

Background: Currently it is unknown whether Smad7 has a functional role in eye development.
Results: Eye development is defective in Smad7 null mice and is accompanied by alterations in the patterning of BMP signals, periocular mesenchymal genes, and sonic hedgehog signaling.
Conclusion: Smad7 is indispensable for eye development in the mouse.
Significance: Smad7 is required for eye development.
Keywords: Bone Morphogenetic Protein (BMP), Development, Eye, Mouse, SMAD Transcription Factor, Transforming Growth Factor β (TGFβ)
Abstract
Smad7 is an intracellular inhibitory protein that antagonizes the signaling of TGF-β family members. Deletion of Smad7 in the mouse leads to an abnormality in heart development. However, whether Smad7 has a functional role in the development of other organs has been elusive. Here we present evidence that Smad7 imparts a role to eye development in the mouse. Smad7 is expressed in both the lens and retina in the developing embryonic eye. Depletion of Smad7 caused various degrees of coloboma and microphthalmia with alterations in cell apoptosis and proliferation in eyes. Smad7 was implicated in lens differentiation but was not required for the induction of the lens placode. The development of the periocular mesenchyme was retarded with the down-regulation of Bmp7 and Pitx2 in mutant mice. Retinal spatial patterning was affected by Smad7 deletion and was accompanied by altered bone morphogenetic protein (BMP) signaling. At late gestation stages, TGF-β signaling was up-regulated in the differentiating retina. Smad7 mutant mice displayed an expanded optic disc with increasing of sonic hedgehog (SHH) signaling. Furthermore, loss of Smad7 led to a temporal change in retinal neurogenesis. In conclusion, our study suggests that Smad7 is essential for eye development. In addition, our data indicate that alterations in the signaling of BMP, TGF-β, and SHH likely underlie the defects in eye development caused by Smad7 deletion.
Introduction
The development of vertebrate eyes is a complex process, proceeding through specification of the anterior neural plate, invagination of the optic vesicle, and differentiation of the lens and retina. This process involves a series of reciprocal interaction between the neuroepithelium, surface ectoderm, and extraocular mesenchyme (1–3), which requires coordination of multiple signaling pathways, such as retinoic acid, fibroblast growth factors, sonic hedgehog (SHH),3 WNTs, and the transforming growth factor β (TGF-β) superfamily (4–7).
Members of the TGF-β superfamily are essential in a wide spectrum of eye development. At the early stage, bone morphogenetic proteins (BMPs) affect early lens induction and the invagination of the optic vesicle (8–10). After formation of the optic cup, BMP signaling patterns the dorsal-ventral axis and maintains the asymmetrical gene expression such as Tbx family genes (11), which are critical for the establishment of correct topographic mapping of retinal ganglion cells (RGC) in axon guidance (12–14). TGF-β also controls ocular cell apoptosis, proliferation and differentiation of the lens, initiation of the neurogenesis, retina pigmentation, and retinal progenitor cell competence (13, 15–17). Different levels of TGF-β superfamily signals regulate the distinct developmental programs of the eye (18). In addition, TGF-β family members are important in controlling the migration and specification of the neural crest-derived periocular mesenchyme (POM), and implicated in the epithelial-mesenchymal transition, all of which contribute to the formation of ocular tissues and the appendages of the eye (19, 20).
Smad proteins are the basic intracellular transducers mediating the signal cascade after TGF-β family ligands bind to their cognate receptors. They are classified into three groups, receptor-regulated Smad (R-Smad), common partner Smad (Co-Smad), and inhibitory Smad (I-Smad). Smad1, -5, -8 are R-Smad activated by the BMP subfamily; whereas Smad2 and Smad3 are activated by TGF-β, activin, and Nodal. Upon activation, R-Smad are phosphorylated, form a complex with Smad4, accumulate in the nucleus, and consequently modulate the expression of target genes. Smad6 and Smad7 are components of the inhibitory Smad family that negatively regulate the signaling of TGF-β family members. Smad6 preferentially inhibits BMP signaling, whereas Smad7 antagonizes both TGF-β/activin- and BMP-mediated signaling by interfering with R-Smad binding to the receptors and the phosphorylation/activation of R-Smad (21).
As a potential central effector in an autoregulatory feedback loop in TGF-β signaling, Smad7 plays a critical role in various physiological processes. Aberrant expression of Smad7 is associated with the pathogenesis of many diseases in humans, including renal fibrosis and inflammation, scleroderma formation, and chronic inflammatory bowel diseases (22–24). In vivo functional study has demonstrated that partial loss of Smad7 function by deletion of the MH1 domain results in some change of B cell response (25). However, deletion of the MH2 domain of Smad7 in mice led to a complete loss of Smad7 function, presenting as multiple defects in cardiovascular development (26). These studies suggest that Smad7 is required for organogenesis during embryonic development. Previous studies on the functions of Smad7 in eyes were focused mainly on the regeneration and pathology of the mature organ. Smad7 is considered an important factor in cornea healing and regeneration of the lens epithelium (27–29). In vitro analysis demonstrates that Smad7 functions as a key molecular switch in balancing the inhibitory effects of BMP7 on TGF-β2 in trabecular meshwork cells (30). Smad7 is also a potential clinical target to prevent and treat proliferative vitreoretinopathy (31). Overexpression of Smad7 in the epithelial cells in mice leads to abnormal eyelid closure and thin corneal stroma (32), indicating that dyregulation of Smad7 in the adjacent ocular tissue may cause pathological alterations in the eye formation. However, currently it is unknown whether Smad7 affects eye development. In this study, we found that deletion of Smad7 led to multiple defects in eye development including dysregulation of lens differentiation, alteration of periocular mesenchyme, disruption of the retina patterning, and changes in the optic stalk. Smad7 may trigger the proper temporal and spatial change of the BMP level for the developing eye, meanwhile regulating SHH signaling at certain stages. Here, we have shown that Smad7 is essential for the timely regulation of the distinct signaling pathways during embryonic eye development.
EXPERIMENTAL PROCEDURES
Mice
All animal procedures and protocols were approved by the Institutional Animal Care and Use Committee of the Institute for Nutritional Sciences, Chinese Academy of Sciences(Approval Number 2010-AN-8). Mice carrying a null allele of Smad7 have been described previously (26). The mice were crossed with the C57BL/6 strain for at least six generations. The presence of the Smad7-null allele was detected by primer 1 (5′-CAGAGCAGCCGATTGTCTGTTGTGC-3′) and primer 2 (5′-ACTTGGACTGGATTGATGTACCAGG-3′), resulting in a 500-bp PCR product. The wild type allele was detected by primers A and B (5′-TGTCCCGCTTGTCTTGTTCTTTGAG-3′ and 5′-TGCTGACTCTCGTTTCCTGTCTTCG-3′, respectively), giving rise to a 154-bp product. To define the embryonic stage, noon of the day that a vaginal plug was found was recorded as embryonic day 0.5 (E0.5).
