

Background: The mechanism by which SHP2 mutations cause LEOPARD syndrome is poorly understood.
Results: LEOPARD syndrome mutations impair SHP2 activity but increase its propensity for an open and active conformation.
Conclusion: LEOPARD syndrome SHP2 mutants bind preferentially to upstream activators to prolong substrate turnover, thus engendering gain-of-function phenotypes.
Significance: The study provides a framework for understanding how individual SHP2 mutations cause diseases.
Keywords: Allosteric Regulation, Enzyme Structure, ERK, Phosphotyrosine Signaling, Tyrosine Protein Phosphatase (Tyrosine Phosphatase), LEOPARD Syndrome
Abstract
SHP2 is an allosteric phosphatase essential for growth factor-mediated Ras activation. Germ-line mutations in SHP2 cause clinically similar LEOPARD and Noonan syndromes, two of several autosomal-dominant conditions characterized by gain-of-function mutations in the Ras pathway. Interestingly, Noonan syndrome SHP2 mutants are constitutively active, whereas LEOPARD syndrome SHP2 mutants exhibit reduced phosphatase activity. How do catalytically impaired LEOPARD syndrome mutants engender gain-of-function phenotypes? Our study reveals that LEOPARD syndrome mutations weaken the intramolecular interaction between the N-SH2 and phosphatase domains, leading to a change in SHP2 molecular switching mechanism. Consequently, LEOPARD syndrome SHP2 mutants bind upstream activators preferentially and are hypersensitive to growth factor stimulation. They also stay longer with scaffolding adapters, thus prolonging substrate turnover, which compensates for the reduced phosphatase activity. The study provides a solid framework for understanding how individual SHP2 mutations cause diseases.
Introduction
The Src homology 2 (SH2)3 domain-containing protein-tyrosine phosphatase-2 (SHP2), encoded by the Ptpn11 gene, is an essential signal transducer downstream of growth factor and cytokine receptors (1). Biochemical and genetic evidence places SHP2 upstream of Ras, an essential component of the signaling pathway that underlies growth factor/cytokine-induced cell proliferation and survival. Importantly, SHP2 phosphatase activity is required for full activation of the Ras extracellular signal-regulated kinase (ERK1/2) cascade. The critical role of SHP2 in cell physiology is underscored by germ-line mutations within SHP2, which are linked to several human diseases including 50% of Noonan syndrome (NS) and 90% of LEOPARD (an acronym for its clinical features of multiple lentigines, ECG conduction abnormalities, ocular hypertelorism, pulmonic stenosis, abnormalities of genitalia, retardation of growth, and deafness) syndrome (LS) (2, 3). Both disorders cause short statures, facial dysmorphism, heart defects, and predisposition to hematologic abnormalities including juvenile myelomonocytic leukemia. In addition, somatic mutations in SHP2 also occur in other types of leukemia (4) as well as solid tumors (5).
SHP2 is a ubiquitously expressed cytoplasmic protein-tyrosine phosphatase (PTP) bearing two tandemly arranged SH2 domains at its N-terminal end (Fig. 1A). Under basal conditions, SHP2 is inactive due to autoinhibition of the PTP domain by the N-SH2 domain (6–8). Via its SH2 domains, SHP2 directly associates with tyrosine-phosphorylated motifs in growth factor receptors (e.g. EGF receptor) or, more commonly, in scaffolding proteins. These binding interactions guide SHP2 subcellular localization to its physiological substrates. Moreover, recruitment of SHP2 to these docking sites is also intimately coupled to SHP2 activation (6, 7, 9).
FIGURE 1.

Structures of the wild-type SHP2 and Y279C mutant. A, shown is a schematic diagram of SHP2 structure. B, shown is the overall structure of the wild-type SHP2. N-SH2, C-SH2, and PTP domain are, respectively, colored in yellow, green, and light blue. The catalytic P-loop is highlighted in red, and the other four important loops are depicted in magenta. The dashed lines represent three undetermined disordered loops in the crystal structure. C, the structure of SHP2/Y279C mutant shows that it is in the “closed” conformation with D′E-loop (spheres) blocking the active site. D, 2Fo − Fc (contoured at 1Σ, blue mesh) and Fo − Fc map (contoured at −3Σ, red mesh) around Tyr-279 validate the Y279C mutation. These density maps were calculated after the refinement of wild-type structure using diffraction data of the Y279C mutant. E, Y279C mutation alters the substrate recognition surface constituted by Tyr-279, Lys-364, and Lys-366, which is represented in mesh for the wild type and solid surface for Y279C, respectively. The bound Tyr(P) (shown in spheres) was modeled by superimposing the SHP1-phosphopeptide complex (PDB ID 1FPR) onto the wild-type SHP2 structure. F, Y279C mutation decreases the interactions of the N-SH2 with the PTP domain. The important interface residues experiencing decreased interactions are shown in stick (yellow for wild-type SHP2 and green for Y279C mutant).
The mechanism of the N-SH2 domain-mediated SHP2 autoinhibition was revealed by the crystal structure of SHP2 (residues 1–527) lacking the C-terminal 66 residues (10). The structure shows that the inactive form of SHP2 adopts a closed conformation with the N-SH2 domain interacting extensively with the PTP domain, directly blocking the phosphatase catalytic site. Thus, the N-SH2 domain has two nonoverlapping ligand binding sites: an intermolecular interaction with phosphopeptides from docking proteins and an intramolecular interaction with the PTP domain. A comparison of the SHP2 structure with those of the isolated N-SH2 domain both in the absence of and in complex with a Tyr(P) peptide suggests that these two sites may communicate with negative cooperativity and are linked via an allosteric change between two different SH2 domain conformations: an active Tyr(P) peptide-bound state and an inactive PTP domain interacting state. Engagement of the SH2-domain to specific Tyr(P)-based docking sequences is suggested to diminish its inhibitory interaction with the PTP domain, leading to SHP2 catalytic activation in the open conformation.
Although SHP2 mutations are associated with a number of developmental and neoplastic disorders, the precise mechanism by which mutations in SHP2 cause diseases remains poorly understood. The vast majority of NS or neoplasia-associated mutations alter amino acid residues clustering at the interface between the N-SH2 and PTP domains. The PTP activity of either NS- or leukemia-associated SHP2 mutants is greater than that of wild type, indicating that these substitutions disrupt the autoinhibitory interface between the N-SH2 and catalytic domains to favor the open, active conformation, resulting in gain-of-function (GOF) effects (11–15). In contrast, mutations associated with LS reside in the PTP domain, cause reduced phosphatase activity, and are, therefore, deemed loss-of-function defects (14, 16, 17). These genetic and biochemical findings generated an enigma; How do mutations that provoke opposite effects on SHP2 phosphatase activity cause phenotypically similar disorders? To address this question, we hypothesize that pathogenic mutations alter not only SHP2 phosphatase activity but also its molecular switching mechanism to drive disease outcomes, and thus detailed understanding of the structure and function of SHP2 will yield new insights into the molecular features that underlie the diseases. Our results reveal that although LS mutants are catalytically impaired and adopt a closed conformation, they have an increased propensity for the open conformation. As a result, the LS mutants bind upstream activators preferentially and stay longer with the scaffolding adapters thus prolonging specific substrate turnover, which compensate for the reduced phosphatase activity. Thus, catalytically impaired LS-associated SHP2 mutants may engender GOF phenotypes.
EXPERIMENTAL PROCEDURES
Materials
Pepsin was obtained from Sigma. Mass Spectrometry grade H2O and acetonitrile were from Burdick and Jackson. D2O (99.9 atom %D) was form Aldrich. Antibodies for ERK1/2, phospho-ERK1/2, and Gab1 were from Cell Signaling Technology.
