-tagged MYXV or VACV bound to cells by flow cytometry. We had already observed that both Venus-tagged viruses bind to BSC-40 and HeLa cells via HS (Fig. 4A). We predicted that if the binding determinants could be saturated, then primary prebinding of nonfluorescently tagged MYXV or VACV to BSC-40 cells would inhibit or reduce the binding of either of the Venus-tagged viruses. We included the BSC-40 cell line to serve as a positive control for this competition assay. As predicted, binding of cold MYXV to BSC-40 cells as a primary virus comparably inhibited the binding of either vMyx-Venus/M093 (homologous) or vVac-Venus/A4 (heterologous) (Fig. 6B). Similarly, nonfluorescently tagged VACV blocked the binding of either of the Venus-tagged viruses to BSC-40 cells (Fig. 6B). Interestingly, binding of either of the Venus-tagged viruses to CCRF-CEM cells was reduced only when the homologous nontagged virus was used as the primary competing virus (Fig. 6B). Thus, nontagged MYXV reduced the binding of vMyx-Venus/M093 without affecting the binding of vVac-Venus/A4, and vice versa (Fig. 6B). This indicates that MYXV and VACV utilize different cellular binding determinants to attach to CCRF-CEM cells. As for the Jurkat cells, either of the nontagged competing viruses reduced the binding of both vMyx-Venus/M093 and vVac-Venus/A4 (Fig. 6B). This result was less surprising for Jurkat cells because we had already shown that binding of both viruses was comparably inhibited by soluble heparin (Fig. 6A). The reduced binding of Venus-tagged virus was somewhat greater when nontagged MYXV was used as the primary competing virus (Fig. 6B).
Binding of Venus-tagged MYXV or VACV to primary human leukocytes is completely independent of heparan sulfate or laminin.
Since MYXV is currently being developed as a purging agent to selectively delete contaminating cancerous cells in an autograft ex vivo prior to reinfusion and oncolytic VACV is being administered intravenously to treat a variety of cancers in vivo, it is very important to determine the types of human leukocytes that are susceptible to infection by these poxviruses. Previous studies have examined certain types of human leukocytes that can be infected by VACV (40–42). Here, we examined if MYXV and VACV could bind or infect similar types of human leukocytes found in primary human blood or marrow samples. Therefore, enriched primary human leukocytes derived from source leukocytes (LifeSouth) were exposed to vMyx-Venus/M093 or vVac-Venus/A4. The human leukocyte lineages that were capable of binding these Venus-tagged viruses were then determined by flow cytometry. Our results showed that a similar spectrum of primary leukocyte types that were susceptible to Venus-tagged VACV binding after 1 h on ice was also comparably susceptible to Venus-tagged MYXV binding (Fig. 7A and C) and that this binding correlated reasonably well with subsequent viral infection after 24 h at 37°C (Fig. 7B and D). However, we noticed that the absolute numbers of infected granulocytes identified as Venus positive by 24 h postinfection were sometimes higher than the numbers of virus-bound cells detected after 1 h of adsorption on ice. This is most likely because the density of cell surface binding determinants recognized by MYXV and VACV on granulocytes is probably variable between cells, and thus, the absolute number of Venus-positive cells detectable by flow cytometry after the 1-h adsorption step was sometimes underestimated. We observed that monocytes were the most susceptible to both of the viruses, while T cells were the least susceptible. Both MYXV and VACV bound and infected B cells and granulocytes, although not as efficiently as monocytes (Fig. 7). In addition to determining the types of human leukocytes that were susceptible to the two poxviruses, we also examined if virus binding was dependent on heparan sulfate or the ECM protein laminin. Therefore, we pretreated purified MYXV or VACV virions with soluble heparin or laminin prior to adsorption to enriched human leukocytes. As shown in Fig. 7, neither soluble heparin nor laminin reduced the binding or subsequent infection by either virus, indicating that the binding of both MYXV and VACV to human leukocytes is independent of either HS or laminin. Unexpectedly, we noted that for both viruses heparin pretreatment actually increased the relative levels of binding to or infection of certain cell types, such as granulocytes and B cells (Fig. 7A and C). However, the mechanism of the modest increase in Venus-tagged virus binding/infection caused by heparin is unknown. In any event, both viruses bind to primary human leukocytes using attachment determinants that are quite distinct from the GAGs and extracellular matrix components utilized on cultured fibroblastic cell lines, such as HeLa and BSC-40 cells.