Antibodies and Reagents
The antibodies used in immunohistochemistry were as follows. The antibody for phosphorylated Smad1/5/8 was from Millipore (Billerica, MA), for Smad7 from Santa Cruz Biotechnology, for phosphorylated Smad2 from Cell Signaling Technology (Danvers, MA), for BrdU from Sigma-Aldrich, and for TUJ1 from Beyotime (Shanghai, China). Alexa 488- or Alexa 546-conjugated secondary antibodies were from Invitrogen. The TSATM Plus fluorescence system was from PerkinElmer Life Sciences, and the Vectastain ABC kit was purchased from Vector Laboratories (Burlingame, CA).
Histology, Immunohistochemistry, and BrdU Labeling
Mouse embryos were harvested by cesarean section and fixed in 4% paraformaldehyde in PBS at 4 °C overnight. After dehydration through a series of graded ethanol baths, the embryos were infiltrated, embedded in wax, and then sectioned at 10 μm. The sections were dehydrated and subjected to standard hematoxylin and eosin (H&E) staining. Antibody staining was performed as described previously (33). Briefly, the sample slides were heated in 10 mm citrate buffer (pH 6.0) under sub-boiling conditions for 30 min. 3% H2O2 was used to quench endogenous peroxidase for 10 min. The section was blocked in 10% goat serum in PBS at room temperature for 1 h and then incubated with primary antibody at 4 °C overnight in a humidified chamber. The sections were treated sequentially with a biotin-conjugated secondary antibody and ABC reagent for the next day and subjected to color reaction with a diaminobenzidine solution (DAB, Sigma-Aldrich). The image was visualized with an Olympus BX51 microscope. For BrdU staining, pregnant mice were injected with BrdU 2 h prior to sacrifice at 0.1 mg/1 gram body weight. The embryos were collected and treated as described above. The section was retrieved with citrate buffer followed by treatment with 1 n HCl for 90 min and then blocked and incubated in anti-BrdU antibody overnight. The secondary antibody was applied to the section, and the nuclei were stained with Hoechst 33342 (Invitrogen). Smad7 staining was performed according to the instruction of the TSATM Plus fluorescence system. The images were observed under an Olympus BX61 fluorescence microscope.
RNA in Situ Hybridization
RNA whole mount in situ hybridization was carried out according to the standard protocol (34). RNA in situ hybridization was performed on sections as described (35). Briefly, mouse embryos were fixed in 4% paraformaldehyde overnight, cryoprotected in 30% sucrose/PBS overnight, embedded in OCT compound, and then sectioned at 10 μm on a Leica CM3050S cryostat. The RNA probes were denatured and applied to the cryosections in a humidified chamber at 65 °C for overnight incubation. The slides were then rinsed in wash buffer (1× SSC, 50% formamide, 0.1% Tween 20) at 65 °C and MABT buffer (100 mm maleic acid, 150 mm NaCl (pH 7.5), 0.1% Tween 20) at room temperature and then blocked in 20% heat-inactivated sheep serum, 2% blocking reagent (Roche Diagnostics, Indianapolis, IN) in MABT buffer for 1 h at room temperature. The sections were incubated in an anti-DIG antibody (Roche Diagnostics) at 4 °C for overnight and then washed in MABT buffer and stained with NBT/BCIP solution (Promega Corp., Madison, WI). The following probes were used in the study: Sox2, Bmp7, shh, and Bmp4 (from Xin Zhang, Indiana University School of Medicine, Indianapolis), Pax2 (nucleotides 210–834, GenBankTM accession number NM_011037), Pax6 (nucleotides 1330–2074, GenBankTM accession number NM_001244202), Fgf15 (nucleotides 43–848, GenBankTM accession number NM_008003), Vax2 (nucleotides 295–933, GenBankTM accession number NM_011912), Vax1 (nucleotides 236–907, GenBankTM accession number NM_009501), Mitf (nucleotides 209–921, GenBankTM accession number NM_008601), Cx40 (nucleotides 899–1770, GenBankTM accession number NM_008121), Rx (nucleotides 404–1150, GenBankTM accession number NM_013833), Math5 (nucleotides 1–450, GenBankTM accession number NM_016864), Brn3b (727–1720, GenBankTM accession number NM_001039353), α-crystallin (313–1034, GenBankTM accession number NM_013501), β-crystallin (19–804, GenBankTM accession number NM_023695), γ-crystallin (34–605, GenBankTM accession number NM_007776), and Tbx5 (11).
TUNEL Assay
TUNEL assays were performed with an in situ cell death detection kit (Roche Diagnostics). Briefly, cryosections were processed for antigen retrieval as described above, incubated with blocking buffer (0.1 m Tris-HCl (pH 7.5), 3% BSA, 20% serum) for 30 min at room temperature and then with TUNEL reaction mixture for 2 h at 37 °C. The signal was detected under an Olympus BX61 fluorescence microscope.
Statistical Analysis
Statistical significance was assessed by Student's t test for the analysis of cell apoptosis and cell proliferation, and p < 0.05 was considered statistically significant.
RESULTS
Ablation of Smad7 Leads to Coloboma and Microphthalmia
To determining the potential role of Smad7 during eye development, we examined the expression of Smad7 in the developing eyes at both mRNA and protein levels (Fig. 1A). Smad7 transcript was detectable in the eye through E10.5 to E14.5 by RT-PCR but not in Smad7 null mice (data not shown). At E10.5, Smad7 protein was detected in the distal portion of the optic vesicle. Smad7 was strongly localized in the lens placode and the adjacent surface ectoderm. The signal was also visible in the surrounding mesenchymal cells (Fig. 1A, arrow). At E11.5, although still expressed in the lens vesicle, Smad7 protein expanded into the middle neural retina. At E14.5, Smad7 was distributed ubiquitously in the whole retina. Meanwhile, in the lens, Smad7 protein was restricted to the epithelial cells and the equatorial zone (Fig. 1A). Smad7 expression was present in the whole retina until at least the first day of birth (data not shown). These results, collectively, suggest that Smad7 is expressed in various eye regions during embryonic development.
FIGURE 1.

Deletion of Smad7 leads to ocular defects. A, Smad7 protein was examined by immunohistochemistry in the developing eyes of mouse embryos at E10.5, E11.5, and E14.5 using the frontal sections. In addition to the lens and retina, Smad7 protein was detected in the surrounding mesenchyme of the eye (indicated by an arrow) at E10.5. B, whole mount view of ocular phenotype in Smad7 null embryos. Pigmentation is deficient in the ventral retina at E11.5 compared with the control (marked by arrow). Coloboma and microphthalmia are observed at E14.5 and at E18.5 in Smad7-deficient embryos (marked by arrows). Only eyeballs are shown for E18.5. C, transverse section stained with H&E shows pigmentation in the optic stalk (marked by arrow), absence of the lens (marked by asterisk), hyperplastic primary vitreous (marked by # sign), and discontinuity of the retina (marked by arrowhead) in Smad7-deficient embryos. Scale bars: 50 μm (A, first and second panels); 100 μm (A, third panel; B, first and third panels; C, all images); 400 μm (B, second panels).