Protein Expression and Purification
SHP2/1–528 was cloned into pET-21a+ vector with His6 tag at the C terminus. Mutant SHP2 was generated using the QuikChange mutagenesis kit (Stratagene). Wild-type and mutant SHP2s were expressed in Escherichia coli BL21(DE3) and purified by nickel-nitrilotriacetic acid-agarose (Qiagen) followed by sequential chromatography of HiPrep 26 desalting column (GE Healthcare), cation exchange column packed with SP-Sepharose (GE Healthcare), and Superdex 75 gel filtration column (GE Healthcare). The purities were determined to be greater than 95% by SDS-PAGE and Coomassie staining.
Crystallization, Data Collection, and Structure Determination
The crystal were grown at 20 °C in the hanging drops containing 1.5 ml of protein solution (8 mg/ml in 20 mm Tris-HCl (pH 7.8), 50 mm NaCl, 2 mm DTT, and 1 mm EDTA) and 1.5 ml of reservoir solution (20% PEG3350, 300 mm KCOOH). The addition of spermidine (100 mm, 0.3 ml) or CaCl2 (100 mm, 0.3 ml) to the drops improved the growth of well ordered crystals for wild-type SHP2 and the Y279C mutant, respectively. The crystals were transferred into the cryoprotectant buffer (30% PEG3350, 200 mm KCOOH, 20 mm Tris-HCl (pH 7.8), 50 mm NaCl, 2 mm DTT, and 1 mm EDTA) and flash-frozen by liquid nitrogen. Data were collected at 19-BM beamline at the Advanced Photon Source (APS) and were processed with HKL3000 (18). The data were collected to 2.3 Å resolution in the P21212 space group. The wild-type SHP2 structure was solved by molecular replacement with Molrep (19) using the coordinates of chain A in the reported SHP2 structure (PDB ID 2SHP) (10) as the search model, and the Y279C structure was solved by molecular replacement using the refined SHP2 structure. The structure refinement was carried out iteratively with phenix.refine in the PHENIX software suite (20). The refined structure had none (for the wild type) or 0.2% (for the Y279C mutant) of the residues in the disallowed regions of the Ramachandran plot.
Molecular Dynamics Simulation and Interface Interaction Energy Calculation
The crystal structure of WT and Y279C as well as the structure of E76K generated in Coot by in silico mutation of WT were used to set up three simulation systems with xleap program in Amber9 software package (21). Missing atoms and hydrogens were added, then the protein was neutralized by Na+ or Cl− counterions and solvated in a truncated octahedral box of TIP3P water with 9 Å buffer between the solute and box edge, and the amber ff03 force field was employed to generate the topology and parameter files. Each system was relaxed by two successive energy minimizations in sander program and followed by heating from 0 to 300 K for 50 ps with a restrain force constant of 10.0 kcal/mol·Å in the pmemd program. A 30-ns MD simulation in NPT ensemble (p = 1 atm, T = 300 K) was performed with the pmemd program. The SHAKE algorithm was employed, and the time step was set to 2.0 fs. Particle mesh Ewald (PME) method was used to treat the long range electrostatic interactions with a default cutoff of 10.0 Å. Both energies and coordinates were saved every 2 ps.
Combined MD simulations and continuum solvation model MM-GBSA (molecular mechanics-generalized born surface area) (22) was utilized to decompose the free energy into individual residue pairs, and the interdomain interaction energy was calculated by adding up the energy of all interface residue pairs. In detail, the energy decomposition was performed on a pairwise per-residue basis (DCTYPE = 3); the gas-phase energy and polar component of solvation free energy were calculated using molecular mechanics (MM = 1) and Onufriev's Generalized Born (GB) method (IGB = 2); the non-polar component was determined through the solvent-accessible surface area (GBSA = 2); the interior and exterior dielectric constants were set to 1 and 80, respectively. The entropy contribution was ignored (NM = 0) because it should be almost identical among these three systems. The calculations were performed on 1461 conformational snapshots extracted from MD trajectory from 0.8 to 30 ns at a time interval of 20 ps.
H-D Exchange Mass Spectrometry
Stock solutions of SHP2-WT (650 μm), E76K (568 μm), and Y279C (568 μm) (1–528) were prepared in a 1H2O-based buffer (pH 7.8). Deuterium exchange was initiated by dilution of each enzyme 20-fold in a similar deuterium 2H2O buffer (pD 7.8) held at ambient temperature. At set deuterium exchange time points (from 10 s to 1 h) the reaction was quenched by the addition of a cold 1H2O-based 100 mm sodium phosphate buffer (pH 2.3) at equal volume. For “peptide-based” mass spectrometry analysis, the quench solution contained solvated pepsin endoproteinase (Sigma) that would make a (1.5:1 (w/w) pepsin:SHP2 enzyme) ratio during digestion. Quenched sample was allowed to digest on ice for 4 min before injection.
Sample peptides were loaded by autosampler onto a XBridge C18 2.5-Micron 2.1 × 50 mm (Waters) column that was submerged under ice. A Surveyor MS pump (Finnigan) was used to generate the chromatographic gradients. Peptides were separated in time using a steep gradient of acetonitrile (10–35% in 7.5 min) and electrosprayed into an LTQ mass spectrometer (Finnigan). Sequest (Thermo) was used to identify SHP2 peptic peptides. Peptides were accepted for analysis based upon XCorr value significance and identification in multiple sample runs. All peptide-based samples were manually prepared and run in triplicate with a general standard deviation per time point of <0.2Da. Unbiased peptide precursor ion peak envelope centroiding was performed using HX-express software (23). Significant deuterium exchange differences in SHP2 mutants relative to WT were mapped onto the SHP2 crystal structure in a “heat map” format.
Kinetic Analysis of SHP2-catalyzed Reaction
Initial rate measurements for the enzyme-catalyzed hydrolysis of para-nitrophenyl phosphate (pNPP) were conducted at 25 °C in a pH 7.0 buffer of 50 mm 3,3-dimethylglutarate containing 1 mm DTT and 1 mm EDTA with an ionic strength of 0.15 m adjusted by the addition of NaCl. Assay mixtures of 200 μl in total volume were set up in a 96-well plate. A substrate concentration range from 0.2 to 5 Km was used to determine the kcat and Km. Reactions were started by the addition of an appropriate amount of wild-type or mutant SHP2. The reaction mixtures were quenched with 50 μl of 5 m sodium hydroxide, and the absorbance at 405 nm was read using a plate reader. The steady state kinetic parameters were determined from a direct fit of the data to the Michaelis-Menten equation using SigmaPlot.
Inhibition of the SHP2 PTP Domain by the N-SH2 Domain
PTP activity was assayed using pNPP as a substrate in a pH 7.0 buffer containing 50 mm 3,3-dimethylglutarate, 1 mm EDTA, 150 mm NaCl at 25 °C. The assays were performed in 96-well plates with a final reaction volume of 0.2 ml. The reaction was initiated by the addition of enzyme (catalytic domain of wild-type SHP2 or Y279C) to a reaction mixture containing pNPP (2 mm for the wild-type or 10 mm for Y279C) with various concentrations of the N-SH2 domain. For Ki determination, pNPP concentration was varied, whereas the N-SH2 domain was fixed at three different concentrations. The reaction rate was measured using a SpectraMax Plus 384 Microplate Spectrophotometer (Molecular Devices). Data fitting was performed using SigmaPlot Kinetics module.
