

Background: The TGF-β pathway is critical for angiogenesis.
Results: Endothelial miR-29a is up-regulated by TGF-β in a Smad4-dependent way to promote angiogenesis via targeting PTEN.
Conclusion: TGF-β promotes angiogenesis by up-regulating miR-29a.
Significance: We demonstrate how TGF-β signaling exerts its angiogenic function by up-regulating proangiogenic miRNA.
Keywords: Angiogenesis, MicroRNA, PTEN, SMAD Transcription Factor, Transforming Growth Factor β (TGF-β)
Abstract
The TGF-β pathway plays an important role in physiological and pathological angiogenesis. MicroRNAs (miRNAs) are a class of 18- to 25-nucleotide, small, noncoding RNAs that function by regulating gene expression. A number of miRNAs have been found to be regulated by the TGF-β pathway. However, the role of endothelial miRNAs in the TGF-β-mediated control of angiogenesis is still largely unknown. Here we investigated the regulation of endothelial microRNA-29a (miR-29a) by TGF-β signaling and the potential role of miR-29a in angiogenesis. MiR-29a was directly up-regulated by TGF-β/Smad4 signaling in human and mice endothelial cells. In a chick chorioallantoic membrane assay, miR-29a overexpression promoted the formation of new blood vessels, and miR-29a suppression completely blocked TGF-β1-stimulated angiogenesis. Consistently, miR-29a overexpression increased tube formation and migration in endothelial cultures. Mechanistically, miR-29a directly targeted the phosphatase and tensin homolog (PTEN) in endothelial cells, leading to activation of the AKT pathway. PTEN knockdown recapitulated the role of miR-29a in endothelial migration, whereas AKT inhibition completely attenuated the stimulating role of miR-29a in angiogenesis. Taken together, these results reveal a crucial role of a TGF-β-regulated miRNA in promoting angiogenesis by targeting PTEN to stimulate AKT activity.
Introduction
Angiogenesis refers to the formation of mature vasculature from a pre-existing primary plexus, occurring during embryonic development and adult life (1). Upon stimulation of angiogenic signals, endothelial cells (ECs)3 turn to be activated. ECs detach adhesions from their neighbors, sprout toward proangiogenic factors, proliferate to form temporary tubes, recruit pericytes, and finally remold and prune to form a functional network. Various signaling pathways, including TGF-β and phosphatase and tensin homolog (PTEN)/AKT signaling, sophisticatedly regulate distinct cellular processes involved in angiogenesis, of which endothelial migration is an essential event (2).
The TGF-β superfamily contains more than 30 members such as TGF-βs, bone morphogenetic proteins, and activins. Members of the TGF-β superfamily transduce their signals initially via binding specific transmembrane serine/threonine kinases receptors and then through intracellular Smad proteins. Receptor-regulated Smad (R-Smad), Smad2, and Smad3, are activated by the TGF-β receptor II-ALK5 complex, whereas Smad1, Smad5, and Smad8 are activated by the TβRII-ALK1 complex. Activated R-Smads associate with Smad4, the unique central mediator of TGF-β signaling, to translocate into the nucleus, where they participate in the transcriptional regulation of downstream target genes (3).
The crucial roles of TGF-β in angiogenesis have been revealed by genetic studies in humans and mice. Mutations in TGF-β signaling pathway components, including ENDOGLIN, ALK1, and SMAD4, account for most hereditary hemorrhagic telangiectasia clinical cases (4). In mice, deletion of various TGF-β signaling members, including TGF-β1, Tgfbr2, Alk5, Alk1, endoglin, Smad1, Smad4, and Smad5, leads to embryonic lethality because of severe vascular abnormalities, including a vascular remodeling defect and the absence of mural cell formation (5–13). In vitro, TGF-β differentially modulates endothelial migration and proliferation through distinct TβRI pathways, emphasizing the complexity of TGF-β signaling in endothelial function. Furthermore, effects of TGF-β during different stages of angiogenesis are usually dose-dependent and largely depend on its cellular context (14, 15).
Recent studies are starting to reveal posttranscriptional mechanisms underlying cellular responses of ECs to known angiogenic pathways. MicroRNAs (miRNAs) are a class of endogenous, 18- to 25-nucleotide, small, noncoding RNAs that function by negatively regulating target mRNAs either through translational inhibition or destabilization of mRNA (16). A number of miRNAs have been demonstrated to have pro- or antiangiogenic effects through regulating endothelial migration, survival, or the cell cycle. MiR-126, the miR-23∼27∼24 cluster, miR-424, miR-130a, miR-296, the miR-30 family, and miR-210 promote angiogenesis (17–25), whereas the miR-17∼92 cluster, miR-214, miR-200b, miR-1, miR-206, miR-221, and miR-222 block angiogenesis (26–32). Increasing evidence has shown that specific miRNAs can modulate the endothelial responses to blood flow, hypoxia, serum, or VEGF and exert their pro- or antiangiogenic effects. miR-126 regulates angiogenesis by activating VEGF signaling in response to blood flow (18, 33). MiR-130a promotes angiogenesis in response to fetal bovine serum by down-regulating antiangiogenic homeobox genes (23). miR-424 and miR-210 induced by hypoxia stimulate angiogenesis via regulating hypoxia-induced factor α isoforms as well as VEGF-driven cell migration (20, 21, 34). On the other hand, miR-125b induced by VEGF or ischemia inhibits angiogenesis through translational suppression of VE-cadherin (35).
miRNAs have been found to mediate TGF-β signaling and participate in TGF-β-regulated biological processes. MiR-155 is induced by TGF-β and promotes epithelial-mesenchymal transformation by targeting RhoA (36). TGF-β inhibits myogenic differentiation by down-regulating miR-24 (37). TGF-β also protects cardiomyocytes from hypertrophic growth by reducing miR-27b (38). TGF-β and bone morphogenetic protein signaling promotes the differentiation of human vascular smooth muscle cells by up-regulating miR-21 (39), and miR-21 accelerates re-epithelialization during wound healing in mice (40) and also participates in TGF-β-induced endothelial-to-mesenchymal transformation (41). Several studies have shown that miR-29 reduces fibrotic response and is down-regulated by TGF-β1 in cultured fibroblasts, tubular epithelial cells, and myogenic C2C12 cells. Nevertheless, the role of TGF-β-regulated miRNAs involved in angiogenesis is poorly investigated (42, 43). In this study, we revealed that endothelial miR-29a is up-regulated by TGF-β1 in a Smad4-dependent way to promote angiogenesis via targeting PTEN.