We next investigated the eye phenotype with the embryos that had homozygous deletion of the Smad7 gene as reported previously (26). The eye defect was detected first at E11.5, when RPE in the wild type embryos was well developed with only the optic fissure open on the ventral side. In contrast, the pigmentation in the Smad7-deleted embryos was overgrown in the dorsal region but absent on the ventral side (Fig. 1B, left panels). Normally, the optic fissures are closed at the ventral side at E12.5, but the optic fissure remained open in Smad7 mutant embryos even at E14.5 and pointed toward the ventral temporal direction (Fig. 1B, middle panels). At E18.5, Smad7 null embryos exhibited various degree of microphthalmia, with 50% of them appearing as unilateral or bilateral coloboma (4 of 8 embryos at E18.5) (Fig. 1B, right panels). A histologic examination of the transverse sections at E14.5 revealed that the pigmentation was invaded into the optic nerve in the mutant embryos (Fig. 1C, second panel), whereas discontinuity was observed at the distal region of the retina (Fig. 1C, fourth panel). In severe cases in Smad7-deleted embryos, the lens was completely absent (Fig. 1C, third panel). We also observed persistent hyperplastic primary vitreous, a dense cell membrane between the lens and the retina in Smad7 mutant embryos (Fig. 1C, fourth panel). Collectively, these results indicate that Smad7 is required for eye development in mice. It is noteworthy that although Smad7 was expressed in the 129sv mice and shared a pattern similar to that of the C57BL/6 mouse (data not shown), the eye abnormality in Smad7 null embryos was distinct when Smad7-deficient mice were backcrossed onto the C57BL/6 mouse strain but was barely seen on the 129/FVB hybrid background (data not shown).
Altered Cell Apoptosis and Proliferation in the Eyes of Smad7 Mutant Embryos
As deletion of Smad7 is associated with defects in eye development, we next addressed whether alterations in cell apoptosis and proliferation may underlie the observed defects. A TUNEL assay revealed cell apoptosis in the optic vesicle at E10.5 (Fig. 2A). The number of apoptotic cells in the retina was significantly elevated in Smad7-deleted embryos as compared with the controls (Fig. 2A). At the same time, we also noticed obvious cell death in POM in Smad7 mutant embryos (Fig. 2A). Meanwhile, we assessed the cell proliferation rate by BrdU incorporation assay. We found that fewer BrdU-positive cells were detected in both the retina and lens in Smad7 mutants than in the wild type controls (Fig. 2B). However, no significant change in the cell proliferation profile was found in the mesenchyma around the eye (data not shown). At the same time, lens pitting was found to be reduced in the mutant embryos in comparison with the controls (Fig. 2B). Based on these data, we postulate that the decreased eye size observed in Smad7-depleted embryos was mainly because of alterations in cell apoptosis and proliferation during eye development.
FIGURE 2.

Altered cell apoptosis and proliferation in Smad7 null mice. A, cell apoptosis in the retina and lens. Representative images of TUNEL assay are shown in the upper panel. The quantification of TUNEL-positive nuclei is shown in the lower panel as mean ± S.D. (n = 3 for control and 4 for Smad7 null embryos). The percentage was calculated as TUNEL-positive cells/total cells in both retina and lens. The single layer cells contacting the optic vesicle were used in counting the TUNEL-positive cells in the lens. The dashed outline indicates the retina cells. B, assessment of cell proliferation by the BrdU incorporation assay. Representative images are shown in the upper panel, and BrdU-positive cells are labeled in red. The quantitation of BrdU incorporation is shown in the lower panel as mean ± S.D. (n = 4 for control and 3 for Smad7 mutant embryos). The percentage was calculated as BrdU-positive cells/total cells in both retina and lens. The nuclei were stained with Hoechest 33342 (blue). * and ** represent p < 0.05 and p < 0.01, respectively, between the two groups of embryos. Scale bar: 50 μm.
Defect in Lens Differentiation in Smad7 Null Mutant Mice
BMP signals are important for the induction and subsequent differentiation of the lens (8–10). To test the potential role of Smad7 in the lens formation, we examined whether the loss of Smad7 would affect the induction and differentiation of lens. Pax6 and Sox2 function cooperatively as activation factors for the lens induction. It has been reported that BMP4 up-regulates Sox2 in the prospective lens ectoderm, whereas BMP7 is upstream of both genes during lens induction (8, 9). In Smad7 mutant mice, we did not observe any change in Pax6 and Sox2 (Fig. 3, A–D), indicating that lens induction was not affected by Smad7 loss. However, Prox1, a gene implicated in differentiation of the primary lens cells (36), was obviously down-regulated in Smad7 null mice (Fig. 3, E and F). Consistently, the lens differentiation markers, α-, β-, and γ-crystallin, were all dramatically reduced in Smad7 mutants at E12.5 (Fig. 3, G–L). Consistently, we also observed that the elongation of lens fiber cells was defective in the mutant mice (Fig. 3, G–L). All of these data suggest that Smad7 is not required for lens induction but is essential for lens differentiation.
FIGURE 3.

Loss of Smad7 causes the defect of lens differentiation. The lens sections of the control and mutant embryos are analyzed by in situ hybridization to examine the pattern of genes involved in lens induction and differentiation, including Pax6 (A and B), Sox2 (C and D), Prox1 (E and F), α-crystallin (G and H), β-crystallin (I and J), and γ-crystallin (K and L). Scale bar: 50 μm.
Altered Gene Expression in the Periocular Mesenchyme in Smad7 Mutant Embryos
It is known that POM affects differentiation of the optic neuroepithelium and anterior ocular tissue (19, 37). Noticing the unusually increased rate of cell death in POM in Smad7 mutant embryos (Fig. 2A), we investigated whether morphogenesis of the periocular mesenchyme would be affected by Smad7 deletion. We investigated the expression of Pitx2, as it is crucial for development of neural crest-derived periocular mesenchyme (38, 39). At E10.5, Pitx2 was noticeably reduced in the mutant embryos in comparison with the wild type controls. Fewer populations of cells surrounding the optic cup were labeled positive (Fig. 4, A and B). The decreased expression of Pitx2 was still observed at E11.5 (Fig. 4, C and D). Bmp7 is expressed in both the ocular and the periocular region and regulates the formation of the optic fissure (9, 40). We found that in the mutant mice, the Bmp7 transcript level was distinguishably lower in the surrounding periocular mesenchyme, indicating that the BMP signal was reduced in this region (Fig. 4, E and F). As periocular mesenchyme is necessary for the development of pericytes and smooth muscle in the eye vessel (41), we used the gap conjunction gene Cx40 to examine blood vessel formation. The blood vessel surrounding the eye was clearly reduced in Smad7 mutant embryos at E12.5, although the hyaloid blood vessels were comparable with those of the controls (Fig. 4, G and H). Furthermore, we also noticed some Mitf-positive cells dissociating from the pigment layer of the retina and migrating into the POM in Smad7 null mice, with some of them accumulating in the anterior of the eyes (Fig. 4, I and J), likely leading to invasion of pigment cells into the optic stalk and inside the hyaloid cavity (Fig. 1C and data not shown). Overall, the changes in Pitx2 and Bmp7 in Smad7 mutant mice indicate abnormal development of POM, an event that may lead to defects in the patterning of the optic neuroepithelium and the development of the POM-related ocular structure.