Activation of the Full-length SHP2 by the SH2 Domain Ligand
The activation of the full-length SHP2 was carried out under the kcat/Km condition with pNPP as a substrate (with the concentration of pNPP more than 10-fold lower than the respective Km values). For wild-type SHP2, pNPP concentration was 0.2 mm, and the protein concentration was 1.75 μm. The full activation control was performed using 160 nm E76K mutant and 0.2 mm pNPP. For the Y279C SHP2 mutant, pNPP concentration was 0.5 mm, and the protein concentration was 3.5 μm. The full activation control was performed using 1.88 μm E76K/Y279C double mutant and 0.5 mm pNPP. Various amounts of the phosphonodifluoromethyl phenylalanine (F2pmp)-derived IRS1 peptide SLN(F2Pmp)IDLDLVK up to 100 μm final concentration were added to the reaction. The percent activity was calculated with normalized linear reaction rates (rate per μm protein) with respect to the normalized control rate. Kd was obtained by fitting the data to Equation 1,
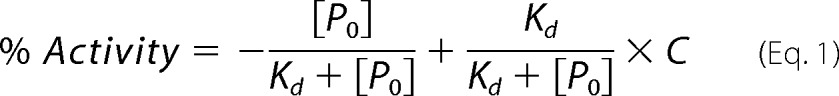 |
where P0 is the total concentration of the activating peptide, Kd is the dissociation constant of the SHP2·peptide complex, and C is the ratio of the basal and fully activated SHP2 activity.
Cell Culture Studies
HEK293 cells were cultured at 37 °C and 5% CO2 in Dulbecco's modified Eagle's medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen) and 1% penicillin-streptomycin. WT or mutant SHP2 (E76K, D61Y, Y279C, T468M) cloned in the mammalian expression vector pCN-HA, a modified version of pcDNA3.1 that generated proteins with N-terminal HA tag, were transfected into HEK293 cells using Lipofectamine 2000 (Invitrogen). Twenty-four hours post-transfection cells were serum-starved for an additional 6 h and then either left unstimulated or stimulated with EGF (5 or 50 ng/ml) for various times (0, 5, 10, 30 min). All growth factor were obtained from Calbiochem. Cells were lysed in 1 ml of lysis buffer (1.0% Nonidet P-40, 50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 5 mm EDTA, 2 mm NaVO3) plus a protease inhibitor mixture (Roche Applied Science) and clarified in a microcentrifuge. Protein concentrations were measured by using Coomassie protein reagent. Proteins were resolved by SDS-PAGE.
RESULTS
LS-associated SHP2 Mutants Are Catalytically Impaired
We determined the kinetic parameters for the hydrolysis of pNPP by the full-length (residues 1–528) and catalytic domain (residues 246–547) of wild-type SHP2, the most representative GOF mutants D61Y and E76K, and 2 of the most recurrent LS mutants, Y279C and T468M. As shown in Table 1, the activity of the full-length SHP2 was less than 5% that of the catalytic domain, in accord with the structural observation that the full-length SHP2 is in an autoinhibited, closed conformation. Consistent with the expectation that the GOF mutants are in an open, activated state, full-length D61Y and E76K exhibit kinetic parameters indistinguishable from those of the wild-type catalytic domain. In contrast, the kcat values for the catalytic domains of Y279C and T468M were 26- and 46-fold lower than that of the wild-type counterpart, indicating that LS-associated SHP2 mutants are catalytically impaired. Interestingly, the kcat values for the full-length Y279C and T468M were still 5- and 13-fold lower than those of their corresponding catalytic domains. These results suggest that full-length Y279C and T468M exist in a closed, autoinhibited conformation. Consistent with this prediction, full activity was restored to SHP2/Y279C when Glu-76 was replaced by a Lys, which likely disrupts the autoinhibitory mechanism.
TABLE 1.
Kinetic parameters of wild-type and mutant SHP2 with pNPP as a substrate
FL, full-length; CD, catalytic domain.
| SHP2 | kcat | Km |
|---|---|---|
| s−1 | mm | |
| WT/CD | 6.4 ± 0.4 | 3.0 ± 0.3 |
| WT/FL | 0.31 ± 0.02 | 2.7 ± 0.2 |
| E76K/FL | 6.8 ± 0.3 | 3.4 ± 0.2 |
| D61Y/FL | 6.0 ± 0.5 | 2.7 ± 0.1 |
| Y279C/CD | 0.25 ± 0.05 | 11.3 ± 0.5 |
| Y279C/FL | 0.052 ± 0.003 | 7.6 ± 0.4 |
| E76K/Y279C/FL | 0.34 ± 0.04 | 8.4 ± 0.6 |
| T468M/CD | 0.14 ± 0.002 | 2.4 ± 0.1 |
| T468M/FL | 0.011 ± 0.001 | 2.1 ± 0.2 |
Y279C Exists in a Closed Conformation, Albeit with Decreased Intramolecular Interaction between N-SH2 and PTP Domains
To gain insight into LS mutations, we solved the structures of both wild-type and Y279C SHP2 (residues 1–528) at 2.3 Å resolution. Data collection and structure refinement statistics are summarized in Table 2. Both structures belong to the P21212 space group with one molecule per asymmetric unit. The wild-type SHP2 structure (Fig. 1B) was very similar to that reported previously (10). The D′E loop and the adjacent D′ and E β strands of the N-SH2 domain insert deeply into the catalytic cleft of the PTP domain, interacting with residues of the active site loops (i.e. the phosphate binding or P-loop, the Tyr(P) recognition loop, and the Q loop) to maintain SHP2 in the closed, autoinhibited conformation. Specifically, Asp-61 and Tyr-62 in the D′E loop mimic the interactions of a Tyr(P) substrate with the catalytic domain in which the side chain of Asp-61 forms water-mediated hydrogen bonds to the main chain amides of the P-loop and the side chains of the catalytic Cys-459, Arg-362, and Arg-465, whereas the side chain of Tyr-62 interacts with Tyr-279 within the Tyr(P)-recognition loop. These interactions sterically block access of substrates to the catalytic site and inhibit SHP2 phosphatase activity.
TABLE 2.
Data collection and refinement statistics
Each dataset was collected from a single crystal. Values in parentheses are for the highest resolution shell.
