

Background: The protease BACE2 regulates β-cell function by acting on an unknown substrate repertoire.
Results: Analysis of the islet β-cell sheddome reveals novel BACE2 and BACE1 targets.
Conclusion: BACE2 and its homologue BACE1 target a diverse substrate repertoire, but naturally only share a small set of substrates.
Significance: The identification of BACE2 and 1 substrates is crucial for understanding pancreatic β-cell function.
Keywords: Diabetes, Pancreatic Islets, Protease, Protein Degradation, Proteomics, Shedding, Secretome, Surface Proteases, Surface Proteome
Abstract
Expansion of functional islet β-cell mass is a physiological process to compensate for increased insulin demand. Deficiency or pharmacological inhibition of the plasma membrane protease BACE2 enhances pancreatic β-cell function and proliferation, and therefore BACE2 is a putative target for the therapeutic intervention under conditions of β-cell loss and dysfunction. To gain a molecular understanding of BACE2 function, we performed a systematic and quantitative proteomic analysis to map the natural substrate repertoire of BACE2 and its homologue BACE1 in β-cells. Loss- and gain-of-function studies of in vitro and in vivo models identified specific and functionally heterogeneous targets. Our analysis revealed non-redundant roles of BACE1/2 in ectodomain shedding with BACE1 regulating a broader and BACE2 a more distinct set of β-cell-enriched substrates including two proteins of the seizure 6 protein family (SEZ6L and SEZ6L2). Lastly, our study provides insights into the global β-cell sheddome and secretome, an important prerequisite to uncover novel mechanisms contributing to β-cell homeostasis and a resource for therapeutic target and biomarker discoveries.
Introduction
The pancreatic β-cell is responsible for maintaining normoglycemia by secreting appropriate amounts of insulin according to blood glucose levels. In healthy individuals, β-cells sense blood glucose levels and adjust their function and mass to meet metabolic needs, ensuring that plasma glucose concentrations remain within a physiological range. During pregnancy, child growth, and obesity, insulin demand is augmented and responded to by increased insulin secretion and β-cell mass (1, 2). These adaptive processes require the communication of β-cells with other tissues (via circulating factors) and their microenvironment that is composed of endocrine cells of the islet of Langerhans and non-endocrine cells (3, 4).
Cell-cell interactions and communication are transmitted via the cell surface proteome, and many proteins pivotal for insulin secretion and mass expansion are plasma membrane proteins, including for example, growth factor and cytokine receptors, transporters, ion channels, and enzymes (5). Many of these proteins are regulated by proteolytic cleavage through surface proteases which control abundance, localization, and activity of their targets and therefore have been implicated in important homeostatic mechanisms of the β-cell (6–9). The pancreatic β-cell-enriched β-site amyloid precursor protein cleaving enzyme 2 (BACE2)3 belongs, together with its homologue BACE1 and other membrane proteases of the “a disintegrin and metalloprotease” (ADAM) family, to the so-called sheddases or secretases. This class of proteases performs ectodomain shedding, a type of proteolysis by which the extracellular part (the ectodomain) of a membrane-bound substrate is cleaved and released into the pericellular milieu where it may enter the circulation (10). The initial cleavage step by a sheddase also generates a membrane-bound fragment of a substrate, which is frequently subject to a further processing step termed regulated intramembrane proteolysis (RIP) (11). The functional consequences of ectodomain shedding and subsequent RIP are diverse and depend on the substrate protein and the generated cleavage fragments, which may have bioactivity on their own.
In the β-cell, BACE2 cleaves the pro-proliferative type I transmembrane protein 27 (TMEM27), the only physiological substrate reported so far apart from the BACE2 prodomain itself (12). Functionally, genetic inactivation of BACE2 leads to increased β-cell mass and improved glucose tolerance due to increased insulin secretion in vivo (8). In addition, treatment with a BACE2 selective inhibitor termed compound J improves glycemic control and increases β-cell mass in insulin-resistant ob/ob mice. The molecular mechanisms by which BACE2 deficiency and inhibition promote β-cell function and proliferation are unknown, but likely involve the stabilization of TMEM27 as well as other yet to be identified BACE2 substrates that synergistically contribute to these effects. Therefore, a key to understanding BACE2 function in pancreatic β-cells is to identify its substrate repertoire. Furthermore, the homologous enzyme BACE1 is particularly known for its role in Alzheimer disease (AD) pathogenesis, while its substrate repertoire in pancreatic β-cell cells, despite the high abundance in the pancreatic islet (8, 13), is still undetermined. Also, whether or not these two enzymes have common targets has major pharmacological implications, as either non-selective or highly BACE1/2-selective inhibitors may be required.
Apart from ectodomain shedding of plasma membrane proteins the β-cell also secretes various peptide hormones, most notably insulin, that enter the bloodstream or exhibit local autocrine or paracrine functions. The collective set of proteins that is secreted via the classical (signal peptide-based), non-classical, or exosomal pathways by a cell, tissue, or organism at a specific circumstance or moment is referred to as the secretome (14). These bioactive proteins are particularly interesting since they are frequently targetable by small molecules or biologicals and constitute a set of potential biomarkers that can be measured in plasma.
In this study, we have performed a systematic quantitative proteomic screen to identify the natural substrate repertoire of BACE2 and its homologue BACE1 in pancreatic β-cells. Loss- and gain-of-function studies identified functionally heterogenous targets of BACE2 and BACE1. Importantly, we demonstrate that BACE2 exhibits substrate specificity. Lastly, we report on the global β-cell sheddome and secretome of pancreatic islets and their corresponding peptides that can be used for quantitative, sensitive, and selective analyses of proteins released from pancreatic β-cells using selected/multiple reaction monitoring (SRM/MRM) mass spectrometry.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following antibodies were used: BACE1 (Epitomics; 2882-1), BACE2 (Santa Cruz; sc-10049), the BACE2 antibody used in immunofluorescence staining was kindly provided by Roche, γ-Tubulin (Sigma; T6557), Glucagon (Millipore; 4031–01F), Insulin (Sigma-Aldrich; I8510), Pancreatic polypeptide (Sigma-Aldrich; SAB2500747), SEZ6L (R&D; AF4804), SEZ6L2 (R&D; AF4916), Somatostatin (Dako; A0566), Synaptophysin (Millipore; MAB5258), antibodies against the shed N-terminal TMEM27 ectodomain and full-length TMEM27 were as previously described (15), V5 (Invitrogen; R960-25) and VAMP2 (Synaptic Systems, 104 211). Compound J (CpdJ) was synthesized by Roche as previously described (8), β-secretase inhibitor IV (BI IV) was purchased from Calbiochem (Merck Chemicals Ltd) and N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT) was purchased from Sigma-Aldrich. All protease inhibitors were dissolved in DMSO (the final concentration of DMSO was ≤ 0.01%).
Experimental Animals
Mice were housed in cages under controlled environment on a 12 h light-dark schedule (temperature 20 ± 2 °C, humidity 45%, lights on from 6 a.m. to 6 p.m.) in a pathogen-free facility at the Institute for Molecular Systems Biology, ETH Zürich (Switzerland). All animal experiments were approved by the Kantonale Veterinäramt Zürich. BACE1 knock-out mice (B6.129-Bace1tm1Pcw/J; stock number 004714) and BACE2-deficient mice (B6;129P2 Bace2tm1Bdes/J; stock number 005618) were purchased from The Jackson Laboratory (Bar Harbor, ME) and were crossed at the ETH Zürich to obtain BACE DKO (double-deficient) mice.
Pancreatic Islet Isolation and Culture
Pancreatic islets were isolated by collagenase digestion and gradient centrifugation using Histopaque (Sigma-Aldrich) according to a standard mouse islet-isolation protocol. The islets were handpicked in RPMI medium 1640 (Invitrogen) supplemented with 10% heat-inactivated FBS, 2 mmol/liter l-glutamine, 100 IU/ml penicillin, and 100 μg/ml streptomycin. The experiments were performed the next day. Batches of 100 size-matched islets were washed once in serum-free RPMI medium and cultured in non-coated 6-well plates (Falcon) for 48 h. For protease inhibition studies, the islets were incubated in 100 nmol/liter CpdJ and DMSO, respectively.