EXPERIMENTAL PROCEDURES
Vector Construction
For the promoter assay, a 1.7-kb genomic fragment upstream of the transcriptional start site of the miR-29a precursor (EU154353) was amplified by PCR using the primer pairs reported previously (44) and cloned into the PGL3-basic vector to obtain the PGL3–29a promoter vector. To delete potential Smad-binding sites in the promoter region, appropriate primer sets were used to amplify several deletion mutant fragments. WT/mut vector (−433 to −420 bp deleted) was obtained by putting fragments (−1706 to −433 and −420 to +1) together with the KpnI restriction enzyme site. Mut/WT (−1327 to −1309 deleted) was constructed by linking fragments (−1706 to −1327 and −1309 to +1) together with KpnI sites. Both the −1327 and −433 regions were deleted in the mut/mut vector. The amplicon was cloned into pGL3. The correct sequence was confirmed by sequencing. Sequences for Smad4 and PTEN RNA interference (RNAi) were cloned into the pSuperRetropuro vector described previously (6).
Small RNA Transfection
ECs were transfected with 20 nmol/liter miR-29a mimic, antagomir, or Scrambled oligonucleotides (GenePharma) using Lipofectamine 2000 (Invitrogen).
Cell Culture
The bEnd.3 cell line was purchased from ATCC. Human umbilical vein endothelial cell (HUVEC) and mice primary ECs were isolated and cultured as described previously (6). Mice were anesthetized with an intraperitoneal injection of sodium pentobarbital (25–50 mg/kg body weight) and then killed by cervical dislocation before isolation of tissues (brain). The depth of anesthesia was confirmed by lack of tail pinch response. All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health after securing the approval of the Committee of Animal Care of the Beijing Institute of Biotechnology.
Reporter Assay
bEnd.3 cells in 24-well plates were transfected with wild-type or mutant pGL3–29a promoters cotransfected with or without the HA-Smad4 plasmid. 24 h later, the cultures were treated with 5 ng/ml TGF-β1 for another 8 h. Luciferase activities were detected with dual luciferase reporter assay reagents (Promega). We used the PGL3-basic plasmid for normalization of luciferase values.
ChIP
bEnd.3 cells were pretreated with 5 ng/ml TGF-β1 for 4 h. ChIP assays were performed according to the instructions of the manufacturer with a SimpleChIP enzymatic chromatin immunoprecipitation kit (Cell Signaling Technology, Inc.). Antibodies used for ChIP were purchased from Cell Signaling Technology, Inc. DNA fragments of the miR-29a promoter region flanking the potential Smad4 binding site were amplified with the following primers: −551 to −392 bp, 5′-TGACTGGAGCATTAACCCTTGCA-3′ and 5′-TGTCCCATAAACGGCTCTGA-3′; −1385 to −1219 bp, 5′-ACTGAGAAAGGACGGCTGTTGGG-3′ and 5′-TCATGGCGTGTCATCTGGATTG-3′). The distal regions of the miR-29a promoter were amplified as a control with primers 5′-CATGACCAGTCTCCTCGTGAAAG-3′ and 5′-AGTCACAGGAAGTGGGACTCGGT-3′.
In Vitro Tube Formation Assay
ECs were planted onto 48-well plates precoated with a thin layer of Matrigel (BD Biosciences) in culture medium containing 5% fetal calf serum and allowed to form tube-like structures for 12 h. Measurements were performed as described previously (11).
Wound Healing Assay
The confluent cell monolayer in a 12-well plate was wounded by manually scraping the cells with a white pipette tip. The cells were treated with 5 ng/ml TGF-β1 in serum-free medium. Cell migration into the wound surface was monitored at various times. Quantitation was done by measuring the distance of the wound edge of the migrating cells from the start point to the migrated point in three independent experiments.
Chick Chorioallantoic Membrane (CAM) Assay
Fertilized eggs were incubated at 37 °C and 60% humidity for 10 days. A square window was made on the air sac to expose the CAM. Sterile 0.25 cm-diameter filter papers were applied onto the surfaces of the CAM, and 3 μl of drugs was added to the filter immediately. The windows were sealed, and the eggs were incubated for another 3 days. The CAMs were fixed with methanol:acetone (1:1, v/v) for 15 min, and the blood vessels around the filter papers within 1 mm were counted.
MTS Assay
bEND3 cells were seeded at a concentration of 5000 cells per well in 96-well plates. Relative cell numbers were quantified every day via (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) assay. For each well, medium was removed, and 20 μl of 5 mg/ml MTS was added. After 4 h of incubation at 37 °C, 150 μl of dimethyl sulfoxide was added to each well, and the absorbance was measured at 492 nm on a multifunction microplate reader.
Cell Cycle Analysis
bEnd.3 were transfected with miR-29a mimic or scramble oligo for 24h. Then cells were harvested and fixed in 70% alcohol for 30 min on ice. Cells were then stained with propidium iodide and RNase A at 37 °C for 30 min. Cell cycle was assessed by flow cytometry and the data were analyzed.
Cytoskeleton Labeling
bEnd.3 cells were transfected with miR-29a antisense oligos as described above. 24 h later, cells were fixed with 2% paraformaldehyde in PBS for 30 min, permeabilized with 0.2% bovine serum albumin in PBS for 30 min, and incubated with 5 μg/ml of phalloidin-TRITC and 2 μg/ml of DAPI for 30 min. Pictures were obtained using a fluorescence microscope and a digital camera.
Real-time RT-PCR
RNA was extracted from pretreated cells with TRIzol reagent (Invitrogen) and reverse-transcribed by using an mRNA-selective PCR kit (TaKaRa). A Taqman miRNA RT kit with multiplex RT rodent primer (Applied Biosystem) was used to quantitate miR-29a. Real-time PCR was performed with a Roche LightCycler 2.0 system. Primers were purchased from Invitrogen Corp. Primers for PTEN were as follows: 5′-TGGATTCGACTTAGACTTGACCT-3′ and 5′-GCGGTGTCATAATGTCTCTCAG-3′. Primers for VEGF and Smad4 were as described previously (6).
Northern Blot Analysis
Total RNAs were isolated using TRIzol reagent (Invitrogen) on the basis of the suggested protocol. Northern blot analysis was performed as described using 20 μg of total RNA from each sample. Probes were purchased from Invitrogen Corp. as follows: miR-29a, 5′-TAACCGATTTCAGATGGTGCTA-3′; miR-106a, 5′-CTACCTGCACTGTAAGCACTTTT); miR-222, 5′-ACCCAGTAGCCAGATGTAGCT-3′; and miR-21, 5′-TCAACATCAGTCTGATAAGCTA-3′.
Western Blot Analysis
20 μg of protein was electrophoresed on 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Immunoblotting was performed according to the instructions of the manufacturer using the following antibodies: PTEN, Smad4, AKT, phosphorylated AKT, and GAPDH (Abcam).
Statistical Methods
Data were evaluated using Student's two-tailed t test. p < 0.05 and p < 0.01 were taken to be statistically significant. The error bars on the graphs represent the mean ± S.D.
