

Background: Matriptase promotes intestinal epithelial barrier formation.
Results: Matriptase and prostasin constitute a common proteolytic pathway where matriptase is indirectly activated by prostasin and functions on the cell surface to regulate barrier formation.
Conclusion: A prostasin → matriptase protease cascade regulates barrier formation in simple intestinal epithelia.
Significance: Cell surface serine proteases have complex cascade relationships that are context- and tissue-specific.
Keywords: Epithelial Cell, Permeability, Protease, Proteolytic Enzymes, Serine Protease, TTSP, Protease Cascade, Zymogen
Abstract
The type II transmembrane serine protease matriptase is a key regulator of epithelial barriers in skin and intestine. In skin, matriptase acts upstream of the glycosylphosphatidylinositol-anchored serine protease, prostasin, to activate the prostasin zymogen and initiate a proteolytic cascade that is required for stratum corneum barrier functionality. Here, we have investigated the relationship between prostasin and matriptase in intestinal epithelial barrier function. We find that similar to skin, matriptase and prostasin are components of a common intestinal epithelial barrier-forming pathway. Depletion of prostasin by siRNA silencing in Caco-2 intestinal epithelium inhibits barrier development similar to loss of matriptase, and the addition of recombinant prostasin to the basal side of polarized Caco-2 epithelium stimulates barrier forming changes similar to the addition of recombinant matriptase. However, in contrast to the proteolytic cascade in skin, prostasin functions upstream of matriptase to activate the endogenous matriptase zymogen. Prostasin is unable to proteolytically activate the matriptase zymogen directly but induces matriptase activation indirectly. Prostasin requires expression of endogenous matriptase to stimulate barrier formation since matriptase depletion by siRNA silencing abrogates prostasin barrier-forming activity. Active recombinant matriptase, however, does not require the expression of endogenous prostasin for barrier-forming activity. Together, these data show that matriptase and not prostasin is the primary effector protease of tight junction assembly in simple columnar epithelia and further highlight a spatial and tissue-specific aspect of cell surface proteolytic cascades.
Introduction
The membrane-anchored serine proteases are a large family of serine proteases that are anchored directly to the plasma membrane, providing focal proteolysis for the generation of biologically active proteins important for diverse cellular and developmental processes (1, 2). The family shares a high degree of amino acid sequence identity in their serine protease catalytic domains, and in particular, they share the histidine-aspartate-serine residues that are necessary for catalytic activity. The multidomain type II transmembrane serine protease, matriptase, was one of the originally identified members of this family and has emerged to play an important role in epithelial development and homeostasis (3). The generation and analysis of mice with complete deficiency of the matriptase gene (St14) originally uncovered a critical role for matriptase in development of skin barrier function. Matriptase-deficient mice die shortly after birth due to severe skin dehydration defects, altered processing of profilaggrin, and generation of defective tight junctions (4). In addition, mutations in the human matriptase gene ST14 are associated with an autosomal recessive skin ichthyosis, including compromised barrier function and hair follicle defects (5).
Serine protease cascades, in which one active serine protease mediates the activation cleavage of another downstream precursor serine protease or zymogen, play an important role in the regulation of a range of physiological and pathological processes. It has been shown that in the skin, matriptase functions as part of a serine protease proteolytic cascade in which it acts upstream of the glycosylphosphatidylinositol-anchored serine protease, prostasin (CAP1/PRSS8) (6, 7). Genetic loss of the prostasin gene (Prss8) from the epidermis of mice causes skin permeability defects identical to those observed in matriptase-deficient mice (8). Moreover, the onset of expression of these two proteases during embryonic development is coordinated and correlated with acquisition of skin barrier functionality (6). Both matriptase and prostasin are synthesized as zymogens and require cleavage at a conserved activation motif between their pro- and catalytic domains to gain catalytic activity. Prostasin is activated by a single cleavage event at Arg44-Ile45 (9), generating a disulfide-linked active form that remains glycosylphosphatidylinositol-linked to the plasma membrane. The matriptase zymogen is capable of intermolecular autoproteolytic activation (10), and the active enzyme is also an effective activator of the prostasin zymogen through direct proteolytic cleavage at its zymogen activation site (6). In the skin, matriptase appears to activate the prostasin zymogen because epidermal tissues from matriptase-deficient mice do not contain the active form of prostasin, whereas both active and zymogen prostasin forms are present in normal epidermis (6, 7). Activated prostasin appears to target several downstream effector proteins, including the epithelial sodium channel (ENaC)2 (8, 11, 12) and the G protein-coupled protease activated receptor-2 (13), which are both matriptase substrates (14–16); however, the involvement of these effector proteins in epidermal barrier formation remains unclear.
Matriptase plays a wider role in the maintenance of epithelial barrier functionality outside of the epidermis (17). Intestinal epithelial-specific ablation of matriptase in mice causes severe postnatal intestinal dysfunction and poor survival, which is associated with increased intestinal permeability, loss of barrier function, and mislocation of tight junction-associated proteins (17). Matriptase hypomorphic mice, which express very low levels of matriptase, are able to survive (18) but display abnormally increased intestinal permeability (19), which renders them highly susceptible to experimental colitis and the development of chronic persistent disease (17, 20, 21). The loss of in vivo intestinal epithelial barrier function and increased permeability due to matriptase deficiency is modeled in vitro using human intestinal epithelial Caco-2 cells (19). When grown on porous Transwell filters, Caco-2 monolayers undergo spontaneous differentiation, become highly polarized, and assemble zonula occludens-type tight junction containing epithelial barriers (22–24). Matriptase is expressed at lateral intercellular adherens junctions and along the basolateral membrane of polarized Caco-2 epithelia (19, 25). siRNA knockdown of matriptase or specific inhibition of matriptase activity causes an increase in Caco-2 barrier permeability as evidenced by increased flux of macromolecules through the paracellular pathway and impaired development of transepithelial electrical resistance (TEER). Abnormally increased expression of the tight junction protein claudin-2, which is associated with the formation of cation-selective ion channels (19) is observed at intercellular junctions of matriptase-depleted Caco-2 epithelia, a permeability associated change also observed in matriptase hypomorphic mice (19, 21). Molecular analyses show that matriptase enhances the rate of claudin-2 protein turnover, which appears to be mediated indirectly through an atypical PKCζ-dependent signaling pathway (19).
The extent to which prostasin contributes to barrier formation in the intestine is unclear. Prostasin is found in abundance on the apical side and not the basolateral side of polarized Caco-2 epithelium (20, 26), although it has been reported to co-localize briefly with matriptase at basolateral membranes prior to being activated by matriptase and transcytosed to the apical membrane where it accumulates (20, 26). Unlike epidermal tissues (6, 18), active forms of prostasin are present in intestinal epithelial cultures depleted of matriptase (19). Prostasin hypomorphic mice (mouse frizzy (fr) mutation) display skin defects (27) with no overt intestinal abnormalities; however, detailed analysis of intestinal barrier function has not been reported. Therefore, we sought to determine the role of prostasin and the proteolytic pathway in intestinal epithelial barrier formation. We demonstrate that prostasin together with matriptase comprise a single common proteolytic pathway that is required for barrier formation. However, in contrast to the protease activation cascade that promotes barrier function in stratified epithelia of the epidermis, we found that prostasin functions upstream of matriptase in simple columnar epithelia of intestine, with matriptase being the critical effector of intestinal epithelial barrier formation and tight junction assembly.