| SHP2/WT | SHP2/Y279C | |
|---|---|---|
| Data collection | ||
| Space group | P21212 | P21212 |
| Cell dimensions | ||
| a, b, c (Å) | 54.85, 221.10, 40.36 | 55.14, 225.12, 40.91 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 2.30 (2.34-2.30) | 2.30 (2.34-2.30) |
| Rmerge | 0.087 (0.628) | 0.081 (0.658) |
| I/σI | 25.3 (2.9) | 31.1 (1.9) |
| Completeness (%) | 95.6 (92.1) | 99.8 (100) |
| Redundancy | 4.9 (4.9) | 7.3 (4.2) |
| Refinement | ||
| Resolution (Å) | 2.30 | 2.30 |
| No. reflections | 21,761 | 23,522 |
| Rwork/Rfree | 0.196/0.255 | 0.209/0.266 |
| No. atoms | ||
| Protein | 4024 | 4040 |
| Water | 150 | 183 |
| B-factors | ||
| Protein | 48.1 | 49.4 |
| Water | 46.0 | 45.7 |
| Root mean square deviations | ||
| Bond lengths Å) | 0.008 | 0.008 |
| Bond angles (°) | 1.143 | 1.115 |
Consistent with the prediction from our kinetic analyses, Y279C also existed in the closed, autoinhibited form (Fig. 1C). This is contrary to early molecular modeling predictions that LS mutants are in an open state (17). Although the Y279C mutation can be unambiguously identified by the strong negative difference map around the phenol side chain (Fig. 1D), the overall Y279C structure was almost identical to that of the wild-type SHP2, with a root mean square deviation of 0.44 Å for 498 Cα atoms. A closer examination of both the wild-type and Y279C structures uncovered the molecular basis of the impaired activity for the Y279C mutant. Tyr-279 was located in the Tyr(P) recognition loop and was well conserved among the PTPs. In PTP1B, the cognate Tyr-46 sets the depth of the catalytic cleft, and its side chain π-π stacks with the benzene ring of Tyr(P) to assist substrate recognition and dephosphorylation (24). Removal of the tyrosine side chain would diminish the interaction between SHP2 and Tyr(P) substrate. To illustrate how Y279C mutation disrupts the catalytic pocket, we superimposed the structures of SHP1-phosphopeptide complex (PDB ID 1FPR) and Y279C onto the wild-type SHP2 structure. As shown in Fig. 1E, the mesh surface, formed by Tyr-279, Lys-364, and Lys-366 in SHP2, perfectly complements the surface of Tyr(P), holding it in position for dephosphorylation. The solid surface, formed by Cys-279, Lys-364, and Lys-366 in the Y279C mutant, had little interaction with Tyr(P). Thus, consistent with the kinetic observations, the Y279C mutation altered the molecular surface of the active site, diminished substrate binding, and impaired catalytic activity. Indeed, a similar decrease in kcat and increase in Km was observed when Tyr-46 in PTP1B was replaced by an Ala (25).
The structures also unveil that substitution of Tyr-279 by a Cys abrogates the interaction of the phenol side chain with Asp-61 and Tyr-62 in the D′E loop of N-SH2. Moreover, the substitution also affected the positioning of several nearby residues (Lys-364, Lys-366, Arg-362, His-426) involved in binding the N-SH2 domain (Fig. 1F). The loss of the phenol group dismissed the van der Waals and polar interactions with Lys-364 and Lys-366, shifting the terminal amino group of Lys-364 ∼2.7 Å away from the D′E loop, which further displaced the guanidinium of Arg-362 ∼2.4 Å and the adjacent imidazole ring of His-426 ∼2.2 Å away from the D′E loop. The combined effect was decreased van der Waals and polar interactions between the D′E loop of N-SH2 and the Tyr(P) loop, WPD loop, and residues 362–366 in the PTP domain.
To provide further support for the prediction that the interaction between the N-SH2 and PTP domains is weakened in Y279C, MM-GBSA energy calculations based on molecular dynamics simulation were employed to calculate the interface interaction energy for both the wild-type SHP2 as well as the Y279C and E76K mutants. This combined computation method has been widely used in free energy calculations and identification of key interactions during complex formation or conformation changes (22). In the current study the free energy of each system was calculated using MM-GBSA and decomposed into pairwise residues. The interdomain interaction energy was then determined by adding up the energy of all interface residue pairs. E76K was included in the calculation as a positive control because it is believed to be in the open conformation. As expected, the interaction strength for the whole N-SH2/PTP interface followed the order of E76K < Y279C < WT (Fig. 2A). The N terminus of N-SH2/PTP and βB-loop-βC/PTP had the same interaction energy among all three systems. The differences for the total energy arise primarily from βD′- D′E -loop-βE/PTP (Fig. 2B) and βF-FB-loop-αB/PTP (Fig. 2C) sub-interfaces. Of note, the Y279C mutation alone decreased the interaction energy by ∼2 kcal/mol (Fig. 2D), and this perturbation further resulted in ∼18 kcal/mol interaction energy loss at the βD′-loop-βE/PTP sub-interface, agreeing well with the decreased interactions revealed by the crystal structures. Thus, interface interaction energy calculation supports the conclusion that the intramolecular interaction between the N-SH2 and PTP domain in Y279C is weakened compared with the wild-type SHP2. Collectively, the structural and computational data reveal why Y279C possesses intrinsically lower catalytic activity and suggests that the interaction between the N-SH2 and PTP domains is weakened in Y279C such that it may have a higher propensity to transition from a closed, autoinhibited conformation to an open, activated form.
FIGURE 2.

Calculated interface interaction energies using the MM-GBSA method. The interaction energy for wild-type SHP2, E76K, and Y279C mutants are represented respectively in black, red, and blue lines in each panel. A, shown is the interaction energy for the whole N-SH2/PTP interface. B, shown is the interaction energy for the βD′-loop-βE/PTP sub-interface. C, shown is the interaction energy for the βF-loop-αB/PTP sub-interface. D, shown is the interaction energy of residue 279 with neighboring interface residues in N-SH2 domain.
LS Mutants Exhibit Increased Propensity for the Open Conformation
To provide additional evidence for the decreased intramolecular interaction between the N-SH2 domain and the PTP domain in Y279C, we measured the ability of the isolated N-SH2 to inhibit the phosphatase activity of the wild-type and Y279C catalytic domains. The N-SH2 domain acts as a competitive inhibitor of the SHP2 catalytic domain-catalyzed pNPP hydrolysis (Fig. 3, A and B). The competitive nature is consistent with the structural observation that the N-SH2 domain binds the PTP domain and prevents substrate access to the active site. Consistent with the observed weaker interaction between N-SH2 and the PTP domain in Y279C, the N-SH2 domain inhibited the Y279C catalytic domain with a Ki (30.6 ± 0.2 μm) that was 10-fold higher than that for the wild-type catalytic domain (2.9 ± 0.2 μm). As a control, the N-SH2 domain containing the E76K substitution displays no inhibitory activity against SHP2 catalytic domain even at 100 μm N-SH2/E76K concentration, which is in line with the expectation that the full-length E76K is in the open conformation.
FIGURE 3.
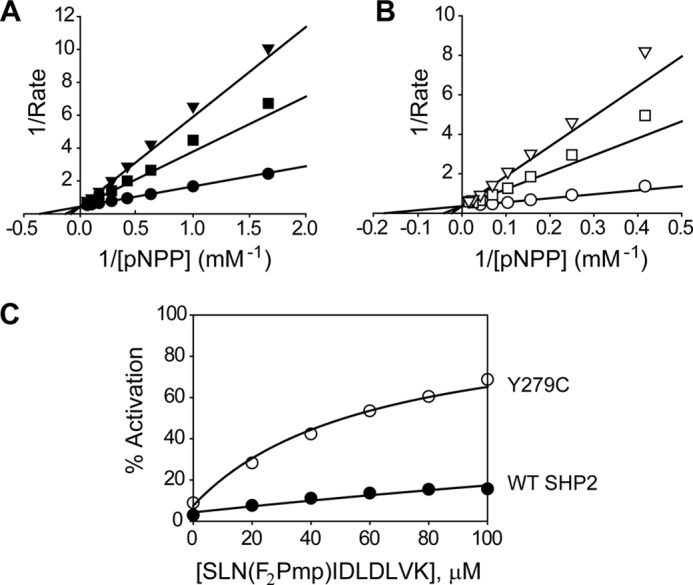
Kinetic properties of wild-type SHP2 and the Y279C mutant. A, the isolated N-SH2 domain acts as a competitive inhibitor against the wild-type SHP2 catalytic domain with a Ki of 2.9 ± 0.2 μm. The concentrations of the N-SH2 domain were 0 (●), 5 (■), and 10 (▾) μm. B, the isolated N-SH2 domain acts as a competitive inhibitor against the catalytic domain of Y279C with a Ki of 30.6 ± 0.2 μm. The concentrations of the N-SH2 domain were 0 (○), 100 (□), and 200 (▿) μm, respectively. To obtain the Ki values, data were fitted to the competitive inhibition mode using the Kinetic module of SigmaPlot. C, shown is activation of full-length wild-type SHP2 (●) and the Y279C mutant (○) by the nonhydrolyzable N-SH2 domain ligand, SLN(F2Pmp)IDLDLVK. To obtain the Kd values, the data were fitted to Equation 1 described under “Experimental Procedures” using SigmaPlot.