Fig 7.

Binding of Venus-tagged MYXV or VACV to primary human leukocytes is not mediated by either cell surface heparan sulfate or the extracellular matrix protein laminin. Buffy coat preparations of fresh primary human leukocytes from healthy donors were incubated with mock-, HP-, or LN-treated vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 10.0 on ice for 1 h. (A) For binding, cells were washed after incubation with virus on ice. (B) For infection, cells were further incubated at 37°C for 24 h after virion adsorption. After binding or infection, cells were stained with phycoerythrin-conjugated anti-CD45 and allophycocyanin-conjugated anti-CD14, anti-CD15, anti-CD19, or anti-CD3 lineage markers. The percentage of Venus-positive cells within each population was determined by flow cytometry. A representative of four independent experiments is shown. (C and D) Summary of the types of primary human cells from donor PBMCs susceptible to MYXV or VACV binding and infection. Average percentages of different cell populations bound (C) or infected (D) by vMyx-Venus/M093 or vVac-Venus/A4 after 1 h at 0°C or 24 h at 37°C, respectively, as detected by flow cytometry under various treatments, were determined. Averages of four independent samples and standard deviations are shown. Statistical analyses between the untreated and HP- or LN-treated groups for each virus were performed using the Student t test. * and **, P ≤ 0.05 and P ≤ 0.005, respectively, which are considered significant; a, P > 0.05, which is considered insignificant.
Venus-tagged MYXV discriminates normal CD34+ stem cells from CD138+ multiple myeloma cells in primary myeloma patient bone marrow more efficiently than VACV.
We have recently shown that MYXV can selectively bind CD138+ multiple myeloma cells that contaminate primary patient bone marrow samples ex vivo, whereas very little or no virus binding to normal CD34+ hematopoietic stem and progenitor cells was detected in the same samples (23). We were interested in whether Venus-tagged VACV would also discriminate normal CD34+ stem cells from CD138+ multiple myeloma cells at the level of virus attachment. Therefore, we compared the relative levels of binding of vMyx-Venus/M093 and vVac-Venus/A4 to CD138+ versus CD34+ cells in individual primary myeloma patient bone marrow samples. Our results in three patient samples showed that Venus-tagged MYXV quantitatively bound and infected essentially all of the CD138+ cells detectable by flow cytometry (Fig. 8A). In contrast, Venus-tagged VACV bound to fewer of the potential target CD138+ cells (Fig. 8A). As for the normal CD34+ hematopoietic stem and progenitor cells in the same samples, neither MYXV nor VACV bound these cells very efficiently (Fig. 8B), and the infection levels for both viruses were consistently less than 10% of the total CD34+ populations, as detected by flow cytometry. We did note that in a minority of patient samples, such as patient 3 in Fig. 8B, there was a small but detectable fraction of CD34+ cells that could adsorb Venus-tagged MYXV, but in these cases, virion binding was transient and did not progress on to a productive infection. At this point, the reason that some patient CD34+ cells could bind MYXV in a nonproductive fashion is unclear, but this binding was uniquely transient and did not lead to significant levels of productive virus infection of these cells. These results indicate that MYXV can selectively bind and eliminate contaminating primary CD138+ multiple myeloma cells more efficiently than VACV, although both viruses are apparently unable to bind or infect the majority of normal CD34+ hematopoietic stem cells present in primary human bone marrow.
Fig 8.

Venus-tagged MYXV or VACV differentially bind CD138+ multiple myeloma cells in primary patient bone marrow samples. Human leukocytes were enriched from the primary patient bone marrow samples through a Ficoll gradient. vMyx-Venus/M093 or vVac-Venus/A4 was bound to leukocytes at an MOI of 10.0 on ice for 1 h. After binding for 1 h on ice or infection for 24 h at 37°C, cells were stained with allophycocyanin-conjugated anti-CD138 (A) or anti-CD34 (B), fixed, and analyzed by flow cytometry. Virion binding was determined by flow cytometry.