MS Loss- and Gain-of-Function Assays in MIN6 Cells
Stable knockdown of BACE2 and BACE1 in MIN6 cells was achieved using lentiviral-mediated shRNA interference according to standard procedures of the RNAi Consortium (protocols are available online). Two different shRNA lentiviral plasmids (pLKO.1-puro) per protease, obtained as frozen bacterial glycerol stocks (Sigma-Aldrich), were used (supplemental Table S1). Loss-of-function (LOF) cells lines and control cell lines expressing the pLKO.1-puro non-mammalian shRNA control plasmid were selected with 2 μg/ml puromycin dihydrochloride (Sigma-Aldrich) and were used for experiments following three to five passages after infection. For gain-of-function (GOF) cell lines MIN6 cells were electroporated with 5 μg of DNA of the corresponding vector in AMAXA nucleofector V solution using the AMAXA Nucleofector device and program G-016 for MIN6 cells (Lonza Group Ltd). Cells were selected for 3 weeks with 380 μg/ml G418 sulfate (Invitrogen). N-Linked glycopeptide capture of MIN6 cells was performed as described previously (9). For N-glycocapture of supernatants, LOF and GOF cell lines (eight 145 × 20 mm dishes per sample) were washed three times with PBS and incubated in phenol red-free OptiMEM (Invitrogen) for 24 h. The medium was collected and centrifuged at 200 × g to remove cell debris. The supernatant was concentrated 240-fold using Centricon plus-70 centrifugal filter units with a 5,000-molecular weight cutoff (Millipore) and purified by five washing steps with 2 mol/liter urea, 0.1 mol/liter ammonium bicarbonate buffer. Non-glycocapture experiments were performed as described previously (8).
LC-MS/MS Analysis
Peptides were identified and quantified on a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific) essentially as described (16). Full-scan MS spectra were acquired with a resolution of 60,000 in the Orbitrap analyzer. For every full scan, the five most intense ions were sequentially selected and fragmented in the linear ion trap. Raw data were processed and searched against a decoy database (consisting of forward and reverse protein sequences) of the predicted proteome from Mus Musculus (SwissProt, Version 57.12, in total 33,182 protein sequences) using the SORCERER-SEQUEST v4.0.3 algorithm (17). The database search results were further processed using the PeptideProphet (18) and ProteinProphet (19) programs and the peptide false discovery rate (FDR) was set to 1% on the peptide and 2% on the protein level. Peptides and proteins were quantified by label-free quantification using the SuperHirn software (20) with default settings. See supplemental Experimental Procedures for more details.
Targeted Proteomics Approach
The SRM spectral library was generated as previously described (21), where N-glycosite and non-glycosite peptides were selected from previous shotgun proteomic screens performed in MIN6 cell lines and from the Mouse Glyco Atlas (www.mrmatlas.org/). For proteins where no glycopeptide was observed in proteomics experiments, the bioinformatic transmembrane prediction tool Phobius (22) was used to select the N-glycosite peptides located on the extracellular domain of the protein. Tryptic digests were prepared from 500 size-matched islets and their 48-h supernatant. To remove N-glycosylations, the dried peptides were solubilized in 50 μl of 100 mmol/liter disodium-hydrogenphosphate buffer, 25 mmol/liter EDTA, pH 7.1 supplemented with 100 units of PNGase F (New England Biolabs), incubated for 4 h at 37 °C, and purified for mass spectrometric analysis. The samples were analyzed in SRM mode as previously described (23), and SRM data were processed using the SRM skyline software (24). Protein significance analysis was performed using SRMstats (25) where a constant normalization was performed to all runs to equalize the median peak intensities of the heavy transitions from all the peptides between runs. All proteins with a p value ≤0.05 and a 1.25-fold change in islet lysates and/or a 25% reduction in islet supernatant versus controls were considered significant.
RESULTS
Shotgun Proteomic Identification of BACE2 and BACE1 Targets in the Pancreatic β-Cell Line MIN6
Among challenges in applying proteomic techniques to β-cells are limitations in the isolation of functional β-cells from pancreatic tissue, due to the small amount of islet β-cell mass. In contrast, immortalized rodent β-cell lines provide unlimited material while retaining many physiological β-cell characteristics (64). As a first step toward the identification of the BACE2 substrate repertoire, we conducted a proteomic screen using loss-of-function (LOF) and gain-of-function (GOF) assays in MIN6 cells (Fig. 1A). This cell line expresses high endogenous levels of BACE2 and BACE1 and was previously successfully used in the discovery of BACE2 as the principle protease cleaving TMEM27 (8). MIN6 cells were either stably infected with two different recombinant lentiviruses expressing shRNAs targeting BACE1/2 and non-targeting shRNA or BACE1 and 2 were overexpressed following transfection of catalytic inactive BACE1/2 control vectors. To enrich for the putative sheddome, we analyzed changes in the abundance of N-glycosylated substrate proteins in cells and cell supernatants by a label-free quantitative proteomics method that takes advantage of the fact that most cell surface proteins are also N-glycosylated (26, 27). Because the supernatants are less complex than whole cell extracts, they were also analyzed without N-glycopeptide enrichment in gain-of-function studies.
FIGURE 1.

Proteomic screen for BACE2 and BACE1 target identification. A, schematic overview summarizing the workflow of proteomic shotgun analysis and the expected changes in substrate protein abundance upon protease loss- and gain-of-function (LOF and GOF). The loss- and gain-of-function phenotype of the used cell lines was validated by immunoblotting and MS (supplemental Fig. S1). B and C, Venn diagrams describing the distribution of identified substrate candidates in LOF (B) and GOF assays (C). D, topology of β-secretase targets. Protein candidates were analyzed by the UniProtKB/Swiss-Prot database and Phobius (22). Among the 145 identified targets were 116 membrane substrate candidates in addition to 29 soluble, putative indirect targets. All candidate BACE2/BACE1 targets and the corresponding abundance ratios in LOF and GOF assays can be found in supplemental Table S2.
The loss- and gain-of-function phenotypes of the generated cell lines were confirmed by Western blot and mass spectrometry, with TMEM27 serving as a positive control (supplemental Fig. S1). In analogy to these effects, we then performed an unbiased screen in which we analyzed accumulation (stabilization) of the full-length protein in protein extracts followed by a decrease of the shed ectodomain in supernatants (LOF assays) or an elevated shed ectodomain in the cell supernatant (GOF assays). All quantitative measurements were performed with biological replicates and based on the fold-changes of known substrates (e.g. TMEM27), proteins with a ratio of at least 2-fold were considered significant to select substrate candidates of BACE2 or BACE1.
In LOF assays, a total of 482 N-linked glycoproteins were quantified in cell lysates of BACE2 and BACE1 knockdown cell lines, 332 of which were repeatedly identified in all five established cell lines. Of these, 30 BACE2 and 29 BACE1 targets were enriched in the cell lysate upon single BACE2 and BACE1 knockdown, including eight potential common targets (see supplemental Table S2 for the corresponding proteins). In double knockdown cell line lysates, 18 of the candidates obtained by the single protease knockdown were confirmed and 26 additional candidates were identified for which a compensating effect of either BACE2 or BACE1 might play a role. The cell supernatants of these cell lines revealed similar but also additional N-glycoprotein targets, which were reduced upon BACE2 and/or BACE1 silencing versus control cell supernatant and were therefore validated independently of their regulation in cell lysates. Together 107 potential β-cell secretase substrates were identified including several putative BACE2 and BACE1 specific as well as common targets (Fig. 1B).