To provide more direct evidence for the increased “openness” for the SHP2 mutants, we examined the overall solution dynamic properties of the wild-type, E76K, and Y279C mutant SHP2 using hydrogen/deuterium exchange mass spectrometry (H/DX-MS). H/DX-MS allows the dynamics and conformation of a protein to be investigated by measuring the exchange of backbone amide hydrogens with the bulk solvent (26). The results showed that in comparison with the wild-type, a number of peptides in E76K, located in the interface between the N-SH2 domain and the PTP domain, display a significant increase in deuterium incorporation (Fig. 4). Within the N-SH2 domain, these peptides reside in the D′E loop and adjacent B, C, D, D′, E, and F β strands, structural elements known to participate in binding the PTP domain. Within the PTP domain, these peptides represent the catalytic loops at the active site, including the P-loop, the Tyr(P) recognition loop, loop-βF-loop, and the Q loop. Given the lack of inhibition of the PTP domain by N-SH2/E76K, the observed increase in deuterium uptake surrounding the N-SH2 and PTP binding site suggests that the binding interface is solvent-exposed, providing the first direct evidence that E76K exists in an open, active conformation. Hydrogen exchange data showed that Y279C also displays enhanced deuterium uptake in the interface region between the N-SH2/PTP domains, albeit to a lesser degree than the peptides observed in E76K (Fig. 4). This result indicates that the Tyr-279-to-Cys mutation causes dynamic perturbations and increased conformational flexibility to residues in the N-SH2/PTP binding interface and supports our finding that the N-SH2 domain of Y279C has a lower affinity for its PTP domain. Moreover, the result also validates our hypothesis that the Y279C mutant has an increased tendency to adopt the open conformation.
FIGURE 4.

H/DX-MS experiments reveal that, relative to the WT SHP2, E76K, and Y279C display increased conformational dynamics in solution especially within the interface region between the N-SH2 and PTP domains. For each protein, a total of 40 peptides were identified that cover 91% of the sequence of SHP2. A, shown is a representative time course for deuterium uptake for peptides located at the interface of N-SH2/PTP domain. The sequence with start and end numbers as well as corresponding secondary structure elements in panel B were shown for each peptide. E76K and Y279C peptides attain significantly more deuterium exchange than those in wild-type SHP2 at different time point. B, shown is a heat map mapped on the WT structure showing significantly increased deuterium exchange for both the E76K and Y279C mutants relative to the WT at the 1 min time point of deuterium labeling. The heat map was generated by color coding the peptides in E76K or Y279C based on the extent of increase in deuterium exchange (green for a 0.4–0.7 Da increase, yellow for a 0.7–1.0 Da increase, orange for a 1.0–1.3 Da increase, and red for a >1.3 Da increase) relative to those in WT, which are depicted as all gray. The color scale was shown internally in the figure. The catalytic cysteine (Cys-459) is labeled for active site orientation. The secondary structure elements were labeled to easily locate those peptides shown in panel A.
LS-associated SHP2 Mutants Exhibit Higher Affinity for Tyr(P) Motifs in Scaffolding Proteins
Our structural and biochemical observations suggest that whereas E76K exists in an open and active state, Y279C is catalytically impaired and adopts a closed conformation. However, the data also show that the Y279C mutation weakens the interaction between the N-SH2 domain and the PTP domain. Thermodynamic and negative cooperativity considerations require that the diminished interaction of the N-SH2 domain with the PTP domain must lead to an increase in its binding affinity for Tyr(P) ligands. The prediction is that the Y279C mutant would be more easily activated by engagement of its N-SH2 domain with Tyr(P) motifs in growth factor receptors and scaffolding proteins. To test this hypothesis, we measured the ability of an N-SH2 domain ligand to activate the wild-type SHP2 as well as the Y279C mutant. To avoid phosphopeptide hydrolysis by the PTP domain, a nonhydrolyzable N-SH2 ligand (SLNpYIDLDLVK, from IRS1 Tyr(P)-1172) was utilized in which the Tyr(P) was replaced by F2Pmp. SLN(F2Pmp)IDLDLVK displays no inhibitory activity against the SHP2 catalytic domain at 100 μm and has a Kd of 2.7 μm for the isolated N-SH2 domain, measured by isothermal titration calorimetry. From the concentration dependence of the N-SH2 ligand-induced SHP2 activation, the affinity of SLN(F2Pmp)IDLDLVK for the N-SH2 domain of Y279C (60.5 ± 4.2 μm) was found more than 10-fold higher than that for the wild-type SHP2 (631 ± 51 μm) (Fig. 3C). Thus, Y279C has a higher affinity for the upstream Tyr(P) motifs and is preferentially (compared with wild-type SHP2) activated by the scaffolding proteins.
LS-associated SHP2 Mutants Engender GOF Phenotypes
SHP2 is required for EGF signaling that controls semilunar valvulogenesis of the heart (27). Interestingly, NS and LS caused by SHP2 mutations share multiple phenotypic features, including cardiac defects (3). A major SHP2 signaling partner in EGF pathway is Grb2-associated binder-1 (Gab1), a docking protein that contains an N-terminal pleckstrin homology domain and several SH3 and SH2 domain binding sites. Upon EGF stimulation, Gab1 is recruited to the cell membrane, becomes tyrosyl phosphorylated, and acts as docking sites for several signaling proteins, including the p85 regulatory subunit of phosphatidylinositol 3-kinase, Shc, Grb2, and SHP2. The SHP2/Gab1 interaction, through the SHP2 N-SH2 domain and a Tyr(P)-based activation motif encompassing residues Tyr-627 and Tyr-659 in Gab1, is necessary for full activation of the Ras-ERK1/2 cascade upon EGF stimulation (28, 29).
A higher affinity of the N-SH2 domain for Tyr(P) motifs may translate into longer resident time of LS mutants on the scaffolding adapters and prolong substrate turnover by the LS mutants, which could compensate for the reduced phosphatase activity. Furthermore, increased affinity of the N-SH2 domain for Tyr(P) sequences within SHP2 docking proteins should also make the LS mutants hypersensitive to growth factor stimulation. Therefore, we hypothesized that catalytically impaired LS-associated SHP2 mutants may engender GOF phenotypes. To test this hypothesis, we examined the effects of the wild-type SHP2 as well as GOF (D61Y and E76K) and LS (Y279C and T468M) mutants on EGF signaling in HEK293 cells, a well established system for studying the role of SHP2 in receptor-tyrosine kinase signaling (17, 30, 31). Consistent with the observation that the N-SH2 domain of LS mutants has higher affinity for Tyr(P) ligands, the Y279C and T468M mutants as well as the D61Y and E76K mutants, which are known to favor the open conformation, more readily formed complexes with Gab1 in the presence of EGF than did the wild-type SHP2 (Fig. 5A). Importantly, the LS mutants, like D61Y and E76K, exhibit prolonged binding to Gab1, lasting at least 30 min after EGF treatment. Thus the LS-associated mutant SHP2 proteins remain in the Gab1 complex and propagate signals at times when wild-type SHP2 does not. Given the absolute requirement of the SHP2/Gab1 interaction for full activation of the Ras-ERK1/2 cascade, we then investigated whether the LS mutants are capable of activating the ERK1/2 pathway. Compared with wild-type SHP2-expressing cells, ERK1/2 activity was higher and sustained up to 30 min after EGF stimulation in cells expressing GOF (E76K and D61Y) as well as the LS (Y279C and T468M) mutants (Fig. 5B). Moreover, increased ERK1/2 activation was also observed upon IGF stimulation in HEK293 cells expressing these disease-associated SHP2 mutants (data not shown).