EXPERIMENTAL PROCEDURES
Antibodies
Antibodies used were rabbit anti-matriptase (Calbiochem), mouse anti-prostasin antibody (BD Transduction Laboratories), rabbit anti-claudin-1 (Invitrogen), rabbit anti-human claudin-2 (Invitrogen), rabbit anti-GAPDH (Cell Signaling Technologies), rabbit anti-p-PKCζ (Santa Cruz Biotechnology), rabbit anti-PKCζ (Cell Signaling Technologies), and goat anti-HAI-1 (R&D Systems). HRP-conjugated anti-mouse and anti-rabbit antibodies were obtained from Jackson ImmunoResearch Laboratories.
Cell Culture, siRNA, and Plasmid Transfections
Human Caco-2 cells (passage 35–50, ATCC) were cultured, and siRNA transfections were performed using 100 nm siRNA and Dharmafect 1 transfection reagent (Dharmacon) as described (19). Twenty-four hours post-transfection, cells were plated in triplicate at high density (5 × 105 cells/well) onto 12-well Transwell filters, for experimental analyses. StealthTM siRNAs targeting matriptase (siM2, ST14-HSS110268 as published (19)), two independent siRNAs targeting prostasin (siP1, PRSS8-HSS108631; and siP2, PRSS8-HSS108633) and % GC-matched negative control (control siRNA) were from Invitrogen. HEK 293T cells were transiently transfected with pcDNA3.1 expression vector, pcDNA3.1-Matriptase (28), alone or in combination with pcDNA3.1-HAI-1 (28), or pcDNA3.1-HAI-1 and pIRES2-enhanced GFP-prostasin (6) using Lipofectamine 2000 (Invitrogen). Cell lysates were analyzed 24 h post-transfection.
Measurement of TEER
Cells were plated in triplicate onto polycarbonate 12-well Transwell® filters (0.4 μm pore size, 1.1 cm2, Costar Corning, Inc.) at 5 × 105 cells/well as described (19). Transwell chambers containing media alone were used to determine the intrinsic resistance of the membrane. TEER was measured using an EVOMTM Voltohmmeter with “chopstick” probes (World Precision Instruments, Sarasota, FL). TEER measurements from wells containing media alone were subtracted from test values. Final values, expressed as ohms·cm2, were obtained by multiplying TEER values by the surface area of the filter.
Measurement of Paracellular Flux Using Labeled Dextran
Paracellular flux of 4-kDa FITC-conjugated dextran (Sigma-Aldrich) across Caco-2 monolayers was assayed according to Refs. 19 and 29). Briefly, 3 mm 4-kDa FITC-dextran was added to the apical compartment of monolayers grown on Transwell filters. After incubation for 3 h at 37 °C, samples of the media from the apical and basal chambers was collected, and the FITC-dextran concentrations were measured at an excitation of 492 nm and an emission of 520 nm to calculate the apparent permeability coefficient (Papp) (19).
Treatment of Caco-2 Monolayers with Recombinant Serine Proteases
Recombinant human matriptase (amino acids 596–855), comprising the carboxyl-terminal end of the fourth LDLRA, the activation, and the catalytic domains, and the catalytically inactive matriptase mutant (G827R-matriptase) in which Gly827 has been replaced with Arg, were expressed in Escherichia coli and purified as described (30, 31). The catalytically active serine protease domains of other human membrane-anchored serine proteases were purchased as follows: prostasin, hepsin, human airway trypsin-like protease, spinesin (R&D Systems), and trypsin (Sigma-Aldrich). Control or siRNA-silenced Caco-2 monolayers cultured on Transwell filters were carefully washed, prior to incubation with the recombinant proteases added to either the apical or basal chambers of Transwells in serum-free DMEM.
Cell Lysis and Immunoblotting
Whole cell lysates were prepared using either LDS Sample Buffer (NuPAGE) or radioimmune precipitation assay buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% Triton X-100, 1 mm EDTA, 0.1% SDS, and 0.5% sodium deoxycholate) containing Complete protease inhibitor mixture (Roche Applied Science). Protein concentrations were estimated using the Bio-Rad protein assay kit. Proteins were resolved by SDS-PAGE under reducing conditions, and immunoblots were probed with the indicated primary antibody and detected with HRP-conjugated secondary antibody using standard techniques. Signals are normalized for protein loading by reprobing blots for GAPDH. Quantitation was performed by densitometry and analyzed using ImageJ software. All results are the average of at least two independent transfection experiments performed in triplicate.
Cleavage of the Matriptase Zymogen
To assess activation cleavage of the matriptase zymogen by recombinant prostasin, the catalytically inactive matriptase zymogens S805A-matriptase or G827R-matriptase mutants (100 nm), which are unable to autoactivate (31), were incubated with 50, 100, or 500 nm active recombinant prostasin or matriptase, in serum-free DMEM for 1 h at 37 °C. Reactions were stopped with LDS sample buffer, and proteins were resolved under reducing conditions by SDS-PAGE and then immunoblotted with the anti-matriptase antibody. The gel was stripped and reprobed using the anti-prostasin antibody.
Statistics
Results are expressed as mean ± S.E. from at least triplicate Transwells for Caco-2 TEER and FITC-dextran permeability measurements. The two-tailed Student's t test was used for statistical analysis and calculated using SigmaPlot software.
RESULTS
Prostasin Is Co-expressed with Matriptase and Increases During Human Caco-2 Cell Differentiation and Barrier Formation
Prostasin expression is relatively low in subconfluent Caco-2 cells (Fig. 1A, day 1) (26) but is increased by 10-fold as Caco-2 monolayers differentiate and develop TEER (Fig. 1A, day 14), as assessed by densitometric analysis after normalizing for protein loading using the GAPDH signal. Prostasin appears as a single band at ∼40 kDa, although Caco-2 cultures produce both the zymogen and activated forms of prostasin, which differ by ∼3 kDa (19). The increase in prostasin is accompanied by a corresponding increase in matriptase expression over the 14-day period (Fig. 1A). Full-length matriptase is initially synthesized as a 95-kDa zymogen that undergoes spontaneous cleavage at Gly149 in its SEA (sea urchin, enterokinase, agrin) domain, prior to a second activation inducing cleavage at its canonical activation motif (between Arg614 and Val615) that generates a disulfide-linked active protease, which remains tethered to the plasma membrane (32). Using an antibody that detects the serine protease domain, the unprocessed full-length matriptase form is not detected in Caco-2 cultures; the predominant form present is the SEA-processed 70-kDa matriptase zymogen (Fig. 1A). The amount of activated matriptase, detected directly by the released 30-kDa catalytic domain in reducing SDS-PAGE gels, also increases as the cells differentiate and develop TEER (Fig. 1A). The co-induction of prostasin and matriptase expression during tight junction assembly and the development of TEER are consistent with a cooperative role for these proteases in intestinal epithelial barrier formation. The mRNA levels of both of these proteases are not significantly changed during this time period (data not shown) (19), indicating that their induction occurs predominantly at a post-transcriptional level.
FIGURE 1.