We also determined the abundance of shed BACE2 and BACE1 substrates in the supernatant of cell lines that overexpress BACE2 and BACE1 and in addition to proteins that were at least 2-fold enriched in these samples, seven protein candidates below this threshold were selected, which, like TMEM27 were consistently increased in supernatant upon “low” to “high” co-expression of BACE2 (see supplemental Table S2 for the corresponding proteins). Together, 55 potential BACE2 and 33 BACE1 and targets were identified. In contrast to LOF assays, a large proportion of candidates (23 proteins, 35% of the detected targets) were targeted by both proteases (Fig. 1C). This discrepancy is likely due to the loss of substrate fidelity as a result of overexpression of the respective protease.
An overview of all the identified putative BACE2 and BACE1 targets in LOF and GOF MIN6 cell assays is shown in Fig. 1D. About 40% of the 145 identified candidate proteins were identified as putative targets of both proteases. The majority of the proteins (101) were type I single-pass transmembrane proteins, although type II single-pass transmembrane proteins and GPI-linked proteins were observed as well. In addition, we detected non-membrane bound proteins such as secreted proteins or proteins located in cytoplasmic vesicles, including endosomes and lysosomes, which likely represent indirect targets of BACE1/2.
To gain more insights into the biological processes that may be regulated by BACE2 and BACE1, we performed functional annotation analysis using the DAVID Bioinformatics Resources (28). Four major pathways were identified, including “Cell adhesion molecules” (21 proteins), “Lysosome” (14 proteins), “Axon guidance” (8 proteins), and “Type I diabetes mellitus” (5 proteins) (supplemental Table S3). Interestingly, eight proteases were identified as putative BACE2 and BACE1 targets (ECE-1, TLL1, CTSF, PCSK2, MPTBS1, REELN, CPD, ENPEP), demonstrating that the abundance of these enzymes in cells, and cell supernatants is associated with BACE2 and BACE1 activity.
Validation of β- and γ-Secretase Substrates in MIN6 Cells
The combination of loss- and gain-of-function screens provided a higher coverage of the putative BACE2 and BACE1 sheddome, as each experiment contributed to the identification of possibly specific and common BACE2 and BACE1 targets. To further validate our screen, and to investigate whether putative substrate proteins are preferentially shed by endogenous BACE2 or BACE1, we transfected MIN6 cells with siRNAs pools targeting each protease and analyzed the levels of the shed proteins in supernatants and cell lysates 48 h after transfection by immunoblotting (Fig. 2A). Five proteins were chosen for which antibodies against the ectodomain preexisted. Silencing of BACE2 but not BACE1 resulted in a marked reduction in supernatant levels of seizure 6-like protein (SEZ6L), seizure 6-like protein 2 (SEZ6L2), cation-independent mannose-6-phosphate receptor (also known as insulin-like growth factor 2 receptor, IGF2R), sortilin (SORT1), and semaphoring-4B (SEMA4B) similar to the regulation pattern of TMEM27. Conversely, full-length protein levels were increased in cells in which BACE2 was silenced. Double knockdown of BACE2 and BACE1 did not further increase the effects observed upon BACE2 silencing demonstrating that the relevant protease cleaving these five substrates in MIN6 cells is BACE2. Likewise, pharmacological inhibition by β-secretase inhibitor IV (BI IV) resulted in a dose-dependent reduction of the shed ectodomains in supernatants and in the expected prodomain shift of BACE2 (Fig. 2B). This was again accompanied by an enrichment of the full-length protein for all five BACE2 substrates tested.
FIGURE 2.

Validation of representative β-secretase substrates in MIN6 cells. A, Western blot for indicated shed substrates in MIN6 lysates and supernatant 48 h after transfection of siRNA pools. The lysates and concentrated 24-h supernatants were separated by SDS-PAGE and blotted with antibodies against the ectodomain (N terminus) using E-Cadherin (CDH1) as negative control. Lys-TMEM27 was blotted with an antibody against the C terminus (Coll4). B, Western blot for five established BACE2 targets on supernatant and lysate of cells treated for 24 h with DMSO and BI IV at indicated concentrations. SN, supernatant; Lys, cell lysate.
In addition to the detection of shed fragments in the supernatant we validated protease targets by monitoring the presence of intracellular C-terminal fragments (CTFs), which are generated upon ectodomain shedding and are frequently further processed by RIP. Specifically, we investigated to which extent the CTF of the identified substrates undergo a proteolytic cascade involving the action of β- and γ-secretases (Fig. 3A). Thirty-eight human potential targets with a C-terminal V5–6× His tag were overexpressed in MIN6 cells and incubated with BI IV or γ-secretase inhibitor (DAPT) followed by immunoblot analysis. Changes in the full-length protein and the CTF were monitored in the cell lysate using an anti-V5 antibody. The known BACE1 substrates APLP1, APLP2, PSGL-1, and the BACE2 substrate TMEM27 served as controls (Fig. 3B). The prototypic regulation pattern of CTFs by β- and γ-secretase inhibition was observed for eight type I single-pass transmembrane proteins (Fig. 3C). The size of the observed CTFs of these proteins matched the predicted length of the protein fragment generated upon ectodomain shedding close to the juxtamembrane region (≈10–20 amino acids N-terminal from the transmembrane domain). These data show that the CTFs of at least eight of the in MIN6 cell identified BACE2 and BACE1 substrates are further processed by γ-secretase. Furthermore, CTFs of a GPI-anchored cell adhesion molecule, limbic system-associated membrane protein (LSAMP), and the type I single-pass transmembrane protein disulfide-isomerase TMX3 did not accumulate upon incubation with DAPT, suggesting that they are not processed by γ-secretase (supplemental Fig. S2). In addition, β-secretase inhibition affected a number of membrane and secretory proteins leading to both an increased (ITGB1, LMAN1, SEMA4B, TTR) or decreased (NEGR1, OLFM3, SMPD1) abundance of overexpressed full-length protein.
FIGURE 3.

CTF assay for the validation of β- and γ-secretase substrates. A, schematic drawing showing substrate protein processing by β- and γ-secretase. Human forms of putative substrate proteins were co-expressed in MIN6 cells with a C-terminal V5–6× His epitope. B, CTF assay of known β- and γ-secretase substrates. Inhibitor of β-secretase (BI IV, 5 μmol/liter), γ-secretase (DAPT, 10 μmol/liter), or DMSO was added for 6 h, and cell lysates were separated on 14% polyacrylamide gels 48 h post-transfection. Full-length proteins and CTFs were blotted using a V5 antibody, and sizes in kDa are shown for full-length protein forms (FL) and CTFs. Note that for SEZ6L an additional band of 36 kDa was detected with the V5 antibody, which may correspond to yet another C-terminal cleavage/degradation product. ICD, intracellular domain.
Validation of the Physiological Substrate Repertoire by Targeted MS Analysis in Isolated Murine Islets
While the proteomic screen and validation experiments in MIN6 cells identified the putative BACE2 and BACE1 sheddome, it does not necessarily reflect the in vivo regulation of BACE1/2 substrates in primary β-cells. Loss-of-function assays in cells from protease-deficient mice have generally been considered the gold-standard for identifying proteases-substrate pairs as they exclude potential overexpression artifacts (29) and were therefore also applied in the present study. Whereas the enrichment of the sheddome using N-glycocapture had proven to be efficient when working with β-cell lines, it was not applicable for the validation in isolated islets because the achievable sample amounts of these “mini-organs” are limited. We therefore used a targeted proteomics approach and established SRM assays that provided the sensitivity to validate a large number of substrate candidates in mouse islets. In contrast to conventional shotgun proteomic studies, SRM measurements target a predetermined set of peptides in a complex sample and consistently quantify them in sample sets. In the current study these were mostly N-glycosite peptides, since most candidates had in fact be identified via these peptides in the first place, which were thereby already validated in their suitability for MS analysis. The assays were carried out in isolated islets and islet supernatant of BACE1 knock-out mice (Bace1−/−), BACE2-deficient mice (Bace2ΔE6/ΔE6), which carry an in-frame deletion of exon 6 that encodes the catalytic active aspartic acid residue D303 and thus produce a shortened and catalytic inactive protein, BACE1 and BACE2 double-deficient (BACE DKO) mice and wildtype controls. In addition, isolated islets of wildtype mice were cultured in the presence of the BACE2 inhibitor compound J (CpdJ) or its vehicle DMSO.