FIGURE 5.

LS SHP2 mutants promote EGF-induced ERK1/2 pathway activation. A, LS SHP2 mutants preferentially associate with and stay longer with Gab1. HEK293 cells expressing HA-tagged wild-type SHP2, E76K, D61Y, and LS mutants Y279C and T468M were starved overnight then were left unstimulated (0 min) or stimulated with EGF (50 ng/ml) for different times (5, 10, and 30 min), lysed, and immunoprecipitated (IP) with either anti-HA or anti-Gab1 antibodies. The immunoprecipitates were immunoblotted with either anti-Gab1 or anti-HA antibodies, respectively. B, shown is sustained up-regulation of the ERK1/2 activation by the LS SHP2 mutants. HEK293 cells expressing wild-type or mutant SHP2 were either left unstimulated (0 min) or stimulated with EGF (5 ng/ml) for 5, 10, or 30 min. ERK1/2 activation was assessed by anti-phospho-ERK1/2 immunoblotting. C, LS SHP2 mutants displayed higher activity than the WT in dephosphorylating its physiological substrate paxillin (Tyr(P)-118 (pY118)). D, LS mutant SHP2 cells display higher sensitivity to EGF stimulation. HEK293 cells expressing wild-type or mutant SHP2 were stimulated with EGF (0, 0.04, and 0.2 ng/ml) for 5 min. ERK1/2 activation was assessed by anti-phospho-ERK1/2 immunoblotting. All results shown in Fig. 5 were representative of two to three independent experiments. Band intensity was quantified using the ImageJ program. Protein binding or phosphorylation level was normalized to total control protein. The numbers below the panels represent ratios of immunoprecipitated Gab1/SHP2 or SHP2/Gab1 (A), pERK/total ERK (B and D), and pPaxillin/total paxillin (C) for the mutants, which are normalized to a value of 1.0 set for wild-type SHP2.
How can phosphatase activity-impaired SHP2 mutants increase Ras signaling? One possibility is that the prolonged association of the LS mutants with Gab1 enables effective dephosphorylation of target SHP2 substrates leading to Ras activation. SHP2-mediated paxillian/Tyr-118 dephosphorylation is required for EGF-stimulated ERK1/2 activation (32). Fig. 5C shows that although the intrinsic activity of Y279C and T468M toward pNPP is 26- and 46-fold lower than that of the wild-type SHP2, both mutants can more efficiently dephosphorylate paxillin (Tyr(P)-118), especially at the later time points. Moreover, the biophysical consequence of the weakened interaction between the N-SH2 domain and the PTP domain in the LS-associated SHP2 mutants is the ability of the N-SH2 domain to bind Tyr(P) motifs present on its physiological interacting proteins preferentially over the wild-type SHP2 and to do so under much less pronounced stimulatory conditions, thus lowering the threshold for Ras/ERK1/2 activation. Indeed, less EGF is required to activate the LS mutant-mediated ERK1/2 activation (Fig. 5D), which should confer growth advantage during development when growth factors are limiting. Furthermore, increased paxillin dephosphorylation by the LS mutants were also observed under low EGF (0.2 ng/ml) concentration (data not shown). Collectively, the results established that LS SHP2 mutants are capable of promoting the EGF induced-ERK1/2 pathway activation.
DISCUSSION
Approximately 50% of NS and 90% of LS cases are caused by missense mutations in SHP2. Despite the opposing effects of NS and LS mutations on SHP2 activity, NS and LS patients exhibit many similar clinical features (3). The NS SHP2 mutations give rise to a constitutively active state of the enzyme and are classified as GOF. These GOF mutants promote sustained activation of ERK1/2 in transfected cells and in animal models (11, 33–35). In addition, GOF mutations in other components of the Ras-ERK1/2 pathway, such as Ras, Raf, and SOS1, have been identified in NS patients lacking SHP2 mutations (36–39). Moreover, other phenotypically overlapping Costelo and cardio-facio-cutaneous syndromes are also caused by GOF Ras/Raf/MEK1/2 mutations (40–42). These observations collectively indicate that aberrant activation of the Ras-Raf-MEK1/2-ERK1/2 pathway plays a key role in these overlapping developmental disorders (43).
Unlike NS, the LS-associated SHP2 mutants are catalytically impaired. From a genetic perspective, it is worth noting that the LS-associated SHP2 alleles are specific missense mutations. If their sole effect in transducing their phenotype was to reduce or eliminate SHP2 phosphatase activity, the existence of haploinsufficient alleles such as nonsense mutations might be anticipated, but none has been observed (2, 44). In addition, loss of one SHP2 allele in mouse has no phenotype, suggesting that the effect of LS mutations on SHP2 function may be more complex than a simple loss-of-function. More recent studies show that ubiquitous expression of LS-causing SHP2 alleles (Y279C and T468M) in Drosophila results in GOF phenotypes through increased EGFR-Ras-ERK1/2 signaling (45), which are similar to those of the NS-causing SHP2 mutant transgenic flies (13). Importantly, the residual phosphatase activity of the LS mutants is required for the GOF developmental effects. In concordance with observations in the Drosophila LS model, phosphorylation of MEK1, the upstream kinase of ERK1/2, and basal pERK1/2 levels are increased in induced pluripotent stem cells from LS patients (46). Finally, the discovery of LS-associated Raf1 GOF alleles provides more direct evidence that enhanced Ras-ERK1/2 signaling might be the cause of LS as well (47).
A major unresolved question in the field is how SHP2 mutations with opposite effects on phosphatase activity elicit overlapping developmental disorders? Specifically, it is not understood how catalytically impaired LS-associated SHP2 mutants produce GOF phenotypes? To address this question, we determined the crystal structures of the wild-type SHP2 as well as the most common LS-causing mutant, Y279C. The structures reveal why Y279C has reduced phosphatase activity. Moreover, the structures show that although the Y279C mutant adopts a closed conformation, the Y279C mutation weakens the inhibitory interaction between the N-SH2 domain and the PTP domain, leading to a change in SHP2 molecular switching mechanism. Further computational and biophysical studies suggest that compared with wild-type SHP2, the LS-associated SHP2 mutants exhibit an increased propensity to transition from a basal autoinhibited closed state to a catalytically activated open conformation. We demonstrate that the diminished interaction between the N-SH2 domain and the PTP domain in the LS-associated SHP2 mutants results in increased binding between the N-SH2 domain and its Tyr(P) ligands. Consequently, the LS-associated SHP2 mutants are hypersensitive to growth factor stimulation and stay longer with the scaffolding adapters, thus prolonging specific substrate turnover, which could compensate for the reduced phosphatase activity. We provide evidence that the LS-associated SHP2 mutants promote sustained ERK1/2 activation by enhanced specific substrate dephosphorylation. Collectively, this study creates a solid framework for understanding the molecular basis of disease-associated SHP2 mutations and provides insight into how catalytically impaired LS-associated SHP2 mutants can engender GOF phenotypes.
This work was supported, in whole or in part, by National Institutes of Health Grant CA69202.
