

Abstract
The direct measurement of mitochondrial [Ca2+] with highly specific probes demonstrated that major swings in organellar [Ca2+] parallel the changes occurring in the cytosol and regulate processes as diverse as aerobic metabolism and cell death by necrosis and apoptosis. Despite great biological relevance, insight was limited by the complete lack of molecular understanding. The situation has changed, and new perspectives have emerged following the very recent identification of the mitochondrial Ca2+ uniporter, the channel allowing rapid Ca2+ accumulation across the inner mitochondrial membrane.
Keywords: Calcium, Calcium Channels, Calcium Transport, Mitochondria, Mitochondrial Transport
The Complexity of Calcium Signaling
The initial concept that Ca2+ ions control physiological events goes back to the seminal observation by Ringer in 1883 that addition of Ca2+ to the perfusion buffer of isolated hearts triggered their contraction (1). The development of techniques to monitor changes in the cytoplasmic Ca2+ concentration ([Ca2+]c) (2, 3) allowed the concept that Ca2+ ions act as second messengers to be to extended to virtually all cells. The advent of powerful probes allowing single cell analysis (the intracellularly trappable fluorescent indicators (4, 5)) revealed that Ca2+ increases can be highly localized (e.g. the synaptic region or the secretory pole of an exocrine cell) or diffused across the cell as a Ca2+ wave and elicit an effect at a distant site (e.g. activate gene transcription in the nucleus of the cell). Moreover, in most cell types, the [Ca2+]c increases are oscillatory (6), and the frequency of oscillation is differentially decoded by the cell (7, 8). It is beyond the scope of this minireview to discuss the molecular and cellular basis of this spatiotemporal complexity. It suffices to mention two key requirements. The first is the cooperation of different sources of Ca2+ in the generation of the [Ca2+] rise: the extracellular medium, a virtually unlimited reservoir with a [Ca2+] of ∼1 mm, and intracellular pools, generally referred to as Ca2+ stores, which are endowed with a [Ca2+] (>100 μm) that allows rapid release through resident channels (9, 10). Although recent work has highlighted a role also for other membrane-bound compartments (e.g. the Golgi apparatus (11) and endo/lysosomes (12)), the most important intracellular stores are the endoplasmic reticulum (ER)2 and its specialized counterpart of muscle cells, the sarcoplasmic reticulum (SR). The second requirement for such a carefully orchestrated signal is the existence of a broad number of molecules generating and decoding [Ca2+] variations and their defined positioning within the cell (13). Thus, specific pumps, channels, and buffering proteins finely tune the spatiotemporal pattern of the [Ca2+]c rises, and specific targets, located in both the cytoplasm and different intracellular organelles, are specifically affected by the ionic change.
The Contribution of Mitochondria to Cellular Calcium Handling
Mitochondrial Ca2+ Uptake: A Historical Perspective
Mitochondria were the first intracellular organelles to be associated with calcium handling. Indeed, Ca2+ uptake by energized isolated mitochondria was directly measured half a century ago (14–16). It is remarkable that this experimental observation anticipated the chemiosmotic theory that provides the thermodynamic basis for rapid accumulation of a positively charged ion into the mitochondrial matrix (17). In the following 2 decades, the process of mitochondrial Ca2+ accumulation was thoroughly investigated. Although the outer membrane is known to be permeable to ions and small solutes, transfer across the inner membrane requires the presence of specific transporters. Accordingly, a rapid electrogenic pathway (denoted the “mitochondrial calcium uniporter” (MCU)) was described that rapidly transports Ca2+ into the matrix, driven by the negative charge of the membrane potential (ΔΨ) established by the respiratory chain. Ca2+ accumulation partially dissipates the electrochemical gradient, closely agreeing with the early observation that Ca2+ stimulates respiration transiently, in contrast with the classical mitochondrial uncouplers. Ca2+ accumulation by MCU does not proceed until electrochemical equilibrium (which would lead to a 106 concentration difference) due to the activity of two main efflux pathways, the Na+/Ca2+ (mNCX) and H+/Ca2+ (mHCX) exchangers, expressed mainly in excitable and non-excitable tissues, respectively. The existence of a sophisticated machinery for Ca2+ handling supported the general consensus that mitochondria could actively and rapidly change their [Ca2+] and participate in cellular homeostasis.
However, this idea was strongly questioned in the 1980s by two key observations. The first was the demonstration by Berridge and co-workers (18) that inositol 1,4,5-trisphosphate (IP3), generated upon stimulation of receptors coupled to phospholipase C, mobilizes Ca2+ from a “non-mitochondrial” Ca2+ store, the ER. Furthermore, another seminal study described the relative affinities for calcium uptake by mitochondria and the ER (19). Quite appropriately, the sarco/endoplasmic reticulum became the main research focus in calcium signaling, leading to the identification of resident molecules involved in Ca2+ transport, the IP3- and ryanodine receptor (RyR)-sensitive channels, and the sarco/endoplasmic reticulum ATPase (10). The second key observation was the accurate [Ca2+]c measurement obtained, in virtually all cell types, with fluorescent indicators. It became clear that given the [Ca2+]c measured under resting (∼0.1 μm) or stimulated (1–3 μm at the peak) conditions, MCU would not allow substantial Ca2+ uptake into the organelle. The general consensus thus changed, and mitochondria gradually faded away from the general picture of cellular Ca2+ homeostasis, as significant Ca2+ uptake was predicted to occur only under conditions of massive Ca2+ overload.