Prostasin expression is co-induced with matriptase during Caco-2 barrier development and is required for closure of the paracellular pathway. A, prostasin expression increases during Caco-2 differentiation and barrier formation. Caco-2 cells were seeded at a subconfluent density and allowed to differentiate forming a highly polarized monolayer associated with increased TEER development. Plot shows development of TEER over time. Values represent mean ± S.E. from triplicate wells measured on the indicated days. Lysates prepared at different times were analyzed by immunoblot for prostasin, matriptase, and GAPDH expression. Using an anti-matriptase antibody that recognizes the matriptase serine protease domain, matriptase is detected as a 70-kDa form that represents the SEA-processed matriptase zymogen, and a 30-kDa form that represents the serine protease domain released after reduction of the single disulfide bridge that links the two peptide chains following proteolytic activation of matriptase (32). An asterisk indicates that blot was overexposed to detect the liberated serine protease domain as a measure of active matriptase. B, Caco-2 cells were transfected with siRNAs against matriptase (siM2), two different siRNAs against prostasin (siP1, siP2), or %GC matched control siRNA (siCtl), and cells plated onto Transwell filters. Cell lysates immunoblotted for prostasin and matriptase show that each siRNA efficiently and specifically suppresses expression of the respective protease for up to 6 days. An asterisk indicates that blot was overexposed to visualize active matriptase. C, TEER measurements show that prostasin suppression (siP1, siP2) inhibits TEER development to the same extent as suppression of matriptase (siM2), whereas control cells efficiently develop high TEER over 6 days (control siRNA (siCtl)). Graph shows mean ± S.E. from triplicate wells. D, prostasin-deficient cultures also exhibit increased permeability to macromolecular FITC-dextran. The paracellular permeability of siRNA transfected cultures to the passage of 4-kDa FITC-dextran was measured on days 2–4 after plating onto Transwell filters. The apparent permeability (Papp) of siM2, siP1, and siP2 cultures is significantly increased compared with control siRNA cells on all 3 days (*, p < 0.05; **, p < 0.001).
Depletion of Prostasin Impairs Caco-2 Barrier Formation
We have previously shown that siRNA silencing of matriptase expression or inhibition of matriptase activity impairs development of TEER and increases the permeability of Caco-2 epithelial barriers (19). When prostasin expression was silenced in Caco-2 monolayers using two independent prostasin siRNAs (siP1, siP2) that each reduced prostasin expression by >90% (Fig. 1B), the development of TEER was significantly impaired compared with control cultures (control siRNA) and was strikingly similar to inhibition mediated by matriptase silencing (siM2) (Fig. 1C) (19). In addition, depletion of prostasin was accompanied by a significant increase in matriptase zymogen levels and a decrease in active matriptase (Fig. 1B), which was further investigated in studies described below. The permeability of the prostasin-silenced Caco-2 monolayers to the nonionic macromolecular 4-kDa FITC-dextran tracer, which can only traverse the monolayer via the paracellular route, was also increased by 6- to 15-fold by day 4 relative to control cultures (siP1, siP2 versus control siRNA) and was comparable with the increase in permeability of matriptase silenced cultures (siM2) (Fig. 1D). These data implicate prostasin, together with matriptase, in a proteolytic pathway that regulates barrier closure during human Caco-2 epithelial differentiation.
Prostasin Depletion Induces Molecular Changes in Tight Junction Assembly Associated with Matriptase Depletion
We showed previously that depletion of matriptase from barrier forming Caco-2 monolayers induces increased incorporation of the permeability-associated, “leaky” tight junction protein claudin-2 at intercellular junctions and decreased activation of the atypical protein kinase C isoform, PKCζ, which is involved in the formation of epithelial tight junctions and establishment of cell polarity (19). siRNA silencing of prostasin resulted in a similar 6–8-fold increase in claudin-2 protein levels compared with control monolayers by day 6 (p < 0.005) (siP1, siP2, siM2 versus control siRNA) (Figs. 2, A and B). As observed after matriptase depletion, claudin-1 levels were not significantly affected by prostasin depletion (Fig. 2A). Prostasin depletion was likewise associated with a 75% decrease in activation of PKCζ, detected by reduced phosphorylation at Thr410 (p < 0.05) (Fig. 2, A and C), showing that prostasin, similar to matriptase, promotes barrier formation through a PKCζ signaling pathway. Taken together, the similarity of these molecular changes suggests that matriptase and prostasin function in a single, common pathway involved in the regulation of intestinal epithelial barrier formation.
FIGURE 2.
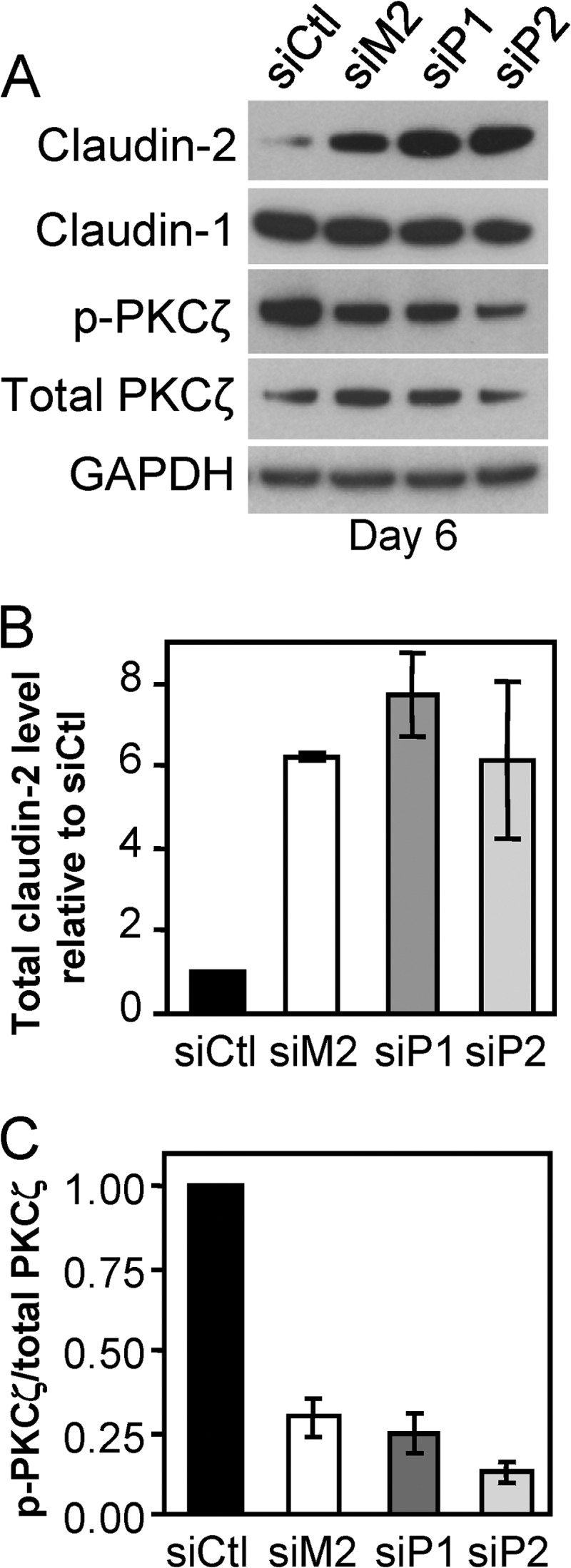
Prostasin depletion induces elevated claudin-2 levels and reduced PKCζ activation. A, representative immunoblot analyses of the tight junction proteins claudin-2 and claudin-1, total PKCζ, and its “active” Thr410 phosphorylated form (p-PKCζ) present in whole cell lysates on day 6 post-transfection. Average TEERs were as follows: 1148 (control siRNA (siCtl)), 477 (siM2), 463 (siP1), and 367 (siP2) ohms·cm2. B, quantification of the relative amount of claudin-2 relative to control siRNA cultures by densitometric scanning of immunoblots. Data are shown as mean ± S.E. C, quantification of the relative ratio of p-PKCζ/total PKCζ by densitometric scanning of immunoblots. Data are shown as mean ± S.E.
