

Background: Accumulating evidence indicates that β receptors (βAR) may be involved in Alzheimer disease (AD) pathology and that amyloid β peptide (Aβ) may interact with β2AR independently of presynaptic activities.
Results: β2AR, PKA, and JNK mediate Aβ-induced phosphorylation of tau in vivo and in vitro.
Conclusion: An Aβ-β2AR signaling is involved in tau pathology in AD.
Significance: This work indicates a potential mechanism for altering AD pathology by blocking β2ARs.
Keywords: Adrenergic Receptor, Alzheimer's disease, Amyloid, Jun N-terminal Kinase (JNK), Protein Kinase A (PKA), Tau
Abstract
Alzheimer disease (AD) is characterized by neurodegeneration marked by loss of synapses and spines associated with hyperphosphorylation of tau protein. Accumulating amyloid β peptide (Aβ) in brain is linked to neurofibrillary tangles composed of hyperphosphorylated tau in AD. Here, we identify β2-adrenergic receptor (β2AR) that mediates Aβ-induced tau pathology. In the prefrontal cortex (PFC) of 1-year-old transgenic mice with human familial mutant genes of presenilin 1 and amyloid precursor protein (PS1/APP), the phosphorylation of tau at Ser-214 Ser-262 and Thr-181, and the protein kinases including JNK, GSK3α/β, and Ca2+/calmodulin-dependent protein kinase II is increased significantly. Deletion of the β2AR gene in PS1/APP mice greatly decreases the phosphorylation of these proteins. Further analysis reveals that in primary PFC neurons, Aβ signals through a β2AR-PKA-JNK pathway, which is responsible for most of the phosphorylation of tau at Ser-214 and Ser-262 and a significant portion of phosphorylation at Thr-181. Aβ also induces a β2AR-dependent arrestin-ERK1/2 activity that does not participate in phosphorylation of tau. However, inhibition of the activity of MEK, an upstream enzyme of ERK1/2, partially blocks Aβ-induced tau phosphorylation at Thr-181. The density of dendritic spines and synapses is decreased in the deep layer of the PFC of 1-year-old PS1/APP mice, and the mice exhibit impairment of learning and memory in a novel object recognition paradigm. Deletion of the β2AR gene ameliorates pathological effects in these senile PS1/APP mice. The study indicates that β2AR may represent a potential therapeutic target for preventing the development of AD.
Introduction
Neurofibrillary tangles composed of hyperphosphorylated tau in the brain is a hallmark of Alzheimer disease (AD)2, and the phosphorylation of tau may be a major pathological cause of the disorder by inducing synapse loss (1–4). Increasing evidence suggests that soluble amyloid β peptide (Aβ) is linked to hyperphosphorylation of tau at serine and threonine residues (5, 6). A recent study has demonstrated that Aβ causes tau to wander into dendrites, leading to loss of synapses, spines, and microtubules (7–9). In 3xTg-AD mice harboring a knockin mutation for presenilin 1 (PS1, M146V) and transgenes for amyloid precursor protein (APPswe) and tau (tauP301L), spine loss occurs exclusively at dystrophic dendrites that accumulate both Aβ oligomers and hyperphosphorylated tau intracellularly (10), and it is the phosphorylation of tau that causes the protein to stray (11). Previous publications have shown that Aβ induces phosphorylation of tau at serine and threonine residues via a myriad of signaling cascades. However, little is known about how Aβ induces tau hyperphosphorylation and AD development.
In a recent epidemiological study, it was found that antihypertensive medication, including β blockers, may reduce the risk of AD (12). Another survey in AD patients indicates that β blockers may be associated with a delay of functional decline in the patients (13). There is also evidence that β2AR may be involved in AD pathogenesis through effects on Aβ production and inflammation (14, 15). Another study has shown recently that polymorphism in β2AR contributes to sporadic late-onset AD, which may be related to the availability and response of β2AR (13, 16). Meanwhile, β2AR also plays an important role in cognition and stress-related behaviors (17, 18).
Recent studies have characterized that Aβ induces activation of β2AR-mediated PKA- and G protein-coupled receptor kinase/arrestin-dependent signal transduction, which is presynaptic activity-independent and requires the N terminus of β2AR (19–22). Although a prolonged treatment with Aβ induces GRK/arrestin 3-dependent internalization and degradation of β2AR, which impairs presynaptic activity-dependent neurotransmission, the intracellular levels of cAMP and PKA activity are partially preserved, reaching a balance between receptor activation and degradation (23). Besides PKA, the internalization-associated arrestin signaling can trigger the phosphorylation of MAPK and JNK that may phosphorylate tau (24), and the activation of the exchange protein activated by cAMP (Epac) may also mediate JNK phosphorylation linked to tau (25). In this study, we aim to understand the significance of β2AR signaling cascades in tau pathology in AD.
EXPERIMENTAL PROCEDURES
Animals
Wild-type, β2AR knockout (β2AR-KO), arrestin-2 knockout (arrestin-2-KO), arrestin-3 knockout (arrestin-3-KO), and presenilin 1/amyloid precursor protein double-transgenic (PS1/APP) and β2-KO/PS1/APP mice in a B6 background were described previously (22, 23, 26). PS1/APP mice were purchased from The Jackson Laboratory (stock number 006554). They overexpress both amyloid precursor protein (695) with Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial AD mutations and human presenilin 1 gene harboring two-familial AD mutations, M146L and L286V (26). PS1/APP mice were cross-bred with β2-KO mice to produce β2-KO/PS1/APP mice. Wild-type and transgenic mice (6 months old and 1 year old) were used for tissue and behavioral studies. All animal experimental procedures were approved by the University of Illinois Animal Care and Use Committee.