RESULTS
TGF-β1 Up-regulates miR-29a Expression in ECs
First we checked the expression of some miRNAs that are mentioned to be regulated by TGF-β and highly expressed in ECs in previous studies (45–47). Northern blot analysis showed that treatment of TGF-β1 at 5 ng/ml induced up-regulation of mature miR-29a in HUVECs dramatically. MiR-21 was increased as reported (39), whereas the expression of miR-106a and miR-222 was hardly changed (Fig. 1A). Through bioinformatic analysis we found that the sequence and seed region of miR-29a were conserved among species, including mouse, human, and chicken (Fig. 1B). We compared the relative expression abundance of mature miR-29a in several kinds of primary cells and normal cell lines, including mouse and human primary ECs (HUVEC), cardiomyocytes, vascular smooth muscle cells, chondrocytes, fibroblasts (3T3), and keratinocytes (HaCaT), showing that miR-29a was highly expressed in ECs and fibroblasts (Fig. 1C). The stimulating effect of TGF-β1 could also be observed in bEnd.3 cells, which is an immortalized mouse brain microvascular EC line, and TGF-β1 increased miR-29a expression in a dose-dependent manner (Fig. 1D). Next we examined the expression of primary miR-29a and mature miR-29a, respectively, in response to TGF-β1 at different time points. After 5 ng/ml TGF-β1 treatment, primary miR-29a increased first, starting at about 15 min. The up-regulation of mature miR-29a was not obvious until 1 h. Both forms of miR-29a expression were at maximums about 2 h after TGF-β1 treatment (Fig. 1E). This result implied that TGF-β1 might regulate miR-29a at the transcriptional level.
FIGURE 1.
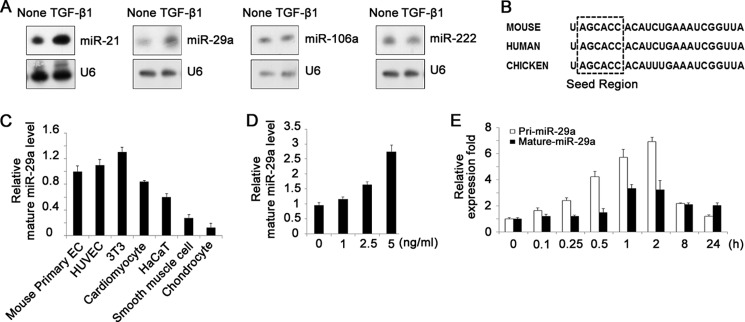
TGF-β1 up-regulates miR-29a expression in ECs. A, Northern blot analysis of miRNAs in HUVECs treated with or without 5 ng/ml TGF-β1, showing up-regulation of miR-21 and miR-29a by TGF-β1. B, sequences of mature miR-29a in mouse, human, and chicken. C, real-time PCR analysis of mature miR-29a expression in several kinds of primary cells and cell lines, including mouse primary ECs, human primary ECs (HUVEC), cardiomyocytes, smooth muscle cells, chondrocytes, a fibroblast cell line (NIH-3T3), and a human keratinocyte line (HaCaT). D, real-time PCR analysis of mature miR-29a expression in bEnd.3 cells. TGF-β1 increased miR-29a expression in a dose-dependent manner (1–5 ng/ml). E, real-time PCR analysis of primary and mature miR-29a expression in response to 5 ng/ml TGF-β1 at different time points.
Smad4 Is Required for the Transcriptional Activation of miR-29a by TGF-β1
As Smad4 is the unique central mediator of canonical TGF-β signaling, we determined the endogenous need of TGF-β signaling for the up-regulation of miR-29a in ECs by using Smad4 knockdown bEnd.3 cells (Fig. 2A). Smad4 RNAi led to down-regulation of miR-29a in bEnd.3 cells and largely blocked the stimulating role of TGF-β1 on miR-29a expression (Fig. 2B). Furthermore, we checked miR-29a expression in primary brain ECs derived from cerebrovascular endothelial-specific Smad4 knockout mice (6), showing that miR-29a was decreased by 50% in the Smad4-deficient cells (Fig. 2C). Next we performed a luciferase assay in the Smad4 knockdown bEnd.3 cells. Knockdown of Smad4 apparently reduced basic miR-29a promoter activity and absolutely abrogated the activating role of TGF-β1 on the miR-29a promoter, strongly suggesting that TGF-β-regulated miR-29a expression is largely dependent on Smad4 (Fig. 2D). Smads regulate gene transcription through their physical association with the Smad binding elements (SBE) in the promoters of target genes. A bioinformatics analysis was carried out to find potential SBEs in the miR-29a promoter with the rVista tool. Sequence analysis of the miR-29a promoter showed that there are several conserved SBEs in the promoter region of miR-29a. As shown in Fig. 2E, we constructed various miR-29a promoter luciferase constructs with SBE isolate or double deletions. TGF-b1 treatment and/or HA-Smad4-cotransfected increased the promoter activity. Moreover, the increases were largely blocked in the absence of either or both SBEs (Fig. 2E), indicating that SBE regions are essential for TGF-β regulation of miR-29a transcription. Next we examined whether Smads physically interact with the miR-29a promoter at these sites. We performed a ChIP assay in bEnd.3 cells that were treated with TGF-β1 for 4 h. DNA that coprecipitated with Smads was analyzed by PCR using primers specific for the two regions containing the −1327- or -433-bp Smad binding regions. Smad1/5/8, Smad2/3, and Smad4 antibodies all pulled down the miR-29a promoter region. TGF-β1 treatment increased amplicon pull-down by Smad4, Smad1/5/8, and Smad2/3 antibodies (Fig. 2F). These data indicate that TGF-β1 up-regulates miR-29a in a Smad4-dependent way.
FIGURE 2.
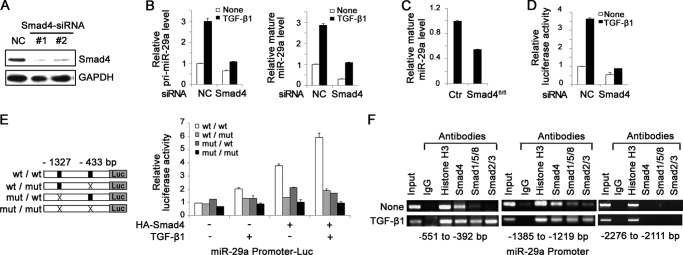
Smad4 is required for the transcriptional activation of miR-29a by TGF-β1. A, Western blot analysis of Smad4 expression in negative control (NC) cells and Smad4-siRNA bEnd.3 cells. B, real-time PCR analysis of primary (pri-miR-29a, left panel) and mature miR-29a (right panel) expression in control and Smad4-siRNA bEnd.3 cells treated with or without 5 ng/ml TGF-β1. C, real-time PCR analysis of mature miR-29a expression in control (Ctr) and Smad4-deficient primary mouse ECs. D, bEnd.3 cells were transfected with pGL3-Luc reporter constructs containing the −1637-bp to +164-bp region of miR-29a cotransfected with or without Smad4-siRNA. Data show that 5 ng/ml TGF-β1 increased the promoter activity of miR-29a, which was absolutely abrogated in the absence of endogenous Smad4. Values are expressed as relative luciferase units. E, bEnd.3 cells were transfected with the indicated plasmids (left panel), cotransfected with or without the HA-Smad4 vector, treated with or without 5 ng/ml TGF-β1, and assayed for luciferase activity (right panel). Data show that the TGF-β1-could not enhance miR-29a promoter activity in the absence SBEs. Values are expressed as relative luciferase units. F, bEnd.3 cells were treated with TGF-β1 for 4 h and subjected to ChIP with anti-histone H3, anti-Smad4, anti-Smad2/3, and anti-Smad1/5/8. The DNA fragments were amplified by PCR using primers for the miR-291 promoter containing Smad binding sites (−552 to −392 bp or −1385 to −1219 bp). A nonbinding region (−2276 to −2111 bp) served as negative control.
