

Background: We previously demonstrated that shear-induced TGF-β1 activation involves thiol-disulfide exchange.
Results: The thiol isomerase binding peptide mastoparan and protein disulfide isomerase inhibit shear-induced TGF-β1 activation.
Conclusion: Thiol isomerase(s) play a role in shear-induced TGF-β1 activation mediated by thiol-disulfide exchange.
Significance: Shear-induced activation of TGF-β1 is a novel mechanism that involves thiol isomerase-mediated thiol-disulfide exchange.
Keywords: Platelets, Redox Regulation, Shear Stress, Thrombosis, Transforming Growth Factor Beta (TGFbeta), Protein Disulfide Isomerases
Abstract
TGF-β1 is a disulfide-bonded homodimeric protein produced by platelets and other cells that plays a role in many physiologic and pathologic processes. TGF-β1 is secreted as an inactive large latent complex (LLC) comprised of TGF-β1, latency-associated peptide, and latent TGF-β binding protein 1. We previously demonstrated that shear force can activate LLC and that thiol-disulfide exchange contributes to the process. We have now investigated the role of thiol isomerases in the activation of LLC in platelet releasates (PR) and recombinant LLC. The wasp venom peptide mastoparan, which inhibits the chaperone activity of PDI, inhibited stirring- and shear-induced activation of latent TGF-β1 by 90 and 75% respectively. To identify the proteins that bind to mastoparan either directly or indirectly, PR were chromatographed on a mastoparan affinity column. Latent TGF-β binding protein 1, latency-associated peptide, TGF-β1, clusterin, von Willebrand factor, multimerin-1, protein disulfide isomerase (PDI), ERp5, ERp57, and ERp72 eluted specifically from the column. Anti-PDI RL90 attenuated the inhibitory effect of mastoparan on LLC activation. Furthermore, reduced PDI inhibited activation of PR LLC, whereas oxidized PDI had no effect. We conclude that thiol isomerases and thiol-disulfide exchange contribute to TGF-β1 activation and identify a number of molecules that may participate in the process.
Introduction
TGF-β1 is a potent cytokine that exerts many physiological and pathological effects (1). Platelets contain 40- to 100-fold more TGF-β1 than other cell types (2, 3), perhaps reflecting a role in wound healing after hemostasis is achieved. The pathological importance of platelet TGF-β1 has been highlighted by the recent discovery that platelet TGF-β1 can confer metastatic potential on tumor cells in synergy with NFκB signaling (4), and our recent report that targeted deletion of platelet and megakaryocyte TGF-β1 can protect mice from developing cardiac hypertrophy, fibrosis, and systolic dysfunction in response to constriction of the transverse aorta (5).
TGF-β1 is synthesized as a precursor peptide (pro-TGF-β1) that undergoes homodimerization. A furin-like convertase cleaves the precursor peptide into an N-terminal latency-associated peptide (LAP)3 and a C-terminal mature 25-kDa TGF-β1 dimer. The TGF-β1 dimer and LAP dimer remain non-covalently associated as a small latent complex, and LAP becomes covalently attached to the 160-kDa latent TGF-β1 binding protein 1 (LTBP-1) via a disulfide linkage involving LAP Cys-33, resulting in a 270-kDa large latent complex (LLC) (6). TGF-β1 is secreted from platelets and other cell types as the LLC. Activation of the LLC in vitro with acid or other treatments leads to the release of the TGF-β1 dimer from the complex (2). In vivo evidence from mouse models and the crystal structure of porcine pro-TGF-β1 support an activation mechanism in which LAP binds to one or more αV-containing integrin receptor via its Arg-Gly-Asp (RGD) sequence, and LTBP-1 binds to the extracellular matrix via its fibrillin-like sequence (7, 8). Traction on the small latent complex as a result of cell contraction is proposed to free the TGF-β1 dimer from the “straitjacket” domains of the LAP, allowing them to interact with TGF-β1 receptors (8). However, other mechanisms of activation may also contribute in vivo, including ones involving thrombospondin-1 (TSP-1) (9), proteases (10), and reactive oxygen species (11). Moreover, the molecular species responsible for TGF-β1 activity in vivo has never been isolated.
We previously demonstrated that shear force can activate TGF-β1 released from platelets in vitro and that TSP-1 partially contributes to the activation process (12, 13). In vivo, we could detect biologically significant amounts of active TGF-β1 in platelet-rich thrombi as soon as 5 min after platelet thrombus formation was induced by FeCl3 in the mouse carotid artery. We also found that stirring or shear decreased the ability of thiol-reactive agents to label proteins in platelet releasates, including TGF-β1 and TSP-1, suggesting an association between thiol oxidation and TGF-β1 activation. A role for thiol-disulfide exchange in TGF-β1 activation was further supported by our finding that multiple thiol-reactive agents inhibited stirring-induced activation. Because thiol isomerases play an important role in catalyzing thiol-disulfide exchange, in this study, we assessed the effects of mastoparan, a peptide that inhibits thiol isomerase chaperone activity, and both oxidized and reduced PDI on shear-dependent TGF-β1 activation.
EXPERIMENTAL PROCEDURES
Materials
Mastoparan peptide (INLKALAALAKKIL) isolated from Vespula lewisii (purity by HPLC >97%), a 17-amino acid control peptide (M17, INLKAKAALAKKLL), and PDI from bovine liver were purchased from Sigma-Aldrich. ProTGF-β1 expression plasmid (IOH4479) was obtained from Source Bioscience (Nottingham, UK). LTBP-1 expression plasmid RC220132 was obtained from OriGene (Rockville, MD). Magnetic streptavidin-coated beads (Dynabeads M-280), 8–16% Tris glycine mini gels (Novex), and 3-(N-maleimidylpropionyl)biocytin (MPB) were obtained from Invitrogen. Anti-TGF-β1 (polyclonal chicken IgY), anti-LAP (polyclonal goat IgG), and anti-LTBP-1 (murine mAb clone 35409) were from R&D Systems (Minneapolis, MN). Anti-clusterin mAb clone 41D was obtained from Millipore (Temecula, CA). Anti-multimerin-1 (goat polyclonal antibody; sc-104426) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-von Willebrand factor (rabbit polyclonal IgG, A0082) was obtained from Dako (Carpinteria, CA). Bacterially expressed human recombinant thioredoxin and a thioredoxin trapping mutant that contains a CXXS motif in place of the CXXC active site was kindly supplied for use as a reductase positive control by professor Phillip Hogg. The sources of all other reagents were described previously (12).
Platelet Releasate Preparation
Platelet releasates were prepared as described previously (12). The research was approved by the Rockefeller University Institutional Review Board, and all human blood donors at Rockefeller University provided written informed consent. Outdated platelet concentrates were obtained from the New York Blood Center (random donor units). Briefly, platelets were isolated from fresh citrated (0.38%) whole blood or the outdated platelets, washed with HBMT buffer (10 mm HEPES, 137 mm NaCl, 12 mm NaHCO3, 2.7 mm KCl, 0.36 mm NaH2PO4, pH 7.4) containing 0.35% bovine serum albumin and 0.1% dextrose, and activated with 10 μm thrombin receptor-activating peptide (SFLLRN) for 5 min at 37 °C without stirring. Samples were then centrifuged at 14,000 × g for 20 min at 4 °C. The platelet releasates were either stored at −80 °C or analyzed immediately.
Expression of Recombinant LLC in Mammalian Cells
Human embryonic kidney cells (HEK-293T) were cultured in DMEM containing 4 mm l-glutamine and 25 mm d-glucose supplemented with 10% fetal calf serum. Cells were plated at 300,000 cells/well in a six-well plate and incubated overnight. After reaching ∼70% confluence, cells were co-transfected with 1 μg each of pro-TGF-β1 plasmid and LTBP-1 plasmid using FuGENE 6 (6:1 ratio) (Invitrogen). After 48 h, the medium was harvested, centrifuged at 1000 × g for 4 min to pellet any non-adherent cells, and subjected to SDS-PAGE on 8–16% gradient Tris-glycine gels. After transfer of proteins to PVDF membranes, the membranes were blocked with 5% BSA in Tris-buffered saline with 0.1% Tween 20 (TBS-T) and incubated with antibodies to TGF-β1 or LTBP-1. Appropriate secondary antibodies conjugated to HRP were used to detect bound antibodies by chemiluminescence (ECL detection system, Amersham Biosciences).