Venus-tagged MYXV, but not VACV, binds and infects HuNS1 myeloma cells.
Since we found that VACV did not appear to bind/infect CD138+ multiple myeloma cells from patient samples as efficiently as MYXV, we next examined the ability of MYXV and VACV to bind and infect established human multiple myeloma cell lines, U266 and HuNS1. Therefore, we infected U266 and HuNS1 cells with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 20.0 and determined the percentage of cells that were Venus labeled by flow cytometry after 4 and 24 h postinfection. At both 4 and 24 h postinfection, more than 90% of U266 and HuNS1 cells infected with Venus-tagged MYXV were positive for Venus fluorescence, indicating that both cell lines are highly susceptible to MYXV binding and infection (Fig. 9A). In contrast, only 70% and 35% of U266 and HuNS1 cells, respectively, were Venus positive following exposure to vVac-Venus/A4 at 4 h postinfection (Fig. 9A), indicating that VACV does not bind to or initiate infection of these established multiple myeloma cell lines, especially HuNS1 cells, as efficiently as MYXV. By 24 h of infection, the number of Venus-positive HuNS1 cells that had been adsorbed with vVac-Venus/A4 increased to about 50% (Fig. 9A). This increase may result from the secondary infection from progeny Venus-tagged viruses produced during the primary VACV infection. Note that whereas our previous data showing vMyx-Venus/M093 binding to HuNS1 cells was not as efficient as that to control BSC-40 cells (Fig. 4A, left), more than 90% of HuNS1 cells became infected by vMyx-Venus/M093 by 4 h postinfection (Fig. 9A). This is most likely because the density of cell surface binding determinants that MYXV recognizes on HuNS1 cells is not as abundant as that on the other tested cells, and hence, the absolute number of Venus-positive cells detectable by flow cytometry was likely underestimated.
Fig 9.

Venus-tagged MYXV uniquely attaches to HuNS1 cells that have little or no surface integrin β1. (A) Differential susceptibility of human multiple myeloma cell lines U266 and HuNS1 to Venus-tagged MYXV versus VACV. U266 or HuNS1 cells were infected with vMyx-Venus/M093 or vVac-Venus/A4 at an MOI of 20.0 at 37°C for 1 h. Unbound virions were removed by washing after adsorption, and cells were further incubated at 37°C. At 4 or 24 h postinfection, cells were fixed and the percentage of virus-infected cells was determined by flow cytometry. Averages of two independent experiments are shown, and the error bars are plotted. Statistical analyses between the two viruses for each cell line were performed using the Student t test. **, P ≤ 0.005, which is considered significant. (B) HuNS1 cells express very little to no integrin β1 on their surface. HeLa, U266, and HuNS1 cells were stained with allophycocyanin-conjugated anti-CD29 at 4°C or left unstained. Unbound antibodies were removed by washing before they were analyzed by flow cytometry. Expression of integrin β1 was examined by measuring the allophycocyanin fluorescence (histograms shown in red). Unstained cells are shown in filled gray histograms.
Venus-tagged MYXV attaches to HuNS1 cells via a novel cellular binding determinant(s).
It was recently reported that binding of VACV to HeLa cells was reduced when the expression of cell surface integrin β1 was knocked down using an siRNA, suggesting that VACV utilizes integrin β1 as a cellular attachment factor for these cells (29). Our results clearly show that, in contrast to HeLa cells, neither MYXV nor VACV utilized cell surface HS or laminin for the attachment to HuNS1 cells since pretreatment of virions with heparin or laminin did not reduce virus binding to these cells (Fig. 4A and data not shown). In addition, we show that HuNS1 cells are not very susceptible to VACV infection compared to their susceptibility to MYXV infection, and this difference is reflected by the differential properties of binding of the two Venus-tagged viruses to HuNS1 cells (Fig. 4A). These results led us to examine the expression levels of integrin β1 on the surfaces of various target cells. We compared the cell surface expression levels of integrin β1 on HeLa, U266, and HuNS1 cells by flow cytometry. Our data showed that HeLa and U266 cells expressed a high level of integrin β1 and were highly infectible by Venus-tagged VACV and MYXV, while HuNS1 cells expressed very little to no integrin β1 on their surface (Fig. 9B). However, HuNS1 cells were still highly susceptible to MYXV but much less so to VACV (Fig. 9A). Taken together, our results confirm the previous conclusion that the susceptibility of many cells to Venus-tagged VACV does indeed correlate with cell surface integrin β1 levels, whereas the binding of Venus-tagged MYXV does not, suggesting that MYXV attaches to HuNS1 cells via a novel cell surface binding determinant(s).