miR-29a Mediates TGF-β1-induced Angiogenesis
As TGF-β1 is an important angiogenic factor and induced miR-29a expression in ECs, we then attempted to study the role of miR-29a in TGF-β1-induced angiogenesis. We performed a CAM assay for in vivo angiogenesis evaluation. We first studied the effect of miR-29a in angiogenesis. Real-time PCR analysis of CAM tissues confirmed efficient overexpression of miR-29a packaged with Lipofectamine (Fig. 3A). Compared with scramble controls, incubation with the miR-29a mimic apparently induced more radial formation of new blood vessels (Fig. 3B). Then TGF-β1 together with miR-29a antagomir were applied onto the CAM surfaces to test whether miR-29a participates in TGF-β1-regulated angiogenesis. Consistent with previous reports, TGF-β1 promoted the formation of new blood vessels on the CAM (48), whereas suppression of miR-29a completely prevented the inducing effect of TGF-β1 on new blood vessel formation (Fig. 3C). These data suggest that miR-29a is involved in TGF-β1-induced angiogenesis.
FIGURE 3.

MiR-29a mediates TGF-β1-induced angiogenesis. A, real-time PCR analysis confirmed miR-29a overexpression with miR-29a mimic transfection (29a M) compared with scrambled sequence transfection (Scr). B, miR-29a mimic promoted new vessel growth in a CAM assay (left panel). Newly formed blood vessels were quantified (right panel). Scale bar = 1 mm. *, p < 0.05. n = 6. Red lines indicate the edge of the filter paper and the newly formed vessels around the filter paper. C, inhibition of miR-29a blocked TGF-β1-induced angiogenesis in the CAM. Filters soaked with TGF-β1 alone, with miR-29a antisense oligonucleotides (29a AS), or with the scrambled sequence were applied onto the CAM (left panel). Newly formed blood vessels were quantified (right panel). Scale bar = 1 mm. *, p < 0.05; **, p < 0.01. n = 6. The red line indicates the vessels around the filter paper.
miR-29a Promotes Endothelial Migration and Tube Formation
We next investigated the influence of miR-29a on endothelial function. By using mature miR-29a mimic or antisense oligonucleotides, we efficiently overexpressed or suppressed miR-29a in ECs, respectively (Fig. 4A). In an in vitro three-dimensional tube forming assay, miR-29a overexpression demonstrated an evidenced increase of tube formation, consistent with its effect on CAM angiogenesis (Fig. 4B). We tested the effect of miR-29a on cell migration in an EC wound healing assay and showed that overexpression of miR-29a increased cell migration considerably (Fig. 4C). Blocking TGF-β signaling by knockdown of Smad4 inhibited cell migration, whereas overexpression of miR-29a largely restored the migrating capacity of ECs (Fig. 4C). Consistently, suppression of miR-29a abrogated TGF-β1-promoted cell migration, suggesting that TGF-β1-induced endothelial migration was mediated by miR-29a (Fig. 4D). The actin cytoskeletal structure was observed by phalloidin staining. MiR-29a knockdown induced a decrease in cytoplasmic extensions into the denuded area in the scratch assay (Fig. 4E), further confirming the endogenous role of miR-29a on promoting EC migration. By MTS assay and flow cytometry analysis, no difference on cell proliferation and apoptosis was detected between miR-29a overexpression and control ECs (Figs. 4, F and G, and data not shown). These data highly implied that the stimulating role of miR-29a on CAM angiogenesis and tube forming capacity was at least partially affected by the migration of ECs.
FIGURE 4.

MiR-29a promotes endothelial migration and tube formation. A, real-time PCR analysis confirmed miR-29a overexpression or down-regulation with mimic (29a M) or antisense oligo (29a AS) in bEnd.3 cells. Scr, scrambled. B, pretreatment with miR-29a mimic increased tube formation of bEnd.3 cells in Matrigel (left panel). Tube lengths of the formed tubes were quantitated (right panel). Scale bar = 0.5 mm. *, p < 0.05; **, p < 0.01. n = 3. C, overexpression of miR-29a rescued the impaired migration ability of Smad4-siRNA bEnd.3 cells in a wound healing assay. Cell migration to the wound surface was monitored from 0–12 h (left panel). The migrated distance of the wound edge was quantified (right panel). Scale bar = 100 μm. *, p < 0.05; **, p < 0.01. n = 6. D, the enhanced migration of bEnd.3 cells induced by 5 ng/ml TGF-β1 was dramatically blocked by miR-29a antisense oligonucleotides in a wound healing assay (left panel). The migrated distance of the wound edge was quantified (right panel). Scale bar = 100 μm. *, p < 0.05; **, p < 0.01. n = 6. E, miR-29a knockdown induced a decrease in cytoplasmic extensions into the denuded area in the scratch assay. The actin cytoskeletal structure was observed by phalloidin staining of control and miR-29a knockdown bEnd.3 cells. F, cell numbers were quantified by MTS assay at the indicated time points. MiR-29a overexpression had no effect on endothelial cell number at 4–96 h of plating. G, cell cycle distribution was assessed using flow cytometry. The percentage cells in S phase were quantified. MiR-29a transfection had no influence on endothelial cell cycle distribution.