SRM assays were developed according to the method based on crude synthetic peptide libraries (21). In total, we generated SRM assays for 527 synthetic peptides corresponding to 149 proteins. Next, the resulting assays were applied to detect and quantify the target proteins in mouse islets and their supernatants by SRM. Overall, 58 (≈40%) of the in MIN6 cells identified BACE2 and 1 targets could be quantified in mouse islets. The targeted proteins, their corresponding peptides and fold changes are listed in supplemental Table S4.
The SRM results revealed that ten membrane proteins exhibited a typical regulation pattern of a substrate protein, i.e. both, stabilization in cell lysates and reduced levels in supernatants of the respective protease-deficient mouse model (Fig. 4A). These comprise the BACE2 substrates SEZ6L and SEZ6L2 that were validated in BACE2-deficient islets, and more numerous BACE1 targets including the voltage-dependent calcium channel subunit α-2/δ-1 (CACNA2D1), IGF2R, glycosyltransferase 8 domain-containing protein 1 (GLT8D1), HEPACAM family member 2 (HEPACAM2), receptor-type tyrosine-protein phosphatase-like N and 2 (PTPRN and PTPRN2), which were validated as targets in BACE1 knock-out islets. The levels of several shed proteins were reduced only in supernatant samples while being unaltered in islet lysates of a respective mouse model, suggesting that analyzing the islet medium proteome, in which shed proteins can accumulate during the 48-h culture period, is a more sensitive readout. In addition, it has been previously shown that attenuated shedding can lead to an accumulation of substrate proteins at the cell surface (8, 30, 31); however, this might not be the case for substrates for which protein turnover is tightly controlled by compensatory proteases/mechanisms or may depend on the detection method (MS versus immunoblotting). These proteins include six additional BACE2 substrates (CD200, IGF2R, LAMP2, MPZL1, SORT1, and TMEM27), 12 BACE1 targets (ALCAM, APLP1, ATP6AP2, CD19, CD200, CNTNAP1, IL6ST, ITFG1, LMAN1, MBTPS1, PAM, and SEZ6L2); and three targets for which a compensating effect of either BACE1 or BACE2 might play a role (LAMP1, NEU1, SIRPA). The pharmacological inhibition of BACE2 and BACE1 by CpdJ confirmed several targets, and additionally revealed two further candidates (CPD and DNER) that were not regulated in islets of mutant mice. In addition, some soluble, putative indirect targets could be established which abundance is affected by BACE2 and/or BACE1 deficiency (Fig. 4B). Furthermore, several protein candidates could be excluded as being specific substrates of BACE2 and BACE1, i.e. substrates for which BACE2 and BACE1 expression is dispensable for cleavage in primary islets (Fig. 4C).
FIGURE 4.

Validation of BACE1/2 targets in islets of β-secretase loss-of-function mouse models. Tryptic digests of islets and 48-h islet supernatants of β-secretase loss-of-function mouse models (Bace1−/−, Bace2ΔE6/ΔE6, BACE DKO) and wildtype controls and of wildtype islets that were cultured in the presence of 100 nmol/l BACE2 inhibitor compound J (CpdJ) or its vehicle DMSO were analyzed by SRM/MRM mass spectrometry. A, only membrane proteins with a significant fold change in at least one condition (enrichment in lysate: 1.25-fold, a 25% reduction in supernatant, p ≤ 0.05) are shown as validated BACE1/2 substrates. The colors represent BACE1/2 targets that were enriched in islet lysate (red), reduced in islet supernatant (SN) (red) or proteins that show inverse regulation patterns (blue). The intensity reflects the corresponding fold-change and/or p value (see supplemental Table S4 for details). White color indicates that the protein concentration was below the detection limit and therefore not quantified. The targets were clustered in BACE2, BACE1, and putative common targets. B, validated soluble proteins representing possible indirect BACE1/2 targets. C, proteins that were excluded as being no major BACE1/2 targets in pancreatic islets. All experiments were performed in technical replicates and protein significance analysis was performed using SRMstats as described in the “Experimental Procedures.” SN, supernatant; Lys, lysate.
In summary, our data demonstrate that most substrates are specific BACE2 or BACE1 targets in vivo while only a relatively small number of proteins may be targeted by both proteases. Furthermore, we identified twice as many BACE1 targets compared with BACE2, suggesting a more specific/restricted role for BACE2 and broader role of BACE1 in ectodomain shedding within the pancreatic islet.
Validation and Characterization of SEZ6L and SEZ6L2 as Specific BACE2 Substrates in Pancreatic β-Cells
From the set of validated putative BACE2 targets, two single-pass type I transmembrane proteins of the seizure 6 protein family were selected for further characterization because of their BACE2 substrate specificity and enrichment in pancreatic islets (supplemental Fig. S3). To further characterize seizure 6-like protein shedding and validate that the proteins are preferably cleaved by BACE2 and/or BACE1, we compared the levels of shed and full-length SEZ6L and SEZ6L2 in islets and islet supernatants of β-secretase loss-of-function mouse models by immunoblotting. The supernatant of Bace2ΔE6/ΔE6 islets and BACE DKO islets contained almost no detectable levels of SEZ6L, SEZ6L2, and TMEM27 after 48h of culture, while the shed ectodomains were found in media of Bace1−/− islets and wildtype control islets (Fig. 5A). Conversely, the full-length forms of the substrate proteins were enriched in islets of Bace2ΔE6/ΔE6 and BACE DKO animals. The fact that loss of BACE1, alone or in combination with BACE2, did not further affect shedding of seizure 6-like proteins indicates that there is no redundancy or compensation in this cleavage process upon permanent deficiency of BACE2 in pancreatic islets. These results propose BACE2 as the rate-limiting enzyme for proteolytic processing of SEZ6L and SEZ6L2 in pancreatic islet β-cells. Moreover, pharmacological inhibition of BACE2 by CpdJ in isolated islets also resulted in an enrichment of full-length SEZ6L and SEZ6L2 (Fig. 5B). Cell surface biotinylation studies in MIN6 cells further indicated that the enrichment of SEZ6L, SORT1 and TMEM27 takes place at the cell surface (Fig. 5C). Additionally, semi-tryptic ectodomain peptides were identified for the BACE2 substrates SEZ6L and TMEM27, which were enriched in the supernatant upon overexpression of BACE2 compared with cells expressing a catalytically inactive mutant or non-transfected MIN6 cells (supplemental Fig. S4), suggesting that they stem from direct BACE2 cleavage. Similar to TMEM27 the potential BACE2 cleavage site in SEZ6L was located 16 amino acids N-terminal from the transmembrane domain. Sequence comparisons between different species showed that the cleavage site is conserved (supplemental Fig. S4C) and that it resembles the prodomain cleavage site of BACE2 and a TMEM27 cleavage site. Taken together, these findings demonstrate that BACE2 is sufficient and required for cleaving SEZ6L and SEZ6L2 in pancreatic β-cells.
FIGURE 5.