FIGURE 4.

Altered gene expressions in the periocular mesenchyme of Smad7 null mice. The eyes of the control and Smad7-deficient embryos were used in in situ hybridization to analyze the expression of genes in the periocular mesenchyme including Pitx2 (A–D), Bmp7 (E and F), Cx40 (G and H), and Mitf (I and J) at different stages of the embryo. Note that the eyes of Smad7 mutant embryos have reduced expression of Pitx2 in the optic cup (marked by an arrow), decreasing BMP7 level in the surrounding mesenchyme (indicated by an arrow), defective blood vessel (Cx40-positive region) surrounding the eye (marked by an arrow), and ectopic Mitf expression (marked by an arrow). Scale bars: 50 μm (A–H) and 100 μm (I and J).
Defects of the Optic Stalk Morphogenesis in Smad7 Mutant Embryos
The observed coloboma in Smad7 mutant mice suggests that the topographic map of the eye might be changed. We also noticed a distortion of the optic nerve (Fig. 1C). We therefore analyzed the key markers for the optic stalk morphogenesis. In the wild type embryos, Pax2 was restricted in the optic stalk at E12.5 (Fig. 5A), consistent with a previous report (42). However, Pax2 expression in Smad7 mutant embryos was broader than in the controls and extended to the temporal neural retina and the outer layer of the optic cup at E12.5 from the transverse view (Fig. 5B); but the alteration of Pax2 expression in the optic stalk in Smad7 mutant embryos was not very obvious at E14.5 (data not shown). However, we observed that some of the Pax2-positive signals were divergent from the trail of the optic stalk in Smad7 mutant embryos (Fig. 5, C and D, and data not shown), suggesting an abnormal trajectory of the optic nerve. Vax1 is another transcriptional factor that is required for the optic stalk formation (43). We found that Vax1 was expressed and distributed in a pattern similar to that of Pax2, with an expanded expression in Smad7 mutant embryos (Fig. 5, E and F). Interestingly, Vax2, a gene closely related to Vax1, was found to have a low and narrow expression in the mutant embryos along the nasal-temporal axis (Fig. 5, G and H). In addition, in contrast to the uniform expression in the retina in the control embryos (Fig. 5I), Rx, a retina marker, was repelled in a broad optic disc region in Smad7 mutant embryos (Fig. 5J). On the other hand, the expression level of Pax6, a master gene for eye development (44), was indistinguishable between the mutant and control embryos (Fig. 5, K and 5L). Although Pax2 was expanded to the presumptive RPE layer and the pigmentation was absent in the outer layer of the optic cup at E12.5 in Smad7 mutant embryos (Fig. 5B), Mitf was still expressed in the presumptive RPE layer in Smad7 mutant embryos with a pattern similar to the controls (Fig. 5, M and N), with the same expression pattern as Otx2, another RPE differentiation marker (data not shown). We also noticed that the presumptive RPE cells in Smad7 mutants stayed cubic at E12.5. In contrast, the RPE cells in the control embryos displayed a thinner and flattened epithelial morphology (Fig. 5, M and N). As Pax2 and Vax1 were both activated by the midline SHH signaling, we examined the expression level of shh and found that the pattern of shh in the ventral telencephalon was expanded in Smad7 mutant mice (Fig. 5, O and P), likely accounting for the broad expression pattern of Pax2 and Vax1. Taken together, all of these data suggest that the boundary of the optic cup and optic stalk is not correctly established in Smad7-deleted mice, thereby leading to alteration in the formation of the optic nerve.
FIGURE 5.
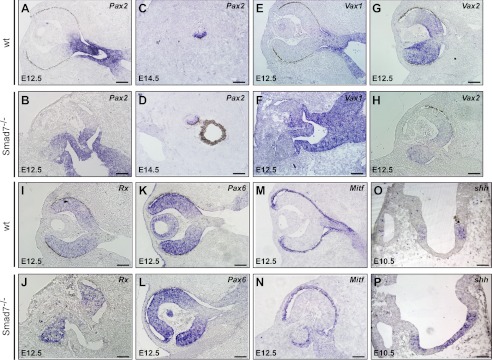
Defect of the optic stalk morphogenesis in Smad7 mutant mice. The eyes of the control and Smad7-deficient embryos were used in in situ hybridization to analyze expression of genes involved in optic stalk morphogenesis, including a transverse view of Pax2 (A and B), a sagittal view of Pax2 in the optic stalk (C and D), and a transverse view of Vax1 (E and F), Vax2 (G and H), Rx (I and J), Pax6 (K and L), Mitf (M and N), and shh (O and P) at different stages of embryonic development. Scale bars: 50 μm (A–N) and 100 μm (O and P).
Disrupted Spatial Retina Patterning in Smad7 Null Mice
A loss of pigmentation in the ventral retina in Smad7 mutant embryos was observed at E11.5, and the optic fissure was not closed normally in these embryos (Fig. 1B). Vax2 was thought to control both the nasal-temporal and the dorsal-ventral axes of the eye (45, 46), but it was reduced at the nasal-temporal gradient in the mutants (Fig. 5H). All of these results suggest that the ocular topography is likely changed by Smad7 deletion. Because BMP signaling is important in retinal patterning and retinotopic mapping, we examined whether Smad7 was able to regulate the spatial patterning of BMP signals at E10.5. In situ hybridization revealed that Bmp4 expression in the retina exhibited a generally high dorsal and low ventral gradient pattern in the control embryos (Fig. 6A). A similar Bmp4 staining pattern was also observed in the dorsal retina in Smad7 mutant embryos (Fig. 6B). However, phosphorylated Smad1/5/8 exhibited a general gradient from the dorsal to ventral axis in the wild type embryos (Fig. 6C) but with an enriched signal at the distal tip of optic cup in Smad7 mutant embryos (Fig. 6D). Meanwhile, the gradient pattern of phosphorylated Smad1/5/8 was absent along the dorsal to ventral axis in the mutant embryos (Fig. 6D). Nevertheless, phosphorylated Smad1/5/8 signal was not altered in the lens of Smad7 mutant compared with the wild type (Fig. 6, C and D). Consistently, a dorsal retina marker, Tbx5, a gene downstream of BMP signaling, was reduced in the dorsal portion but uniformly distributed through the retina at E10.5 in Smad7-deleted embryos (Fig. 6, E and F). In contrast, when further analyzing the molecules involved in spatial patterning along the nasal-temporal at E14.5 on the transverse section, we found that Tbx5 was confined in the differentiated RGCs in the wild type embryos (Fig. 6G). However, Tbx5 was more intense in the retina of Smad7 mutant mice than in the controls and was ectopically expressed at the distal tip of the retina (Fig. 6, H and I), suggesting that topographic defects occurred in Smad7 mutant embryos. Furthermore, the BMP downstream gene COUP-TF1 appeared to be increased in Smad7 mutants (data not shown), and the late patterning marker, EphB2, exhibited reduced expression (data not shown). These results suggest that BMP signaling was increased in the mutant mice at the late gestation stage.