Cell Culture and Aβ Treatment
Newborn wild-type and knockout mice were used to isolate prefrontal cortex (PFC) neurons under a stereomicroscope (22). Isolated neurons were plated on poly-d-lysine-coated dishes at a density of 1.0 × 105-106 cells/ml in DMEM/F12 medium (1:1) containing 10% FBS, 1% insulin-transferrin-selenium supplement, 25 ng/ml nerve growth factor, 1 mm glutamine, 20 nm water-soluble progesterone, and 100 nm putrescine. Three days later, cells were changed to serum-free neurobasal/B-27 medium containing cytosine β-d-arabinofuranoside (2.5 μm, Sigma). Neurons were cultured for 2–3 weeks before the experiment. Aβ1–42 (Biopeptide, CA) stock solutions were made by dissolving Aβ1–42 at 10−3 m in 5% ammonium hydroxide and freshly diluting in dimethyl sulfoxide just before use, which yields mostly monomers, dimers, and trimers with a small amount of higher-order oligomers (22). Cells were treated with soluble Aβ as indicated. In some experiments, inhibitors for kinases and receptors were added as indicated 10 min before administration of Aβ.
Golgi Staining
An FD Rapid GolgiStaintm kit (MTR Scientific, MD) was used to stain dendritic spines of neurons in the deep layer of the PFC of 1-year-old and 6-month-old wild-type, β2-KO, PS1/APP, and β2-KO/PS1/APP mice. Briefly, animals were perfused with heparinized PBS and 2% paraformaldehyde, followed by an additional perfusion with PBS to wash away excessive PFA in the body. Brains were dissected out and stained with Golgi-Cox impregnation solutions. After staining, the brains were sliced at a thickness of 240 μm on a LEICA Vibratome 1000. The slices were dehydrated and mounted on slides. Images were taken using a Carl Zeiss LSM-700 microscope equipped with DIC objective lenses. All spines observable along 100-μm dendritic segments at least 25 μm from the cell soma were counted.
Immunofluorescence Microscopy
Wild-type, β2-KO, PS1/APP, and β2-KO/PS1/APP were perfused consecutively in vivo with heparinized PBS and 2% PFA. The brains were dissected out and post-fixed with 2% PFA overnight. After serial dehydration in sucrose, the brains were frozen in Tissue-Tek O.C.T compound (VWR LabShop, IL), and slices were cut at a thickness of 40 μm on a CM3050 S cryostat (Leica Microsystems, Inc., Germany). Brain slices and fixed primary neurons were blocked and permeabilized with goat serum and Nonidet P-40 in PBS and then incubated with primary antibodies. Alexa Fluor 488- or Alexa Fluor 568-conjugated secondary antibodies (Invitrogen) were used to reveal the primary antibodies. Nuclei were counterstained with DAPI (Thermo Scientific, IL). Quantification of synapsin I positively stained synapses was performed with the Analyze Particles commands of the Fiji software.
Western Blotting
Proteins resolved by SDS-PAGE were transferred to nitrocellulose membranes (Millipore, MA) and blocked with 5% milk in buffer (10 mm Tris-HCl (pH 7.4), 100 mm NaCl, 25 mm NaF, 8 mm NaN3, and 0.1% Tween 20). Then the membranes were incubated with primary antibodies against phospho-tau (phospho-Ser-214 and phospho-Ser-262, Santa Cruz Biotechnology, Inc. and Invitrogen, respectively, and phospho-Thr-181, Abcam, MA) and tau (Sigma-Aldrich, MO); phospho- and total stress-activated protein kinase/JNK, GSK3α/β, Ca2+/calmodulin-dependent protein kinase II, and ERK1/2 (Cell Signaling Technology, Inc.); γ-tubulin (Sigma-Aldrich); or synapsin I (Cell Signaling Technology, Inc.) at 4 °C overnight. Phospho-tau antibodies recognize epitopes of phosphorylated tau of both human and mouse. After washing, membranes were incubated with secondary antibodies for detection with the Li-Cor system (Li-Cor, NE). The optical density of the bands was analyzed with the gel analyzer of the Fiji software.
Novel Object Recognition Test
The task was carried out according to previous publications (27, 28). The experimental apparatus consisted of a Plexiglas open-field box (40 × 40 × 29 cm). The apparatus was placed in a sound-isolated room. The novel object recognition task procedure consisted of three sessions: habituation, training, and retention sessions. Each mouse was habituated individually to the box with 10 min of exploration in the absence of objects. During the training session in the next day, two objects (A and B) were placed in the back corner of the box, 10 cm from the side wall. A mouse was then placed in the middle front of the box, and the total time spent in exploring the two objects was recorded for 10 min by the experimenter with two stopwatches. Exploration of an object was defined as directing the nose to an object at a distance of less than 2 cm and/or touching it with the nose. During the retention session on the third day (24 h after the training session), the animals were placed back into the same box, in which one of the familiar objects was replaced by a novel object, C. The animals were then allowed to explore freely for 10 min, and the time spent exploring each object was recorded. Throughout the experiments, the objects were used in a counterbalanced manner in terms of their physical complexity and emotional neutrality. A preference index, which is the ratio of the amount of time spent in exploration of any one of the two objects (training session) or the novel object (retention session) over the total time spent exploring both objects, was used to measure cognition.
Statistical Analyses
Unpaired Student's t test and one- or two-way analysis of variance was used to compare different groups with Prism software as indicated (GraphPad, CA). p < 0.05 was considered significant.
RESULTS
To explore the role of β2AR signaling in tau pathology in relationship with Aβ in AD, we cross-bred the AD animal model overexpressing the human familial APPswe and PS1 mutants (PS1/APP) with mice lacking the β2AR gene (β2-KO). We found that the phosphorylation of tau at Ser-214, Ser-262, and Thr-181 was increased in the PFC of 6-month-old and 1-year-old PS1/APP mice compared with wild-type mice (Fig. 1, A–C, and data not shown). However, deletion of the β2AR gene abolished the increases in phosphorylation of tau at Ser-214 and Ser-262 and significantly reduced the increase in phosphorylation of tau at Thr-181 in the PFC of PS1/APP mice (Fig. 1, A–C, and data not shown). The phosphorylation of JNK1, GSK3α/β, and CaMK II was also increased in the PFC of 1-year-old PS1/APP animals, but the increases in phosphorylation of these proteins were greatly blunted in β2-KO/PS1/APP mice (Fig. 1, D–F).