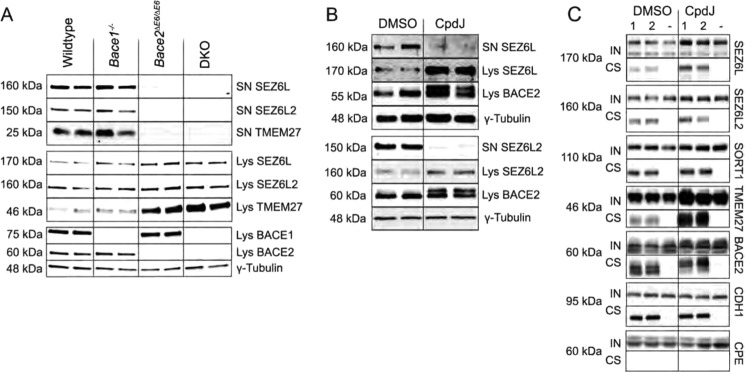
Seizure 6-like protein shedding is governed by BACE2. A, Western blot for SEZ6L and SEZ6L2 in islet lysates and 48-h islet supernatant of β-secretase-deficient mouse models and wildtype controls. Seizure 6-like protein shedding was abolished in BACE2-deficient mice similar to BACE DKO mice indicating BACE2 as the rate-limiting protease. B, Western blot of cultured mouse islets after treatment with 100 nmol/liter CpdJ for 48 h versus DMSO control. BACE2 inhibition was confirmed by the accumulation of the higher molecular weight band of BACE2 in CpdJ-treated murine islets (prodomain shift). C, cell surface expression of BACE2 substrates upon pharmacological inhibition of BACE2. MIN6 cells were cultured in the presence of 200 nmol/l CpdJ or DMSO and then biotinylated with a membrane impermeable biotinylation agent following subsequent isolation of biotinylated plasma membrane proteins by affinity purification. Input (IN) and cell surface (CS) fractions were analyzed by Western blotting. “1′” and “2” are biological duplicates from cells biotinylated on the surface prior to lysis; − is from cells that were not treated with biotin. CDH1 was used as cell surface marker, carboxypeptidase E (CPE) as a non-plasma membrane marker.
Proteomic Mapping of the β-Cell Secretome and Sheddome
The analysis of proteins released in the media of cell lines or primary cultures is a powerful tool to identify biologically active peptides and facilitate their evaluation as biomarker for cancer, neurodegenerative diseases and diabetes (32). We have employed the technique to estimate the fraction of the sheddome that is regulated by BACE2 and BACE1, but also to explore the global secretome and sheddome of MIN6 cells and murine isolated pancreatic islets. To determine the islet secretome and sheddome, primary mouse islets were cultured for 48 h and the conditioned medium was subjected to shotgun MS analysis (see supplemental Experimental Procedures for details). Together 594 proteins were identified in the conditioned medium of MIN6 cells, 240 of which were specifically detected by enrichment of N-linked glycopeptides. The analysis of islet medium proteomes yielded 742 proteins, including 204 proteins that were found in the β-cell line as well (Fig. 6A). Analysis of the conditioned medium proteins by “subcellular location” in the UniProtKB/Swiss-Prot database identified ∼50% of all annotated proteins as putative secreted proteins and membrane proteins, thus representing the putative islet and β-cell secretome and sheddome (Fig. 6B). The other half of the mapped proteins in conditioned media is composed predominantly of intracellular proteins that originate from a minority of cells that die during the culturing/harvesting procedure, however, some of these proteins that lack a typical signal sequence for the classical pathway of protein secretion via the endoplasmic reticulum/Golgi complex may also be released into the extracellular space by non-conventional ways where they fulfill biological functions (33). Since secretory proteins and shed membrane proteins may be present at low levels compared with highly abundant intracellular proteins that appeared in the conditioned media, we ranked the proteins according to their absolute levels (34). Several secreted and shed proteins were found to be more abundant compared with the putative “cytosolic contaminants” (Fig. 6C). Thus, all major islet hormones (insulin, glucagon, somatostatin and pancreatic polypeptide) were detected among other known secretory proteins such as granins (chromogranins and secretogranins), transthyretin (TTR) (35), islet amyloid polypeptide (iAPP) (36), and neuropeptide Y (NPY) (37). In addition several secreted factors were identified of which the roles have yet to be studied in the pancreatic β-cell, including vitamin D-binding protein (DBP), a multifunctional plasma protein of which altered circulating levels are associated with type 1 diabetes (38). The islet sheddome identified here consists predominantly of type I membrane proteins, some of which were previously found to be cleaved in different tissues or cell types. Furthermore, several GPI- or lipid anchored proteins were detected in addition to other membrane-associated proteins, which are putative targets of ectodomain sheddases. 116 of the shed proteins from MIN6 cells were found to be regulated by BACE2 and BACE1 (Fig. 1D), corresponding to ≈53% of the total sheddome, suggesting that the proteases do only regulate a defined protein subset.
FIGURE 6.

Global profiling of the islet and β-cell secretome and sheddome. A, Venn diagram showing the overlap between islet and MIN6-conditioned media proteomes. B, topology of the putative secretome and sheddome. All in islet and MIN6 supernatant identified proteins were analyzed using the UniProtKB/Swiss-Prot database. Approximately 50% of all annotated proteins were identified as secreted proteins and membrane proteins of different topology. C, relative abundance of proteins released from MIN6 cells by ranking them according to absolute levels (non-N-linked glycocapture approach) (34). Secretory and membrane proteins are shown in red, whereas putative intracellular proteins are shown in blue. Insulin as well as chromogranin-A and secretogranin-2 and -3 were the most abundant hits, shed proteins (e.g. TMEM27, SEZ6L, and SEZ6L2) were in a range ∼1000-fold lower. Note that insulin is also contained in plain OptiMEM, in which MIN6 cells were incubated for secretome analysis. D, functional categorization by PANTHER showing the top nine protein classes represented in the islet and MIN6 secretome and sheddome. The corresponding protein information can be found in supplemental Table S5. MP, membrane protein; TMP, transmembrane protein.
The functional consequences of ectodomain shedding and protein secretion can be diverse and depend on the function(s) of the particular protein or of the protein cleavage products. We therefore grouped the islet/β-cell secretome and sheddome into functional categories using the PANTHER protein classification system (39). Nine major categories were identified including receptor proteins (12%), hydrolases (12%), signaling molecules (10%), extracellular matrix proteins (8%), cell adhesion molecules (8%), enzyme modulators (8%), defense/immunity proteins (7%), proteases (6%), and transporters (5%) (Fig. 6D). This heterogeneity of putative shed substrate proteins suggests that diverse biological processes are regulated by the activity of β-cell surface proteases.
DISCUSSION
In this study we used a proteomic approach to screen for BACE2 and BACE1 target proteins using loss- and gain-of-function models. Besides the pancreatic β-cell BACE2 substrate TMEM27, four proteins previously recorded as BACE1 substrates in other systems were identified in MIN6 cells including APP, APLP1, APLP2, and LRP1. In addition, 12 of the BACE1 candidates recently determined in primary neurons (40, 41) and 24 BACE1 substrates found in two epithelial cell lines (42) were here established as BACE2 and BACE1 targets, suggesting a biological significance in processing these proteins in the β-cell. As previously shown for matrix metalloproteinases (43), interfering with the levels of active proteases will not solely change the abundance of substrate protein, but can indirectly alter the expression of an array of other proteins. Importantly, we also identified proteases and other soluble proteins that were regulated in cell lysates and/or supernatants by altered BACE1/2 levels, therefore not representing substrates but rather otherwise interconnected proteins. Thus, our studies do not only indicate putative substrate proteins, but also provide valuable information of indirect targets of a complex BACE1/2-dependent network.
While the proteomic substrate screens in the various MIN6 cell lines suggested that ∼50% of the 116 identified membrane protein candidates are targeted by both proteases, validation in single and combined BACE2- and BACE1-deficient mice showed that the enzymes process mainly specific and selective substrates. This underlines the importance of validating putative in vivo substrates using primary cells, such as primary β-cells that are clustered with other endocrine cells and non-endocrine cells in the functional unit of the islet of Langerhans. The observation that, even in the setting of lifelong BACE2 or BACE1 deficiency, there is no compensation among the proteases suggests that the enzymes are indispensable for the proteolytic processing of their substrates and that they likely fulfill distinct functions.