Stirring of Platelet Releasate or Cell Supernatant
Platelet releasate or HEK 293T cell supernatant containing LLC (200 μl) was incubated in 7.25-mm diameter glass cuvettes with PDI (5 μg), or either mastoparan or M17 peptide (both at 0–100 μm in deionized H2O) for 2 h at 37 °C without or with stirring at 1200 rpm. In some experiments, anti-PDI mAb RL90 was added prior to stirring at a final concentration of 1 μg/ml.
Activation of Latent TGF-β1 by Shear
Platelet releasates (135 μl) were subjected to a shear rate of 1800 s−1 in a cone and plate device (Impact-R; DiaMed Corp.) for 2 h in the presence or absence of mastoparan or M17 peptide (25 μm). Samples were collected and assayed for active and total TGF-β1.
Measurement of Active and Total TGF-β1
Active and total TGF-β1 were measured using a two antibody ELISA specific for active TGF-β1 (R&D Systems) or a mink lung epithelial cell-based assay as described previously (12). Active TGF-β1 was measured in untreated samples, and total TGF-β1 was measured after activating latent TGF-β1 by treating the sample with 0.2 n HCl for 20 min at room temperature. Active TGF-β1 is reported as the percentage of total TGF-β1 in the same sample.
Labeling of Free Thiols
Proteins containing free thiols were labeled with 100 μm MPB as described previously (12). Residual unreacted MPB was quenched by adding 200 μm reduced glutathione. In some studies, MPB-labeled proteins were incubated with streptavidin-coated magnetic beads to isolate the MPB-labeled proteins. The beads were washed and then resuspended in 1× Tris-glycine SDS sample buffer to release the proteins from the beads. Proteins were detected by immunoblotting after SDS-PAGE.
Conjugation of Mastoparan and M17 Peptides to N-Hydroxysuccinimide-Sepharose
Mastoparan or M17 peptides (5 mg/ml in coupling buffer (0.5 m NaCl, 0.2 m NaHCO3, pH 8.3)) were conjugated to N-hydroxysuccinimide-activated Sepharose (HiTrap, GE Healthcare) as per the manufacturer's instructions. Briefly, after washing the column with ice-cold 1 mm HCl, the mastoparan or M17 peptide solution was run into the column and incubated for 4 h at 4 °C. The column was washed with 0.5 m NaCl, 0.5 m ethanolamine, pH 8.3 (buffer A), followed by 0.5 m NaCl, 0.1 m sodium acetate, pH 4 (buffer B), and then again with buffer A. The column was incubated for an additional 4 h at 4 °C and then washed sequentially with buffer B, buffer A, and buffer B. The unconjugated control column was prepared as per the conjugated columns, but without adding peptide to the coupling buffer for the 4-h incubation. The columns were stored at 4 °C in 0.05 m Na2HPO4, 0.1% NaN3, pH 7, until use.
Mastoparan Affinity Chromatography
Platelet releasates in HBMT buffer (10 mm HEPES 137 mm NaCl, 12 mm NaHCO3, 2.7 mm KCl, 0.36 mm NaH2PO4) were chromatographed on the control Sepharose column, the mastoparan-conjugated Sepharose column, or the M17-conjugated Sepharose column. The columns were equilibrated with 5 column volumes of binding buffer A (HBMT with 0.1% dextrose), and after applying 1 ml of platelet releasate, the columns were washed with 10 column volumes of binding buffer. Proteins were then eluted with a step gradient of NaCl in the binding buffer, up to a concentration of 1 m. Protein elution was monitored by absorbance at 280 nm.
In separate experiments, platelet releasates were diluted 1:3 in 10 mm HEPES buffer, pH 7.4, and chromatographed on mastoparan- or M17-conjugated Sepharose columns pre-equilibrated with binding buffer B (10 mm HEPES buffer containing 50 mm NaCl, pH 7.4). After washing with 5 column volumes of binding buffer B, proteins were eluted with a step gradient of 100, 150, 200, and 1000 mm NaCl. Fractions (1 ml) were treated with trichloroacetic acid, and the precipitated proteins were separated by SDS-PAGE under non-reducing condition. Gels were either used for immunoblotting or stained with Coomassie Blue. The higher molecular weight (HMW) band(s) were cut from the gel and analyzed by mass spectroscopy. Total TGF-β1 levels were measured by ELISA.
Thiol Sepharose Chromatography
All buffers were filtered and degassed under vacuum. Control and thiol-Sepharose beads (GE Healthcare) were washed on sintered glass filters and resuspended in binding buffer (0.1 m NaCl, 0.1 m Tris/HCl, pH 7.5). The slurry was degassed under vacuum for 1 h and poured into columns, resulting in a 1-ml bed volume. Columns were equilibrated with 10 ml of binding buffer and capped to prevent exposure to ambient oxygen. Platelet releasate (1 ml) was allowed to run into the column and then incubated for 1 h at room temperature. Columns were washed with 10 ml of the same buffer, and the flow-through fractions were collected. Bound proteins were eluted with 25 mm DTT. Samples were analyzed by SDS-PAGE, and proteins were identified by mass spectrometry.
Expression and Purification of PDI Trapping Mutant
A PDI trapping mutant was expressed in Escherichia coli. BL21(DE3) cells transformed with pET28a-PDI CXXA plasmid (His-tagged human PDI with both C-terminal cysteines of the active site mutated to alanine) were cultured at 37 °C with shaking at 200 rpm until the A600 reached 0.6. Protein expression was then induced with 1 mm isopropyl β-d-1-thiogalactopyranosidase for a further 5 h at 37 °C with shaking at 200 rpm. Cells were harvested, washed, and lysed after freeze-thaw with lysis buffer (300 mm NaCl, 50 mm Tris/HCl, 10 mm imidazole, pH 8.0 containing 1 mm PMSF, 0.06% (w/v) DNaseI, 0.06% (w/v) RNase A, and 0.06% (w/v) lysozyme) at 4 °C overnight. Cell debris was pelleted, and the protein-containing supernatant was purified with nickel-nitrilotriacetic acid beads (Qiagen, Valencia, CA) as per the manufacturer's instructions. The eluted protein was dialyzed against PBS, pH 7.4, before use.
Oxidation and Reduction of PDI
PDI was oxidized or reduced as described previously (14). Briefly, 250 μg of PDI in PBS was incubated overnight at room temperature with either 10 mm GSSG (oxidation) or 10 mm DTT (reduction). Excess GSSG and DTT were removed using a PD-10 Sephadex G-25 column equilibrated with PBS. Free thiol content of oxidized and reduced PDI was determined using 1.25 mm 5,5′-dithiobis-(2-nitrobenzoic acid) with detection at 412 nm. Oxidized PDI contained 0.7 ± 0.4 μmol of free thiol per μmol PDI, whereas reduced PDI contained 2.9 ± 0.9 μmol of free thiol per μmol PDI (mean ± S.D., n = 5, p = 0.001).
Mass Spectrometric Identification of Eluted Proteins
Protein samples (in solution or in gel) were digested with 25 ng/μl trypsin (Promega) in solution at 37 °C overnight, and the resulting peptides were analyzed by liquid chromatography (LC)-MS/MS. Briefly, the peptides were desalted, separated by reverse phase nanoflow liquid chromatography (Dionex), and analyzed in an Orbitrap XL mass spectrometer using tandem MS in ion trap collision-induced dissociation (CID) mode. MS/MS data were extracted using Proteome Discoverer (version 1.3, Thermo, Bremen, Germany) and queried against the Human International Protein Index (version 3.87) using MASCOT (version 2.3, Matrixscience, London, UK). For the search, mass tolerance of 20 ppm and 0.5 Da were used for peptide precursors and peptide fragments, respectively. Matched proteins were filtered to ensure that each protein was matched by a minimum of two rank 1 peptides with a MASCOT score of >20. The four different samples were compared based on the number of peptide spectrum matches (Spectral Counts).
Statistical Analysis
Data were analyzed with GraphPad Prism software (version 5) and are presented as mean ± S.E. unless otherwise stated. All t tests were two-tailed, and p values < 0.05 were considered statistically significant.