FIGURE 1.

Deletion of the β2AR gene ameliorates hyperphosphorylation of tau in AD animal brain. Phosphorylation of tau at Ser-214 (A), Ser-262 (B), Thr-181(C), JNK (D), GSK3α/β (E), and CaMK II (F) in the PFC tissues of 1-year-old WT, β2-KO, PS1/APP, and β2-KO/PS1/APP mice. **, p < 0.01 versus WT mice; #, p < 0.05 versus PS1/APP mice; ##, p < 0.01 versus PS1/APP mice. n = 6.
We then applied primary PFC neurons isolated from wild-type and β2-KO animals to further dissect Aβ-induced β2AR signaling cascades in tau phosphorylation. Aβ (10−6 m) induced tau phosphorylation at Ser-214, Ser-262, and Thr-181 in wild-type PFC neurons, but the increases in tau phosphorylation were almost abolished at Ser-214 and Ser-262 and blunted significantly at Thr-181 in β2-KO neurons (Fig. 2, A–C). A minimal dose of 10−8 m of Aβ was effective to promote tau phosphorylation (data not shown). Meanwhile, a β2AR-selective antagonist, ICI118551, blocked Aβ-induced tau phosphorylation (Fig. 3, A–C). As a control, a general βAR agonist, isoproterenol, also induced robust increases in tau phosphorylation, which was inhibited by a βAR antagonist, alprenolol (Fig. 3, A–C). Moreover, the effects of Aβ on tau phosphorylation at Ser-214 and Ser-262 were blocked by inhibition of adenylyl cyclase with 2,5-dideoxyadenosine-3-triphosphate tetrasodium (10−4 m, Fig. 3G) or inhibition of PKA inhibitor with membrane-permeable myristoylated PKI (10−5 m, D and E). The Aβ-induced tau phosphorylation was not affected by the Epac inhibitor brefeldin A (10−7 m), and treating the cells with the Epac-selective activator 8-CPT-2Me-cAMP (10−7 m) for 5 min did not induce tau phosphorylation (Fig. 3, H and I). These results indicate that tau phosphorylation at Ser-214 and Ser-262 is primarily dependent on β2AR-adenylyl cyclase-PKA signaling. In comparison, Aβ-induced tau phosphorylation at Thr-181 was only blocked partially by inhibition of PKA with PKI (10−5 m, Fig. 3F).
FIGURE 2.

Time course of Aβ-induced phosphorylation of tau in WT and β2-KO PFC neurons. Aβ-induced phosphorylation of tau at Ser-214 (A), Ser-262 (B), and Thr-181 (C) in primary PFC neurons of WT and β2-KO mice was examined. **, p < 0.01 between WT and β2-KO by two-way analysis of variance. n = 6.
FIGURE 3.

Role of β2AR signaling in Aβ-induced phosphorylation of tau. The effects of a general βAR antagonist alprenolol (10−6 m) and a selective β2AR antagonist, ICI118551 (10−6 m) on Aβ-induced (10−6 m) or a general βAR agonist-induced (isoproterenol, 10−7 m) phosphorylation of tau at Ser-214 (A), Ser-262 (B), and Thr-181 (C) in primary WT PFC neurons were examined. The role of PKA in Aβ-induced phosphorylation of tau at Ser-214 (D), Ser-262 (E), and Thr-181 (F) in primary PFC neurons was examined. The roles of the adenylyl cyclase (AC) inhibitor (G), Epac inhibitor (H), and Epac activator (I) in Aβ-induced tau phosphorylation in primary PFC neurons were examined. Con, control group; Iso, isoproterenol; Alp, alprenolol; ICI, ICI118551; PKI, myristoylated PKA inhibitor. **, p < 0.01, versus control; &&, p < 0.01 versus isoproterenol-treated neurons; ##, p < 0.01 versus Aβ-treated neurons. n = 6.
We then attempted to define intracellular signaling cascades involved in Aβ-induced phosphorylation of tau via β2AR activation. A JNK inhibitor, SP600125 (2 × 10−7 m, IC50 = 4∼9 × 10−8 m), blocked the phosphorylation at Thr-181 and significantly blunted the phosphorylation of tau at Ser-214 and Ser-262 (Fig. 4, A–C). In comparison, the CaMK II inhibitor KN-93 (10−6 m, IC50 = 3 × 10−7 m), the PKC inhibitor calphostin (10−7 m, IC50 = 5 × 10−8 m), and the GSK inhibitor (2′Z,3′E)-6-bromoindirubin-3′-oxime (BIO) (2 × 10−8 m, IC50 = 7 × 10−9 m) minimally affected these phosphorylation events (Fig. 4, A–C). These data suggest a major role of JNK in the Aβ-induced β2AR signaling for tau phosphorylation in PFC neurons. Accordingly, Aβ (10−6 m) induced phosphorylation of JNK in wild-type PFC neurons, and the Aβ-induced JNK phosphorylation was blunted in β2-KO PFC neurons (Fig. 4D). Moreover, Aβ-induced (10−6 m) phosphorylation of JNK was partially inhibited by PKI (10−5 m) but not by the Epac inhibitor brefeldin A (10−7 m, Fig. 4E). These data suggest that a β2AR-PKA-JNK pathway contributes to the Aβ-induced tau phosphorylation. In comparison, acute Aβ treatment for 5 min induced minimal change in the phosphorylation of CaMK II in either wild-type or β2-KO neurons (Fig. 4, F and G), indicating that the increase in the phosphorylation of CaMK II observed in vivo, shown in Fig. 1, likely requires additional activation of other receptors and ion channels under chronic conditions.