miR-29a Targets PTEN in Endothelial Cells
To identify the targets of miR-29a in ECs, we used three algorithms (Pictar, miRanda, and Targetscan) to predict potential direct targets. PTEN is one of the candidates predicted by all three algorithms that has two potential binding regions in the 3′ UTR completely complementary to the seed region of miR-29a. We confirmed that miR-29a down-regulated PTEN in ECs at both mRNA and protein levels (Fig. 5A). Supportively, miR-29a targets PTEN via binding to its 3′ UTR in other cell types (49). PTEN is a multifunctional phosphatase, and its major substrate is phosphatidylinositol-3,4,5,-trisphosphate, a lipid second messenger molecule. Phosphatidylinositol-3,4,5,-trisphosphate activates numerous downstream molecules, including the serine-threonine kinase PKB/AKT (50). Because TGF-β/Smad4 signaling up-regulated miR-29a and miR-29a directly targeted PTEN, we further examined PTEN/AKT signaling in the absence of Smad4. We confirmed that without endogenous TGF-β/Smad4 activity, PTEN was increased, and phosphorylated AKT was reduced concomitantly (Fig. 5B). Consistently, TGF-β1 activated AKT signaling in ECs (Fig. 5C). Importantly, miR-29a antagomir blocked the effect of TGF-β1 on AKT phosphorylation, implying that TGF-β1-stimulated AKT activity was largely mediated by miR-29a (Fig. 5C). We examined the effect of PTEN on the migrating ability of ECs. As shown in Fig. 5, D and E, knockdown of PTEN caused an obvious increase in AKT phosphorylation and apparently accelerated endothelial migration, recapitulating the role of miR-29a in promoting migration. Most importantly, inhibition of miR-29a had no effect on EC migration in the absence of PTEN (Fig. 5E) in contrast to the negative role of miR-29a antagomir in EC migration, as shown in Fig. 4D. These results suggest that miR-29a promotes endothelial migration by reducing PTEN expression and that PTEN is a specific target of miR-29a in ECs to exert its proangiogenic function.
FIGURE 5.
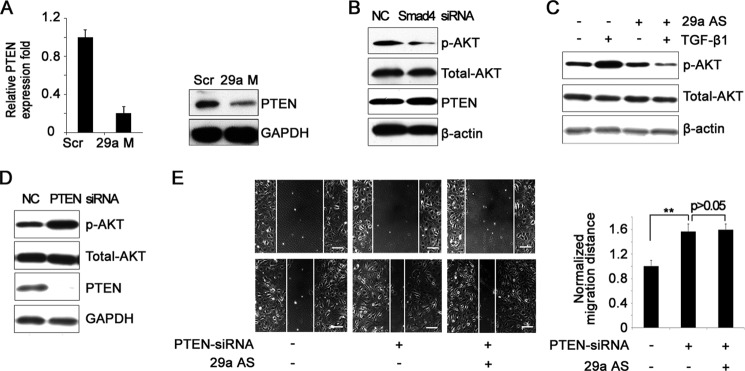
MiR-29a targets PTEN in ECs. A, real-time PCR (left panel) and Western blot analysis (right panel) of PTEN expression in scrambled (Scr) and miR-29a mimic-transfected (29a M) bEnd.3 cells. B, Western blot analysis of extracts from negative control (NC) and Smad4-siRNA bEnd.3 cells with the indicated antibodies. C, Western blot analysis of p-AKT in bEnd.3 cells with the indicated treatments. 29a AS, antisense oligo. D, Western blot analysis of p-AKT and PTEN expression in NC and PTEN-siRNA bEnd.3 cells. E, knockdown of PTEN increased endothelial migration of bEnd.3 cells in a wound healing assay. MiR-29a knockdown did not block this effect (left panel). The migrated distance of the wound edge was quantified (right panel). Scale bar = 100 μm. *, p < 0.05; **, p < 0.01. n = 6.
MiR-29a Promotes Angiogenesis by Activating AKT Signaling
We supposed that miR-29a promoted angiogenesis by activating AKT signaling through targeting PTEN. We used PI3K inhibitor LY294002 to evaluate the role of miR-29a-stimulated AKT activation on endothelial migration and tube formation. Western blotting assured that LY294002 inhibited miR-29a-induced AKT phosphorylation (Fig. 6A). A number of studies have shown that PI3K/AKT signaling induces angiogenesis and can increase VEGF expression (50). We checked the expression of the VEGFA164 isoform in b.End3 cells and showed an increase by miR-29a over-expression, which was completely attenuated by LY294002 treatment (Fig. 6B). Functionally, treatment with LY294002 completely blocked the effect of miR-29a in promoting EC migration in the wound healing assay and three-dimensional tube formation in Matrigel (Fig. 6, C and D). In the CAM angiogenesis assay, addition of LY294002 neutralized the stimulating role of miR-29a (Fig. 6E). Taken together, these in vitro and in vivo EC functional assays supported the notion that miR-29a promoted angiogenesis largely through activating AKT signaling.
FIGURE 6.

MiR-29a promotes angiogenesis by activating AKT signaling. A, Western blot analysis of p-AKT in bEnd.3 cells with the indicated treatments, showing that pretreatment with LY294002 (LY) abrogates the stimulating role of miR-29a overexpression (29a M) on p-AKT expression. B, real-time PCR analysis of VEGF164 expression in bEnd.3 cells with the indicated treatments, showing that pretreatment with LY294002 abrogates the stimulating role of miR-29a overexpression on VEGF164 expression. C, treatment with LY294002 blocked the effect of miR-29a in promoting EC migration of bEnd.3 cells (left panel). The migrated distance of the wound edge was quantified (right panel). Scale bar = 100 μm. *, p < 0.05; **, p < 0.01. n = 6. D, treatment with LY294002 blocked the effect of miR-29a in promoting tube formation in Matrigel (left panel). The lengths of newly formed tubes were quantified (right panel). Scale bar = 0.5 mm. *, p < 0.05; **, p < 0.01. n = 3. E, LY294002 neutralized the stimulating role of miR-29a overexpression on CAM angiogenesis (left panel). The newly formed vessels were counted (right panel). Scale bar = 1 mm. *, p < 0.05; **, p < 0.01. n = 3. The red lines indicate the vessels around the filter paper.
DISSCUSION
Here we showed that TGF-β-regulated miR-29a promoted angiogenesis, demonstrating a novel epigenetic mechanism of TGF-β signaling in controlling endothelial function. Canonical TGF-β selectively induces the transcription of downstream molecules through distinct type I receptors. Previous studies suggest that the balance between TGF-β/ALK1 versus TGF-β/ALK5 determines the effects of TGF-β on angiogenesis. Activation of ALK5 by TGF-β induces PAI-1 and inhibits migration and proliferation, whereas TGF-β-induced activation of ALK1 up-regulates Id1 expression and stimulates migration and proliferation (14). We provided the first evidence to prove that TGF-β signaling could up-regulate proangiogenic miRNA to exert its angiogenic function.