DISCUSSION
Two distinctly different poxviruses, namely, MYXV and VACV, are currently being developed as novel oncolytic therapeutics to treat a variety of human cancers (2–4, 10, 17). Despite their similarities as members of the poxvirus family, the two viruses exhibit very divergent host tropisms, and they can differ dramatically in terms of which specific human cancer cells are permissive for infection and oncolysis (32). Although most poxviruses bind to a wide spectrum of mammalian cells in culture (15), there is growing evidence that the initial binding/entry step can be quite different among poxviruses (43, 44). In fact, even different strains of VACV can enter target cells by distinctive mechanisms (31, 45). This suggests that the binding/entry stage of poxviruses might be an important discriminatory stage that mediates tropism specificity for oncolytic virotherapy.
We have recently shown that ex vivo treatment with MYXV can selectively infect leukemic stem and progenitor cells and eliminate them from acute myeloid leukemia (AML) patient samples, while sparing the normal human stem and progenitor cells needed for immune reconstitution following autologous transplantation (22). In addition, we have reported the ability of MYXV to specifically infect contaminating CD138+ multiple myeloma cells and eliminate them from patient bone marrow samples (23). Using MYXV virions tagged with Cy5, we demonstrated that the reason that CD34+ stem cells are unaffected by ex vivo MYXV treatment is because the virus does not bind to this cell population (23). Collectively, these data demonstrate that MYXV has genuine potential to be exploited as an ex vivo cancer cell-purging agent prior to autologous stem cell transplantation, and hence, MYXV is now being developed as a candidate for use for this purpose as part of an oncolytic virotherapeutic approach.
However, we still do not understand why MYXV specifically binds and therefore infects the various diverse contaminating cancerous cell types without binding the normal CD34+ human stem and progenitor cells. Although many studies have investigated the binding determinants needed for VACV infection of cultured mammalian cells, detailed studies on the cellular attachment factors required for MYXV binding to target cells have not been conducted. This study investigated the binding of MYXV and VACV constructs for which the virions were tagged with a structural protein fused to the Venus fluorescent protein. These Venus-tagged viruses were then used to investigate the properties of virion binding to various human cancer cell lines, primary human leukocytes from healthy donors, and cells from the bone marrow of multiple myeloma patients. We examined if MYXV utilized the same cell surface attachment factors used by VACV to bind various classes of target cells and observed some similarities but also some notable differences.
We report that, similarly to VACV, MYXV binds to cell surface glycosaminoglycans, particularly HS, for attachment to some adherent cultured cell lines, such as HeLa and BSC-40, and this binding can be partially competed by exogenous heparin. Unlike VACV, however, MYXV binding cannot be competed with the exogenous cell surface ECM protein laminin for attachment to the same target cells. We also found that the binding of MYXV and VACV to many of the adherent established human cancer cell lines tested is mainly mediated by cell surface HS. In stark contrast, however, the binding of both of the viruses to virtually all of the normal human leukocytes tested is completely independent of either cell surface HS or laminin. Therefore, our study suggests that the cell attachment factor(s) utilized by both poxviruses to bind to primary human leukocytes is in fact very different from the previous cellular binding determinants identified from the studies conducted on established cell lines. Indeed, the existence of a still unidentified cellular attachment factor(s) utilized by poxviruses for the attachment to primary human leukocytes is strongly suggested by our studies.