Prostasin Modulates Matriptase Zymogen Levels During Caco-2 Barrier Formation
We consistently observed that the failure in barrier formation caused by prostasin depletion (Fig. 1) was accompanied by the cell associated accumulation of the 70-kDa matriptase zymogen (Fig. 1B; matriptase zymogen, siP1, siP2 versus control siRNA), and loss of active matriptase, as evidenced by the disappearance of the 30-kDa matriptase under reducing conditions (Fig. 1B; matriptase active, siP1, siP2 versus control siRNA). Quantitation of matriptase protein levels revealed that prostasin depletion induced a ∼2-fold increase in matriptase zymogen levels by day 6 (p < 0.01) that was accompanied by a 75% reduction in active 30-kDa active form of matriptase (p < 0.001) (siP1, siP2 versus control siRNA) (Fig. 3A). These data suggested that prostasin could regulate matriptase zymogen activation during intestinal epithelial barrier formation. To test this directly, prostasin was co-expressed with matriptase in HEK293T cells (Fig. 3B). Effective expression of matriptase in HEK293T cells requires co-transfection with the Kunitz-type inhibitor, hepatocyte growth factor activator inhibitor 1 (HAI-1), which functions both as a chaperone and a reversible inhibitor (10, 28, 32). Expression of matriptase/HAI-1 in the absence of prostasin resulted exclusively in the presence of the matriptase precursors, full-length matriptase, and the 70-kDa (SEA-processed) matriptase zymogen, whereas co-expression with prostasin induced significant conversion of the 70-kDa matriptase zymogen to the active protease, as evidenced by the appearance of the 30-kDa matriptase serine protease domain under reducing conditions (Fig. 3B).
FIGURE 3.

Prostasin induces matriptase zymogen activation and stimulates TEER development. A, prostasin depletion increases matriptase zymogen accumulation and decreases matriptase zymogen activation. Quantification of the relative amounts of zymogen and active matriptase in prostasin silenced (siP1 and siP2) Caco-2 monolayers compared with control siRNA (siCtl) cultures on day 6 determined by densitometric analysis of immunoblots from three independent transfection experiments. Data are shown as mean ± S.E. B, prostasin expression induces matriptase activation. Prostasin was co-expressed in HEK293T cells along with matriptase and HAI-1. Immunoblots of cell lysates reveal that prostasin cleaves the matriptase zymogen to liberate the 30-kDa active form. An asterisk indicates that the blot was overexposed to detect active matriptase. C, exogenous addition of recombinant prostasin induces matriptase activation. Confluent Caco-2 cultures grown on Transwell filters for 4 days were treated with 5 nm recombinant prostasin (rProstasin), which was added to either the basal (rProstasin-Basal) or apical (rProstasin-Apical) chamber of triplicate wells. Lysates were prepared at the indicated times and analyzed by immunoblot for matriptase zymogen, active matriptase, and GAPDH. D, TEER development after the addition of recombinant prostasin in C. The plot shows the average change in TEER over time (mean ± S.E. from triplicate wells), relative to the initial TEER (∼400 ohms·cm2). E, stimulation of TEER development is specific to prostasin. Caco-2 monolayers cultured on transwell filters for 4 days were treated with 5 nm of the indicated recombinant proteases. Plot shows average change in TEER over time (mean ± S.E.), relative to the initial TEER (initial TEER ∼ 580 ohms·cm2).
Likewise, recombinant prostasin effectively induces activation of the matriptase zymogen (Fig. 3C). When added to the basal side of day 4 Caco-2 monolayers, 5 nm prostasin stimulated the conversion of the cell-associated 70-kDa matriptase zymogen to the two-chain active form, as evidenced by the concomitant appearance of 30-kDa matriptase serine protease domain over time (Fig. 3C). This prostasin-induced activation of matriptase zymogen was associated with a concomitant rapid enhancement of barrier formation, as evidenced by the development of TEER (Fig. 3D). When added apically to polarized monolayers, recombinant prostasin did not induce matriptase zymogen activation (Fig. 3C) and did not enhance TEER development (Fig. 3D). The stimulation of barrier forming activity by prostasin appears to be specific because other recombinant proteases, hepsin, human airway trypsin-like protease, spinesin, or trypsin, either applied basally (Fig. 3E) or apically (data not shown) at a similar concentration could not stimulate TEER development.
Prostasin Stimulation of Caco-2 Barrier Formation Requires Endogenous Matriptase
To address whether prostasin stimulation of TEER development was dependent on matriptase zymogen activation or whether prostasin could enhance barrier formation independent of matriptase, recombinant prostasin was added to matriptase siRNA-depleted Caco-2 monolayers. Recombinant prostasin could not stimulate development of TEER (Fig. 4A), indicating that the presence of endogenous matriptase is required for prostasin-induced barrier formation. However, when the recombinant prostasin was added to Caco-2 monolayers depleted of endogenous prostasin, TEER development was efficiently restored to control levels within 3 h (Fig. 4B), correlating with increased activation of cell-associated endogenous matriptase (Fig. 4C). These data show that prostasin barrier-forming activity requires expression of endogenous matriptase, strongly indicating that prostasin acts upstream of matriptase in the barrier-forming pathway.
FIGURE 4.

Prostasin induced barrier formation requires endogenous matriptase. A, matriptase (siM2) depleted Caco-2 monolayers were treated on the basal side on day 4 with 5 nm recombinant prostasin (rProstasin) or left untreated, and TEER development was measured in comparison with control (control siRNA (siCtl)) cultures over the course of 5 h. Immunoblot of cell lysates prepared on day 5 shows that effective depletion of matriptase is sustained. The plot shows TEER development over time (mean ± S.E.). The low TEER of matriptase-depleted cultures is not enhanced by treatment with 5 nm recombinant prostasin. B, prostasin (siP1) depleted cultures were treated with 5 nm recombinant prostasin as in (A) or left untreated, and TEER was measured in comparison with control (control siRNA (siCtl)) cultures over the course of 5 h. The low TEER of prostasin-depleted cultures is restored to the levels of the control cultures within 3 h. C, immunoblots of cell lysates prepared from parallel cultures at the indicated times in B show that the addition of recombinant prostasin stimulates matriptase zymogen activation in prostasin-depleted cells.
The Matriptase Zymogen Is Not Activated by Prostasin through Direct Proteolytic Activation Cleavage
To be processed to a catalytically active form, the matriptase zymogen must undergo proteolytic activation by cleavage after Arg614 within the activation cleavage site motif, R↓VVGG, present in the serine protease domain (10). Mutations that affect the catalytic activity of matriptase such as the active site serine mutant (S805A-matriptase), or the human genetic mutant G827R-matriptase, which has been reported in individuals with autosomal recessive skin ichthyosis (5), result in zymogen forms that are unable to autoactivate (31, 33). Therefore, recombinant forms of these mutant proteins can be used to test whether prostasin is capable of direct activation cleavage of the matriptase zymogen. Cleavage of S805A-matriptase or the G827R-matriptase (30 kDa) at the zymogen activation site is expected to produce a faster migrating disulfide-linked two-chain catalytically inactive protease, which is detected as a 26-kDa peptide under reducing conditions after immunoblotting, due to separation of the disulfide linked 4-kDa prodomain (Fig. 5A). To determine whether prostasin is capable of proteolytically cleaving the matriptase zymogen at the zymogen activation site, S805A-matriptase and G827R-matriptase were each exposed to increasing concentrations of recombinant prostasin. Prostasin was unable to catalyze the cleavage of either S805A-matriptase or G827R-matriptase zymogens to the two-chain form (Fig. 5B), demonstrating that the matriptase zymogen activation site is not a prostasin substrate. Because matriptase can catalyze its own activation, the matriptase zymogens were also exposed to recombinant active matriptase as a control for direct activation cleavage of the mutant protein, and both mutants were readily cleaved to the two-chain form by recombinant matriptase (Fig. 5C). These data demonstrate that the matriptase zymogen is not a prostasin substrate, and prostasin therefore cannot activate the matriptase zymogen by direct proteolytic cleavage, but instead, likely triggers matriptase zymogen activation through an indirect mechanism.
FIGURE 5.