MiR-29a has been reported as a miRNA that can be regulated by TGF-β. TGF-β down-regulates miR-29a in skeletal muscle cells to influence TGF-β-mediated control of myogenic differentiation (43). Furthermore, TGF-β reduces the level of miR-29a in fibroblast and HK-2 cells to stimulate collagen expression (51, 52). In contrast to most previous studies, we found that TGF-β1 up-regulated miR-29a at transcriptional level in ECs in a Smad4-dependent manner. Importantly, miR-29a expression was reduced 2-fold in primary isolated Smad4-deficient ECs, confirming the positive regulation of miR-29a by TGF-β signaling under physiological conditions. We demonstrated that suppression of miR-29a significantly inhibited TGF-β1-induced CAM angiogenesis. In Smad4-deficient ECs, down-regulation of miR-29a (Fig. 2C) correlated with defective angiogenesis, evidenced by compromised tube-forming capacity (11). These results indicate that TGF-β1 promotes angiogenesis at least partially via up-regulating miR-29a. MiR-29a may not be the only miRNA mediating TGF-β induced angiogenesis. We found that miR-21 was also significantly up-regulated upon treatment of TGF-β1 on HUVECs (Fig. 1A). Previous studies have revealed that miR-21 induces angiogenesis through AKT and ERK activation and hypoxia-induced factor α expression (53). We also showed that similar to miR-29a, miR-21 overexpression promoted EC migration and tube formation. Notably, endothelial PTEN was not regulated by miR-21 overexpression4. Thus, miR-21 must execute its function via other targets in ECs. It would be interesting to explore the synergistic role of miR-29a and miR-21 in TGF-β-regulated angiogenesis in future studies. It is highly possible that TGF-β signaling regulates groups of proangiogenic miRNAs as well as anti-angiogenic miRNAs to elicit their pleiotropic and complex effects on angiogenesis. How ECs choose to activate or inhibit specific miRNAs in response to TGF-β signaling under certain physiological or pathologic conditions needs to be investigated further.
For the first time, we deciphered the role of miR-29a in endothelial cells. We identified miR-29a as a proangiogenic miRNA by positively regulating EC migration and tube formation. It has been reported that the TGF-β/Smad and PTEN/AKT pathways could regulate each other reciprocally in many other systems (54–56). In this study, we showed that TGF-β signaling could up-regulate the miRNA targeting PTEN to activate AKT signaling in ECs. The inhibitor of AKT could significantly diminish miR-29a-promoted angiogenesis, demonstrating that the function of TGF-β-regulated proangiogenic miR-29a is largely dependent on activated AKT signaling. PTEN knockdown or LY294002 treatment had no effect on mature miR-29a expression (data now shown). Thus, it is unlikely that PTEN/AKT play a role upstream of miR-29a to regulate angiogenesis. Previous studies have revealed that PTEN inhibits vascular sprouting and endothelial tube formation induced by VEGF (57). Sustained endothelial activation of AKT1 has been shown to induce the formation of structurally and functionally abnormal blood vessels (58). Notably, embryos deficient for the endothelial p110α catalytic subunit of PI3K develop severe vascular sprouting and remodeling defects, leading to embryonic lethality at mid-gestation that highly resembles the phenotype observed in endothelial-specific Smad4 deleted mice (59). Furthermore, p110α promotes endothelial migration and tube formation, similar to the role of TGF-β/Smad4 signaling in ECs (11). MiR-29a has been shown to have an antifibrotic effect by directly targeting a set of extracellular matrix genes in the heart, kidney, and other organs (51, 60, 61). We also found that miR-29a could down-regulate collagen genes in ECs (data not shown). Considering that extracellular matrix degradation is another key step in angiogenesis, we could not exclude the possibility that the stimulating effect of miR-29a in CAM angiogenesis might be partially caused by reduced expression of extracellular matrix-related genes.
In summary, our results suggest a novel mechanism by which TGF-β/Smad4 signaling promote angiogenesis, thwarting PTEN by up-regulating miR-29a and thus activating AKT to promote EC migration and tube formation. Dysregulation of miR-29a has been shown to occur in some types of cancers (62). Whether the dysregulation is mediated by TGF-β signaling and whether miR-29a plays a role in tumor angiogenesis are worth further investigation.
This work was supported by Chinese National Key Program on Basic Research Grants 2012CB945103, 2011CB964803, and 2011CB504202; by National Natural Science Foundation of China Grants 31030040 and 31171410; and by State Key Laboratory of Proteomics Grant SKLP-K201102.
J. Wang, Y. Wang, Y. Wang, Y. Ma, Y. Lan, and X. Yang, unpublished data.
- EC
- endothelial cell
- PTEN
- phosphatase and tensin homolog
- miRNA
- microRNA
- HUVEC
- human umbilical vein endothelial cell
- CAM
- chick chorioallantoic membrane
- MTS
- 3-(4,5-dimethylthiazol-2yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- SBE
- Smad binding element
- VE-Cadherin
- vascular endothelial cadherin
- TRITC
- Tetramethylrhodamine-5-(and-6)-isothiocyanate
- PKB
- Protein Kinase B.
REFERENCES
- 1. Carmeliet P., Jain R. K. (2011) Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weis S. M., Cheresh D. A. (2011) Tumor angiogenesis. Molecular pathways and therapeutic targets. Nat. Med. 17, 1359–1370 [DOI] [PubMed] [Google Scholar]
- 3. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 4. Lenato G. M., Guanti G. (2006) Hereditary haemorrhagic telangiectasia (HHT). Genetic and molecular aspects. Curr. Pharm. Des. 12, 1173–1193 [DOI] [PubMed] [Google Scholar]
- 5. Lechleider R. J., Ryan J. L., Garrett L., Eng C., Deng C., Wynshaw-Boris A., Roberts A. B. (2001) Targeted mutagenesis of Smad1 reveals an essential role in chorioallantoic fusion. Dev. Biol. 240, 157–167 [DOI] [PubMed] [Google Scholar]
- 6. Li F., Lan Y., Wang Y., Wang J., Yang G., Meng F., Han H., Meng A., Wang Y., Yang X. (2011) Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev. Cell. 20, 291–302 [DOI] [PubMed] [Google Scholar]
- 7. Dickson M. C., Martin J. S., Cousins F. M., Kulkarni A. B., Karlsson S., Akhurst R. J. (1995) Defective haematopoiesis and vasculogenesis in transforming growth factor-β 1 knock out mice. Development 121, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 8. Oshima M., Oshima H., Taketo M. M. (1996) TGF-β receptor type II deficiency results in defects of yolk sac hematopoiesis and vasculogenesis. Dev. Biol. 179, 297–302 [DOI] [PubMed] [Google Scholar]