Our study also identified SEZ6L and SEZ6L2 as BACE2 specific substrates, two pancreatic islet-enriched cell surface proteins. Interestingly, the Seizure 6 protein family, has recently been shown to be cleaved by BACE1 in primary neurons (40). These distinct observations point toward a tissue-specific action of the protease-substrate pairs. The reasons why SEZ6L and SEZ6L2 are preferred substrates of BACE2 over BACE1 are currently unknown, but likely include organ-dependent protease abundance, subcellular compartmentalization of protease, and target and cleavage site/substrate isoform-related preferences of each protease. In line with the results of the present study, SEZ6L2 has been described as a putative cell surface protein expressed in endocrine cells of pancreatic islets and has been suggested as a useful marker for endocrine precursor islet cells during pancreatic development (44). Because of the selective abundance of BACE2 in pancreatic β-cells, shed BACE2 substrates could be particularly useful to monitor β-cell BACE2 activity or to evaluate pancreatic β-cell mass. Monitoring the dynamics of β-cell mass and function in vivo could have major benefits for facilitating early diabetes diagnosis, indicating the stages and progression of the disease, or help in predicting and monitoring a patient response to an individualized therapy. The molecular function and the role of cleavage of seizure 6-like proteins in the islet are unknown. BACE2 cleavage of TMEM27 has been suggested to inactivate TMEM27 since only the full-length protein is capable in stimulating β-cell proliferation (15). In contrast, other proteins have been shown to be activated by ectodomain shedding following intramembrane proteolysis (45, 46). Interestingly, a recent study identified opposing actions of two isoforms of SEZ6 on neuron-branching, a transmembrane form and a secreted form lacking the transmembrane domain (47), suggesting that the proteolytically released cleavage products of this protein family may also have functions of their own.
In addition to SEZ6L, SEZ6L2, and TMEM27, we identified further BACE2 targets that were, rather than being stabilized, characterized by the absence in islet medium proteomes of loss-of-function mouse models. The IGF2R was mapped here as an islet substrate for both BACE1 and BACE2. A major function of this receptor is to bind and transport mannose-6-phosphate-bearing glycoproteins (e.g. lysosomal enzymes) from the trans-Golgi network to lysosomes (reviewed by Refs. 48, 49). In addition, the IGF2R plays a major role in binding of insulin growth factor 2 (IGF2), which attenuates the hormone activity by targeting it for lysosomal degradation. IGF2 is a mitogenic factor with important functions for islet growth (50) and insulin exocytosis at non-stimulatory glucose concentrations through binding of the IGF2R (51). The soluble IGF2R also acts as negative regulator of growth by sequestering IGF2 and other ligands (52). Together these findings are consistent with BACE2 and BACE1 as regulators of extracellular IGF2 levels that act by producing a soluble IGF2R, which complexes IGF2 and thereby attenuates IGF2 signaling.
Another protein that was identified as a putative BACE2 substrate with recognized functions in protein trafficking is sortilin (SORT1). SORT1 is a multifunctional protein that binds various ligands known to participate in a diverse range of cellular processes (reviewed by (53)). For example, SORT1 acts as a receptor for neurotensin, a neuropeptide that has been shown to influence insulin, glucagon, and somatostatin secretion (54, 55). Recent evidence suggests that ADAM10 is the principal protease that cleaves SORT1 in neurons and fibroblasts (56). Although both ADAM10 and BACE2 are expressed in β-cells (8, 9), our findings indicate that BACE2 is the major SORT1 sheddase in primary pancreatic β-cells. Whether BACE1 plays a role in SORT1 shedding in islets still needs to be established, since the substrate peptides were not detected in islet supernatants of BACE1-deficient mice. Together, these data provide further insight into how BACE2 impacts β-cell function: by regulating trafficking of functionally heterogeneous proteins through acting on two key antiproliferative/transport and sorting proteins, IGF2R and SORT1.
We have previously demonstrated that BACE2 is selectively expressed in pancreatic β-cells within human and mouse islets (8), while BACE1 has also been detected in α-cells (57). Given that the substrates determined here were initially mapped in a β-cell line and that an islet consists of 50–80% of insulin-secreting β-cells (58), it is very likely that the ectodomains are released from β-cells, although in the case of BACE1, they may be cleaved simultaneously in α-cells. While it will be crucial to further elucidate the involved substrates and mechanisms by which BACE2 deficiency promotes β-cell proliferation and function, it will be also important to characterize BACE1 and the here identified targets in pancreatic islets and other tissues where BACE1 is expressed. The fact that a higher number of putative BACE1 substrates than BACE2 targets was identified may implicate a broader role of BACE1 in ectodomain shedding and supports the notion that therapeutic inhibition of BACE1 in attempts to prevent AD could have multiple side effects. Some of the BACE1 targets identified herein, including the α2δ1 subunit (CACNA2D1) of the voltage-dependent calcium channel, IGF2R, and the β-cell autoantigens of type 1 diabetes PTPRN and PTPRN2, have been previously linked to important functions in pancreatic β-cells, such as insulin secretion, insulin gene transcription, and β-cell proliferation (7, 51, 59–61). It will be interesting to study if and how BACE1 affects the molecular function of these proteins in pancreatic islets and whether β-cell-specific BACE1 deficiency, similar to BACE2 deficiency, leads to augmented functional β-cell mass.
The BACE2 and BACE1 candidate proteins mapped in MIN6 cells and pancreatic islets thus provide a basis to further elucidate the molecular function of these enzymes. The analysis of the amino acid sequence of these substrates also reveals that such empirical approaches are essential, as there is no clear consensus motif that will allow for unambiguous bioinformatics screening of databases for substrate identification. Moreover, the analysis of the sheddome under circumstances of elevated protease levels has the potential to map targets that may normally not or only to a minor degree be processed by BACE2 and/or BACE1, but their specific cleavage may still be of biological significance. In addition, the protease activity per se might be subject to regulation and could predispose to certain pathologies, one example being BACE1 in AD (62, 63). The SRM assays established here can be used to systematically screen BACE2 and BACE1 substrates in different cell types, to monitor changes in the protease degradome and to test the specificity and impact of novel selective BACE2 and BACE1 inhibitors. The proteins determined here thus represent a resource for the evaluation of BACE2 and BACE1 and their substrates as potential therapeutic targets or biomarkers, and facilitate the assessment of protease inhibitors and potential side effects.
Taken together, our study provides a detailed and global map of specific and common BACE1/2 substrates in pancreatic β-cells and offers molecular insights of how these proteases regulate β-cell growth and function. Furthermore, we report the global secretome and sheddome repertoire of pancreatic β-cells that will facilitate future analysis of the released cell surface proteome components to systematically assess if the determined proteins harbor diagnostic and therapeutic potential.
Acknowledgments
We thank Ulrich Omasits and Bernd Wollscheid for discussions, bioinformatic analysis of N-linked glycosite peptides, and for providing SRM assay coordinates for the targeted proteomics workflow, Nora Lieske for technical help, and Rebekka Park for helpful inputs during the preparation of the manuscript.
This work was funded in part by a grant from the Juvenile Diabetes Research Foundation and EU Grant CEED3.

This article contains supplemental Tables S1–S5, Figs. S1–S4, and Experimental Procedures.
- BACE
- β-site amyloid precursor protein cleaving enzyme
- AD
- Alzheimer disease
- FL
- full-length
- GOF
- gain-of-function
- LOF
- loss-of-function
- Lys
- lysate
- MP
- membrane protein
- MRM
- multiple reaction monitoring
- nd
- not determined
- RIP
- regulated intramembrane proteolysis
- SN
- supernatant
- SRM
- single reaction monitoring
- TMP
- transmembrane protein
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester
- CTF
- C-terminal fragment.