Prostasin is not capable of direct activation cleavage of the matriptase zymogen. A, schematic representing the recombinant wild type and mutant matriptase serine protease domains. Matriptase is capable of intermolecular autoactivation by proteolytic cleavage between the pro- and catalytic domains, and recombinant matriptase (rMatriptase) possesses full catalytic activity. It is resolved under reducing conditions as a ∼26-kDa protein that is detected using an antibody generated against the serine protease domain. The matriptase mutants S805A-matriptase and G827R-matriptase are single chain zymogens that under reducing conditions retain the 4-kDa prodomain and are resolved at ∼30 kDa. S-S represents the disulfide link between the pro- and catalytic domains. B, the matriptase zymogen is not a prostasin substrate. 100 nm S805A-matriptase or G827R-matriptase were incubated with increasing concentrations of recombinant prostasin (50, 100, 500 nm) for 1 h at 37 °C. Reactions were immunoblotted (IB) using anti-matriptase antibodies. No change in the S805A-matriptase or G827R-matriptase molecular weight is observed, indicating lack of proteolytic cleavage. The blot was reprobed for prostasin to show its presence in the reactions (B, lower panels). C, the matriptase zymogen is a matriptase substrate. 100 nm S805A-matriptase or G827R-matriptase were incubated with increasing concentrations of recombinant matriptase (50, 100, 500 nm) for 1 h at 37 °C. Reactions were immunoblotted (IB) using anti-matriptase antibodies. Loss of the 30-kDa zymogen forms are observed and are accompanied by appearance of the 26-kDa catalytic domain. The lower immunoblot is of a control assay to show the amounts of recombinant matriptase added to the reaction mix because the added recombinant matriptase overlays the released 26kDa catalytic domain. D, amiloride does not inhibit prostasin-induced matriptase cleavage and activation. Immunoblots of cell lysates prepared at 4 h show that activation of matriptase by recombinant prostasin is not inhibited by the presence of amiloride. An asterisk indicates that the blot was overexposed to detect the liberated serine protease domain as a measure of active matriptase.
Prostasin is established as a proteolytic activator of ENaCs, which regulate sodium currents in epithelial cells (34, 35). Prostasin catalyzed activation of ENaC induces an open channel conformation associated with sodium influx that results in membrane depolarization (14, 35), which could potentially trigger matriptase autoactivation. This does not appear to be the case however because prostasin-induced matriptase zymogen activation (Fig. 5D) was unaffected by the ENaC inhibitor amiloride.
Matriptase Is the Crucial Activator of Caco-2 Barrier Closure
Our data implicate a prostasin→matriptase activation pathway in intestinal epithelial barrier formation. To investigate the importance of matriptase activity to barrier formation, recombinant matriptase was added to the basal side of confluent Caco-2 monolayers. A rapid, dose-dependent increase in TEER was induced (Fig. 6A) that was similar to the development of TEER stimulated by recombinant prostasin (Fig. 3C). Barrier formation was observed when matriptase was added basally; apical application of active matriptase did not enhance TEER development nor was TEER development stimulated by the catalytically inactive G827R-matriptase mutant protease (Fig. 6B). Similar results were found when recombinant matriptase was added to the basal side but not the apical side of polarized MDCK II canine kidney epithelial monolayers (data not shown), showing that matriptase stimulation of barrier formation is not specific to human Caco-2 intestinal epithelium. Prostasin siRNA depletion did not affect the ability of recombinant matriptase to stimulate TEER development (Fig. 6C), again demonstrating that prostasin is upstream of matriptase in the proteolytic cascade and emphasizing the importance of matriptase as a crucial activator of tight junction assembly in simple columnar epithelia.
FIGURE 6.

Matriptase is the crucial activator of Caco-2 barrier closure. A, stimulation of TEER development by recombinant matriptase. Four day Caco-2 monolayers on Transwell filters (TEER ∼ 250 ohms·cm2) were treated on the basolateral side with the indicated concentrations of recombinant matriptase, and TEER was monitored over the course of 5 h. The graph shows the change in TEER relative to the TEER measured at time 0. B, basolateral matriptase is required for TEER development. Four day Caco-2 monolayers on (TEER ∼ 650 ohms·cm2) were treated on the apical or basal sides with 5 nm recombinant matriptase or inactive G827R-matriptase. The graph shows the change in TEER relative to the TEER measured at time 0. C, endogenous prostasin is not required for the induction of TEER development by recombinant matriptase. Prostasin (siP1)-depleted Caco-2 monolayers were treated on the basal side on day 4 with 5 nm recombinant matriptase (siP1 + rMatriptase) or left untreated, and TEER was measured relative to control cultures (control siRNA (siCtl)) over the course of 5 h. Graphs show mean ± S.E. from triplicate wells.
DISCUSSION
Intestinal epithelial barrier function is frequently compromised in inflammatory bowel diseases and is a critical factor in disease development (36, 37). Barrier function is controlled by intercellular tight junction multiprotein complexes, which seal the paracellular space and regulate barrier permeability. The state of assembly of epithelial tight junctions must be modified rapidly and in a coordinated fashion to meet diverse physiological challenges. It has only recently been appreciated that serine proteases play key roles in the regulation and maintenance of epithelial barrier function (8, 17, 19, 38–40); however, the molecular mechanisms by which these enzymes mediate these processes are poorly understood. In the present study, we provide evidence for the coordinate activities of two membrane-anchored serine proteases, prostasin and matriptase, in promoting intestinal epithelial barrier closure using the Caco-2 model of intestinal epithelium. We find that prostasin stimulation of barrier assembly in this model of simple epithelia absolutely requires endogenous matriptase and that prostasin stimulates barrier formation by modulating matriptase zymogen activation. Matriptase is therefore the critical proteolytic activator of barrier closure.
Studies using experimental monolayers (Caco-2, MDCK) have been instrumental in providing valuable mechanistic insight into the endogenous processes important for barrier closure and tight junction assembly. These studies reveal that epithelial barrier integrity is regulated by the trans-tight junction flux of small charged ions (pore pathway), which is measured by the development of TEER, and also by the paracellular permeability to larger uncharged macromolecules (leak pathway), measured by FITC-dextran flux (41, 42). We previously showed that intestinal epithelial barrier formation via both the pore and leak pathways are matriptase-dependent (19). The data presented here demonstrate that matriptase and prostasin are components of a common pathway that promotes intestinal barrier closure. Similar to matriptase (19), prostasin depletion in barrier-forming Caco-2 intestinal epithelia increases permeability, resulting in impaired TEER development and increased paracellular permeability to macromolecular FITC-dextran. The molecular changes associated with prostasin depletion, including the reduced activation of PKCζ and enhanced expression of claudin-2, mimic those observed in matriptase silenced epithelial monolayers (19). Prostasin has been reported previously to regulate TEER development in renal M-1 epithelial cells through a protease activity-dependent mechanism, although this activity was reportedly confined to the apical surface (39). Although prostasin and matriptase are co-expressed in the intestinal epithelium in both humans and mice (43–46), matriptase is localized to basolateral membranes and the adherens junctions of polarized intestinal epithelium, whereas prostasin is predominantly found on the apical surfaces (17, 19, 39, 44, 47). Co-transfection of matriptase and prostasin in HEK293T cells and basolateral addition of recombinant prostasin to Caco-2 monolayers were both remarkably effective in inducing matriptase zymogen activation. Apical addition of prostasin was not effective in inducing matriptase zymogen activation, suggesting that prostasin may translocate to the basolateral surface to indirectly induce matriptase activation. Support for a protease signaling axis involving prostasin stimulation of matriptase activation comes from recent in vivo studies of neural tube closure in the developing embryo (15) and placental labyrinth formation (48), using HAI-1- or HAI-2-deficient mice. Although it appears that prostasin and matriptase cooperate to maintain the homeostasis of different organs, these findings illustrate that complex and context-specific zymogen activation mechanisms exist for these membrane serine proteases, possibly analogous to the well recognized local protease networks of secreted trypsin-like serine proteases in the coagulation, fibrinolytic, and digestive systems.