RESULTS
Mastoparan Inhibits Stirring-induced TGF-β1 Activation in a Dose- and Shear-dependent Manner
Platelet releasates were incubated in glass cuvettes at 37 °C and either unstirred or stirred. In accordance with our previous reports, stirring platelet releasates for 120 min at 37 °C significantly increased TGF-β1 activity (from 1.2 ± 0.6% to 8.9 ± 1.1% of total TGF-β1; n = 3; p = 0.02), as measured by a mink lung epithelial cell-based assay (Fig. 1A). Adding mastoparan (100 μm) to the platelet releasates prior to stirring inhibited TGF-β1 activation by more than 90% (1.9 ± 0.6% of total TGF-β1; n = 3, p = 0.004). No inhibition was observed with an inactive mastoparan-like control peptide (M17) (100 μm; 8.6 ± 1.0% of total TGF-β1; n = 3, p = 0.66 compared with untreated sample). To eliminate the possibility that the inhibition by mastoparan was due to an effect on the mink lung epithelial cells rather than the activation process, we used a TGF-β1 ELISA detection system for all subsequent experiments. When added before stirring, mastoparan inhibited TGF-β1 activation in a dose-dependent manner with an IC50 of 1.64 ± 0.87 μm (Fig. 1B), with maximal inhibition of TGF-β1 activation achieved with 25 μm, the dose chosen for subsequent experiments. The inhibition of mastoparan was evident at every time point up to 120 min (n = 3; p = 0.023 for control versus mastoparan at 120 min) (Fig. 1C). In contrast, adding mastoparan after stirring for a further 120 min did not affect the active TGF-β1 level, indicating that mastoparan was not interfering with the detection of active TGF-β1 by the assay (Fig. 1D). We also tested the effect of mastoparan on stirring-induced activation of recombinant LLC expressed by HEK293T cells. TGF-β1 and LTBP-1 could be detected by immunoblotting and ELISA of the conditioned medium of cells co-transfected with pro-TGF-β1 and LTBP-1 (Fig. 1E). Adding mastoparan (25 μm) to the conditioned medium prior to stirring at 1200 rpm for 120 min completely inhibited stirring-induced activation of TGF-β1 when compared with the M17 control peptide (n = 4; p = 0.001 for stirred versus stirred with mastoparan) (Fig. 1F).
FIGURE 1.

A, mastoparan inhibits stirring-induced TGF-β1 activation. Platelet releasate was incubated without (US) or with (S) stirring for 2 h at 37 °C, in the absence or presence of 100 μm mastoparan (S + M) or M17 control peptide (S + M17). Total and active TGF-β1 activity was measured by the mink lung epithelial cell-based assay and active TGF-β1 was expressed as a percentage of total TGF-β1. **, p = 0.001; ns, non-significant. B, mastoparan inhibits stirring-induced TGF-β1 activation in a concentration-dependent manner. Total and active TGF-β1 in B–G were measured by ELISA, and active TGF-β1 was expressed as a percentage of total TGF-β1 (n = 7). C, time-dependent, stirring-induced TGF-β1 activation is inhibited by 25 μm mastoparan peptide. Platelet releasates were stirred for 0, 10, 30, 60, and 120 min at 37 °C. **, p = 0.02 for control versus mastoparan at 120 min; p = 0.07 for mastoparan versus M17 peptide at 120 min. D, addition of mastoparan to platelet releasates after stirring has no effect on TGF-β1 activity. Platelet releasates were stirred in the absence (None) or presence of 25 μm mastoparan, which was added either before or after stirring for 120 min. E, transfection of HEK 293T cells with human pro-TGF-β1 and LTBP-1 results in secretion of LLC into the supernatant (48 h). Supernatants were immunoblotted to detect TGF-β1 (25 kDa) and LTBP-1·LAP complex (270 kDa). Results shown are representative of three separate experiments. F, mastoparan inhibits stirring-induced activation of recombinant LLC. Supernatant (200 μl) from HEK 293T cells containing LLC was either unstirred (US) or stirred (S) in the absence or presence of 25 μm mastoparan (M) or M17 at 1200 rpm for 120 min at 37 °C. **, p = 0.001. G, platelet releasates were incubated (US) or subjected to shear (S) in the absence or presence of 25 μm mastoparan (S+M) or M17 peptide (S+M17) for 2 h. Data are presented as % of activated TGF-β1 with shear-activated TGF-β1 set at 100%. Data presented as mean ± S.E.
We previously demonstrated that latent TGF-β1 released from platelets can also be activated by shear force (12), so we tested the effect of mastoparan on shear-induced TGF-β1 activation by subjecting platelet releasates to a shear rate of 1800 s−1 for 2 h at 22 °C in a cone and plate device. To normalize for variability in shear-induced activation between experiments, the shear-induced activity was set at 100% (expressed as a percentage of total) for each separate experiment (n = 9). TGF-β1 activity in the absence of shear was 2.3 ± 0.8% of the value in the presence of shear. The addition of mastoparan (25 μm) before shear reduced TGF-β1 activation to 26.1 ± 8.2% (p < 0.0001). In sharp contrast, addition of the M17 peptide had minimal effect on shear-induced TGF-β1 activation (90.6 ± 7.6%; p = 0.26; n = 7) (Fig. 1G).
Mastoparan Does Not Disrupt the Covalent Association of LTBP-1 and LAP That Is Necessary for Shear-induced Activation of TGF-β1
Because we previously found that LTBP-1 must be covalently bound to LAP for TGF-β1 to be activatable by shear (12), we assessed whether mastoparan affected the covalent association of LTBP-1 with LAP. We observed no change in the immunoblot pattern of either LTBP-1 or LAP when the releasates were stirred in the presence of 25 μm mastoparan (Fig. 2A). LTBP-1 was present as free LTBP-1 (160 kDa), LTBP-1 disulfide-bonded to LAP as part of the LLC (240 kDa), LTBP-1 disulfide-bonded to LAP as part of a HMW complex (>270 kDa), and as part of one or more additional HMW complexes that do not include LAP (∼280 kDa). LAP was disulfide-bonded to LTBP-1 in both LLC (240 kDa) and HMW (>250 kDa) complexes; no free LAP dimer or uncleaved LAP/TGF-β1 was detected.
FIGURE 2.

A, mastoparan does not alter the covalent structure of LLC. Mastoparan (25 μm) or M17 peptide (25 μm) was added to platelet releasates that were then stirred for 120 min at 37 °C. The samples were analyzed by non-reduced SDS-PAGE and immunoblotting with antibodies to LTBP-1 and LAP. No change in the Mr of LAP or LTBP-1 was observed in the presence of mastoparan. The immunoblot is representative of three separate experiments. B, mastoparan alters the thiol labeling of proteins with MPB after stirring compared with control M17 peptide. Platelet releasates were stirred in the absence (control, C) or presence of 25 μm mastoparan (M) or M17 for 120 min. The samples were then labeled with MPB (100 μm), and the proteins captured with streptavidin-coupled beads. Proteins were eluted with SDS sample buffer, electrophoresed on SDS-PAGE, and transferred to PVDF membrane. MPB-labeled proteins were identified with streptavidin (SA) HRP. Black arrows indicate protein bands with increased MPB labeling in the presence of mastoparan, and red arrows indicate protein band with decreased MPB labeling. A representative blot from three separate experiments is shown. US, unstirred; S, stirred.
Free Thiol Labeling of Proteins in Platelet Releasates Is Altered after Stirring in the Presence of Mastoparan
Neither mastoparan nor M17 altered the thiol labeling of unstirred platelet releasates (Fig. 2B and data not shown). Consistent with our previous observations, there was a loss of free thiols in platelet releasate proteins upon stirring as judged by a decrease in MPB labeling (Fig. 2B). Incubation of platelet releasates with mastoparan before stirring, however, altered the post-stirring thiol labeling pattern of proteins with MPB, with some proteins showing increased labeling (black arrows) and others showing reduced labeling (red arrows) (Fig. 2B, right panel; high exposure lanes). Protein levels were not altered by exposure to stirring prior to pulldown with streptavidin beads as observed with Coomassie protein stain (data not shown).
Identification of Proteins in Platelet Releasates, including TGF-β1, LTBP-1, and Thiol Isomerases, That Bind Specifically to a Mastoparan Affinity Column
When platelet releasates were chromatographed on the control, mastoparan-conjugated, or M17-conjugated Sepharose columns, and then the columns were washed and eluted with a stepwise salt gradient, significantly more protein and more TGF-β1 eluted from the mastoparan column compared with the control columns at 200 mm NaCl (Fig. 3, A and B; n = 3; p = 0.02 for mastoparan versus M17 peptide, and p = 0.01 mastoparan versus control). (Supplemental Fig. 2 shows elution with 50 mm NaCl buffer.) SDS-PAGE and Coomassie Blue staining revealed depletion of a HMW band in the flow-through fraction from the mastoparan column and a much stronger band in this region in the eluate of the mastoparan column compared with the control and M17 columns. In addition, several other proteins eluted from the mastoparan column but not the unconjugated or M17 peptide columns (Fig. 3C, left panel). Many of the proteins eluted from the mastoparan column labeled with MPB, indicating that they contain free thiols, including a major HMW complex(es) (Fig. 3C, right panel).