FIGURE 4.

Roles of JNK, GSK3β, CaMK II, and PKC in Aβ-induced phosphorylation of tau in primary PFC neurons. The effects of protein kinase inhibitors on Aβ-induced phosphorylation of tau at Ser-214 (A), Ser-262 (B), and Thr-181 (C) were investigated. The Aβ-induced phosphorylation of JNK at Thr-183/185 (D) and CaMK II at Thr-286 (F) in primary PFC neurons of WT and β2-KO was examined. The effects of a PKA inhibitor, myristoylated PKI, and an Epac inhibitor, brefeldin A (BFA), on Aβ-induced phosphorylation of JNK at Thr-183/185 (E) and CaMK II at Thr-286 (G) were investigated. Con, control group; SP600125, JNK inhibitor; BIO, GSK3β inhibitor; KN-93, CaMK II inhibitor; Calphostin C, PKC inhibitor. *, p < 0.05; **, p < 0.01 versus control; #, p < 0.05 versus Aβ-treated neurons. n = 6.
Activation of β2AR also induces MAPK signaling via arrestins. Here, Aβ (10−6 m) induced a β2AR-dependent activation of ERK1/2 (Fig. 5A). However, Aβ failed to increase phosphorylation of ERK1/2 in arrestin-3-KO neurons, indicating that arrestin-3 is required for Aβ-induced activation of ERK1/2 (Fig. 5, B and C). As a control, the βAR agonist isoproterenol (10−7 m) increased the phosphorylation of ERK1/2 in both arrestin-2- and arrestin-3-KO neurons (Fig. 5, B and C). Because MAP kinases can also promote phosphorylation of tau, we tested the role of the arrestin-ERK1/2 pathway in Aβ-induced phosphorylation of tau in PFC neurons. In PFC neurons lacking arrestin-2 and/or arrestin-3, Aβ (10−6 m) increased the phosphorylation of tau at Ser-214, Ser-262, and Thr-181 (Fig. 6, A–C and E). These data essentially rule out a role of arrestins in Aβ-induced phosphorylation of tau. However, inhibition of MEK, an upstream enzyme of ERK1/2, with U0126 (2 × 10−7 m, IC50 = 6∼7 × 10−8 m) partially blocked the phosphorylation of tau at Thr-181, but not at Ser-214 and Ser-262 (Fig. 6D). Together, these data indicate that Aβ induces activation of MEK, which phosphorylates tau at Thr-181 and ERK1/2 through different signaling machineries, and only the ERK1/2 phosphorylation is dependent on arrestin-3 (Fig. 7).
FIGURE 5.
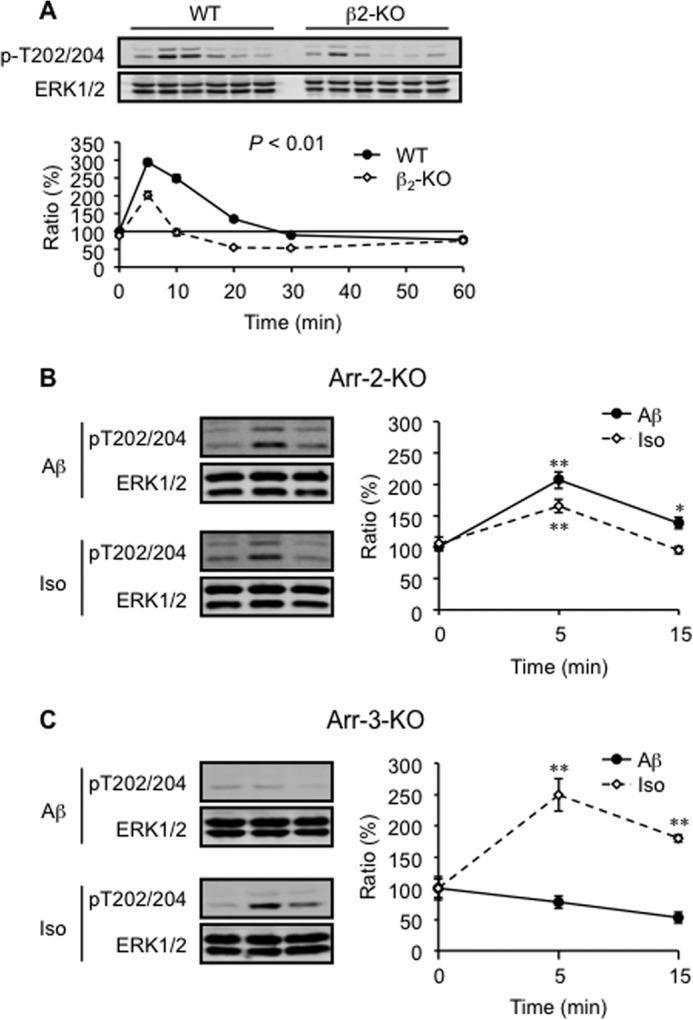
Aβ induces arrestin-3-dependent activation of the ERK1/2 pathway. A, Aβ-induced (10−6 m) phosphorylation of ERK1/2 in primary PFC neurons isolated from WT and β2-KO mice was investigated. Aβ-induced ERK1/2 phosphorylation in primary Arr-2-KO (B) and Arr-3-KO (C) PFC neurons was examined. Iso, isoproterenol; Arr, arrestin. *, p < 0.05; **, p < 0.01 versus basal level. n = 6.
FIGURE 6.