An important finding from this study is that prostasin functions upstream of matriptase to stimulate intestinal epithelial barrier formation, rather than downstream of matriptase, as reported in skin. We and others (6, 18) have established previously that prostasin is a matriptase substrate and matriptase directly cleaves and activates the prostasin zymogen in the epidermis. Studies in matriptase-depleted mouse epidermis or matriptase-silenced human epidermal cultures demonstrate an absence of activated prostasin (6, 18). In contrast, matriptase depletion does not alter prostasin zymogen activation in intestinal epithelial cultures (19), and we show here that prostasin induced intestinal epithelial barrier formation requires the presence of endogenous matriptase (Fig. 4A). Using the respective purified recombinant proteases, we found that prostasin cannot activate the matriptase zymogen by direct proteolytic cleavage. Matriptase is well known to catalyze its own zymogen activation (10) and therefore does not necessarily require an upstream protease for activation site cleavage. Although prostasin is unable to catalyze the activation cleavage of matriptase directly, prostasin activity appears to trigger matriptase zymogen activation through an indirect pathway, potentially through stimulating the autoactivation of matriptase. The factors involved in autoactivation of matriptase are not well defined, but autoactivation can be induced by a decrease in extracellular pH (49), by the serum sphingolipid sphingosine 1-phosphate and the anionic, sulfide-rich small molecule suramin (50) through a process that requires multiple elements of the matriptase stem domains (10, 51). It is possible that prostasin stimulates these autoactivation pathways or may act as a cofactor that interacts directly with matriptase stem domains to stimulate autoactivation. Alternatively prostasin may inhibit the release of active matriptase from the cell surface, resulting in the accumulation of active cell associated enzyme. After activation matriptase is thought to be rapidly (but reversibly) inhibited by HAI-1, and the matriptase-HAI-1 complex may then be shed from the cell surface (25, 52). It is possible that expression of prostasin, which is also inhibited by HAI-1 (53), could compete with matriptase for HAI-1 binding, therefore allowing increased activated matriptase to remain on the cell surface.
Spontaneous mutation in the prostasin gene in mice (V170D frizzy (fr)) and in the rat (hairless (frCR), Gly54-Pro57 deletion) show skin abnormalities and aberrant hair phenotypes (12). The rat Gly54-Pro57 deletion provokes hyperplastic skin, hyperkeratosis, increased transepithelial water loss, and diarrhea with increased stool hydration, possibly indicating an effect of prostasin deletion on tight junctions and/or on sodium transport and therefore water reabsorption. On the apical surface of polarized epithelium, prostasin is a potent physiological activator of amiloride-sensitive ENaCs (12, 34, 35). ENaC activity was found to be reduced in the distal colon in frizzy and hairless prostasin mutant animals (12); however, the data presented here, which is supported by studies by others (39, 54), suggest that the effects of prostasin on tight junctions and barrier integrity are independent of ENaC activation. Furthermore, our data show no effect of ENaC inhibition on prostasin stimulation of matriptase zymogen activation, suggesting other mechanisms are likely to be involved.
The matriptase catalytic domain was shown to rapidly and efficiently stimulate barrier closure, and is capable of rescuing the barrier defect of Caco-2 monolayers depleted of endogenous prostasin. This molecular insight into the mechanism of matriptase barrier forming activity suggests that the noncatalytic domains of matriptase, i.e. the cytoplasmic amino terminus and the conserved domains comprising the stem region (55), are involved in additional biological functions. Candidate biological substrates of matriptase, including hepatocyte growth factor, prostasin, or protease-activated receptor-2, have not been identified to mediate barrier forming activity in intestinal epithelia, and matriptase has not been demonstrated to proteolytically process tight junction proteins directly (19). Additional studies will be required to elucidate the downstream effectors of matriptase activity that contribute to tight junction assembly and barrier maintenance.
In summary, this study has identified a prostasin-matriptase axis that regulates intestinal epithelial barrier closure. Many diseases, including inflammatory bowel disease, celiac disease, diabetes, ischemic disease, and graft versus host disease are associated with leaky intestinal epithelium and increased intestinal permeability (37, 56–58). It will be important to further define this membrane-anchored serine protease axis and how its dysregulation may contribute to the development and pathogenesis of diseases of the gastrointestinal tract that are associated with increased intestinal permeability, specifically Crohn disease and ulcerative colitis as matriptase expression is significantly down-regulated in human intestinal tissues from these patients (21). A better understanding of these processes will be of fundamental importance for achieving a wider understanding of gastrointestinal physiology and mechanisms of gastrointestinal pathologies.
Acknowledgments
Human matriptase and HAI-1 expression vectors were provided by Dr. C.-Y. Lin (Georgetown University), and the human prostasin expression vector was provided by Dr. T. H. Bugge (NIDCR, National Institutes of Health).
This work was supported in part by National Institutes of Health Grants CA098369 and HL084387 (to T. M. A.), Department of Defense Grants PR110378 (to T. M. A.), and by the Canadian Institutes of Health Research (to R. L.).
- ENaC
- epithelial sodium channel
- TEER
- transepithelial electrical resistance
- HAI
- hepatocyte growth factor activator inhibitor.