REFERENCES
- 1. Bouwens L., Rooman I. (2005) Regulation of pancreatic beta-cell mass. Physiol. Rev. 85, 1255–1270 [DOI] [PubMed] [Google Scholar]
- 2. Kahn S. E., Hull R. L., Utzschneider K. M. (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 [DOI] [PubMed] [Google Scholar]
- 3. Eberhard D., Lammert E. (2009) The pancreatic beta-cell in the islet and organ community. Curr. Opin. Genet. Dev. 19, 469–475 [DOI] [PubMed] [Google Scholar]
- 4. Prentki M., Nolan C. J. (2006) Islet beta cell failure in type 2 diabetes. J. Clin. Investig. 116, 1802–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stützer I., Esterházy D., Stoffel M. (2012) The pancreatic beta cell surface proteome. Diabetologia 55, 1877–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buteau J., Foisy S., Joly E., Prentki M. (2003) Glucagon-like peptide 1 induces pancreatic beta-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 52, 124–132 [DOI] [PubMed] [Google Scholar]
- 7. Trajkovski M., Mziaut H., Altkrüger A., Ouwendijk J., Knoch K. P., Müller S., Solimena M. (2004) Nuclear translocation of an ICA512 cytosolic fragment couples granule exocytosis and insulin expression in {beta}-cells. J. Cell Biol. 167, 1063–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Esterházy D., Stützer I., Wang H., Rechsteiner M. P., Beauchamp J., Döbeli H., Hilpert H., Matile H., Prummer M., Schmidt A., Lieske N., Boehm B., Marselli L., Bosco D., Kerr-Conte J., Aebersold R., Spinas G. A., Moch H., Migliorini C., Stoffel M. (2011) Bace2 is a beta cell-enriched protease that regulates pancreatic beta cell function and mass. Cell Metabolism 14, 365–377 [DOI] [PubMed] [Google Scholar]
- 9. Danzer C., Eckhardt K., Schmidt A., Fankhauser N., Ribrioux S., Wollscheid B., Müller L., Schiess R., Züllig R., Lehmann R., Spinas G., Aebersold R., Krek W. (2012) Comprehensive description of the N-glycoproteome of mouse pancreatic beta-cells and human islets. J. Proteome Res. 11, 1598–1608 [DOI] [PubMed] [Google Scholar]
- 10. Arribas J., Merlos-Suárez A. (2003) Shedding of plasma membrane proteins. Current Topics in Dev. Biol. 54, 125–144 [DOI] [PubMed] [Google Scholar]
- 11. Lichtenthaler S. F., Steiner H. (2007) Sheddases and intramembrane-cleaving proteases: RIPpers of the membrane. Symposium on regulated intramembrane proteolysis. EMBO Rep. 8, 537–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hussain I., Christie G., Schneider K., Moore S., Dingwall C. (2001) Prodomain processing of Asp1 (BACE2) is autocatalytic. J. Biol. Chem. 276, 23322–23328 [DOI] [PubMed] [Google Scholar]
- 13. Figueroa D. J., Shi X. P., Gardell S. J., Austin C. P. (2001) Abetapp secretases are co-expressed with Abetapp in the pancreatic islets. J. Alzheimer's Disease : JAD 3, 393–396 [DOI] [PubMed] [Google Scholar]
- 14. Makridakis M., Vlahou A. (2010) Secretome proteomics for discovery of cancer biomarkers. J. Proteomics 73, 2291–2305 [DOI] [PubMed] [Google Scholar]
- 15. Akpinar P., Kuwajima S., Krützfeldt J., Stoffel M. (2005) Tmem27: a cleaved and shed plasma membrane protein that stimulates pancreatic beta cell proliferation. Cell Metabolism 2, 385–397 [DOI] [PubMed] [Google Scholar]
- 16. Schmidt A., Beck M., Malmström J., Lam H., Claassen M., Campbell D., Aebersold R. (2011) Absolute quantification of microbial proteomes at different states by directed mass spectrometry. Mol. Systems Biol. 7, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 18. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 19. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 20. Mueller L. N., Rinner O., Schmidt A., Letarte S., Bodenmiller B., Brusniak M. Y., Vitek O., Aebersold R., Müller M. (2007) SuperHirn - a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 21. Picotti P., Rinner O., Stallmach R., Dautel F., Farrah T., Domon B., Wenschuh H., Aebersold R. (2010) High-throughput generation of selected reaction-monitoring assays for proteins and proteomes. Nature Methods 7, 43–46 [DOI] [PubMed] [Google Scholar]
- 22. Käll L., Krogh A., Sonnhammer E. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 23. Selevsek N., Matondo M., Sanchez Carbayo M., Aebersold R., Domon B. (2011) Systematic quantification of peptides/proteins in urine using selected reaction monitoring. Proteomics 11, 1135–1147 [DOI] [PubMed] [Google Scholar]
- 24. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang C. Y., Picotti P., Huttenhain R., Heinzelmann-Schwarz V., Jovanovic M., Aebersold R., Vitek O. (2012) Protein significance analysis in selected reaction monitoring (SRM) measurements. Mol. Cell Proteomics 11, M111 014662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schiess R., Mueller L. N., Schmidt A., Mueller M., Wollscheid B., Aebersold R. (2009) Analysis of cell surface proteome changes via label-free, quantitative mass spectrometry. Mol. Cell. Proteomics 8, 624–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang H., Li X. J., Martin D. B., Aebersold R. (2003) Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nature Biotechnol. 21, 660–666 [DOI] [PubMed] [Google Scholar]
- 28. Huang da W., Sherman B. T., Lempicki R. A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols 4, 44–57 [DOI] [PubMed] [Google Scholar]
- 29. Overall C. M., Blobel C. P. (2007) In search of partners: linking extracellular proteases to substrates. Nat. Rev. Mol. Cell Biol. 8, 245–257 [DOI] [PubMed] [Google Scholar]
- 30. McDermott M. F., Aksentijevich I., Galon J., McDermott E. M., Ogunkolade B. W., Centola M., Mansfield E., Gadina M., Karenko L., Pettersson T., McCarthy J., Frucht D. M., Aringer M., Torosyan Y., Teppo A. M., Wilson M., Karaarslan H. M., Wan Y., Todd I., Wood G., Schlimgen R., Kumarajeewa T. R., Cooper S. M., Vella J. P., Amos C. I., Mulley J., Quane K. A., Molloy M. G., Ranki A., Powell R. J., Hitman G. A., O'Shea J. J., Kastner D. L. (1999) Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 97, 133–144 [DOI] [PubMed] [Google Scholar]
- 31. Peiretti F., Canault M., Bernot D., Bonardo B., Deprez-Beauclair P., Juhan-Vague I., Nalbone G. (2005) Proteasome inhibition activates the transport and the ectodomain shedding of TNF-α receptors in human endothelial cells. J. Cell Science 118, 1061–1070 [DOI] [PubMed] [Google Scholar]
- 32. Dowling P., Clynes M. (2011) Conditioned media from cell lines: a complementary model to clinical specimens for the discovery of disease-specific biomarkers. Proteomics 11, 794–804 [DOI] [PubMed] [Google Scholar]
- 33. Butler G. S., Overall C. M. (2009) Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 8, 935–948 [DOI] [PubMed] [Google Scholar]
- 34. Silva J. C., Gorenstein M. V., Li G. Z., Vissers J. P., Geromanos S. J. (2006) Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol. Cell Proteomics 5, 144–156 [DOI] [PubMed] [Google Scholar]
- 35. Dowling P., Shields W., Rani S., Meleady P., Henry M., Jeppesen P., O'Driscoll L., Clynes M. (2008) Proteomic analysis of conditioned media from glucose responsive and glucose non-responsive phenotypes reveals a panel of secreted proteins associated with beta cell dysfunction. Electrophoresis 29, 4141–4149 [DOI] [PubMed] [Google Scholar]