- SH2
- Src homology 2
- SHP2
- SH2 domain-containing protein-tyrosine phosphatase-2
- NS
- Noonan syndrome
- GOF
- gain-of-function
- MM-GBSA
- molecular mechanics-generalized born surface area
- pNPP
- p-nitrophenyl phosphate
- F2pmp
- phosphonodifluoromethyl phenylalanine
- PTP
- protein-tyrosine phosphatase
- H/DX-MS
- hydrogen/deuterium exchange mass spectrometry
- LS
- LEOPARD Syndrome.
REFERENCES
- 1. Neel B. G., Gu H., Pao L. (2003) The “Shp”ing news. SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem. Sci. 28, 284–293 [DOI] [PubMed] [Google Scholar]
- 2. Digilio M. C., Conti E., Sarkozy A., Mingarelli R., Dottorini T., Marino B., Pizzuti A., Dallapiccola B. (2002) Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet. 71, 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tartaglia M., Gelb B. D. (2005) Noonan syndrome and related disorders. Genetics and pathogenesis. Annu. Rev. Genomics Hum. Genet. 6, 45–68 [DOI] [PubMed] [Google Scholar]
- 4. Tartaglia M., Niemeyer C. M., Fragale A., Song X., Buechner J., Jung A., Hählen K., Hasle H., Licht J. D., Gelb B. D. (2003) Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes, and acute myeloid leukemia. Nat. Genet. 34, 148–150 [DOI] [PubMed] [Google Scholar]
- 5. Bentires-Alj M., Paez J. G., David F. S., Keilhack H., Halmos B., Naoki K., Maris J. M., Richardson A., Bardelli A., Sugarbaker D. J., Richards W. G., Du J., Girard L., Minna J. D., Loh M. L., Fisher D. E., Velculescu V. E., Vogelstein B., Meyerson M., Sellers W. R., Neel B. G. (2004) Activating mutations of the Noonan syndrome-associated SHP2/PTPN11 gene in human solid tumors and adult acute myelogenous leukemia. Cancer Res. 64, 8816–8820 [DOI] [PubMed] [Google Scholar]
- 6. Sugimoto S., Wandless T.J., Shoelson S.E., Neel B.G., Walsh C.T. (1994) Activation of the SH2-containing protein tyrosine phosphatase, SH-PTPs by phosphotyrosine-containing peptides derived from insulin receptor substrate-1. J. Biol. Chem. 269, 13614–13622 [PubMed] [Google Scholar]
- 7. Pluskey S., Wandless T.J., Walsh C.T., Shoelson S.E. (1995) Potent stimulation of SHPTPs phosphatase activity by simultaneous occupancy of both SH2 domains. J. Biol. Chem. 270, 2897–2900 [DOI] [PubMed] [Google Scholar]
- 8. Pei D., Wang J., Walsh C.T. (1996) Differential functions of the two Src homology 2 domains in protein tyrosine phosphatase SHPTP1. Proc. Natl. Acad. Sci. U.S.A. 93, 1141–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lechleider R.J., Sugimoto S., Bennett A.M., Kashishian A.S., Cooper J.A., Shoelson S.E., Walsh C.T., Neel B.G. (1993) Activation of the SH2-containing phosphotyrosine phosphatase SH-PTPs by its binding site, phosphotyrosine 1009, on the human platelet-derived growth factor receptor. J. Biol. Chem. 268, 21478–21481 [PubMed] [Google Scholar]
- 10. Hof P., Pluskey S., Dhe-Paganon S., Eck M. J., Shoelson S. E. (1998) Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450 [DOI] [PubMed] [Google Scholar]
- 11. Fragale A., Tartaglia M., Wu J., Gelb B. D. (2004) Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum. Mutat. 23, 267–277 [DOI] [PubMed] [Google Scholar]
- 12. Keilhack H., David F. S., McGregor M., Cantley L. C., Neel B. G. (2005) Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J. Biol. Chem. 280, 30984–30993 [DOI] [PubMed] [Google Scholar]
- 13. Oishi K., Gaengel K., Krishnamoorthy S., Kamiya K., Kim I. K., Ying H., Weber U., Perkins L. A., Tartaglia M., Mlodzik M., Pick L., Gelb B. D. (2006) Transgenic Drosophila models of Noonan syndrome causing PTPN11 gain-of-function mutations. Hum. Mol. Genet. 15, 543–553 [DOI] [PubMed] [Google Scholar]
- 14. Tartaglia M., Martinelli S., Stella L., Bocchinfuso G., Flex E., Cordeddu V., Zampino G., Burgt I. v., Palleschi A., Petrucci T. C., Sorcini M., Schoch C., Foa R., Emanuel P. D., Gelb B. D. (2006) Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet. 78, 279–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Martinelli S., Torreri P., Tinti M., Stella L., Bocchinfuso G., Flex E., Grottesi A., Ceccarini M., Palleschi A., Cesareni G., Castagnoli L., Petrucci T. C., Gelb B. D., Tartaglia M. (2008) Diverse driving forces underlie the invariant occurrence of the T42A, E139D, I282V, and T468M SHP2 amino acid substitutions causing Noonan and LEOPARD syndromes. Hum. Mol. Genet. 17, 2018–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hanna N., Montagner A., Lee W. H., Miteva M., Vidal M., Vidaud M., Parfait B., Raynal P. (2006) Reduced phosphatase activity of SHP-2 in LEOPARD syndrome. Consequences for P13K binding on Gab1. FEBS Lett. 580, 2477–2482 [DOI] [PubMed] [Google Scholar]
- 17. Kontaridis M. I., Swanson K. D., David F. S., Barford D., Neel B. G. (2006) PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J. Biol. Chem. 281, 6785–6792 [DOI] [PubMed] [Google Scholar]
- 18. Otwinowski Z., Minor W. (1997) in Macromolecular Crystallography, Part A, pp 307–326, Academic Press Inc., San Diego, CA [Google Scholar]
- 19. Vagin A., Teplyakov A. (1997) MOLREP. an automated program for molecular replacement. J. Appl. Crystallogr. 30, 1022–1025 [Google Scholar]
- 20. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX. a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kollman P. A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D. A., Cheatham T. E., 3rd (2000) Calculating structures and free energies of complex molecules. Combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 [DOI] [PubMed] [Google Scholar]
- 23. Weis D. D., Engen J. R., Kass I. J. (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 17, 1700–1703 [DOI] [PubMed] [Google Scholar]
- 24. Jia Z., Barford D., Flint A. J., Tonks N. K. (1995) Structural basis for phosphotyrosine peptide recognition by protein tyrosine phosphatase 1B. Science 268, 1754–1758 [DOI] [PubMed] [Google Scholar]
- 25. Sarmiento M., Zhao Y., Gordon S. J., Zhang Z. Y. (1998) Molecular basis for substrate specificity of protein-tyrosine phosphatase 1B. J. Biol. Chem. 273, 26368–26374 [DOI] [PubMed] [Google Scholar]
- 26. Hoofnagle A. N., Resing K. A., Ahn N. G. (2003) Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 32, 1–25 [DOI] [PubMed] [Google Scholar]
- 27. Chen B., Bronson R. T., Klaman L. D., Hampton T. G., Wang J. F., Green P. J., Magnuson T., Douglas P. S., Morgan J. P., Neel B. G. (2000) Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat. Genet. 24, 296–299 [DOI] [PubMed] [Google Scholar]