Previous studies on the binding of VACV to various mammalian cells have indicated that VACV can utilize cell surface glycosaminoglycans such as HS and CS, the ECM protein laminin, and/or integrin β1 as a cellular attachment factor(s) (24, 25, 29). We found that the sensitivity of Venus-tagged MYXV and VACV binding to inhibition by soluble heparin varied considerably among the adherent cell lines that we tested (Fig. 4 and 6). It appears that cell surface HS is the main cellular attachment factor utilized by Venus-tagged MYXV to bind HeLa cells, for example, since soluble heparin inhibited this binding by more than 80% (Fig. 4A). However, only about a 20% reduction was observed with Venus-tagged VACV binding to the same cells, suggesting that VACV binds to cell surface determinants in addition to HS for the attachment to HeLa cells. It has also been reported that the knockdown of integrin β1 expression reduced VACV binding to HeLa cells (29). In addition, we confirm previous reports that VACV binding to HeLa cells was also reduced by the exogenous ECM protein laminin (Fig. 4B) (24). However, MYXV does not encode an ortholog of the VACV laminin-binding protein A26 (11). Indeed, laminin did not inhibit MYXV binding to HeLa cells at all (Fig. 4B), indicating that MYXV uniquely does not utilize the cell surface ECM protein laminin for virion attachment.
Importantly, our binding studies with two human T cell lymphoblastoid lines, Jurkat and CCRF-CEM, revealed that MYXV and VACV could differentially bind/infect these cell types (Fig. 5). While Jurkat cells were highly susceptible to MYXV binding and infection, CCRF-CEM cells were very poorly infected by MXYV because of a failure of virions to attach (Fig. 5). In contrast, VACV bound/infected Jurkat cells relatively poorly and CCRF-CEM cells much more efficiently (Fig. 5). In addition, the sensitivity of the two viruses to inhibition by soluble heparin was the opposite of what was observed with HeLa cells; i.e., binding of Venus-tagged VACV to Jurkat cells was reduced to a level greater than that for Venus-tagged MYXV (Fig. 6A). In fact, there was very little reduction in binding of MYXV to CCRF-CEM cells when virions were pretreated with soluble heparin (Fig. 6A), indicating that MYXV does not use cell surface HS to any significant degree for the attachment to CCRF-CEM cells. These differences in MYXV and VACV binding to and infection of each of the two human T cell lines suggest that the cell surface attachment factors utilized by these poxviruses for entry into the target leukocytes can be quite dissimilar, depending on the target cells.
Our studies on the binding of MYXV and VACV to established human multiple myeloma cell lines, U266 and HuNS1, demonstrate further that the cellular determinants for the attachment of these two viruses on cancerous leukocytes can be quite different. Our data show that HuNS1 cells express very little cell surface integrin β1, an attachment factor for VACV (Fig. 9B). This provides one possible explanation as to why VACV did not bind or infect HuNS1 cells very efficiently (Fig. 4A and 9A). However, despite the low integrin β1 expression level, HuNS1 cells were highly susceptible to MYXV binding and infection (Fig. 4A and 9A). Additionally, we found that the attachment of both MYXV and VACV to HuNS1 cells was completely independent of HS (Fig. 4B). Hence, our results indicate that MYXV utilizes a previously unidentified cellular determinant(s) different from the integrin β1 utilized by VACV for entry into HuNS1 cells.
Studies on the susceptibility of primary human leukocytes from peripheral blood mononuclear cells of healthy donors to VACV and MYXV infection were also conducted. It was previously reported that human monocytes are very susceptible to VACV (40–42). On the other hand, resting human T cells have been reported to be not susceptible to VACV, although they became susceptible once the cells were activated (40). It was reported that a cell surface receptor(s) expressed on the activated T cells is responsible for the increased susceptibility to VACV and the virion attachment to activated T cells is mediated by a cell surface protein(s) other than HS (40). However, the specific cellular receptor(s) that allowed VACV binding and therefore infection has not yet been identified. In addition, primary human natural killer cells and B cells have been shown to be moderately susceptible to VACV infection (40, 42). Our study indicates that similar overall types of human leukocytes infected by VACV were also generally susceptible to MYXV (Fig. 7). We also found that both MYXV and VACV could bind and infect CD15+ granulocytes to a comparable degree (Fig. 7).