FIGURE 3.

A, proteins bind specifically to a mastoparan affinity column compared with an M17 affinity column. Unstirred platelet releasates were loaded onto columns containing unconjugated, mastoparan-conjugated, or M17-conjugated Sepharose. After washing, proteins were eluted with buffer containing 200 mm NaCl. UV absorbance at 280 nm of unconjugated, mastoparan-conjugated, and M17-conjugated column eluates is shown (average of three experiments shown; error bars omitted for clarity). More protein eluted from the mastoparan affinity column than from either of the other columns (n = 3; ***, p = 0.001). B, TGF-β1 binds specifically to the mastoparan-conjugated column. Total TGF-β1 protein in the eluates from the columns was detected by ELISA (n = 3; *, p = 0.02). C, many proteins eluted from mastoparan affinity column contain free thiols. Left panel: Coomassie blue staining of platelet releasates (PR), flow-through fractions from the unconjugated (C), mastoparan-conjugated (M), and M17-conjugated Sepharose columns, and the eluates (concentrated 30-fold) from the same columns reacted with MPB after elution. Right panel, MPB-labeled proteins were detected by streptavidin HRP and demonstrated dramatic enrichment in the mastoparan eluate.
Identification of Platelet Releasate Proteins That Bound to the Mastoparan Affinity Column and the Thiol-Sepharose Column
Proteins eluted from the control, mastoparan, or M17 columns were digested with trypsin, and the mass spectra of the peptides were obtained. Table 1 lists the proteins that were identified in the eluate from the mastoparan column, but not the eluates from the other two columns. LTBP-1 was detected in all experiments and TGF-β1 precursor (which includes peptides from both LAP and mature TGF-β1) was detected in three of four experiments. Clusterin, multimerin-1, von Willebrand factor (vWF), nidogen-1, and protein 14-3-3 ζ/δ also bound specifically to the mastoparan affinity column in three of four experiments. Supplemental Table 1 contains a full list of the specifically bound proteins. We confirmed the selective presence of TGF-β1, LAP, LTBP-1, clusterin, multimerin-1, and vWF in the eluate from the mastoparan affinity column by immunoblotting (Fig. 4A). TSP-1 bound non-specifically to all three columns as judged by both mass spectrometry and immunoblotting (Fig. 4A and supplemental Table 1). Fibrinogen was present in the eluate from all the columns but was enriched in the eluate from the mastoparan column (supplemental Fig. 2). Furthermore, we specifically targeted the HMW complex(es) as seen in Fig. 3C for identification of its component protein(s). The mass spectrometry analysis of the HMW band(s) identifies TSP-1, fibrinogen, multimerin-1, LTBP-1, and TGF-β1, which were also validated by immunoblotting (supplemental Fig. 2).
TABLE 1.
Proteins identified by mass spectrometry as specifically binding to the mastoparan-conjugated Sepharose column as compared to the M17-conjugated or unconjugated Sepharose columns
All of the proteins have been previously identified as being in platelets and released upon platelet activation (n = 4).
| Protein | Detection frequencya |
|---|---|
| Latent TGFβ-binding protein isoform 1 | 4/4 |
| TGF-β1 precursor | 3/4 |
| Clusterin | 3/4 |
| Multimerin-1 | 3/4 |
| 14-3-3 protein ζ/δ | 3/4 |
| von Willebrand factor | 3/4 |
| Nidogen-1 | 3/4 |
a No. of experiments in which detected/No. of experiments in which tested.
FIGURE 4.

A, immunoblots of proteins eluted from affinity columns. Top row, non-reduced SDS-PAGE. LTBP-1 and TGF-β1 bound specifically to the mastoparan-conjugated column (M), whereas TSP-1 bound non-specifically to all three columns (C, control; M, mastoparan peptide; M17, M17 control peptide). Bottom row, non-reduced SDS-PAGE of LAP and reduced SDS-PAGE of clusterin, vWF, and multimerin-1. All bound specifically to the mastoparan-conjugated column. Immunoblots shown are representative of four separate experiments. B, thiol isomerase enzymes bind specifically to the mastoparan affinity column. The thiol isomerase enzymes PDI (60 kDa), ERp5 (48 kDa), ERp57 (57 kDa), and ERp72 (72 kDa) bound specifically to the mastoparan-conjugated column. Immunoblots shown are representative of four separate experiments. C, anti-PDI RL90 attenuates the inhibition of stirring-induced TGF-β1 activation by mastoparan. Platelet releasate (200 μl) was stirred alone, in the presence of 1 μg/ml RL90 (S+RL90), 25 μm mastoparan (S+M), or mastoparan and RL90 (S+M+RL90). TGF-β1 activation was detected by ELISA, and data were normalized to the stirring control (100%). RL90 attenuated the inhibitory effect of mastoparan on TGF-β1 activation (n = 8; *, p = 0.02). ns, not significant.
We also investigated whether thiol isomerase enzymes were selectively present in the mastoparan column eluates. PDI, ERp5, ERp57, and ERp72 were all identified by immunoblotting in the mastoparan column eluates but not the control columns (Fig. 4B).
Because mastoparan is known to bind to the thiol-containing enzyme PDI, and thus may potentially bind to other thiol-containing proteins, we assessed proteins containing free thiols in platelet releasates by performing mass spectroscopy on samples eluted by DTT from a thiol-Sepharose column, but not a control Sepharose column. Several of the molecules that bound to the mastoparan column also bound to the thiol-Sepharose column, including TSP-1, vWF, fibrinogen, and multimerin-1 (supplemental Table 2 and supplemental Fig. 1).
The Inhibitory Effect of Mastoparan on Stirring-induced TGF-β1 Activation Can Be Partially Attenuated with Anti-PDI
To define better the role of PDI in stirring-induced TGF-β1 activation, we tested the effect of mAb RL90, which inhibits PDI activity, on stirring-induced activation of TGF-β1 in the absence and presence of mastoparan. We found no significant effect of mAb RL90 on shear-dependent TGF-β1 activation in the absence of mastoparan (n = 8; p = 0.52 for stirring versus stirring with RL90) (Fig. 4C). In contrast, when RL90 was present, mastoparan produced significantly less inhibition of TGF-β1 activation than in its absence (n = 8; p = 0.02 for stirring with mastoparan versus stirring with mastoparan and RL90).
Reduced PDI, but Not Oxidized PDI, Decreases Stirring-induced TGF-β1 Activation
To assess whether the thiol isomerase enzyme, PDI, can affect shear-induced TGF-β1 activation, 5 μg of purified oxidized PDI or reduced PDI was added to the platelet releasates before stirring. Adding oxidized PDI (containing 0.7 ± 0.4 μmol of free thiol per μmol PDI) had no significant effect on stirring-induced TGF-β1 activation, whereas partially reduced PDI (containing 2.9 ± 0.9 μmol of free thiol per μmol PDI) inhibited activation by ∼36% (n = 11, p = 0.03 for S versus S + PDI red) (Fig. 5A). We also tested the effect of adding a PDI trapping mutant that lacks oxidizing activity but retains reducing activity, and it also inhibited stirring-induced TGF-β1 activation (n = 4; p = 0.007 for S versus CXXA) (Fig. 5B). Exposing the trapping mutant to either oxidizing or reducing conditions had no impact on the inhibition, which was expected because its CXXA motif cannot be oxidized (n = 4; p = 0.049). Because the PDI trapping mutant can react with protein thiols but cannot catalyze oxidation or release of the protein, its substrate(s) becomes covalently bound to the mutant PDI. Similarly, reduced PDI can form covalent complexes with its substrate(s) by forming mixed disulfides. Fig. 5, B and C, show that both reduced PDI and the trapping mutant formed stable disulfide complexes with other proteins as shown by the shift of the anti-PDI immunoblotted bands to higher Mr values and the loss of these higher Mr bands with reduction by 100 mm DTT. (We also detected a band at ∼150 kDa in the presence of 100 mm DTT, and we suggest that this may be a nonspecific band and not PDI).
FIGURE 5.