- 28. Cunnick J. M., Dorsey J. F., Munoz-Antonia T., Mei L., Wu J. (2000) Requirement of SHP2 binding to Grb2-associated binder-1 for mitogen-activated protein kinase activation in response to lysophosphatidic acid and epidermal growth factor. J. Biol. Chem. 275, 13842–13848 [DOI] [PubMed] [Google Scholar]
- 29. Cunnick J. M., Mei L., Doupnik C. A., Wu J. (2001) Phosphotyrosines 627 and 659 of Gab1 constitute a bisphosphoryl tyrosine-based activation motif (BTAM) conferring binding and activation of SHP2. J. Biol. Chem. 276, 24380–24387 [DOI] [PubMed] [Google Scholar]
- 30. Deb T. B., Wong L., Salomon D. S., Zhou G., Dixon J. E., Gutkind J. S., Thompson S. A., Johnson G. R. (1998) A common requirement for the catalytic activity and both SH2 domains of SHP-2 in mitogen-activated protein (MAP) kinase activation by the ErbB family of receptors. A specific role for SHP-2 in MAP, but not c-jun amino-terminal kinase activation. J. Biol. Chem. 273, 16643–16646 [DOI] [PubMed] [Google Scholar]
- 31. Qu C. K., Yu W. M., Azzarelli B., Feng G. S. (1999) Genetic evidence that Shp-2 tyrosine phosphatase is a signal enhancer of the epidermal growth factor receptor in mammals. Proc. Natl. Acad. Sci. U.S.A. 96, 8528–8533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ren Y., Meng S., Mei L., Zhao Z. J., Jove R., Wu J. (2004) Roles of Gab1 and SHP2 in paxillin tyrosine dephosphorylation and Src activation in response to epidermal growth factor. J. Biol. Chem. 279, 8497–8505 [DOI] [PubMed] [Google Scholar]
- 33. Araki T., Mohi M. G., Ismat F. A., Bronson R. T., Williams I. R., Kutok J. L., Yang W., Pao L. I., Gilliland D. G., Epstein J. A., Neel B. G. (2004) Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat. Med. 10, 849–857 [DOI] [PubMed] [Google Scholar]
- 34. Mohi M. G., Williams I. R., Dearolf C. R., Chan G., Kutok J. L., Cohen S., Morgan K., Boulton C., Shigematsu H., Keilhack H., Akashi K., Gilliland D. G., Neel B. G. (2005) Prognostic, therapeutic, and mechanistic implications of a mouse model of leukemia evoked by Shp2 (PTPN11) mutations. Cancer Cell 7, 179–191 [DOI] [PubMed] [Google Scholar]
- 35. Krenz M., Gulick J., Osinska H. E., Colbert M. C., Molkentin J. D., Robbins J. (2008) Role of ERK1/2 signaling in congenital valve malformations in Noonan syndrome. Proc. Natl. Acad. Sci. U.S.A. 105, 18930–18935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Carta C., Pantaleoni F., Bocchinfuso G., Stella L., Vasta I., Sarkozy A., Digilio C., Palleschi A., Pizzuti A., Grammatico P., Zampino G., Dallapiccola B., Gelb B. D., Tartaglia M. (2006) Germline missense mutations affecting KRAS isoform B are associated with a severe Noonan syndrome phenotype. Am. J. Hum. Genet. 79, 129–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schubbert S., Zenker M., Rowe S. L., Böll S., Klein C., Bollag G., van der Burgt I., Musante L., Kalscheuer V., Wehner L. E., Nguyen H., West B., Zhang K. Y., Sistermans E., Rauch A., Niemeyer C. M., Shannon K., Kratz C. P. (2006) Germline KRAS mutations cause Noonan syndrome. Nat. Genet. 38, 331–336 [DOI] [PubMed] [Google Scholar]
- 38. Roberts A. E., Araki T., Swanson K. D., Montgomery K. T., Schiripo T. A., Joshi V. A., Li L., Yassin Y., Tamburino A. M., Neel B. G., Kucherlapati R. S. (2007) Germline gain-of-function mutations in SOS1 cause Noonan syndrome. Nat. Genet. 39, 70–74 [DOI] [PubMed] [Google Scholar]
- 39. Tartaglia M., Pennacchio L. A., Zhao C., Yadav K. K., Fodale V., Sarkozy A., Pandit B., Oishi K., Martinelli S., Schackwitz W., Ustaszewska A., Martin J., Bristow J., Carta C., Lepri F., Neri C., Vasta I., Gibson K., Curry C. J., Siguero J. P., Digilio M. C., Zampino G., Dallapiccola B., Bar-Sagi D., Gelb B. D. (2007) Gain-of-function SOS1 mutations cause a distinctive form of Noonan syndrome. Nat. Genet. 39, 75–79 [DOI] [PubMed] [Google Scholar]
- 40. Aoki Y., Niihori T., Kawame H., Kurosawa K., Ohashi H., Tanaka Y., Filocamo M., Kato K., Suzuki Y., Kure S., Matsubara Y. (2005) Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat. Genet. 37, 1038–1040 [DOI] [PubMed] [Google Scholar]
- 41. Niihori T., Aoki Y., Narumi Y., Neri G., Cavé H., Verloes A., Okamoto N., Hennekam R. C., Gillessen-Kaesbach G., Wieczorek D., Kavamura M. I., Kurosawa K., Ohashi H., Wilson L., Heron D., Bonneau D., Corona G., Kaname T., Naritomi K., Baumann C., Matsumoto N., Kato K., Kure S., Matsubara Y. (2006) Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat. Genet. 38, 294–296 [DOI] [PubMed] [Google Scholar]
- 42. Rodriguez-Viciana P., Tetsu O., Tidyman W. E., Estep A. L., Conger B. A., Cruz M. S., McCormick F., Rauen K. A. (2006) Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311, 1287–1290 [DOI] [PubMed] [Google Scholar]
- 43. Gelb B. D., Tartaglia M. (2006) Noonan syndrome and related disorders. Dysregulated RAS-mitogen activated protein kinase signal transduction. Hum. Mol. Genet. 15, R220–R226 [DOI] [PubMed] [Google Scholar]
- 44. Legius E., Schrander-Stumpel C., Schollen E., Pulles-Heintzberger C., Gewillig M., Fryns J. P. (2002) PTPN11 mutations in LEOPARD syndrome. J. Med. Genet. 39, 571–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Oishi K., Zhang H., Gault W. J., Wang C. J., Tan C. C., Kim I. K., Ying H., Rahman T., Pica N., Tartaglia M., Mlodzik M., Gelb B. D. (2009) Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum. Mol. Genet. 18, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Carvajal-Vergara X., Sevilla A., D'Souza S. L., Ang Y. S., Schaniel C., Lee D. F., Yang L., Kaplan A. D., Adler E. D., Rozov R., Ge Y., Cohen N., Edelmann L. J., Chang B., Waghray A., Su J., Pardo S., Lichtenbelt K. D., Tartaglia M., Gelb B. D., Lemischka I. R. (2010) Patient-specific induced pluripotent stem cell-derived models of LEOPARD syndrome. Nature 465, 808–812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pandit B., Sarkozy A., Pennacchio L. A., Carta C., Oishi K., Martinelli S., Pogna E. A., Schackwitz W., Ustaszewska A., Landstrom A., Bos J. M., Ommen S. R., Esposito G., Lepri F., Faul C., Mundel P., López Siguero J. P., Tenconi R., Selicorni A., Rossi C., Mazzanti L., Torrente I., Marino B., Digilio M. C., Zampino G., Ackerman M. J., Dallapiccola B., Tartaglia M., Gelb B. D. (2007) Gain-of-function RAF1 mutations cause Noonan and LEOPARD syndromes with hypertrophic cardiomyopathy. Nat. Genet. 39, 1007–1012 [DOI] [PubMed] [Google Scholar]