FIGURE 6.

Altered spatial patterning in Smad7 mutant mice. In situ hybridization was used to analyze the expression of genes involved in spatial patterning in the eyes, including Bmp4 (A and B) and Tbx5 (E–I). Immunohistochemistry was used to analyze phosphorylated Smad1/5/8 (C and D) and phosphorylated Smad2 (J–L). The arrowheads (K and L) mark phospho-Smad2-positive cells in the neuroblast layer in Smad7 mutant embryos. Scale bars: 50 μm (A–H and J–L); 100 μm (G–I).
We also analyzed the phosphorylation status of Smad2, which is downstream of TGF-β/activins. We found that phosphorylated Smad2 was restrained in the inner half of the retina, the region containing the first postmitotic neurons (Fig. 6J). In the Smad7 null embryos, phosphorylated Smad2 was extensively up-regulated in comparison with the littermate controls (Fig. 6, K and L). More phosphorylated Smad2-positive cells were detected in the neuroblast layer in Smad7 mutant embryos (Fig. 6, K and L). We also noticed some ectopic phospho-Smad2 staining in the distal margin retina (Fig. 6L). Taken together, all of these data suggest that retina patterning was affected by Smad7 deletion. These data also indicate that the eye axis was likely rotated along both the dorsal-ventral and nasal-temporal axes upon Smad7 deletion.
Retarded Retinal Neurogenesis in Smad7 Mutant Mice
The BMP signal is essential for the retinal growth and neurogenesis. Deletion of Bmpr1a;Bmpr caused a reduction of the retinal predifferentiation markers, Chx10 and Fgf15, with an obvious neurogenesis defect (18). Math5 is an early transcriptional factor that determines neural retinal progenitor cell competence (47). A defect in Math5 affects RGC formation and differentiation. In wild type embryos, Math5 was initiated in the center of the retina (Fig. 7A) and then radiantly extended into the marginal region, reaching a strong expression at E14.5 (Fig. 7C) followed by the obviously down-regulated Math5 in the central neuroblast layer at E17.5 (Fig. 7E). In the retina of Smad7 mutant embryos, the expression of Math5 was obviously delayed at E12.5 (Fig. 7B). Only sparse Math5-positive cells were detected in the mutant mice at this stage (Fig. 7B). At E14.5, Math5 reached a comparable expression level and expanded to a region similar to that of the control embryos (Fig. 7, C and D). At E17.5, the expression of Math5 was still evident in the central neuroblast layer in Smad7 mutant mice (Fig. 7F), which was different from the obviously reduced expression in this area in the wild type embryos (Fig. 7E). However, we did not observe any change in the expression pattern of Chx10 and Fgf15 (Fig. 7, G and H, and data not shown). Consistent with the detention of Math5 expression, the downstream gene Brn3b was absent in Smad7 null mice at E12.5 (Fig. 7, I and J), whereas the neural cell marker Tuj1 was only detected in the retinal center in the wild type but not in Smad7-deficient embryos (Fig. 7, K and L). Collectively, these data indicate that neurogenesis was not executed properly in Smad7 mutant mice, an event likely leading to an alteration in retina differentiation at later stages.
FIGURE 7.

Retarded retinal neurogenesis in Smad7 null mice. In situ hybridization analysis of genes involved in retinal growth and differentiation, including Math5 (A–F), Fgf15 (G and H), and Brn3b (I and J). Neural marker Tuj1 was examined by immunohistochemistry (K and L). Note that no Tuj1 signal was detected in the central retina in the mutant mice (marked by arrow), where the neural cell was initiated in the wild type control (marked by arrowhead). Scale bar: 50 μm.
DISCUSSION
In the present study, we have demonstrated for the first time that Smad7, a key negative regulator of TGF-β family members, is essential for the development of the lens and retina, as it affects multiple signaling pathways including those of the BMPs and TGF-β/activins, as well as midline SHH signaling at distinctive developmental stages. Our results suggest that Smad7 is required for the development of periocular mesenchyme and patterning of the optic cups and optic stalk, as well as the timely initiation of neurogenesis.
Proper eye morphogenesis relies on orchestration among the neural ectoderm, surface ectoderm, and periocular mesenchyme. POM, which originates from the neural crest and mesoderm, gives rise to various highly specialized structures in the anterior segment of the eye (48). A recent study has revealed that POM serves as an organizer for the formation of the eye by activation of TGF-β and Wnt signaling (49). It also acts as a signaling source for the specification of RPE and the differentiation of the optic stalk (19). Pitx2 is considered a key regulator for patterning the anterior ocular segment. Neural crest expression of Pitx2 is required not only for the derivation of anterior ocular tissue but also for the partitioning of the neural retina, optic stalk, and RPE (38). Ap2α is expressed in POM and is crucial for correct positioning of the optic vesicle. The Ap2α mutant mouse has defective POM with a reduction of Pitx2 (50). The optic vesicle in the Ap2α mutant mouse cannot receive the correct combination of signals from POM, thus leading to improper optic vesicle/optic stalk regionalization (50). In the Smad7 null mice, Pitx2 is down-regulated in the surrounding ocular region, indicating defective patterning of POM. Consistently, we noticed that the optic disc was expanded, as marked by broad expression of the optic stalk markers Pax2 and Vax1, an event that may lead to repression of RPE specification. Furthermore, in Smad7 mutant embryos, the orientation of the eye is frequently rotated with the ventral and temporal side of the optic cup being partially retracted toward the midline and embedded in a mount of mesenchymal cells (Figs. 4H and 5L). All of those defects could be due in part to impropriate signaling induction in POM. Our data also suggest that BMP7 transcript is reduced in POM, indicating a reduction of BMP signaling in this region. It has been proposed that TGF-β signaling can regulate the specification of the cranial neural crest and the establishment of the normal ocular anterior segments. Reduction of TGF-β signaling is linked to the human disorder Axenfeld-Rieger anomaly by regulating Pitx2 at the late embryonic stage (20, 51–53). However, in Smad7 mutant embryos, the broadened optic disc appears only for a short time. At E14.5, the expression of Pitx2 speeds up and reaches the normal level in the Smad7 mutant embryos (data not shown), and Pax2 and Vax1 are as restricted as in the control embryos. But in some cases, we can still detect the optic stalk deviating away from the trajectory, as marked by Pax2 staining (Fig. 5D and data not shown).