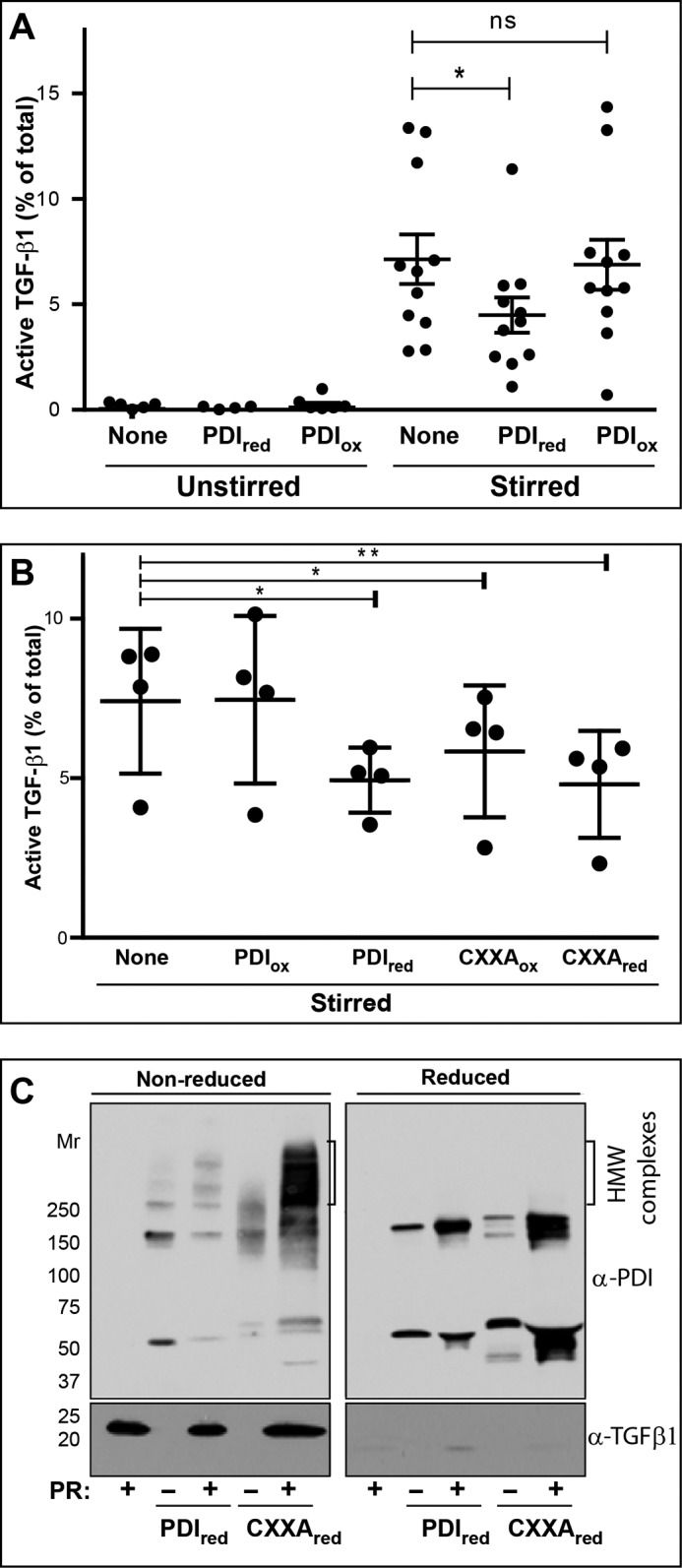
A, reduced PDI inhibits stirring-dependent TGF-β1 activation. Platelet releasate (200 μl) was unstirred or stirred at 1200 rpm for 120 min at 37 °C in the absence or presence of reduced (red) or oxidized (ox) PDI (5 μg). Active and total TGF-β1 levels were measured by ELISA (n = 11; *, p = 0.03). Data are presented as median ± interquartile range. B, trapping mutant PDI inhibits stirring-dependent TGF-β1 activation. The experiment described in Fig. 1A was repeated along with samples containing mutant PDI (5 μg) in which alanines were substituted for the C-terminal cysteines in the two CXXC sites (n = 4). *, p = 0.049; **, p = 0.007. C, reduced PDI is incorporated into higher molecular weight disulfide-bonded complexes with its substrates in platelet releasate. Left panel, non-reduced SDS-PAGE. Shown are immunoblots of samples in the absence (−) or presence (+) of PR with anti-PDI (top panel) and anti-TGF-β1 (bottom panel). Reduced PDI and PR shows some high molecular weight bands containing PDI, whereas the samples containing PDI trapping mutant (CXXA) shows much more intense staining of high molecular weight bands. TGF-β1 at 25 kDa is not incorporated into these disulfide-bonded complexes. Right panel (reduced SDS-PAGE), after DTT reduction, all of the high molecular weight complexes containing PDI or PDI trapping mutant are lost. ns, not significant.
DISCUSSION
The activation of latent TGF-β1 by stirring or shear is a novel mechanism with potential implications for understanding the physiologic and pathologic roles of this cytokine in human biology. In this study, we extended our previous findings by showing that stirring could also activate recombinant LLC. Because we previously demonstrated that this activation is partially thiol-dependent (12), in this study, we analyzed the role of thiol isomerases in this process by measuring the effects exerted by PDI and by mastoparan, a peptide that binds to the peptide-binding site of PDI and inhibits its chaperone activity. Mastoparan has also been reported to produce similar effects on other thiol isomerase enzymes (15). Partially reduced PDI and trapping mutant PDI inhibited shear-dependent TGF-β1 activation, whereas oxidized PDI had no significant effect. Mastoparan, but not a related control peptide (M17), dramatically decreased stirring-induced activation of TGF-β1 in platelet releasates, and this same inhibitory effect was observed with recombinant LLC secreted from mammalian cells. In addition, we found that TGF-β1, LAP, and LTBP-1, as well as several thiol isomerase enzymes and disulfide-containing proteins in platelet releasates bound selectively to a mastoparan affinity column, indicating that they interacted with mastoparan either directly or indirectly via intermediary proteins. Finally, we found that the anti-PDI RL90 attenuated the inhibitory effects of mastoparan, suggesting that mastoparan elicits its effects at least partially through PDI.
We previously demonstrated that passing platelet releasate through a thiol-Sepharose column strongly inhibits stirring-induced activation of TGF-β1, suggesting that free thiol-containing proteins are required for this activation process (13). Many proteins in platelet releasates, including one or more in the LLC complex itself, contain free thiols as evidenced by their binding to thiol-Sepharose and labeling with thiol-reactive agents. Our previous studies showed that the labeling of many of the thiols in platelet releasate proteins is lost upon stirring or shear (12), suggesting that the thiols are oxidized to disulfides and/or become inaccessible to thiol-reactive reagents. Our observations are in agreement with other studies demonstrating that thiol-disulfide exchange contributes to changes in the structure and function of several proteins released from platelets in vitro, including vWF and TSP-1 (16, 17). In the case of vWF, shear-induced changes in its structure are also associated with oxidation of free thiols (16).
To further probe the thiol dependence of shear-dependent TGF-β1 activation, we assessed the effect of mastoparan on this process. Thiol isomerase enzymes can catalyze oxidation, reduction, and/or isomerization of disulfide bonds in proteins. Mastoparan is an amphiphilic wasp venom peptide that directly binds the thiol isomerase enzymes PDI and ERp5, inhibiting their chaperone function (15, 18–20). Because mastoparan can also produce many diverse biological effects on cells and platelets (21) (e.g. activates phospholipase A2 in vitro (22), inhibits calmodulin activity (23), affects GTP hydrolysis in G protein-coupled receptors (24), facilitates mitochondrial permeability transition (25), degranulates mast cells (26), and initiates exocytosis in many cell types, including platelets (27, 28)), to avoid any confounding cell-based effects, we removed both cells and platelets from the platelet releasate before testing the effect of mastoparan.
PDI has four thioredoxin-like domains; the a and a′ domains contain CXXC motifs, characteristic of enzymes that catalyze thiol-disulfide exchange, oxidation, and/or reduction, and the non-catalytic b and b′ domains (reviewed by Hatahet and Ruddock (29)). Oxidation of the active site a′ domain cysteines induces a looser PDI conformation and exposes the hydrophobic substrate-binding surface of the b′ domain. As substrate binds to PDI, the disulfide bond in a′ is transferred to thiols in the substrate, followed by the release of the oxidized substrate (30). The conformational change that accompanies the oxidation of the active site a′ domain cysteines also activates the chaperone activity of PDI (31). Mastoparan binds to the hydrophobic surface within the b′ domain of PDI exposed in oxidized PDI and thereby prevents its interaction with its many substrates (18, 32). For example, addition of mastoparan to purified PDI inhibits the refolding of RNase A (19). This effect of mastoparan on PDI, and/or perhaps other thiol isomerases, probably accounts for our finding that mastoparan both inhibits TGF-β1 activation and alters the free thiol content of proteins detected after stirring. Because thiol-reacting molecules only partially inhibit shear-induced activation, however, the profound inhibitory effect of mastoparan suggests that it operates on both thiol-dependent and thiol-independent TGF-β1 activation mechanisms.