- 36. Kahn S. E., D'Alessio D. A., Schwartz M. W., Fujimoto W. Y., Ensinck J. W., Taborsky G. J., Jr., Porte D., Jr. (1990) Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes 39, 634–638 [DOI] [PubMed] [Google Scholar]
- 37. Whim M. D. (2011) Pancreatic beta cells synthesize neuropeptide Y and can rapidly release peptide co-transmitters. PloS one 6, e19478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blanton D., Han Z., Bierschenk L., Linga-Reddy M. V., Wang H., Clare-Salzler M., Haller M., Schatz D., Myhr C., She J. X., Wasserfall C., Atkinson M. (2011) Reduced serum vitamin D-binding protein levels are associated with type 1 diabetes. Diabetes 60, 2566–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thomas P. D., Kejariwal A., Campbell M. J., Mi H., Diemer K., Guo N., Ladunga I., Ulitsky-Lazareva B., Muruganujan A., Rabkin S., Vandergriff J. A., Doremieux O. (2003) PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31, 334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuhn P. H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., Volbracht C., Schepers U., Imhof A., Hoffmeister A., Haass C., Rossner S., Brase S., Lichtenthaler S. F. (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J 31, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou L., Barão S., Laga M., Bockstael K., Borgers M., Gijsen H., Annaert W., Moechars D., Mercken M., Gevaert K., De Strooper B. (2012) The neural cell adhesion molecules L1 and CHL1 are cleaved by BACE1 protease in vivo. J. Biol. Chem. 287, 25927–25940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hemming M. L., Elias J. E., Gygi S. P., Selkoe D. J. (2009) Identification of beta-secretase (BACE1) substrates using quantitative proteomics. PloS one 4, e8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Butler G. S., Overall C. M. (2007) Proteomic validation of protease drug targets: pharmacoproteomics of matrix metalloproteinase inhibitor drugs using isotope-coded affinity tag labelling and tandem mass spectrometry. Curr. Pharm. Des. 13, 263–270 [DOI] [PubMed] [Google Scholar]
- 44. Hald J., Galbo T., Rescan C., Radzikowski L., Sprinkel A. E., Heimberg H., Ahnfelt-Rønne J., Jensen J., Scharfmann R., Gradwohl G., Kaestner K. H., Stoeckert C., Jr., Jensen J. N., Madsen O. D. (2012) Pancreatic islet and progenitor cell surface markers with cell sorting potential. Diabetologia 55, 154–165 [DOI] [PubMed] [Google Scholar]
- 45. Schroeter E. H., Kisslinger J. A., Kopan R. (1998) Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393, 382–386 [DOI] [PubMed] [Google Scholar]
- 46. Rawson R. B., Zelenski N. G., Nijhawan D., Ye J., Sakai J., Hasan M. T., Chang T. Y., Brown M. S., Goldstein J. L. (1997) Complementation cloning of S2P, a gene encoding a putative metalloprotease required for intramembrane cleavage of SREBPs. Mol. Cell 1, 47–57 [DOI] [PubMed] [Google Scholar]
- 47. Gunnersen J. M., Kim M. H., Fuller S. J., De Silva M., Britto J. M., Hammond V. E., Davies P. J., Petrou S., Faber E. S., Sah P., Tan S. S. (2007) Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron 56, 621–639 [DOI] [PubMed] [Google Scholar]
- 48. Ghosh P., Dahms N. M., Kornfeld S. (2003) Mannose 6-phosphate receptors: new twists in the tale. Nature Reviews. Mol. Cell Biol. 4, 202–212 [DOI] [PubMed] [Google Scholar]
- 49. Brown J., Jones E. Y., Forbes B. E. (2009) Interactions of IGF-II with the IGF2R/cation-independent mannose-6-phosphate receptor mechanism and biological outcomes. Vitam. Horm. 80, 699–719 [DOI] [PubMed] [Google Scholar]
- 50. Hill D. J., Petrik J., Arany E. (1998) Growth factors and the regulation of fetal growth. Diabetes Care 21, Suppl. 2, B60–B69 [PubMed] [Google Scholar]
- 51. Zhang Q., Tally M., Larsson O., Kennedy R. T., Huang L., Hall K., Berggren P. O. (1997) Insulin-like growth factor II signaling through the insulin-like growth factor II/mannose-6-phosphate receptor promotes exocytosis in insulin-secreting cells. Proc. Natl. Acad. Sci. U. S.A. 94, 6232–6237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zaina S., Squire S. (1998) The soluble type 2 insulin-like growth factor (IGF-II) receptor reduces organ size by IGF-II-mediated and IGF-II-independent mechanisms. J. Biol. Chem. 273, 28610–28616 [DOI] [PubMed] [Google Scholar]
- 53. Willnow T. E., Petersen C. M., Nykjaer A. (2008) VPS10P-domain receptors-regulators of neuronal viability and function. Nature Reviews. Neuroscience 9, 899–909 [DOI] [PubMed] [Google Scholar]
- 54. Dolais-Kitabgi J., Kitabgi P., Brazeau P., Freychet P. (1979) Effect of neurotensin on insulin, glucagon, and somatostatin release from isolated pancreatic islets. Endocrinology 105, 256–260 [DOI] [PubMed] [Google Scholar]
- 55. Beraud-Dufour S., Abderrahmani A., Noel J., Brau F., Waeber G., Mazella J., Coppola T. (2010) Neurotensin is a regulator of insulin secretion in pancreatic beta-cells. Int. J. Biochem. Cell Biol. 42, 1681–1688 [DOI] [PubMed] [Google Scholar]
- 56. Evans S. F., Irmady K., Ostrow K., Kim T., Nykjaer A., Saftig P., Blobel C., Hempstead B. L. (2011) Neuronal brain-derived neurotrophic factor is synthesized in excess, with levels regulated by sortilin-mediated trafficking and lysosomal degradation. J. Biol. Chem. 286, 29556–29567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Casas S., Casini P., Piquer S., Altirriba J., Soty M., Cadavez L., Gomis R., Novials A. (2010) BACE2 plays a role in the insulin receptor trafficking in pancreatic ss-cells. Amer. J. Physiol. Endocrinol. Metab. 299, E1087–E1095 [DOI] [PubMed] [Google Scholar]
- 58. In't Veld P., Marichal M. (2010) Microscopic anatomy of the human islet of Langerhans. Adv. Exp. Med. Biol. 654, 1–19 [DOI] [PubMed] [Google Scholar]
- 59. Braun M., Ramracheya R., Bengtsson M., Zhang Q., Karanauskaite J., Partridge C., Johnson P. R., Rorsman P. (2008) Voltage-gated ion channels in human pancreatic beta-cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628 [DOI] [PubMed] [Google Scholar]
- 60. Mziaut H., Trajkovski M., Kersting S., Ehninger A., Altkrüger A., Lemaitre R. P., Schmidt D., Saeger H. D., Lee M. S., Drechsel D. N., Müller S., Solimena M. (2006) Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nature Cell Biol. 8, 435–445 [DOI] [PubMed] [Google Scholar]
- 61. Torii S., Saito N., Kawano A., Hou N., Ueki K., Kulkarni R. N., Takeuchi T. (2009) Gene silencing of phogrin unveils its essential role in glucose-responsive pancreatic beta-cell growth. Diabetes 58, 682–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fukumoto H., Cheung B. S., Hyman B. T., Irizarry M. C. (2002) Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 59, 1381–1389 [DOI] [PubMed] [Google Scholar]
- 63. Holsinger R. M., McLean C. A., Beyreuther K., Masters C. L., Evin G. (2002) Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Annals Neurol. 51, 783–786 [DOI] [PubMed] [Google Scholar]
- 64. Miyazaki J., Araki K., Yamato E., Ikegami H., Asano T., Shibasaki Y., Oka Y., Yamamura K. (1990) Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132 [DOI] [PubMed] [Google Scholar]