Another potential pathway implicated in the formation of eye defects in Smad7 null mice is the midline SHH signaling, which regulates the optic neuroepithelium patterning along the proximal-distal axis (54). It works as ventral signaling to set up dorsal-ventral regionalization in the optic vesicle (55). SHH promotes the optic stalk specification and establishes the boundary of the optic stalk-optic cup by modulating the expression of Pax2 and Vax1 (55). In our study, we detected increased shh signal in Smad7-deleted embryos, an event that may result in expanded expression of Pax2 but inhibition of Rx in the optic disc. In sporadic severe cases, we observed complete loss of the eyes in Smad7 mutant embryos (data not shown). Therefore, the alteration of SHH signaling may also account for the defects in the optic stalk formation in Smad7 mutant embryos.
Establishment of the dorsal-ventral and nasal-temporal patterning is key to eye development and is orchestrated by multiple transcriptional factors expressed in the discrete compartment with distinct cell type distribution. Correct patterning along the two axes is the prerequisite for the appropriate outgrowth of RGC along the visual pathway and correct connections with their neuronal targets in the brain. BMP is a known dorsal molecule family that plays an essential role in patterning the dorsal ventral axis by countering the ventral SHH signaling. In addition, BMP also maintains the nasal-temporal gradient in the retina. Differences in the BMP signaling threshold are considered as specifying the spatial patterning (18). Such a process requires the appropriate input and maintenance of BMP signaling along the spatial axes. In our study, the gradient distribution pattern of BMP downstream markers, including phosphorylated Smad1/5/8 and Tbx5, was largely lost throughout the retina at E10.5 in Smad7 null mice (Fig. 6). Consistently, the ventral retina pigmentation in Smad7 null mice lagged in comparison with the controls, likely causing detainment of the partition of the optic vesicle and optic stalk. At E14.5, the aberrant BMP signaling in Smad7 mutant embryos may also affect the nasal-temporal patterning, as ectopic expression of Tbx5 is detected in the retina tip along the anterior-posterior axis (Fig. 6I), indicating an aberrantly up-regulated BMP signaling in this area during the late stage. Consistently, another BMP target gene, COUP-TF, was up-regulated at late stage (data not shown). Meanwhile, a duel regulator in the dorsal-ventral and nasal-temporal axes, Vax2, was down-regulated along the nasal-temporal axis at E12.5 in Smad7 mutant embryos (Fig. 5H). Thus, our data collectively indicate that Smad7 is required for the maintenance of BMP signaling along both the dorsal-ventral and nasal-temporal axes. Furthermore, our findings suggest that how BMP signaling is affected by Smad7 is dependent on the stage of the embryos.
Loss of Smad7 activates the TGF-β/activin signaling pathway in the retinal ganglion cells and neuroblast cells at late embryonic stage (Fig. 6, K and L). It is reported that a canonical TGF-β/activin ligand, GDF11, is expressed in RGC layers and controls the genesis and competence of retinal ganglion cell (16). The initiation of RGC starts in the center of the retina. Elusive cues from the neighboring structure, such as the optic stalk, may be required. We speculate that the delay in RGC generation in Smad7 mutant mice is due in part to the detainment in the morphogenesis of the optic stalk. However, whether Smad7 is also involved in retinal lamination in the later stages needs to be addressed in the future.
Numerous studies have identified the fact that dysregulation of transcriptional factors and extracellular signaling molecules may lead to ocular abnormalities, such as Pax2 (56), Vax (45), Pitx2 (38), AP2α (50), BMPs (57), and SHH (57). These factors are expressed in different ocular tissues and cooperate to regulate the formation of the eye. The ocular phenotype in the Smad7 mutant mice might be caused by dysregulation of some or all of those factors. Deletion of Smad7 alters multiple molecules required for eye patterning in different ocular regions. The eye defects in the Smad7 null mice could be caused by up-regulation of SHH signaling abnormality of POM, altered patterning of optic cup/optic stalk, and dysregulation in lens differentiation. By regulating distinct signal molecules in a short time window, Smad7 may harmonize different aspects of ocular development. Therefore, the Smad7 mutant mouse may stand as a useful model for dissecting the complex roles of TGF-β superfamily members in eye development and in their cross-talk with multiple signaling pathways.
This work was supported by Research Grants 2012CB524900 (to Y. C.) and 2010CB529506 (to Y. P. and Z. W.) from the Ministry of Science and Technology of China, 81021002 and 81130077 (to Y. C.) and 30971660 (to Y. P.) from the National Natural Science Foundation of China, and KSCX2-EW-R-08 (to Y. C.) from the Chinese Academy of Sciences and by a grant from the SA-SIBS Scholarship Program (to Y. P.).
- SHH
- sonic hedgehog
- BMP
- bone morphogenetic protein
- POM
- periocular mesenchyme
- RGC
- retinal ganglion cell(s)
- RPE
- retinal pigmented epithelium.