Mastoparan is known to bind to both PDI (18) and ERp5 (15), and it is possible that it also reacts with the other thiol isomerase enzymes in the platelet releasate. Previous studies demonstrated the release of PDI (33), ERp5 (34), ERp57, ERp72, ERp29, and ERp44 from platelets (35); our studies confirmed the presence of PDI, ERp57, ERp72, and ERp5 in platelet releasate and showed that all of these bound specifically to the mastoparan column. To test whether one or more of these thiol isomerases contributed to stirring-induced activation, we analyzed the effects of antibodies to PDI, ERp57, ERp72, or ERp5. We did not observe inhibition of TGF-β1 activation by any one of these antibodies (data not shown), which may reflect redundancy in this system. We considered mixing all of the antibodies, but previous studies demonstrated that even when mixed, the antibodies are unable to completely inhibit thiol isomerase activity (36).
Thiol isomerase enzymes can catalyze oxidation, reduction, and/or isomerization of disulfide bonds in proteins and the redox status of the enzymes determine the nature of the reaction. Because oxidized thiol isomerases are expected to catalyze oxidation of thiols and reduced thiol isomerases are expected to catalyze reduction of disulfide bonds, we focused on whether an oxidized thiol isomerase may be contributing to shear-induced activation of TGF-β1. Activated platelets secrete PDI (33), which has been shown to play a role in mediating conformational changes in GPIbα (37), integrin-mediated adhesion (38), and platelet aggregation (39). Platelet releasate in the absence of platelets has been previously shown to oxidize free thiols in recombinant soluble tissue factor, suggesting that platelet-released PDI behaves as an oxidant (40). Our data are also consistent with platelet releasate PDI being oxidized because the thiol-reactive reagent MPB did not label PDI in platelet releasates (supplemental Fig. 3). We assessed the effect of PDI on shear-dependent TGF-β1 activation. We were able to fully oxidize the CXXC motifs as judged by titration of the residual thiols but were able to only partially reduce the PDI, presumably because some of the reduced disulfide bonds became oxidized as the reducing agent was separated from PDI by size exclusion gel filtration. Addition of oxidized PDI had no significant effect, suggesting either that oxidized PDI does not participate, that there is sufficient endogenous oxidized PDI available for full activation or that proteins involved are already oxidized. In contrast, the partially reduced PDI inhibited shear-dependent TGF-β1 activation. The partially reduced PDI may exert its inhibitory effect by reducing disulfide bonds in platelet releasate proteins, potentially including oxidized PDI itself, and/or by preventing the formation or isomerization of disulfide bonds in TGF-β1 or other proteins in platelet releasate upon exposure to shear. We also tested a “trapping mutant” PDI, which contains two CXXA motifs instead of two CXXC active sites, and thus is incapable of being oxidized. As a result, it has no oxidase activity, but it can still reduce disulfide bonds as it forms a mixed disulfide with its substrates. The trapping mutant, similar to the partially reduced PDI, inhibited shear-dependent TGF-β1 activation. Because the N-terminal cysteine of the PDI active sites has a pKa value of 6.7 (41), we expect this cysteine to be deprotonated in our experiments, all of which are performed at pH 7.4. Both reduced PDI and the CXXA trapping mutant were incorporated into HMW disulfide-bonded complexes when added to platelet releasate and stirred, demonstrating the presence of several PDI substrates, but the immunoblot staining intensity of the HMW complexes was much greater with the trapping mutant, reflecting the irreversible nature of its interactions. To test the generalizability of the findings with PDI, we also tested the effects of reduced thioredoxin and a thioredoxin trapping mutant that we obtained in limited amounts on stirring-induced TGF-β1 activation. In a single experiment, reduced thioredoxin inhibited stirring-induced TGF-β1 activity from 6.1 to 5.2%, whereas the thioredoxin trapping mutant inhibited stirring-induced TGF-β1 activity from 6.1 to 2.7%. As with the PDI trapping mutant, the thioredoxin trapping mutant was incorporated into higher Mr complexes in the presence of platelet releasate (n = 2), supporting the hypothesis that oxidation is required for TGF-β1 activation.
Based on our data, we propose the following working model for the thiol-dependent contribution to shear-induced TGF-β1 activation (Fig. 6). Oxidized PDI and/or another thiol isomerase or protein(s) participates in stirring-induced activation of LLC by oxidizing one or more sulfhydryl groups in either LLC or an interacting protein. Mastoparan inhibits the process by binding to and inactivating oxidized PDI and/or another thiol isomerase(s) and thus prevents it from catalyzing the protein oxidation. Our model is supported by data showing that there is loss of free thiols upon stirring- and shear-induced activation of TGF-β1 (12), that there is loss of shear-dependent TGF-β1 activation when thiol-containing proteins are depleted from platelet releasates (12), and that mastoparan binding produces chemical shift perturbations in oxidized PDI a′ and b′ domains that are not observed when added to reduced PDI (30). Because adding exogenous oxidized PDI did not enhance TGF-β1 activation, our model predicts that platelet releasates contain enough oxidized PDI (and/or another thiol isomerase(s)) to support maximal TGF-β1 activation. Exogenous reduced PDI, which is added in great molar excess to endogenous PDI, reduces platelet releasate proteins (either directly or through intermediate proteins or thiol isomerases), thus interfering with the protein oxidation required for the thiol-dependent mechanism of TGF-β1 activation. The PDI trapping mutant inhibits activation by forming a stable disulfide intermediate complex(es) with substrate(s) of oxidized PDI. The partial inhibition observed with both the reduced PDI and the trapping mutant is consistent with our previous data indicating that thiol-alkylating agents produce only partial inhibition of shear-induced activation (12).
FIGURE 6.

Working model of the thiol-dependent component of shear-induced TGF-β1 activation. Oxidized PDI (PDIox) in platelet releasates contributes to oxidation of substrate proteins during stirring, resulting in activation of TGF-β1. Reduced PDI (PDIred) and trapping mutant PDI (PDITM) inhibit the oxidation by catalyzing reduction of disulfide bonds and/or competing with oxidized PDI for substrate proteins. Mastoparan acts, in part by binding to oxidized PDI, preventing oxidation of platelet releasate proteins. The anti-PDI mAb RL90 attenuates the inhibitory effect of mastoparan by inhibiting mastoparan binding to oxidized PDI.
The effect of RL90, a mAb directed at unknown epitopes on PDI (and possibly ERp57, Ref. 42) that inhibits the reductase activity of PDI, are complex. When RL90 is added to platelet releasates in the absence of mastoparan, it does not affect stirring-induced TGF-β1 activation, whereas it partially attenuates the inhibitory effect of mastoparan on TGF-β1 activation. The former finding could be explained if either other thiol isomerases can compensate for loss of PDI (and ERp57) or if RL90 does not inhibit PDI oxidase activity. The latter finding could be explained if the binding of RL90 prevents the binding of mastoparan to PDI, but does not inhibit PDI oxidase activity, and if mastoparan also blocks the oxidase activity of one or more other thiol isomerases that participates in the process. To test our hypothesis directly, we sought a standardized and validated thiol isomerase oxidase assay but found that the only published assay (43), which was used to compare the oxidase activities of PDI, ERp72, and ERp5, cannot be performed at the pH of our experiments (pH 7.4) because at pH 7 or above the rate of spontaneous oxidation of the substrate confounds the analysis. The greater inhibition of stirring-induced TGF-β1 activation with mastoparan than with reduced PDI and thiol alkylators suggests that mastoparan inhibits both thiol-dependent and thiol-independent activation mechanisms.
We found that TGF-β1, LAP and LTBP-1, as well as several other disulfide-containing proteins in platelet releasates bound selectively to the mastoparan affinity column, indicating that they interacted with mastoparan either directly or indirectly via intermediary proteins. These proteins are thus potential candidates for mediating or facilitating thiol-disulfide exchange. Clusterin, a heterodimeric protein expressed in megakaryocytes and platelets that contains two subunits of 35 kDa connected by five disulfide bridges (44), is known to be a substrate of both ERp57 and PDI (45). It is of note, therefore, that clusterin can promote epithelial-to-mesenchymal transition mediated by TGF-β1, and clusterin gene expression is highly up-regulated in response to TGF-β1 signaling (46). In fact, clusterin has been demonstrated to attenuate renal fibrosis through TGF-β1-dependent mechanisms (47). MMRN1 (multimerin-1) is a large, disulfide-linked protein produced by megakaryocytes and endothelial cells and found in the α-granules of platelets (48). It binds coagulation factor V, which may account for both proteins being identified in the eluate from the mastoparan affinity column. MMRN1 contains 21 cysteine residues, with 16 of these residues postulated to be involved in disulfide bonds (49). Emilin-1, a member of the multimerin protein family, has been shown to bind to uncleaved pro-TGF-β1 in the extracellular space and prevents its cleavage; targeted deletion of the emilin-1 gene increases TGF-β1 signaling (50). Emilin-3 has also been characterized as a pro-TGF-β antagonist, suggesting a role for the larger multimerin-like protein family in regulation of TGF-β1 activity (51). Thus, thiol-containing proteins, such as fibrinogen, multimerin, vWF, and clusterin may form part of the HMW complex that is eluted from the mastoparan affinity column. We identified TSP-1, fibrinogen, multimerin-1, LTBP-1, and TGF-β1 in the HMW complex. Collectively, these data strongly suggest that the thiol alterations seen in HMW platelet releasate proteins with mastoparan may involve these particular proteins.