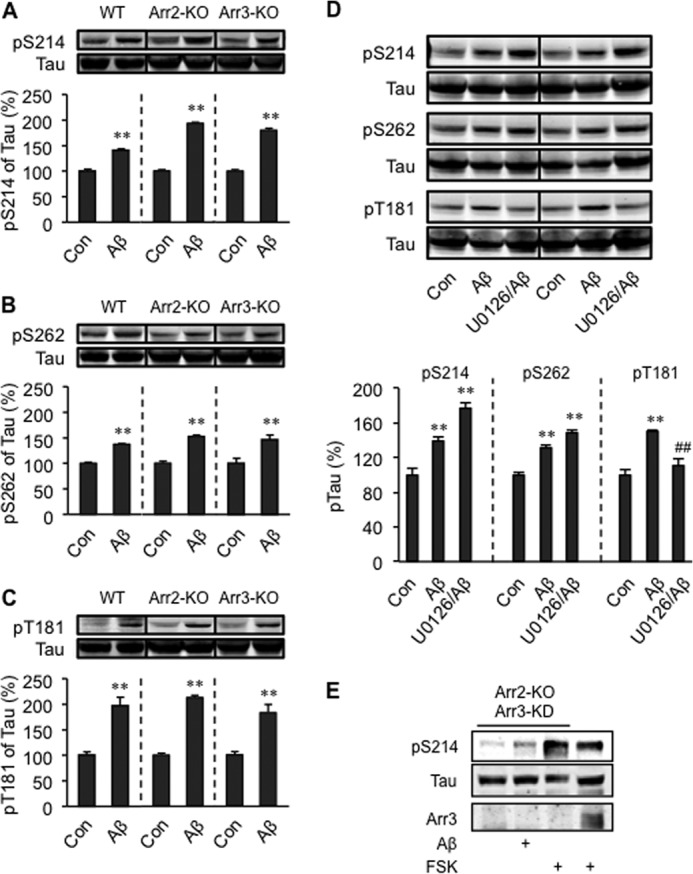
Roles of MAPK and arrestins in tau phosphorylation induced by Aβ. Phosphorylation of tau at Ser-214 (A), Ser-262 (B), and Thr-181 (C) induced by Aβ in primary PFC neurons isolated from WT, arrestin-2 knockout (Arr2-KO), and arrestin-3 knockout (Arr3-KO) mice was examined. D, the effects of MEK inhibitor on tau phosphorylation. E, Aβ- and forskolin-induced phosphorylation of tau at Ser-214 in arrestin-2 knockout PFC neurons that were infected with lentivirus-encoding shRNA for the knockdown of arrestin-3 (Arr3-KD). Con, control group; U0126, MEK inhibitor; FSK, forskolin. **, p < 0.01 versus Con; ##, p < 0.01 versus Aβ-treated neurons. n = 6.
FIGURE 7.

The β2AR-signaling machinery regulates Aβ-induced tau phosphorylation. Aβ, amyloid β peptide; β2, β2 receptor; Arr3, arrestin 3.
In agreement with published literature, we found that the density of dendritic spines in the deep layer of the PFC in 1-year-old and 6-month-old PS1/APP mice was decreased. However, deletion of the β2AR gene reversed the decrease (Fig. 8, A–C). Unlike the relatively even distribution of synapses in the deep layer of the PFC in WT mice, PS1/APP mice displayed regions with a dramatically decreased number of synapses, as indicated by synapsin I staining (Fig. 8, D and E), and surrounding synapses remained in clusters (arrows). Deletion of the β2AR gene in PS1/APP mice yielded a distribution of synapsin I positively stained synapses similar to those in WT or β2-KO mice (Fig. 8, D and E). To assess the cognitive role of β2AR in PS1/APP transgenic AD animals, we tested learning and memory in 1-year-old mice in a novel object recognition paradigm. We found that PS1/APP mice showed impaired learning and memory, whereas β2-KO/PS1/APP mice performed significantly better than PS1/APP mice (Fig. 9A). Knockout of the β2AR gene itself tended to improve learning and memory in 1-year-old mice (Fig. 9A). However, it tended to impair learning and memory in 6-month-old mice (Fig. 9D). In the training session, one-year-old animals in each group showed a similar preference for the reference object (Fig. 9B). The total exploration time in PS1/APP and β2-KO/PS1/APP animals in the training session was similar, indicating similar locomotor activity in these mice (Fig. 9C).
FIGURE 8.

Deletion of the β2AR gene reduces loss of dendritic spines and synapses in the brain of senile mice. A, representative figures show the density of dendritic spines in the deep layer of the PFC of 1-year-old wild-type, β2-KO, PS1/APP, and β2-KO/PS1/APP mice. B, semiquantitative analysis of the density of dendritic spines in A (n = 80). C, representative figures show the density of dendritic spines in deep layer of PFC of 6-month-old WT, β2-KO, PS1/APP, and β2-KO/PS1/APP mice. D, synapsin I staining for synapses in the deep layer of the PFC of 1-year-old WT, β2-KO, PS1/APP, and β2-KO/PS1/APP mice. Synapsin-I positively stained clusters of synapses surrounded by blank areas are indicated with arrows. E, semiquantification of the density of synapses in D. *, p < 0.05; **, p < 0.01 versus WT; ##, p < 0.01 versus PS1/APP mice. n = 8∼15.
FIGURE 9.

Deletion of the β2AR gene rescues learning and memory in senile mice. Learning and memory in 1-year-old and 6-month-old WT, β2-KO, PS1/APP, and β2-KO/PS1/APP mice were examined with the novel object recognition test. A, preference for the novel object in 1-year-old mice in the testing sessions of the novel object recognition test. B, preference for reference object in 1-year-old WT, β2-KO, PS1/APP, and β2-KO/PS1/APP mice in the training sessions of the novel objective recognition test. C, total exploration time for objects in the training sessions of the novel object recognition test in 1-year-old animals. D, preference for the novel object in 6-month-old WT, β2-KO, PS1/APP, and β2-KO/PS1/APP mice in the testing sessions of the novel object recognition test. **, p < 0.01 versus WT mice; ##, p < 0.01 versus PS1/APP mice. n = 12∼27.