REFERENCES
- 1. Chow R. L., Lang R. A. (2001) Early eye development in vertebrates. Annu. Rev. Cell Dev. Biol. 17, 255–296 [DOI] [PubMed] [Google Scholar]
- 2. Streit A. (2004) Early development of the cranial sensory nervous system: from a common field to individual placodes. Dev. Biol. 276, 1–15 [DOI] [PubMed] [Google Scholar]
- 3. Adler R., Canto-Soler M. V. (2007) Molecular mechanisms of optic vesicle development: complexities, ambiguities and controversies. Dev. Biol. 305, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Van Raay T. J., Vetter M. L. (2004) Wnt/frizzled signaling during vertebrate retinal development. Dev. Neurosci. 26, 352–358 [DOI] [PubMed] [Google Scholar]
- 5. Sasagawa S., Takabatake T., Takabatake Y., Muramatsu T., Takeshima K. (2002) Axes establishment during eye morphogenesis in Xenopus by coordinate and antagonistic actions of BMP4, Shh, and RA. Genesis 33, 86–96 [DOI] [PubMed] [Google Scholar]
- 6. Lupo G., Liu Y., Qiu R., Chandraratna R. A., Barsacchi G., He R. Q., Harris W. A. (2005) Dorsoventral patterning of the Xenopus eye: a collaboration of retinoid, Hedgehog, and FGF receptor signaling. Development 132, 1737–1748 [DOI] [PubMed] [Google Scholar]
- 7. Martinez-Morales J. R., Del Bene F., Nica G., Hammerschmidt M., Bovolenta P., Wittbrodt J. (2005) Differentiation of the vertebrate retina is coordinated by an FGF signaling center. Dev. Cell 8, 565–574 [DOI] [PubMed] [Google Scholar]
- 8. Furuta Y., Hogan B. L. (1998) BMP4 is essential for lens induction in the mouse embryo. Genes Dev. 12, 3764–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wawersik S., Purcell P., Rauchman M., Dudley A. T., Robertson E. J., Maas R. (1999) BMP7 acts in murine lens placode development. Dev. Biol. 207, 176–188 [DOI] [PubMed] [Google Scholar]
- 10. Hammerschmidt M., Kramer C., Nowak M., Herzog W., Wittbrodt J. (2003) Loss of maternal Smad5 in zebrafish embryos affects patterning and morphogenesis of optic primordia. Dev. Dyn. 227, 128–133 [DOI] [PubMed] [Google Scholar]
- 11. Chapman D. L., Garvey N., Hancock S., Alexiou M., Agulnik S. I., Gibson-Brown J. J., Cebra-Thomas J., Bollag R. J., Silver L. M., Papaioannou V. E. (1996) Expression of the T-box family genes, Tbx1–Tbx5, during early mouse development. Dev. Dyn. 206, 379–390 [DOI] [PubMed] [Google Scholar]
- 12. McLaughlin T., Hindges R., O'Leary D. D. (2003) Regulation of axial patterning of the retina and its topographic mapping in the brain. Curr. Opin. Neurobiol. 13, 57–69 [DOI] [PubMed] [Google Scholar]
- 13. Sakuta H., Takahashi H., Shintani T., Etani K., Aoshima A., Noda M. (2006) Role of bone morphogenic protein 2 in retinal patterning and retinotectal projection. J. Neurosci. 26, 10868–10878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koshiba-Takeuchi K., Takeuchi J. K., Matsumoto K., Momose T., Uno K., Hoepker V., Ogura K., Takahashi N., Nakamura H., Yasuda K., Ogura T. (2000) Tbx5 and the retinotectum projection. Science 287, 134–137 [DOI] [PubMed] [Google Scholar]
- 15. Müller F., Rohrer H., Vogel-Höpker A. (2007) Bone morphogenetic proteins specify the retinal pigment epithelium in the chick embryo. Development 134, 3483–3493 [DOI] [PubMed] [Google Scholar]
- 16. Kim J., Wu H. H., Lander A. D., Lyons K. M., Matzuk M. M., Calof A. L. (2005) GDF11 controls the timing of progenitor cell competence in developing retina. Science 308, 1927–1930 [DOI] [PubMed] [Google Scholar]
- 17. Liu J., Wilson S., Reh T. (2003) BMP receptor 1b is required for axon guidance and cell survival in the developing retina. Dev. Biol. 256, 34–48 [DOI] [PubMed] [Google Scholar]
- 18. Murali D., Yoshikawa S., Corrigan R. R., Plas D. J., Crair M. C., Oliver G., Lyons K. M., Mishina Y., Furuta Y. (2005) Distinct developmental programs require different levels of Bmp signaling during mouse retinal development. Development 132, 913–923 [DOI] [PubMed] [Google Scholar]
- 19. Cvekl A., Tamm E. R. (2004) Anterior eye development and ocular mesenchyme: new insights from mouse models and human diseases. Bioessays 26, 374–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ittner L. M., Wurdak H., Schwerdtfeger K., Kunz T., Ille F., Leveen P., Hjalt T. A., Suter U., Karlsson S., Hafezi F., Born W., Sommer L. (2005) Compound developmental eye disorders following inactivation of TGFβ signaling in neural-crest stem cells. J. Biol. 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heldin C. H., Miyazono K., ten Dijke P. (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- 22. Dong C., Zhu S., Wang T., Yoon W., Li Z., Alvarez R. J., ten Dijke P., White B., Wigley F. M., Goldschmidt-Clermont P. J. (2002) Deficient Smad7 expression: a putative molecular defect in scleroderma. Proc. Natl. Acad. Sci. U.S.A. 99, 3908–3913 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23. Böttinger E. P., Bitzer M. (2002) TGF-β signaling in renal disease. J. Am. Soc. Nephrol. 13, 2600–2610 [DOI] [PubMed] [Google Scholar]
- 24. Monteleone G., Boirivant M., Pallone F., MacDonald T. T. (2008) TGF-β1 and Smad7 in the regulation of IBD. Mucosal Immunol. 1, Suppl. 1, S50–S53 [DOI] [PubMed] [Google Scholar]
- 25. Li R., Rosendahl A., Brodin G., Cheng A. M., Ahgren A., Sundquist C., Kulkarni S., Pawson T., Heldin C. H., Heuchel R. L. (2006) Deletion of exon I of SMAD7 in mice results in altered B cell responses. J. Immunol. 176, 6777–6784 [DOI] [PubMed] [Google Scholar]
- 26. Chen Q., Chen H., Zheng D., Kuang C., Fang H., Zou B., Zhu W., Bu G., Jin T., Wang Z., Zhang X., Chen J., Field L. J., Rubart M., Shou W., Chen Y. (2009) Smad7 is required for the development and function of the heart. J. Biol. Chem. 284, 292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Funaki T., Nakao A., Ebihara N., Setoguchi Y., Fukuchi Y., Okumura K., Ra C., Ogawa H., Kanai A. (2003) Smad7 suppresses the inhibitory effect of TGF-β2 on corneal endothelial cell proliferation and accelerates corneal endothelial wound closure in vitro. Cornea 22, 153–159 [DOI] [PubMed] [Google Scholar]
- 28. Saika S., Ikeda K., Yamanaka O., Sato M., Muragaki Y., Ohnishi Y., Ooshima A., Nakajima Y., Namikawa K., Kiyama H., Flanders K. C., Roberts A. B. (2004) Transient adenoviral gene transfer of Smad7 prevents injury-induced epithelial-mesenchymal transition of lens epithelium in mice. Lab. Invest. 84, 1259–1270 [DOI] [PubMed] [Google Scholar]