In conclusion, although the full details of the mechanism(s) of TGF-β1 activation by shear force remain to be elucidated, our discovery that mastoparan is a strong inhibitor supports our previous hypothesis that activation involves thiol-disulfide exchange. It also provides a valuable tool for further analysis of TGF-β1 activation and potentially the biochemical basis for other shear-dependent thiol-disulfide exchange phenomena (16). Finally, the growing evidence for a role of platelet-derived TGF-β1 in pathological states in which shear force may play a role raises the possibility that shear force-induced activation of TGF-β1 may be a therapeutic target.
Acknowledgments
We thank Dr. Henrik Molina and Joseph Fernandez at the Rockefeller University Proteomics Resource Center for assistance with mass spectrometry analysis and Dr. Mayte Suarez-Farinas at the Rockefeller University for advice on statistical analysis. We also thank Professor Jonathan Gibbins (University of Reading, U.K.) for providing antibodies to platelet thiol isomerases and Professor Philip J Hogg (University of New South Wales, Australia) for providing PDI trapping mutant plasmid DNA and thioredoxin trapping mutant protein.
This work was supported by Grant HL19278 from the National Heart, Lung, and Blood Institute; a Clinical and Translational Science Award (TR000043) from the National Center for Research Resources and the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health; a New York City Community Trust Grant Award; an Irma T. Hirschl/Monique Weill-Caulier Trust Research Award (to J. A.); and funds from Stony Brook University.

This article contains supplemental Tables 1 and 2 and Figs. 1–3.
- LAP
- latency-associated peptide
- LLC
- large latent complex
- PR
- platelet releasate
- PDI
- protein disulfide isomerase
- LTBP-1
- latent TGF-β1 binding protein 1
- TSP-1
- thrombospondin-1
- MPB
- 3-(N-maleimidylpropionyl)biocytin
- HMW
- higher molecular weight
- vWF
- von Willebrand factor.
REFERENCES
- 1. Blobe G. C., Schiemann W. P., Lodish H. F. (2000) Role of transforming growth factor β in human disease. N. Engl. J. Med. 342, 1350–1358 [DOI] [PubMed] [Google Scholar]
- 2. Assoian R. K., Komoriya A., Meyers C. A., Miller D. M., Sporn M. B. (1983) Transforming growth factor-β in human platelets. Identification of a major storage site, purification, and characterization. J. Biol. Chem. 258, 7155–7160 [PubMed] [Google Scholar]
- 3. Wakefield L. M., Smith D. M., Flanders K. C., Sporn M. B. (1988) Latent transforming growth factor-β from human platelets. A high molecular weight complex containing precursor sequences. J. Biol. Chem. 263, 7646–7654 [PubMed] [Google Scholar]
- 4. Labelle M., Begum S., Hynes R. O. (2011) Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 20, 576–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meyer A., Wang W., Qu J., Croft L., Degen J. L., Coller B. S., Ahamed J. (2012) Platelet TGF-β1 contributions to plasma TGF-β1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 119, 1064–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gleizes P. E., Beavis R. C., Mazzieri R., Shen B., Rifkin D. B. (1996) Identification and characterization of an eight-cysteine repeat of the latent transforming growth factor-β binding protein-1 that mediates bonding to the latent transforming growth factor-β1. J. Biol. Chem. 271, 29891–29896 [DOI] [PubMed] [Google Scholar]
- 7. Munger J. S., Huang X., Kawakatsu H., Griffiths M. J., Dalton S. L., Wu J., Pittet J. F., Kaminski N., Garat C., Matthay M. A., Rifkin D. B., Sheppard D. (1999) The integrin αvβ6 binds and activates latent TGF β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 [DOI] [PubMed] [Google Scholar]
- 8. Shi M., Zhu J., Wang R., Chen X., Mi L., Walz T., Springer T. A. (2011) Latent TGF-β structure and activation. Nature 474, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murphy-Ullrich J. E., Poczatek M. (2000) Activation of latent TGF-β by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev. 11, 59–69 [DOI] [PubMed] [Google Scholar]
- 10. Jenkins G. (2008) The role of proteases in transforming growth factor-β activation. Int. J. Biochem. Cell Biol. 40, 1068–1078 [DOI] [PubMed] [Google Scholar]
- 11. Jobling M. F., Mott J. D., Finnegan M. T., Jurukovski V., Erickson A. C., Walian P. J., Taylor S. E., Ledbetter S., Lawrence C. M., Rifkin D. B., Barcellos-Hoff M. H. (2006) Isoform-specific activation of latent transforming growth factor β (LTGF-β) by reactive oxygen species. Radiat. Res. 166, 839–848 [DOI] [PubMed] [Google Scholar]
- 12. Ahamed J., Burg N., Yoshinaga K., Janczak C. A., Rifkin D. B., Coller B. S. (2008) In vitro and in vivo evidence for shear-induced activation of latent transforming growth factor-β1. Blood 112, 3650–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ahamed J., Janczak C. A., Wittkowski K. M., Coller B. S. (2009) In vitro and in vivo evidence that thrombospondin-1 (TSP-1) contributes to stirring- and shear-dependent activation of platelet-derived TGF-β1. PLoS One 4, e6608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Raturi A., Mutus B. (2007) Characterization of redox state and reductase activity of protein disulfide isomerase under different redox environments using a sensitive fluorescent assay. Free Radic. Biol. Med. 43, 62–70 [DOI] [PubMed] [Google Scholar]
- 15. Kikuchi M., Doi E., Tsujimoto I., Horibe T., Tsujimoto Y. (2002) Functional analysis of human P5, a protein disulfide isomerase homologue. J. Biochem. 132, 451–455 [DOI] [PubMed] [Google Scholar]
- 16. Choi H., Aboulfatova K., Pownall H. J., Cook R., Dong J. F. (2007) Shear-induced disulfide bond formation regulates adhesion activity of von Willebrand factor. J. Biol. Chem. 282, 35604–35611 [DOI] [PubMed] [Google Scholar]
- 17. Huang E. M., Detwiler T. C., Milev Y., Essex D. W. (1997) Thiol-disulfide isomerization in thrombospondin: effects of conformation and protein disulfide isomerase. Blood 89, 3205–3212 [PubMed] [Google Scholar]
- 18. Klappa P., Ruddock L. W., Darby N. J., Freedman R. B. (1998) The b' domain provides the principal peptide-binding site of protein disulfide isomerase but all domains contribute to binding of misfolded proteins. EMBO J. 17, 927–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Denisov A. Y., Määttänen P., Dabrowski C., Kozlov G., Thomas D. Y., Gehring K. (2009) Solution structure of the bb′ domains of human protein disulfide isomerase. FEBS J. 276, 1440–1449 [DOI] [PubMed] [Google Scholar]
- 20. Raturi A., Ruf W. (2010) Effect of protein disulfide isomerase chaperone activity inhibition on tissue factor activity. J. Thromb. Haemost. 8, 1863–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones S., Howl J. (2006) Biological applications of the receptor mimetic peptide mastoparan. Curr. Protein Pept. Sci. 7, 501–508 [DOI] [PubMed] [Google Scholar]
- 22. Argiolas A., Pisano J. J. (1983) Facilitation of phospholipase A2 activity by mastoparans, a new class of mast cell degranulating peptides from wasp venom. J. Biol. Chem. 258, 13697–13702 [PubMed] [Google Scholar]
- 23. Barnette M. S., Daly R., Weiss B. (1983) Inhibition of calmodulin activity by insect venom peptides. Biochem. Pharmacol. 32, 2929–2933 [DOI] [PubMed] [Google Scholar]