Importantly, our study addressed an important question of whether or not MYXV and VACV utilize the same cell attachment factors, particularly HS and laminin, for entry into human cells. Our data clearly show that, in stark contrast to adherent cell lines like HeLa or BSC-40, neither MYXV nor VACV utilizes cell surface HS or laminin for attachment to any primary human leukocytes tested (Fig. 7). In fact, for reasons that are unclear, treatment of Venus-tagged MYXV virions with soluble heparin actually increased the binding to certain cell types, such as granulocytes and B cells. Our observations that neither MYXV nor VACV utilizes a ubiquitously expressed cell surface attachment factor, i.e., heparan sulfate, to bind and infect human leukocytes, whereas only MYXV can infect myeloma cells that are deficient in integrin β1, may help to explain why MYXV is such a selective purging agent against human myeloma cells while at the same time it does not bind or infect normal CD34+ stem and progenitor cells.
As mentioned previously, both MYXV and VACV are being developed as oncolytic virotherapeutic agents for a variety of human cancers. In both cases, the viruses are first exposed primarily to patient leukocytes: MYXV is used ex vivo to treat donor bone marrow or mobilized peripheral blood mononuclear cells (PBMCs) needed for subsequent autologous stem cell transplantation, whereas VACV is injected intravenously into the circulation of cancer patients. Hence, it is important to determine which poxvirus would be optimal for the specific oncolytic virotherapy strategy and whether their relative tropisms for both cancer cells and normal leukocytes are optimal for that therapeutic option. For example, we recently reported that MYXV could selectively bind to and infect CD138+ multiple myeloma cells that contaminate primary patient bone marrow samples but could not even bind to normal CD34+ stem cells, and this discrimination is crucial to the oncolytic specificity of MYXV for myeloma (23). Here, we showed that Venus-tagged MYXV selectively bound and infected CD138+ multiple myeloma cells from patient bone marrow samples, whereas Venus-tagged VACV attached to these primary myeloma cells much less efficiently (Fig. 8A). Neither of the viruses bound or infected the normal CD34+ stem and progenitor cells to any appreciable extent, suggesting that neither virus would perturb the engraftment/differentiation potential of these cells (Fig. 8B). Therefore, our data suggest that MYXV is more discriminating for myeloma cells than VACV as a purging agent to specifically eliminate contaminating CD138+ multiple myeloma cells from the patient's stem cell isograft prior to retransplantation.
In summary, our study indicates that the cellular determinants utilized by MYXV and VACV for attachment to many target cells can be significantly different from each other, particularly for leukocytes and certain human cancer cells, such as multiple myeloma. The binding of both poxviruses to primary human leukocytes is also not mediated by cell surface HS or laminin, and we conclude that an additional poxvirus cell attachment factor(s) remains to be identified. Finally, our data indicate that the oncolytic discrimination potentials of MYXV and VACV are quite different and both viruses can have unique roles to play in cancer virotherapy.
ACKNOWLEDGMENTS
This work was supported in part by NIH R01 CA138541 and AI080607 research grants. W.M.C. was supported in part by NHI Training Grant in Cancer Biology T32 CA009126.