REFERENCES
- 1. Szabo R., Bugge T. H. (2011) Membrane-anchored serine proteases in vertebrate cell and developmental biology. Annu. Rev. Cell Dev. Biol. 27, 213–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Antalis T. M., Buzza M. S., Hodge K. M., Hooper J. D., Netzel-Arnett S. (2010) The cutting edge: membrane-anchored serine protease activities in the pericellular microenvironment. Biochem. J. 428, 325–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Miller G. S., List K. (2013) The matriptase-prostasin proteolytic cascade in epithelial development and pathology. Cell Tissue Res. 351, 245–253 [DOI] [PubMed] [Google Scholar]
- 4. List K., Haudenschild C. C., Szabo R., Chen W., Wahl S. M., Swaim W., Engelholm L. H., Behrendt N., Bugge T. H. (2002) Matriptase/MT-SP1 is required for postnatal survival, epidermal barrier function, hair follicle development, and thymic homeostasis. Oncogene 21, 3765–3779 [DOI] [PubMed] [Google Scholar]
- 5. Basel-Vanagaite L., Attia R., Ishida-Yamamoto A., Rainshtein L., Ben Amitai D., Lurie R., Pasmanik-Chor M., Indelman M., Zvulunov A., Saban S., Magal N., Sprecher E., Shohat M. (2007) Autosomal recessive ichthyosis with hypotrichosis caused by a mutation in ST14, encoding type II transmembrane serine protease matriptase. Am. J. Hum. Genet. 80, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Netzel-Arnett S., Currie B. M., Szabo R., Lin C. Y., Chen L. M., Chai K. X., Antalis T. M., Bugge T. H., List K. (2006) Evidence for a matriptase-prostasin proteolytic cascade regulating terminal epidermal differentiation. J. Biol. Chem. 281, 32941–32945 [DOI] [PubMed] [Google Scholar]
- 7. Chen Y. W., Wang J. K., Chou F. P., Chen C. Y., Rorke E. A., Chen L. M., Chai K. X., Eckert R. L., Johnson M. D., Lin C. Y. (2010) Regulation of the matriptase-prostasin cell surface proteolytic cascade by hepatocyte growth factor activator inhibitor-1 during epidermal differentiation. J. Biol. Chem. 285, 31755–31762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leyvraz C., Charles R. P., Rubera I., Guitard M., Rotman S., Breiden B., Sandhoff K., Hummler E. (2005) The epidermal barrier function is dependent on the serine protease CAP1/Prss8. J. Cell Biol. 170, 487–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen M., Chen L. M., Lin C. Y., Chai K. X. (2010) Hepsin activates prostasin and cleaves the extracellular domain of the epidermal growth factor receptor. Mol. Cell Biochem. 337, 259–266 [DOI] [PubMed] [Google Scholar]
- 10. Oberst M. D., Williams C. A., Dickson R. B., Johnson M. D., Lin C. Y. (2003) The activation of matriptase requires its noncatalytic domains, serine protease domain, and its cognate inhibitor. J. Biol. Chem. 278, 26773–26779 [DOI] [PubMed] [Google Scholar]
- 11. Planès C., Randrianarison N. H., Charles R. P., Frateschi S., Cluzeaud F., Vuagniaux G., Soler P., Clerici C., Rossier B. C., Hummler E. (2010) ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol. Med. 2, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Frateschi S., Keppner A., Malsure S., Iwaszkiewicz J., Sergi C., Merillat A. M., Fowler-Jaeger N., Randrianarison N., Planès C., Hummler E. (2012) Mutations of the serine protease CAP1/Prss8 lead to reduced embryonic viability, skin defects, and decreased ENaC activity. Am. J. Pathol. 181, 605–615 [DOI] [PubMed] [Google Scholar]
- 13. Frateschi S., Camerer E., Crisante G., Rieser S., Membrez M., Charles R. P., Beermann F., Stehle J. C., Breiden B., Sandhoff K., Rotman S., Haftek M., Wilson A., Ryser S., Steinhoff M., Coughlin S. R., Hummler E. (2011) PAR2 absence completely rescues inflammation and ichthyosis caused by altered CAP1/Prss8 expression in mouse skin. Nat. Commun. 2, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vuagniaux G., Vallet V., Jaeger N. F., Hummler E., Rossier B. C. (2002) Synergistic activation of ENaC by three membrane-bound channel-activating serine proteases (mCAP1, mCAP2, and mCAP3) and serum- and glucocorticoid-regulated kinase (Sgk1) in Xenopus Oocytes. J. Gen. Physiol. 120, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Camerer E., Barker A., Duong D. N., Ganesan R., Kataoka H., Cornelissen I., Darragh M. R., Hussain A., Zheng Y. W., Srinivasan Y., Brown C., Xu S. M., Regard J. B., Lin C. Y., Craik C.S., Kirchhofer D., Coughlin S.R. (2010) Local protease signaling contributes to neural tube closure in the mouse embryo. Dev. Cell 18, 25–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Takeuchi T., Harris J. L., Huang W., Yan K. W., Coughlin S. R., Craik C. S. (2000) Cellular localization of membrane-type serine protease 1 and identification of protease-activated receptor-2 and single-chain urokinase-type plasminogen activator as substrates. J. Biol. Chem. 275, 26333–26342 [DOI] [PubMed] [Google Scholar]
- 17. List K., Kosa P., Szabo R., Bey A. L., Wang C. B., Molinolo A., Bugge T. H. (2009) Epithelial integrity is maintained by a matriptase-dependent proteolytic pathway. Am. J. Pathol. 175, 1453–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. List K., Currie B., Scharschmidt T. C., Szabo R., Shireman J., Molinolo A., Cravatt B. F., Segre J., Bugge T. H. (2007) Autosomal ichthyosis with hypotrichosis syndrome displays low matriptase proteolytic activity and is phenocopied in ST14 hypomorphic mice. J. Biol. Chem. 282, 36714–36723 [DOI] [PubMed] [Google Scholar]
- 19. Buzza M. S., Netzel-Arnett S., Shea-Donohue T., Zhao A., Lin C. Y., List K., Szabo R., Fasano A., Bugge T. H., Antalis T. M. (2010) Membrane-anchored serine protease matriptase regulates epithelial barrier formation and permeability in the intestine. Proc. Natl. Acad. Sci. U.S.A. 107, 4200–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kosa P., Szabo R., Molinolo A. A., Bugge T. H. (2012) Suppression of Tumorigenicity-14, encoding matriptase, is a critical suppressor of colitis and colitis-associated colon carcinogenesis. Oncogene 31, 3679–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Netzel-Arnett S., Buzza M. S., Shea-Donohue T., Désilets A., Leduc R., Fasano A., Bugge T. H., Antalis T. M. (2012) Matriptase protects against experimental colitis and promotes intestinal barrier recovery. Inflamm. Bowel Dis. 18, 1303–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simons K., Fuller S. D. (1985) Cell surface polarity in epithelia. Annu. Rev. Cell Biol. 1, 243–288 [DOI] [PubMed] [Google Scholar]
- 23. Cereijido M., Meza I., Martínez-Palomo A. (1981) Occluding junctions in cultured epithelial monolayers. Am. J. Physiol. 240, C96–102 [DOI] [PubMed] [Google Scholar]
- 24. Sambuy Y., De Angelis I., Ranaldi G., Scarino M. L., Stammati A., Zucco F. (2005) The Caco-2 cell line as a model of the intestinal barrier: influence of cell and culture-related factors on Caco-2 cell functional characteristics. Cell Biol. Toxicol. 21, 1–26 [DOI] [PubMed] [Google Scholar]
- 25. Wang J. K., Lee M. S., Tseng I. C., Chou F. P., Chen Y. W., Fulton A., Lee H. S., Chen C. J., Johnson M. D., Lin C. Y. (2009) Polarized epithelial cells secrete matriptase as a consequence of zymogen activation and HAI-1-mediated inhibition. Am. J. Physiol. Cell Physiol. 297, C459–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Friis S., Godiksen S., Bornholdt J., Selzer-Plon J., Rasmussen H. B., Bugge T. H., Lin C. Y., Vogel L. K. (2011) Transport via the transcytotic pathway makes prostasin available as a substrate for matriptase. J. Biol. Chem. 286, 5793–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Spacek D. V., Perez A. F., Ferranti K. M., Wu L. K., Moy D. M., Magnan D. R., King T. R. (2010) The mouse frizzy (fr) and rat “hairless” (frCR) mutations are natural variants of protease serine S1 family member 8 (Prss8). Exp. Dermatol. 19, 527–532 [DOI] [PubMed] [Google Scholar]