- 24. Higashijima T., Uzu S., Nakajima T., Ross E. M. (1988) Mastoparan, a peptide toxin from wasp venom, mimics receptors by activating GTP-binding regulatory proteins (G proteins). J. Biol. Chem. 263, 6491–6494 [PubMed] [Google Scholar]
- 25. Pfeiffer D. R., Gudz T. I., Novgorodov S. A., Erdahl W. L. (1995) The peptide mastoparan is a potent facilitator of the mitochondrial permeability transition. J. Biol. Chem. 270, 4923–4932 [DOI] [PubMed] [Google Scholar]
- 26. Hirai Y., Yasuhara T., Yoshida H., Nakajima T., Fujino M., Kitada C. (1979) A new mast cell degranulating peptide “mastoparan” in the venom of Vespula lewisii. Chem. Pharm. Bull. 27, 1942–1944 [DOI] [PubMed] [Google Scholar]
- 27. Ozaki Y., Matsumoto Y., Yatomi Y., Higashihara M., Kariya T., Kume S. (1990) Mastoparan, a wasp venom, activates platelets via pertussis toxin-sensitive GTP-binding proteins. Biochem. Biophys. Res. Commun. 170, 779–785 [DOI] [PubMed] [Google Scholar]
- 28. Wheeler-Jones C. P., Saermark T., Kakkar V. V., Authi K. S. (1992) Mastoparan promotes exocytosis and increases intracellular cyclic AMP in human platelets. Evidence for the existence of a Ge-like mechanism of secretion. Biochem. J. 281, 465–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hatahet F., Ruddock L. W. (2009) Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid. Redox Signal. 11, 2807–2850 [DOI] [PubMed] [Google Scholar]
- 30. Serve O., Kamiya Y., Maeno A., Nakano M., Murakami C., Sasakawa H., Yamaguchi Y., Harada T., Kurimoto E., Yagi-Utsumi M., Iguchi T., Inaba K., Kikuchi J., Asami O., Kajino T., Oka T., Nakasako M., Kato K. (2010) Redox-dependent domain rearrangement of protein disulfide isomerase coupled with exposure of its substrate-binding hydrophobic surface. J. Mol. Biol. 396, 361–374 [DOI] [PubMed] [Google Scholar]
- 31. Wang C., Yu J., Huo L., Wang L., Feng W., Wang C. C. (2012) Human protein-disulfide isomerase is a redox-regulated chaperone activated by oxidation of domain a′. J. Biol. Chem. 287, 1139–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Byrne L. J., Sidhu A., Wallis A. K., Ruddock L. W., Freedman R. B., Howard M. J., Williamson R. A. (2009) Mapping of the ligand-binding site on the b′ domain of human PDI: interaction with peptide ligands and the x-linker region. Biochem. J. 423, 209–217 [DOI] [PubMed] [Google Scholar]
- 33. Chen K., Lin Y., Detwiler T. C. (1992) Protein disulfide isomerase activity is released by activated platelets. Blood 79, 2226–2228 [PubMed] [Google Scholar]
- 34. Jordan P. A., Stevens J. M., Hubbard G. P., Barrett N. E., Sage T., Authi K. S., Gibbins J. M. (2005) A role for the thiol isomerase protein ERP5 in platelet function. Blood 105, 1500–1507 [DOI] [PubMed] [Google Scholar]
- 35. Holbrook L. M., Watkins N. A., Simmonds A. D., Jones C. I., Ouwehand W. H., Gibbins J. M. (2010) Platelets release novel thiol isomerase enzymes which are recruited to the cell surface following activation. Br. J. Haematol. 148, 627–637 [DOI] [PubMed] [Google Scholar]
- 36. Jordan P. A., Gibbins J. M. (2006) Extracellular disulfide exchange and the regulation of cellular function. Antioxid. Redox Signal. 8, 312–324 [DOI] [PubMed] [Google Scholar]
- 37. Burgess J. K., Hotchkiss K. A., Suter C., Dudman N. P., Szöllösi J., Chesterman C. N., Chong B. H., Hogg P. J. (2000) Physical proximity and functional association of glycoprotein 1bα and protein-disulfide isomerase on the platelet plasma membrane. J. Biol. Chem. 275, 9758–9766 [DOI] [PubMed] [Google Scholar]
- 38. Lahav J., Gofer-Dadosh N., Luboshitz J., Hess O., Shaklai M. (2000) Protein disulfide isomerase mediates integrin-dependent adhesion. FEBS letters 475, 89–92 [DOI] [PubMed] [Google Scholar]
- 39. Essex D. W., Li M. (1999) Protein disulphide isomerase mediates platelet aggregation and secretion. Br. J. Haematol. 104, 448–454 [DOI] [PubMed] [Google Scholar]
- 40. Reinhardt C., von Brühl M. L., Manukyan D., Grahl L., Lorenz M., Altmann B., Dlugai S., Hess S., Konrad I., Orschiedt L., Mackman N., Ruddock L., Massberg S., Engelmann B. (2008) Protein disulfide isomerase acts as an injury response signal that enhances fibrin generation via tissue factor activation. J. Clin. Invest. 118, 1110–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hawkins H. C., Freedman R. B. (1991) The reactivities and ionization properties of the active-site dithiol groups of mammalian protein disulphide-isomerase. Biochem. J. 275, 335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu Y., Ahmad S. S., Zhou J., Wang L., Cully M. P., Essex D. W. (2012) The disulfide isomerase ERp57 mediates platelet aggregation, hemostasis, and thrombosis. Blood 119, 1737–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alanen H. I., Salo K. E., Pirneskoski A., Ruddock L. W. (2006) pH dependence of the peptide thiol-disulfide oxidase activity of six members of the human protein disulfide isomerase family. Antioxid. Redox Signal. 8, 283–291 [DOI] [PubMed] [Google Scholar]
- 44. Tschopp J., Jenne D. E., Hertig S., Preissner K. T., Morgenstern H., Sapino A. P., French L. (1993) Human megakaryocytes express clusterin and package it without apolipoprotein A-1 into α-granules. Blood 82, 118–125 [PubMed] [Google Scholar]
- 45. Jessop C. E., Chakravarthi S., Garbi N., Hämmerling G. J., Lovell S., Bulleid N. J. (2007) ERp57 is essential for efficient folding of glycoproteins sharing common structural domains. EMBO J. 26, 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lenferink A. E., Cantin C., Nantel A., Wang E., Durocher Y., Banville M., Paul-Roc B., Marcil A., Wilson M. R., O'Connor-McCourt M. D. (2010) Transcriptome profiling of a TGF-β-induced epithelial-to-mesenchymal transition reveals extracellular clusterin as a target for therapeutic antibodies. Oncogene 29, 831–844 [DOI] [PubMed] [Google Scholar]
- 47. Jung G. S., Kim M. K., Jung Y. A., Kim H. S., Park I. S., Min B. H., Lee K. U., Kim J. G., Park K. G., Lee I. K. (2012) Clusterin attenuates the development of renal fibrosis. J. Am. Soc. Nephrol. 23, 73–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hayward C. P., Bainton D. F., Smith J. W., Horsewood P., Stead R. H., Podor T. J., Warkentin T. E., Kelton J. G. (1993) Multimerin is found in the α-granules of resting platelets and is synthesized by a megakaryocytic cell line. J. Clin. Invest. 91, 2630–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jeimy S. B., Tasneem S., Cramer E. M., Hayward C. P. (2008) Multimerin 1. Platelets 19, 83–95 [DOI] [PubMed] [Google Scholar]
- 50. Zacchigna L., Vecchione C., Notte A., Cordenonsi M., Dupont S., Maretto S., Cifelli G., Ferrari A., Maffei A., Fabbro C., Braghetta P., Marino G., Selvetella G., Aretini A., Colonnese C., Bettarini U., Russo G., Soligo S., Adorno M., Bonaldo P., Volpin D., Piccolo S., Lembo G., Bressan G. M. (2006) Emilin1 links TGF-β maturation to blood pressure homeostasis. Cell 124, 929–942 [DOI] [PubMed] [Google Scholar]
- 51. Schiavinato A., Becker A. K., Zanetti M., Corallo D., Milanetto M., Bizzotto D., Bressan G., Guljelmovic M., Paulsson M., Wagener R., Braghetta P., Bonaldo P. (2012) EMILIN-3, peculiar member of elastin microfibril interface-located protein (EMILIN) family, has distinct expression pattern, forms oligomeric assemblies, and serves as transforming growth factor β (TGF-β) antagonist. J. Biol. Chem. 287, 11498–11515 [DOI] [PMC free article] [PubMed] [Google Scholar]