DISCUSSION
Recent epidemiological studies suggest that β blockers may reduce the incidence of AD in patients suffering from hypertension and are associated with delay of functional decline in sporadic AD patients (13). Among three subtypes in the βAR family, both β1AR and β2AR play important roles in cognition and stress-dependent behaviors (17, 18). Accumulating evidence suggests that β1AR and β2AR, especially β2AR, may be involved in AD pathogenesis through effects on Aβ production or inflammation (14, 19, 29) and that polymorphisms of β2AR contribute to sporadic late-onset AD, which may be related to the availability and response of β2AR (13, 16). Our previous studies have shown that Aβ can bind to β2AR and induce allosteric activation of the receptor that leads to cAMP/PKA- and GRK/arrestin-mediated cell signaling (19, 22, 23). In this study, we find that β2AR plays a necessary role in Aβ-induced tau phosphorylation at Ser-214, Ser-262, and Thr-181 in vitro and in vivo. Deletion of the β2AR gene prevents tau hyperphosphorylation, loss of dendritic spines and synapses, and impairment of learning and memory in a transgenic AD animal model. This study places β2AR as an essential link between increasing Aβ and tau phosphorylation levels in the brain, which are both hallmarks of AD pathogenesis.
In tauopathies such as AD, frontotemporal dementia, and Parkinson disease, tau is hyperphosphorylated abnormally at multiple serine/threonine sites. In this study, one-year-old PS1/APP transgenic AD animals show hyperphosphorylation of tau at Ser-214, Ser-262, and Thr-181. Deletion of the β2AR gene significantly attenuates the phosphorylation of tau at Thr-181 and completely blocks the phosphorylation of tau at Ser-214 and Ser-262 in vivo and in vitro, suggesting that β2AR is a primary receptor for Aβ-induced phosphorylation of tau at these sites. PKA and CaMK II are downstream from β2AR. Previous studies have shown that both PKA and CaMK II readily phosphorylate tau. However, PKA phosphorylates tau to a significantly greater extent with a broader range of the sites than CaMK II (30, 31). Phosphorylation of tau by PKA also significantly decreases tubulin binding (30). In a tandem mass spectrometry study, CaMK II phosphorylated recombinant human tau at the sites, including Ser-214 and Ser-262, that may produce paired helical filament tau (32). Here, we find that Aβ-induced phosphorylation of tau at Ser-214 and Ser-262 is primarily dependent on PKA, whereas the phosphorylation at Thr-181 is partially inhibited by PKA inhibitor PKI. These data support that β2AR signals through PKA in Aβ-induced tau phosphorylation. In comparison, inhibition of CaMK II does not block the Aβ-induced phosphorylation at these sites in PFC neurons. It has been shown that Aβ may induce hyperactivities in AMPA receptors under electric stimulation in PFC slices (22). Here, acute treatment with Aβ alone for 5 min without electric stimulation does not induce significant phosphorylation of CaMK II in both wild-type and β2KO PFC neurons, probably because of lack of glutamate released from presynapses for activation of AMPA receptors. Nevertheless, one-year-old PS1/APP transgenic animals show an increased phosphorylation of CaMK II that is dependent on expression of β2AR. Thus, a possible role of CaMK II for Aβ-induced and β2AR-mediated tau phosphorylation in vivo remains to be addressed.
In addition, the JNK pathway amplifies and drives subcellular changes in tau phosphorylation (1) and plays key role in tau phosphorylation in AD models (33). GSK3β is a major physiological tau kinase that requires priming phosphorylation at Ser-404 to further phosphorylate tau at paired helical filament 1 (34). In isolated PFC neurons, a JNK inhibitor totally blocks the phosphorylation of tau at Thr-181 and significantly attenuates the phosphorylation of Tau at Ser-214 and Ser-262 induced by Aβ. Although Aβ induces phosphorylation of JNK in isolated PFC neurons via a β2AR-PKA pathway, the phosphorylation of JNK is only partially blunted in the PFC of 1-year-old β2AR-KO/PS1/APP animals. These data indicate that β2AR is a major receptor associated with JNK phosphorylation and that other Aβ-induced receptor pathways or Aβ deposition-induced inflammation can also promote JNK phosphorylation in vivo. Together, Aβ induces JNK phosphorylation through activating β2AR-PKA signaling and other signaling mechanisms in which PKA and JNK independently contribute to tau phosphorylation at Ser-214, Ser-262, and Thr-181.
We also find that Aβ can activate a β2AR-arrestin-MAPK pathway in PFC neurons. Surprisingly, we find that MEK, but not downstream ERK1/2 in the MAPK pathway, contributes to phosphorylation of tau at Thr-181. MEK-mediated tau phosphorylation does not require expression of arrestin-2 or arrestin-3, the non-visual arrestins function downstream of β2AR in the brain. In comparison, arrestin-3 is required for the Aβ-induced ERK1/2 phosphorylation. These data indicate that Aβ-induced MEK phosphorylation leads to two divergent pathways: an arrestin-3-dependent ERK1/2 activation and an arrestin-independent tau phosphorylation.
Genetic data have implied that deranged tau-microtubule interactions induced either by phosphorylation or increased levels of tau, contribute to or even are sufficient to cause synaptic and dendritic degeneration in primary tauopathies (35–37). It has been reported that transfection of tau in mature neurons leads to an improper distribution of tau into the somatodendritic compartment with concomitant degeneration of synapses, as seen by the disappearance of spines and presynaptic and postsynaptic markers (3, 7). In this study, there is a degeneration of synapses shown by synapsin I staining and dendritic spines in the deep layer of the PFC of PS1/APP double-transgenic animals. Deletion of the β2AR gene in PS1/APP animals reverses the degenerative effects. These findings again argue for a potential beneficial role of inhibition of β2AR in altering the pathological course of AD. Conclusive evidence comes from the behavioral experiments. In the novel object recognition test, learning and memory deficits are present in 1-year-old PS1/APP animals. Deletion of the β2AR gene rescues the outcome resulting from overexpressing mutant PS1 and APP genes from human familial AD. It is worth noting that 1-year-old β2-KO animals show a tendency to perform better than wild-type animals in the behavioral test. However, 6-month-old β2-KO animals show a tendency to have slightly decreased learning and memory. In either case, deletion of the β2AR gene in PS1/APP animals has beneficial effects in the test. Taken together, the cellular and behavioral experiments in this study provide evidence that β2AR may represent a potential therapeutic target for preventing the development of AD.