- 9. Yang X., Castilla L. H., Xu X., Li C., Gotay J., Weinstein M., Liu P. P., Deng C. X. (1999) Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 126, 1571–1580 [DOI] [PubMed] [Google Scholar]
- 10. Bourdeau A., Dumont D. J., Letarte M. (1999) A murine model of hereditary hemorrhagic telangiectasia. J. Clin. Invest. 104, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lan Y., Liu B., Yao H., Li F., Weng T., Yang G., Li W., Cheng X., Mao N., Yang X. (2007) Essential role of endothelial Smad4 in vascular remodeling and integrity. Mol. Cell. Biol. 27, 7683–7692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Urness L. D., Sorensen L. K., Li D. Y. (2000) Arteriovenous malformations in mice lacking activin receptor-like kinase-1. Nat. Genet. 26, 328–331 [DOI] [PubMed] [Google Scholar]
- 13. Li D. Y., Sorensen L. K., Brooke B. S., Urness L. D., Davis E. C., Taylor D. G., Boak B. B., Wendel D. P. (1999) Defective angiogenesis in mice lacking endoglin. Science 284, 1534–1537 [DOI] [PubMed] [Google Scholar]
- 14. Goumans M. J., Valdimarsdottir G., Itoh S., Rosendahl A., Sideras P., ten Dijke P. (2002) Balancing the activation state of the endothelium via two distinct TGF-β type I receptors. EMBO. J. 21, 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pardali E., Goumans M. J., ten Dijke P. (2010) Signaling by members of the TGF-beta family in vascular morphogenesis and disease. Trends. Cell. Biol. 20, 556–567 [DOI] [PubMed] [Google Scholar]
- 16. Bartel D. P. (2009) MicroRNAs. target recognition and regulatory functions. Cell. 136, 215–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kuhnert F., Mancuso M. R., Hampton J., Stankunas K., Asano T., Chen C. Z., Kuo C. J. (2008) Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development 135, 3989–3993 [DOI] [PubMed] [Google Scholar]
- 18. Wang S., Aurora A. B., Johnson B. A., Qi X., McAnally J., Hill J. A., Richardson J. A., Bassel-Duby R., Olson E. N. (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell. 15, 261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhou Q., Gallagher R., Ufret-Vincenty R., Li X., Olson E. N., Wang S. (2011) Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23∼27∼24 clusters. Proc. Natl. Acad. Sci. U.S.A. 108, 8287–8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ghosh G., Subramanian I. V., Adhikari N., Zhang X., Joshi H. P., Basi D., Chandrashekhar Y. S., Hall J. L., Roy S., Zeng Y., Ramakrishnan S. (2010) Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-α isoforms and promotes angiogenesis. J. Clin. Invest. 120, 4141–4154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alaiti M. A., Ishikawa M., Masuda H., Simon D. I., Jain M. K., Asahara T., Costa M. A. (2012) Up-regulation of miR-210 by vascular endothelial growth factor in ex vivo expanded CD34+ cells enhances cell-mediated angiogenesis. J. Cell. Mol. Med. 16, 2413–2421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Würdinger T., Tannous B. A., Saydam O., Skog J., Grau S., Soutschek J., Weissleder R., Breakefield X. O., Krichevsky A. M. (2008) miR-296 regulates growth factor receptor overexpression in angiogenic endothelial cells. Cancer Cell. 14, 382–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen Y., Gorski D. H. (2008) Regulation of angiogenesis through a microRNA (miR-130a) that down-regulates antiangiogenic homeobox genes GAX and HOXA5. Blood 111, 1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bridge G., Monteiro R., Henderson S., Emuss V., Lagos D., Georgopoulou D., Patient R., Boshoff C. (2012) The microRNA-30 family targets DLL4 to modulate endothelial cell behavior during angiogenesis. Blood 120, 5063–5072 [DOI] [PubMed] [Google Scholar]
- 25. Urbich C., Kaluza D., Frömel T., Knau A., Bennewitz K., Boon R. A., Bonauer A., Doebele C., Boeckel J. N., Hergenreider E., Zeiher A. M., Kroll J., Fleming I., Dimmeler S. (2012) MicroRNA-27a/b controls endothelial cell repulsion and angiogenesis by targeting semaphorin 6A. Blood 119, 1607–1616 [DOI] [PubMed] [Google Scholar]
- 26. van Mil A., Grundmann S., Goumans M. J., Lei Z., Oerlemans M. I., Jaksani S., Doevendans P. A., Sluijter J. P. (2012) MicroRNA-214 inhibits angiogenesis by targeting Quaking and reducing angiogenic growth factor release. Cardiovasc. Res. 93, 655–665 [DOI] [PubMed] [Google Scholar]
- 27. Doebele C., Bonauer A., Fischer A., Scholz A., Reiss Y., Urbich C., Hofmann W. K., Zeiher A. M., Dimmeler S. (2010) Members of the microRNA-17–92 cluster exhibit a cell-intrinsic antiangiogenic function in endothelial cells. Blood 115, 4944–4950 [DOI] [PubMed] [Google Scholar]
- 28. Bonauer A., Carmona G., Iwasaki M., Mione M., Koyanagi M., Fischer A., Burchfield J., Fox H., Doebele C., Ohtani K., Chavakis E., Potente M., Tjwa M., Urbich C., Zeiher A. M., Dimmeler S. (2009) MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324, 1710–1713 [DOI] [PubMed] [Google Scholar]
- 29. Chen Y., Banda M., Speyer C. L., Smith J. S., Rabson A. B., Gorski D. H. (2010) Regulation of the expression and activity of the antiangiogenic homeobox gene GAX/MEOX2 by ZEB2 and microRNA-221. Mol. Cell. Biol. 30, 3902–3913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wu Y. H., Hu T. F., Chen Y. C., Tsai Y. N., Tsai Y. H., Cheng C. C., Wang H. W. (2011) The manipulation of miRNA-gene regulatory networks by KSHV induces endothelial cell motility. Blood 118, 2896–2905 [DOI] [PubMed] [Google Scholar]
- 31. Fish J. E., Srivastava D. (2009) MicroRNAs. Opening a new vein in angiogenesis research. Sci. Signal 2, pe1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stahlhut C., Suárez Y., Lu J., Mishima Y., Giraldez A. J. (2012) miR-1 and miR-206 regulate angiogenesis by modulating VegfA expression in zebrafish. Development 139, 4356–4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicoli S., Standley C., Walker P., Hurlstone A., Fogarty K. E., Lawson N. D. (2010) MicroRNA-mediated integration of haemodynamics and VEGF signalling during angiogenesis. Nature 464, 1196–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fasanaro P., D'Alessandra Y., Di Stefano V., Melchionna R., Romani S., Pompilio G., Capogrossi M. C., Martelli F. (2008) MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J. Biol. Chem. 283, 15878–15883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Muramatsu F., Kidoya H., Naito H., Sakimoto S., Takakura N. (2012) microRNA-125b inhibits tube formation of blood vessels through translational suppression of VE-cadherin. Oncogene 32, 414–421 [DOI] [PubMed] [Google Scholar]