Footnotes
Published ahead of print 6 February 2013
REFERENCES
- 1. Moss B. 2007. Poxviridae: the viruses and their replication, p 2905–2946 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Guse K, Cerullo V, Hemminki A. 2011. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin. Biol. Ther. 11:595–608 [DOI] [PubMed] [Google Scholar]
- 3. Kirn DH, Thorne SH. 2009. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat. Rev. Cancer 9:64–71 [DOI] [PubMed] [Google Scholar]
- 4. Kochneva GV, Sivolobova GF, Iudina KV, Babkin IV, Chumakov PM, Netesov SV. 2012. Oncolytic poxviruses. Mol. Gen. Mikrobiol. Virusol., p 8–15 (In Russian.) [PubMed] [Google Scholar]
- 5. Lusky M, Erbs P, Foloppe J, Acres RB. 2010. Oncolytic vaccinia virus: a silver bullet? Expert Rev. Vaccines 9:1353–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, Diallo JS, Falls T, Burns J, Garcia V, Kanji F, Evgin L, Hu K, Paradis F, Knowles S, Hwang TH, Vanderhyden BC, Auer R, Kirn DH, Bell JC. 2012. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol. Ther. 20:749–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thorne SH. 2012. Next-generation oncolytic vaccinia vectors. Methods Mol. Biol. 797:205–215 [DOI] [PubMed] [Google Scholar]
- 8. Kerr P, McFadden G. 2002. Immune responses to myxoma virus. Viral Immunol. 15:229–246 [DOI] [PubMed] [Google Scholar]
- 9. Kerr PJ. 2012. Myxomatosis in Australia and Europe: a model for emerging infectious diseases. Antiviral Res. 93:387–415 [DOI] [PubMed] [Google Scholar]
- 10. Liu J, Wennier S, McFadden G. 2010. The immunoregulatory properties of oncolytic myxoma virus and their implications in therapeutics. Microbes Infect. 12:1144–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cameron C, Hota-Mitchell S, Chen L, Barrett J, Cao JX, Macaulay C, Willer D, Evans D, McFadden G. 1999. The complete DNA sequence of myxoma virus. Virology 264:298–318 [DOI] [PubMed] [Google Scholar]
- 12. Barrett JW, Cao JX, Hota-Mitchell S, McFadden G. 2001. Immunomodulatory proteins of myxoma virus. Semin. Immunol. 13:73–84 [DOI] [PubMed] [Google Scholar]
- 13. Zuniga MC. 2002. A pox on thee! Manipulation of the host immune system by myxoma virus and implications for viral-host co-adaptation. Virus Res. 88:17–33 [DOI] [PubMed] [Google Scholar]
- 14. Fenner F. 1959. Myxomatosis. Br. Med. Bull. 15:240–245 [DOI] [PubMed] [Google Scholar]
- 15. McFadden G. 2005. Poxvirus tropism. Nat. Rev. Microbiol. 3:201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lun X, Alain T, Zemp FJ, Zhou H, Rahman MM, Hamilton MG, McFadden G, Bell J, Senger DL, Forsyth PA. 2010. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 70:598–608 [DOI] [PubMed] [Google Scholar]
- 17. Stanford MM, McFadden G. 2007. Myxoma virus and oncolytic virotherapy: a new biologic weapon in the war against cancer. Expert Opin. Biol. Ther. 7:1415–1425 [DOI] [PubMed] [Google Scholar]
- 18. Bartee E, McFadden G. 2009. Human cancer cells have specifically lost the ability to induce the synergistic state caused by tumor necrosis factor plus interferon-beta. Cytokine 47:199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bartee E, Mohamed MR, Lopez MC, Baker HV, McFadden G. 2009. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J. Virol. 83:498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang G, Barrett JW, Stanford M, Werden SJ, Johnston JB, Gao X, Sun M, Cheng JQ, McFadden G. 2006. Infection of human cancer cells with myxoma virus requires Akt activation via interaction with a viral ankyrin-repeat host range factor. Proc. Natl. Acad. Sci. U. S. A. 103:4640–4645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Werden SJ, McFadden G. 2010. Pharmacological manipulation of the Akt signaling pathway regulates myxoma virus replication and tropism in human cancer cells. J. Virol. 84:3287–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim M, Madlambayan GJ, Rahman MM, Smallwood SE, Meacham AM, Hosaka K, Scott EW, Cogle CR, McFadden G. 2009. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia 23:2313–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bartee E, Chan WM, Moreb JS, Cogle CR, McFadden G. 2012. Selective purging of human multiple myeloma cells from autologous stem cell transplantation grafts using oncolytic myxoma virus. Biol. Blood Marrow Transplant. 18:1540–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chiu WL, Lin CL, Yang MH, Tzou DL, Chang W. 2007. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 81:2149–2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Foo CH, Lou H, Whitbeck JC, Ponce-de-Leon M, Atanasiu D, Eisenberg RJ, Cohen GH. 2009. Vaccinia virus L1 binds to cell surfaces and blocks virus entry independently of glycosaminoglycans. Virology 385:368–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hsiao JC, Chung CS, Chang W. 1998. Cell surface proteoglycans are necessary for A27L protein-mediated cell fusion: identification of the N-terminal region of A27L protein as the glycosaminoglycan-binding domain. J. Virol. 72:8374–8379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hsiao JC, Chung CS, Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PW, Liao CL, Chang W. 2012. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 86:6677–6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin CL, Chung CS, Heine HG, Chang W. 2000. Vaccinia virus envelope H3L protein binds to cell surface heparan sulfate and is important for intracellular mature virion morphogenesis and virus infection in vitro and in vivo. J. Virol. 74:3353–3365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bengali Z, Townsley AC, Moss B. 2009. Vaccinia virus strain differences in cell attachment and entry. Virology 389:132–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Villa NY, Bartee E, Mohamed MR, Rahman MM, Barrett JW, McFadden G. 2010. Myxoma and vaccinia viruses exploit different mechanisms to enter and infect human cancer cells. Virology 401:266–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zachertowska A, Brewer D, Evans DH. 2004. MALDI-TOF mass spectroscopy detects the capsid structural instabilities created by deleting the myxoma virus cupro-zinc SOD1 homolog M131R. J. Virol. Methods 122:63–72 [DOI] [PubMed] [Google Scholar]
- 34. Johnston JB, Barrett JW, Chang W, Chung CS, Zeng W, Masters J, Mann M, Wang F, Cao J, McFadden G. 2003. Role of the serine-threonine kinase PAK-1 in myxoma virus replication. J. Virol. 77:5877–5888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Smallwood SE, Rahman MM, Smith DW, McFadden G. 2010. Myxoma virus: propagation, purification, quantification, and storage. Curr. Protoc. Microbiol. Chapter 14:Unit 14A.1. doi:10.1002/9780471729259.mc14a01s17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ward BM. 2005. Visualization and characterization of the intracellular movement of vaccinia virus intracellular mature virions. J. Virol. 79:4755–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mossman K, Upton C, McFadden G. 1995. The myxoma virus-soluble interferon-gamma receptor homolog, M-T7, inhibits interferon-gamma in a species-specific manner. J. Biol. Chem. 270:3031–3038 [DOI] [PubMed] [Google Scholar]
- 38. Cudmore S, Blasco R, Vincentelli R, Esteban M, Sodeik B, Griffiths G, Krijnse Locker J. 1996. A vaccinia virus core protein, p39, is membrane associated. J. Virol. 70:6909–6921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roos N, Cyrklaff M, Cudmore S, Blasco R, Krijnse-Locker J, Griffiths G. 1996. A novel immunogold cryoelectron microscopic approach to investigate the structure of the intracellular and extracellular forms of vaccinia virus. EMBO J. 15:2343–2355 [PMC free article] [PubMed] [Google Scholar]
- 40. Chahroudi A, Chavan R, Kozyr N, Waller EK, Silvestri G, Feinberg MB. 2005. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J. Virol. 79:10397–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hou W, Gibbs JS, Lu X, Brooke CB, Roy D, Modlin RL, Bennink JR, Yewdell JW. 2012. Viral infection triggers rapid differentiation of human blood monocytes into dendritic cells. Blood 119:3128–3131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sanchez-Puig JM, Sanchez L, Roy G, Blasco R. 2004. Susceptibility of different leukocyte cell types to vaccinia virus infection. Virol. J. 1:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Moss B. 2012. Poxvirus cell entry: how many proteins does it take? Viruses 4:688–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schmidt FI, Bleck CK, Mercer J. 2012. Poxvirus host cell entry. Curr. Opin. Virol. 2:20–27 [DOI] [PubMed] [Google Scholar]
- 45. Bengali Z, Satheshkumar PS, Moss B. 2012. Orthopoxvirus species and strain differences in cell entry. Virology 433:506–512 [DOI] [PMC free article] [PubMed] [Google Scholar]