This work was supported by a new investigator research grant from the Alzheimer's Association (to Y. K. X.) and by The National Alliance for Research on Schizophrenia and Depression (NARSAD) Young Investigator Award and Alzheimer's Disease Research Fund from the Illinois Department of Public Health (to D. W.).
- AD
- Alzheimer disease
- Aβ
- amyloid β peptide
- β2AR
- β2-adrenergic receptor
- PKA
- protein kinase A
- Epac
- exchange protein activated by cAMP
- APP
- amyloid precursor protein
- PFC
- prefrontal cortex.
REFERENCES
- 1. Vogel J., Anand V. S., Ludwig B., Nawoschik S., Dunlop J., Braithwaite S. P. (2009) The JNK pathway amplifies and drives subcellular changes in tau phosphorylation. Neuropharmacology 57, 539–550 [DOI] [PubMed] [Google Scholar]
- 2. Hall G. F., Chu B., Lee G., Yao J. (2000) Human tau filaments induce microtubule and synapse loss in an in vivo model of neurofibrillary degenerative disease. J. Cell Sci. 113, 1373–1387 [DOI] [PubMed] [Google Scholar]
- 3. Thies E., Mandelkow E. M. (2007) Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J. Neurosci. 27, 2896–2907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mattson M. P. (2004) Pathways towards and away from Alzheimer's disease. Nature 430, 631–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Giuffrida M. L., Caraci F., Pignataro B., Cataldo S., De Bona P., Bruno V., Molinaro G., Pappalardo G., Messina A., Palmigiano A., Garozzo D., Nicoletti F., Rizzarelli E., Copani A. (2009) β-Amyloid monomers are neuroprotective. J. Neurosci. 29, 10582–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma Q. L., Yang F., Rosario E. R., Ubeda O. J., Beech W., Gant D. J., Chen P. P., Hudspeth B., Chen C., Zhao Y., Vinters H. V., Frautschy S. A., Cole G. M. (2009) β-Amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling. Suppression by omega-3 fatty acids and curcumin. J. Neurosci. 29, 9078–9089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zempel H., Thies E., Mandelkow E., Mandelkow E. M. (2010) Aβ oligomers cause localized Ca(2+) elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 30, 11938–11950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mitsuyama F., Futatsugi Y., Okuya M., Karagiozov K., Peev N., Kato Y., Kanno T., Sano H., Koide T. (2009) Amyloid β. A putative intra-spinal microtubule-depolymerizer to induce synapse-loss or dendritic spine shortening in Alzheimer's disease. Ital. J. Anat. Embryol. 114, 109–120 [PubMed] [Google Scholar]
- 9. Mitsuyama F., Futatsugi Y., Okuya M., Kawase T., Karagiozov K., Kato Y., Kanno T., Sano H., Nagao S., Koide T. (2012) Stimulation-dependent intraspinal microtubules and synaptic failure in Alzheimer's disease. A review. Int. J. Alzheimers Dis. 2012, 519682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bittner T., Fuhrmann M., Burgold S., Ochs S. M., Hoffmann N., Mitteregger G., Kretzschmar H., LaFerla F. M., Herms J. (2010) Multiple events lead to dendritic spine loss in triple transgenic Alzheimer's disease mice. PLoS ONE 5, e15477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoover B. R., Reed M. N., Su J., Penrod R. D., Kotilinek L. A., Grant M. K., Pitstick R., Carlson G. A., Lanier L. M., Yuan L. L., Ashe K. H., Liao D. (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68, 1067–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Violin J. D., DiPilato L. M., Yildirim N., Elston T. C., Zhang J., Lefkowitz R. J. (2008) β2-adrenergic receptor signaling and desensitization elucidated by quantitative modeling of real time cAMP dynamics. J. Biol. Chem. 283, 2949–2961 [DOI] [PubMed] [Google Scholar]
- 13. Rosenberg P. B., Mielke M. M., Tschanz J., Cook L., Corcoran C., Hayden K. M., Norton M., Rabins P. V., Green R. C., Welsh-Bohmer K. A., Breitner J. C., Munger R., Lyketsos C. G. (2008) Effects of cardiovascular medications on rate of functional decline in Alzheimer disease. Am. J. Geriatr. Psychiatry 16, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ni Y., Zhao X., Bao G., Zou L., Teng L., Wang Z., Song M., Xiong J., Bai Y., Pei G. (2006) Activation of β2-adrenergic receptor stimulates γ-secretase activity and accelerates amyloid plaque formation. Nat. Med. 12, 1390–1396 [DOI] [PubMed] [Google Scholar]
- 15. Yu J. T., Wang N. D., Ma T., Jiang H., Guan J., Tan L. (2011) Roles of β-adrenergic receptors in Alzheimer's disease. Implications for novel therapeutics. Brain Res. Bull. 84, 111–117 [DOI] [PubMed] [Google Scholar]
- 16. Yu J. T., Tan L., Ou J. R., Zhu J. X., Liu K., Song J. H., Sun Y. P. (2008) Polymorphisms at the β2-adrenergic receptor gene influence Alzheimer's disease susceptibility. Brain Res. 1210, 216–222 [DOI] [PubMed] [Google Scholar]
- 17. Morilak D. A., Barrera G., Echevarria D. J., Garcia A. S., Hernandez A., Ma S., Petre C. O. (2005) Role of brain norepinephrine in the behavioral response to stress. Prog. Neuropsychopharmacol. Biol. Psychiatry 29, 1214–1224 [DOI] [PubMed] [Google Scholar]