- 36. Kong W., Yang H., He L., Zhao J. J., Coppola D., Dalton W. S., Cheng J. Q. (2008) MicroRNA-155 is regulated by the transforming growth factor β/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol. Cell. Biol. 28, 6773–6784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun Q., Zhang Y., Yang G., Chen X., Zhang Y., Cao G., Wang J., Sun Y., Zhang P., Fan M., Shao N., Yang X. (2008) Transforming growth factor-β-regulated miR-24 promotes skeletal muscle differentiation. Nucleic Acids Res. 36, 2690–2699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J., Song Y., Zhang Y., Xiao H., Sun Q., Hou N., Guo S., Wang Y., Fan K., Zhan D., Zha L., Cao Y., Li Z., Cheng X., Zhang Y., Yang X. (2012) Cardiomyocyte overexpression of miR-27b induces cardiac hypertrophy and dysfunction in mice. Cell. Res. 22, 516–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Davis B. N., Hilyard A. C., Lagna G., Hata A. (2008) SMAD proteins control DROSHA-mediated microRNA maturation. Nature 454, 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yang X., Wang J., Guo S. L., Fan K. J., Li J., Wang Y. L., Teng Y. (2011) miR-21 promotes keratinocyte migration and re-epithelialization during wound healing. Int. J. Biol. Sci. 7, 685–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kumarswamy R., Volkmann I., Jazbutyte V., Dangwal S., Park D. H., Thum T. (2012) Transforming growth factor-β-induced endothelial-to-mesenchymal transition is partly mediated by microRNA-21. Arterioscler. Thromb. Vasc. Biol. 32, 361–369 [DOI] [PubMed] [Google Scholar]
- 42. Qin W., Chung A. C., Huang X. R., Meng X. M., Hui D. S., Yu C. M., Sung J. J., Lan H. Y. (2011) TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 22, 1462–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Winbanks C. E., Wang B., Beyer C., Koh P., White L., Kantharidis P., Gregorevic P. (2011) TGF-β regulates miR-206 and miR-29 to control myogenic differentiation through regulation of HDAC4. J. Biol. Chem. 286, 13805–13814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mott J. L., Kurita S., Cazanave S. C., Bronk S. F., Werneburg N. W., Fernandez-Zapico M. E. (2010) Transcriptional suppression of mir-29b-1/mir-29a promoter by c-Myc, hedgehog, and NF-κB. J. Cell. Biochem. 110, 1155–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Poliseno L., Tuccoli A., Mariani L., Evangelista M., Citti L., Woods K., Mercatanti A., Hammond S., Rainaldi G. (2006) MicroRNAs modulate the angiogenic properties of HUVECs. Blood 108, 3068–3071 [DOI] [PubMed] [Google Scholar]
- 46. McCall M. N., Kent O. A., Yu J., Fox-Talbot K., Zaiman A. L., Halushka M. K. (2011) MicroRNA profiling of diverse endothelial cell types. BMC Med. Genomics 4, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guduric-Fuchs J., O'Connor A., Cullen A., Harwood L., Medina R. J., O'Neill C. L., Stitt A. W., Curtis T. M., Simpson D. A. (2012) Deep sequencing reveals predominant expression of miR-21 amongst the small non-coding RNAs in retinal microvascular endothelial cells. J. Cell. Biochem. 113, 2098–2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ma J., Wang Q., Fei T., Han J. D., Chen Y. G. (2007) MCP-1 mediates TGF-β-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 109, 987–994 [DOI] [PubMed] [Google Scholar]
- 49. Kong G., Zhang J., Zhang S., Shan C., Ye L., Zhang X. (2011) Upregulated microRNA-29a by hepatitis B virus X protein enhances hepatoma cell migration by targeting PTEN in cell culture model. PLoS ONE 6, e19518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jiang B. H., Liu L. Z. (2009) PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv. Cancer Res. 102, 19–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maurer B., Stanczyk J., Jüngel A., Akhmetshina A., Trenkmann M., Brock M., Kowal-Bielecka O., Gay R. E., Michel B. A., Distler J. H., Gay S., Distler O. (2010) MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis. Rheum. 62, 1733–1743 [DOI] [PubMed] [Google Scholar]
- 52. Du B., Ma L. M., Huang M. B., Zhou H., Huang H. L., Shao P., Chen Y. Q., Qu L. H. (2010) High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 584, 811–816 [DOI] [PubMed] [Google Scholar]
- 53. Liu L. Z., Li C., Chen Q., Jing Y., Carpenter R., Jiang Y., Kung H. F., Lai L., Jiang B. H. (2011) MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1α expression. PLoS ONE 6, e19139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chow J. Y., Cabral J. A., Chang J., Carethers J. M. (2008) TGFβ modulates PTEN expression independently of SMAD signaling for growth proliferation in colon cancer cells. Cancer Biol. Ther. 7, 1694–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chow J. Y., Dong H., Quach K. T., Van Nguyen P. N., Chen K., Carethers J. M. (2008) TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am. J. Physiol. Gastrointest Liver Physiol. 294, G899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chow J. Y., Ban M., Wu H. L., Nguyen F., Huang M., Chung H., Dong H., Carethers J. M. (2010) TGF-β downregulates PTEN via activation of NF-κB in pancreatic cancer cells. Am. J. Physiol. Gastrointest Liver Physiol. 298, G275–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Huang J., Kontos C. D. (2002) PTEN modulates vascular endothelial growth factor-mediated signaling and angiogenic effects. J. Biol. Chem. 277, 10760–10766 [DOI] [PubMed] [Google Scholar]
- 58. Perry B., Banyard J., McLaughlin E. R., Watnick R., Sohn A., Brindley D. N., Obata T., Cantley L. C., Cohen C., Arbiser J. L. (2007) AKT1 overexpression in endothelial cells leads to the development of cutaneous vascular malformations in vivo. Arch. Dermatol. 143, 504–506 [DOI] [PubMed] [Google Scholar]
- 59. Graupera M., Guillermet-Guibert J., Foukas L. C., Phng L. K., Cain R. J., Salpekar A., Pearce W., Meek S., Millan J., Cutillas P. R., Smith A. J., Ridley A. J., Ruhrberg C., Gerhardt H., Vanhaesebroeck B. (2008) Angiogenesis selectively requires the p110α isoform of PI3K to control endothelial cell migration. Nature 453, 662–666 [DOI] [PubMed] [Google Scholar]
- 60. Cushing L., Kuang P. P., Qian J., Shao F., Wu J., Little F., Thannickal V. J., Cardoso W. V., Lü J. (2011) miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell. Mol. Biol. 45, 287–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roderburg C., Urban G. W., Bettermann K., Vucur M., Zimmermann H., Schmidt S., Janssen J., Koppe C., Knolle P., Castoldi M., Tacke F., Trautwein C., Luedde T. (2011) Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 53, 209–218 [DOI] [PubMed] [Google Scholar]
- 62. Wang F., Wang X. S., Yang G. H., Zhai P. F., Xiao Z., Xia L. Y., Chen L. R., Wang Y., Wang X. Z., Bi L. X., Liu N., Yu Y., Gao D., Huang B. T., Wang J., Zhou D. B., Gong J. N., Zhao H. L., Bi X. H., Yu J., Zhang J. W. (2012) miR-29a and miR-142–3p down-regulation and diagnostic implication in human acute myeloid leukemia. Mol. Biol. Rep. 39, 2713–2722 [DOI] [PubMed] [Google Scholar]