- 29. Saika S., Ikeda K., Yamanaka O., Miyamoto T., Ohnishi Y., Sato M., Muragaki Y., Ooshima A., Nakajima Y., Kao W. W., Flanders K. C., Roberts A. B. (2005) Expression of Smad7 in mouse eyes accelerates healing of corneal tissue after exposure to alkali. Am. J. Pathol. 166, 1405–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fuchshofer R., Stephan D. A., Russell P., Tamm E. R. (2009) Gene expression profiling of TGFβ2- and/or BMP7-treated trabecular meshwork cells: Identification of Smad7 as a critical inhibitor of TGF-β2 signaling. Exp. Eye Res. 88, 1020–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saika S., Yamanaka O., Nishikawa-Ishida I., Kitano A., Flanders K. C., Okada Y., Ohnishi Y., Nakajima Y., Ikeda K. (2007) Effect of Smad7 gene overexpression on transforming growth factor β-induced retinal pigment fibrosis in a proliferative vitreoretinopathy mouse model. Arch. Ophthalmol. 125, 647–654 [DOI] [PubMed] [Google Scholar]
- 32. He W., Li A. G., Wang D., Han S., Zheng B., Goumans M. J., Ten Dijke P., Wang X. J. (2002) Overexpression of Smad7 results in severe pathological alterations in multiple epithelial tissues. EMBO J. 21, 2580–2590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pan Y., Woodbury A., Esko J. D., Grobe K., Zhang X. (2006) Heparan sulfate biosynthetic gene Ndst1 is required for FGF signaling in early lens development. Development 133, 4933–4944 [DOI] [PubMed] [Google Scholar]
- 34. Zhang X., Friedman A., Heaney S., Purcell P., Maas R. L. (2002) Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev. 16, 2097–2107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pan Y., Carbe C., Powers A., Zhang E. E., Esko J. D., Grobe K., Feng G. S., Zhang X. (2008) Bud specific N-sulfation of heparan sulfate regulates Shp2-dependent FGF signaling during lacrimal gland induction. Development 135, 301–310 [DOI] [PubMed] [Google Scholar]
- 36. Wigle J. T., Chowdhury K., Gruss P., Oliver G. (1999) Prox1 function is crucial for mouse lens-fibre elongation. Nat. Genet. 21, 318–322 [DOI] [PubMed] [Google Scholar]
- 37. Fuhrmann S., Levine E. M., Reh T. A. (2000) Extraocular mesenchyme patterns the optic vesicle during early eye development in the embryonic chick. Development 127, 4599–4609 [DOI] [PubMed] [Google Scholar]
- 38. Evans A. L., Gage P. J. (2005) Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum. Mol. Genet. 14, 3347–3359 [DOI] [PubMed] [Google Scholar]
- 39. Gage P. J., Suh H., Camper S. A. (1999) Dosage requirement of Pitx2 for development of multiple organs. Development 126, 4643–4651 [DOI] [PubMed] [Google Scholar]
- 40. Morcillo J., Martínez-Morales J. R., Trousse F., Fermin Y., Sowden J. C., Bovolenta P. (2006) Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development 133, 3179–3190 [DOI] [PubMed] [Google Scholar]
- 41. Etchevers H. C., Vincent C., Le Douarin N. M., Couly G. F. (2001) The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development 128, 1059–1068 [DOI] [PubMed] [Google Scholar]
- 42. Adler R., Belecky-Adams T. L. (2002) The role of bone morphogenetic proteins in the differentiation of the ventral optic cup. Development 129, 3161–3171 [DOI] [PubMed] [Google Scholar]
- 43. Mui S. H., Kim J. W., Lemke G., Bertuzzi S. (2005) Vax genes ventralize the embryonic eye. Genes Dev. 19, 1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashery-Padan R., Gruss P. (2001) Pax6 lights up the way for eye development. Curr. Opin. Cell Biol. 13, 706–714 [DOI] [PubMed] [Google Scholar]
- 45. Barbieri A. M., Broccoli V., Bovolenta P., Alfano G., Marchitiello A., Mocchetti C., Crippa L., Bulfone A., Marigo V., Ballabio A., Banfi S. (2002) Vax2 inactivation in mouse determines alteration of the eye dorsal-ventral axis, misrouting of the optic fibres and eye coloboma. Development 129, 805–813 [DOI] [PubMed] [Google Scholar]
- 46. Mui S. H., Hindges R., O'Leary D. D., Lemke G., Bertuzzi S. (2002) The homeodomain protein Vax2 patterns the dorsoventral and nasotemporal axes of the eye. Development 129, 797–804 [DOI] [PubMed] [Google Scholar]
- 47. Yang Z., Ding K., Pan L., Deng M., Gan L. (2003) Math5 determines the competence state of retinal ganglion cell progenitors. Dev. Biol. 264, 240–254 [DOI] [PubMed] [Google Scholar]
- 48. Gage P. J., Rhoades W., Prucka S. K., Hjalt T. (2005) Fate maps of neural crest and mesoderm in the mammalian eye. Invest. Ophthalmol. Vis. Sci. 46, 4200–4208 [DOI] [PubMed] [Google Scholar]
- 49. Grocott T., Johnson S., Bailey A. P., Streit A. (2011) Neural crest cells organize the eye via TGF-β and canonical Wnt signalling. Nat. Commun. 2, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bassett E. A., Williams T., Zacharias A. L., Gage P. J., Fuhrmann S., West-Mays J. A. (2010) AP-2α knockout mice exhibit optic cup patterning defects and failure of optic stalk morphogenesis. Hum. Mol. Genet. 19, 1791–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., Doetschman T. (1997) TGFβ2 knock-out mice have multiple developmental defects that are non-overlapping with other TGFβ knock-out phenotypes. Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flügel-Koch C., Ohlmann A., Piatigorsky J., Tamm E. R. (2002) Disruption of anterior segment development by TGF-β1 overexpression in the eyes of transgenic mice. Dev. Dyn. 225, 111–125 [DOI] [PubMed] [Google Scholar]
- 53. Iwao K., Inatani M., Matsumoto Y., Ogata-Iwao M., Takihara Y., Irie F., Yamaguchi Y., Okinami S., Tanihara H. (2009) Heparan sulfate deficiency leads to Peters anomaly in mice by disturbing neural crest TGF-β2 signaling. J. Clin. Invest. 119, 1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang X. M., Yang X. J. (2001) Temporal and spatial effects of Sonic hedgehog signaling in chick eye morphogenesis. Dev. Biol. 233, 271–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yang X. J. (2004) Roles of cell-extrinsic growth factors in vertebrate eye pattern formation and retinogenesis. Semin. Cell Dev. Biol. 15, 91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sanyanusin P., Schimmenti L. A., McNoe L. A., Ward T. A., Pierpont M. E., Sullivan M. J., Dobyns W. B., Eccles M. R. (1995) Mutation of the PAX2 gene in a family with optic nerve colobomas, renal anomalies, and vesicoureteral reflux. Nat. Genet. 9, 358–364 [DOI] [PubMed] [Google Scholar]
- 57. Gregory-Evans C. Y., Williams M. J., Halford S., Gregory-Evans K. (2004) Ocular coloboma: a reassessment in the age of molecular neuroscience. J. Med. Genet. 41, 881–891 [DOI] [PMC free article] [PubMed] [Google Scholar]