- 18. Ramos B. P., Arnsten A. F. (2007) Adrenergic pharmacology and cognition. Focus on the prefrontal cortex. Pharmacol. Ther. 113, 523–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Igbavboa U., Johnson-Anuna L. N., Rossello X., Butterick T. A., Sun G. Y., Wood W. G. (2006) Amyloid β-protein1–42 increases cAMP and apolipoprotein E levels which are inhibited by β1 and β2-adrenergic receptor antagonists in mouse primary astrocytes. Neuroscience 142, 655–660 [DOI] [PubMed] [Google Scholar]
- 20. Prapong T., Uemura E., Hsu W. H. (2001) G protein and cAMP-dependent protein kinase mediate amyloid β-peptide inhibition of neuronal glucose uptake. Exp. Neurol. 167, 59–64 [DOI] [PubMed] [Google Scholar]
- 21. Echeverria V., Ducatenzeiler A., Chen C. H., Cuello A. C. (2005) Endogenous β-amyloid peptide synthesis modulates cAMP response element-regulated gene expression in PC12 cells. Neuroscience 135, 1193–1202 [DOI] [PubMed] [Google Scholar]
- 22. Wang D., Govindaiah G., Liu R., De Arcangelis V., Cox C. L., Xiang Y. K. (2010) Binding of amyloid β peptide to β2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity. FASEB J. 24, 3511–3521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang D., Yuen E. Y., Zhou Y., Yan Z., Xiang Y. K. (2011) Amyloid β peptide-(1–42) induces internalization and degradation of β2 adrenergic receptors in prefrontal cortical neurons. J. Biol. Chem. 286, 31852–31863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo C., Whitmarsh A. J. (2008) The β-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J. Biol. Chem. 283, 15903–15911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hochbaum D., Tanos T., Ribeiro-Neto F., Altschuler D., Coso O. A. (2003) Activation of JNK by Epac is independent of its activity as a Rap guanine nucleotide exchanger. J. Biol. Chem. 278, 33738–33746 [DOI] [PubMed] [Google Scholar]
- 26. Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R. (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations. Potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nagai T., Yamada K., Kim H. C., Kim Y. S., Noda Y., Imura A., Nabeshima Y., Nabeshima T. (2003) Cognition impairment in the genetic model of aging klotho gene mutant mice. A role of oxidative stress. FASEB J. 17, 50–52 [DOI] [PubMed] [Google Scholar]
- 28. Wang D., Noda Y., Zhou Y., Mouri A., Mizoguchi H., Nitta A., Chen W., Nabeshima T. (2007) The allosteric potentiation of nicotinic acetylcholine receptors by galantamine ameliorates the cognitive dysfunction in β amyloid25–35 i.c.v.-injected mice. Involvement of dopaminergic systems. Neuropsychopharmacology 32, 1261–1271 [DOI] [PubMed] [Google Scholar]
- 29. Johnson J. D., Cortez V., Kennedy S. L., Foley T. E., Hanson H., 3rd, Fleshner M. (2008) Role of central β-adrenergic receptors in regulating proinflammatory cytokine responses to a peripheral bacterial challenge. Brain Behav. Immun. 22, 1078–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Johnson G. V. (1992) Differential phosphorylation of tau by cyclic AMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase II. Metabolic and functional consequences. J. Neurochem. 59, 2056–2062 [DOI] [PubMed] [Google Scholar]
- 31. Litersky J. M., Johnson G. V., Jakes R., Goedert M., Lee M., Seubert P. (1996) Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem. J. 316, 655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yoshimura Y., Ichinose T., Yamauchi T. (2003) Phosphorylation of tau protein to sites found in Alzheimer's disease brain is catalyzed by Ca2+/calmodulin-dependent protein kinase II as demonstrated tandem mass spectrometry. Neurosci. Lett. 353, 185–188 [DOI] [PubMed] [Google Scholar]
- 33. Ploia C., Antoniou X., Sclip A., Grande V., Cardinetti D., Colombo A., Canu N., Benussi L., Ghidoni R., Forloni G., Borsello T. (2011) JNK plays a key role in tau hyperphosphorylation in Alzheimer's disease models. J. Alzheimers Dis. 26, 315–329 [DOI] [PubMed] [Google Scholar]
- 34. Sato S., Tatebayashi Y., Akagi T., Chui D. H., Murayama M., Miyasaka T., Planel E., Tanemura K., Sun X., Hashikawa T., Yoshioka K., Ishiguro K., Takashima A. (2002) Aberrant tau phosphorylation by glycogen synthase kinase-3β and JNK3 induces oligomeric tau fibrils in COS-7 cells. J. Biol. Chem. 277, 42060–42065 [DOI] [PubMed] [Google Scholar]
- 35. Jaworski T., Kugler S., Van Leuven F. (2010) Modeling of tau-mediated synaptic and neuronal degeneration in Alzheimer's disease. Int. J. Alzheimers Dis. 2010, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jaworski T., Lechat B., Demedts D., Gielis L., Devijver H., Borghgraef P., Duimel H., Verheyen F., Kügler S., Van Leuven F. (2011) Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am. J. Pathol. 179, 2001–2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jaworski T., Dewachter I., Lechat B., Croes S., Termont A., Demedts D., Borghgraef P., Devijver H., Filipkowski R. K., Kaczmarek L., Kügler S., Van Leuven F. (2009) AAV-tau mediates pyramidal neurodegeneration by cell-cycle re-entry without neurofibrillary tangle formation in wild-type mice. PLoS ONE 4, e7280. [DOI] [PMC free article] [PubMed] [Google Scholar]