- 28. Oberst M. D., Chen L. Y., Kiyomiya K., Williams C. A., Lee M. S., Johnson M. D., Dickson R. B., Lin C. Y. (2005) HAI-1 regulates activation and expression of matriptase, a membrane-bound serine protease. Am. J. Physiol. Cell Physiol. 289, C462-C470 [DOI] [PubMed] [Google Scholar]
- 29. Sanders S. E., Madara J. L., McGuirk D. K., Gelman D. S., Colgan S. P. (1995) Assessment of inflammatory events in epithelial permeability: a rapid screening method using fluorescein dextrans. Epithelial Cell Biol. 4, 25–34 [PubMed] [Google Scholar]
- 30. Désilets A., Longpré J. M., Beaulieu M. E., Leduc R. (2006) Inhibition of human matriptase by eglin c variants. FEBS Lett. 580, 2227–2232 [DOI] [PubMed] [Google Scholar]
- 31. Désilets A., Béliveau F., Vandal G., McDuff F. O., Lavigne P., Leduc R. (2008) Mutation G827R in matriptase causing autosomal recessive ichthyosis with hypotrichosis yields an inactive protease. J. Biol. Chem. 283, 10535–10542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lin C. Y., Tseng I. C., Chou F. P., Su S. F., Chen Y. W., Johnson M. D., Dickson R. B. (2008) Zymogen activation, inhibition, and ectodomain shedding of matriptase. Front Biosci. 13, 621–635 [DOI] [PubMed] [Google Scholar]
- 33. Takeuchi T., Shuman M. A., Craik C. S. (1999) Reverse biochemistry: use of macromolecular protease inhibitors to dissect complex biological processes and identify a membrane-type serine protease in epithelial cancer and normal tissue. Proc. Natl. Acad. Sci. U.S.A. 96, 11054–11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vuagniaux G., Vallet V., Jaeger N. F., Pfister C., Bens M., Farman N., Courtois-Coutry N., Vandewalle A., Rossier B. C., Hummler E. (2000) Activation of the amiloride-sensitive epithelial sodium channel by the serine protease mCAP1 expressed in a mouse cortical collecting duct cell line. J. Am. Soc. Nephrol. 11, 828–834 [DOI] [PubMed] [Google Scholar]
- 35. Bruns J. B., Carattino M. D., Sheng S., Maarouf A. B., Weisz O. A., Pilewski J. M., Hughey R. P., Kleyman T. R. (2007) Epithelial Na+ channels are fully activated by furin- and prostasin-dependent release of an inhibitory peptide from the γ-subunit. J. Biol. Chem. 282, 6153–6160 [DOI] [PubMed] [Google Scholar]
- 36. Mankertz J., Schulzke J. D. (2007) Altered permeability in inflammatory bowel disease: pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 23, 379–383 [DOI] [PubMed] [Google Scholar]
- 37. Salim S. Y., Söderholm J. D. (2011) Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel. Dis. 17, 362–381 [DOI] [PubMed] [Google Scholar]
- 38. Swystun V. A., Renaux B., Moreau F., Wen S., Peplowski M. A., Hollenberg M. D., MacNaughton W. K. (2009) Serine proteases decrease intestinal epithelial ion permeability by activation of protein kinase Cζ. Am. J. Physiol. Gastrointest. Liver Physiol. 297, G60–70 [DOI] [PubMed] [Google Scholar]
- 39. Steensgaard M., Svenningsen P., Tinning A. R., Nielsen T. D., Jorgensen F., Kjaersgaard G., Madsen K., Jensen B. L. (2010) Apical serine protease activity is necessary for assembly of a high-resistance renal collecting duct epithelium. Acta Physiol. (Oxf) 200, 347–359 [DOI] [PubMed] [Google Scholar]
- 40. Vetrano S., Ploplis V. A., Sala E., Sandoval-Cooper M., Donahue D. L., Correale C., Arena V., Spinelli A., Repici A., Malesci A., Castellino F. J., Danese S. (2011) Unexpected role of anticoagulant protein C in controlling epithelial barrier integrity and intestinal inflammation. Proc. Natl. Acad. Sci. U.S.A. 108, 19830–19835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Shen L., Weber C. R., Raleigh D. R., Yu D., Turner J. R. (2011) Tight junction pore and leak pathways: a dynamic duo. Annu. Rev. Physiol. 73, 283–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anderson J. M., Van Itallie C. M. (2009) Physiology and function of the tight junction. Cold Spring Harb. Perspect. Biol. 1, a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. List K., Szabo R., Molinolo A., Nielsen B. S., Bugge T. H. (2006) Delineation of matriptase protein expression by enzymatic gene trapping suggests diverging roles in barrier function, hair formation, and squamous cell carcinogenesis. Am. J. Pathol. 168, 1513–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. List K., Hobson J. P., Molinolo A., Bugge T. H. (2007) Co-localization of the channel activating protease prostasin/(CAP1/PRSS8) with its candidate activator, matriptase. J. Cell Physiol. 213, 237–245 [DOI] [PubMed] [Google Scholar]
- 45. Oberst M. D., Singh B., Ozdemirli M., Dickson R. B., Johnson M. D., Lin C. Y. (2003) Characterization of matriptase expression in normal human tissues. J. Histochem. Cytochem. 51, 1017–1025 [DOI] [PubMed] [Google Scholar]
- 46. Yu J. X., Chao L., Chao J. (1995) Molecular cloning, tissue-specific expression, and cellular localization of human prostasin mRNA. J. Biol. Chem. 270, 13483–13489 [DOI] [PubMed] [Google Scholar]
- 47. Tsuzuki S., Murai N., Miyake Y., Inouye K., Hirayasu H., Iwanaga T., Fushiki T. (2005) Evidence for the occurrence of membrane-type serine protease 1/matriptase on the basolateral sides of enterocytes. Biochem. J. 388, 679–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Szabo R., Uzzun Sales K., Kosa P., Shylo N. A., Godiksen S., Hansen K. K., Friis S., Gutkind J. S., Vogel L. K., Hummler E., Camerer E., Bugge T. H. (2012) Reduced prostasin (CAP1/PRSS8) activity eliminates HAI-1 and HAI-2 deficiency-associated developmental defects by preventing matriptase activation. PLoS. Genet. 8, e1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lee M. S., Tseng I. C., Wang Y., Kiyomiya K., Johnson M. D., Dickson R. B., Lin C. Y. (2007) Autoactivation of matriptase in vitro: requirement for biomembrane and LDL receptor domain. Am. J. Physiol. Cell Physiol. 293, C95–105 [DOI] [PubMed] [Google Scholar]
- 50. Lee M. S., Kiyomiya K., Benaud C., Dickson R. B., Lin C. Y. (2005) Simultaneous activation and hepatocyte growth factor activator inhibitor 1-mediated inhibition of matriptase induced at activation foci in human mammary epithelial cells. Am. J. Physiol. Cell Physiol. 288, C932–941 [DOI] [PubMed] [Google Scholar]
- 51. Inouye K., Tomoishi M., Yasumoto M., Miyake Y., Kojima K., Tsuzuki S., Fushiki T. (2013) Roles of CUB and LDL receptor class A domain repeats of a transmembrane serine protease matriptase in its zymogen activation. J. Biochem. 153, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Godiksen S., Selzer-Plon J., Pedersen E. D., Abell K., Rasmussen H. B., Szabo R., Bugge T. H., Vogel L. K. (2008) Hepatocyte growth factor activator inhibitor-1 has a complex subcellular itinerary. Biochem. J. 413, 251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fan B., Wu T. D., Li W., Kirchhofer D. (2005) Identification of hepatocyte growth factor activator inhibitor-1B as a potential physiological inhibitor of prostasin. J. Biol. Chem. 280, 34513–34520 [DOI] [PubMed] [Google Scholar]
- 54. Verghese G. M., Gutknecht M. F., Caughey G. H. (2006) Prostasin regulates epithelial monolayer function: cell-specific Gpld1-mediated secretion and functional role for GPI anchor. Am. J. Physiol. Cell Physiol. 291, C1258–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hooper J. D., Clements J. A., Quigley J. P., Antalis T. M. (2001) Type II transmembrane serine proteases. Insights into an emerging class of cell surface proteolytic enzymes. J. Biol. Chem. 276, 857–860 [DOI] [PubMed] [Google Scholar]
- 56. Shen L., Su L., Turner J. R. (2009) Mechanisms and functional implications of intestinal barrier defects. Dig. Dis. 27, 443–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Turner J. R. (2009) Intestinal mucosal barrier function in health and disease. Nat. Rev. Immunol. 9, 799–809 [DOI] [PubMed] [Google Scholar]
- 58. Henderson P., van Limbergen J. E., Schwarze J., Wilson D. C. (2011) Function of the intestinal epithelium and its dysregulation in inflammatory bowel disease. Inflamm. Bowel. Dis. 17, 382–395 [DOI] [PubMed] [Google Scholar]