The Resurrection of Mitochondria and the Microdomain Concept
The pendulum swung again in the early 1990s. Although mitochondrial Ca2+ uptake had already been described (20), the development of recombinant targeted probes was crucial for the reappraisal of the role of mitochondria. The pioneer of these probes was the photoprotein aequorin (21), which was followed by GFP-based fluorescent probes allowing the direct visualization of [Ca2+] changes in imaging experiments (4, 22). These highly specific probes clearly revealed that when a cell is stimulated with a [Ca2+]c-increasing agonist, mitochondria always accumulate Ca2+ in the matrix with a speed and an amplitude that greatly exceed those expected from the properties of MCU in isolated mitochondria (23). Indeed, the upstroke of the mitochondrial [Ca2+] ([Ca2+]m) rise closely follows that of the cells (24), and the peak value reaches, in some cell types, values in excess of 100 μm (Fig. 1) (25).
FIGURE 1.

A, schematic representation (upper) and immunolocalization (lower) of the mitochondrial aequorin (mtAEQ) probe (showing correct targeting to mitochondria); HA, haemagglutinin epitope. B, [Ca2+]m (blue traces) and [Ca2+]c (red traces) measurements in HeLa cells upon treatment with 100 μm histamine in the absence (left panel) and presence (right panel) of the uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP). cytAEQ, cytosolic aequorin.
The apparent discrepancy with the low affinity of MCU was solved by the demonstration that mitochondria are located in close proximity to the Ca2+ channels eliciting the Ca2+ rise, i.e. the IP3 receptors (IP3Rs; or RyRs) in the ER/SR (26–29), or different classes of channels on the plasma membrane (e.g. voltage- and store-operated channels) (30–33). Therefore, they sense a microdomain of high [Ca2+] that meets the low affinity of MCU and is then dissipated, thus preventing mitochondrial Ca2+ overload and/or vicious Ca2+ cycling across the mitochondrial membrane. This idea was initially supported by the morphological demonstration of the close proximity of the two organelles and by the evidence that only perfusion of IP3 or of Ca2+ at a 10-fold higher concentration than that measured in the bulk cytosol could induce rapid Ca2+ uptake in permeabilized cells (23). More recently, the [Ca2+] reached on the outer surface of mitochondria was directly measured, thus providing reliable estimates of the [Ca2+] microdomain sensed by mitochondria (34, 35). The concept that mitochondria respond to a pulsatile event, the generation of a rapidly dissipating microdomain, has major functional implications. The first stems from the old observation that both ATP phosphorylation and Ca2+ uptake occur at the expense of mitochondrial ΔμΗ+. During cell stimulation, continuous accumulation (and cycling) of Ca2+ across the mitochondrial membrane would interfere with ATP production, whereas a rapid and transient response allows a functional response with limited energy drain, as discussed below. The second crucial implication of a microdomain-based signaling mechanism is that the shape and positioning of mitochondria within the cell become critical determinants of their responsiveness to Ca2+ signals. Thus, not only in neurons, where localization in synaptic regions was known to underlie rapid Ca2+ uptake (and Ca2+ buffering), but in most cells, mitochondria appear to be docked to dedicated signaling sites known as the mitochondria-associated ER membrane (36). In addition, the highly regulated state of fusion/fission influences the mitochondrial Ca2+ responses, as fragmentation disconnects part of the mitochondria from the signaling sites (37). MFN2 (mitofusin 2), a critical component of the mitochondrial fusion/fission machinery, is enriched in mitochondria-associated ER membranes, where it was shown to tether the ER and mitochondria (38), thus participating in the formation of mitochondria-ER contacts. Indeed, in fibroblasts lacking MFN2, the distance between the ER and mitochondria increases, and mitochondrial Ca2+ uptake is drastically reduced (38).
The Physio(patho)logical Role of Mitochondrial Calcium Handling
The demonstration that mitochondria rapidly accumulate Ca2+ upon stimulation was followed by a large body of work that allowed the physiological (and pathological) role of mitochondrial Ca2+ homeostasis to be established in the following years (Fig. 2). At first, the role in the control of aerobic metabolism was directly investigated. Such a role was predicted based on the biochemical evidence, dating back to the 1970s, that three matrix dehydrogenases are activated by Ca2+: pyruvate dehydrogenase is regulated by a Ca2+-dependent phosphatase, and α-ketoglutarate and isocitrate dehydrogenases is regulated by direct binding of Ca2+ to the enzyme (39, 40). Stimulation of Ca2+-sensitive dehydrogenases increases NADH availability and hence the flow of electrons down the respiratory chain, thus, in principle, enhancing a rate-limiting step for rapid ATP synthesis in stimulated cells.
FIGURE 2.
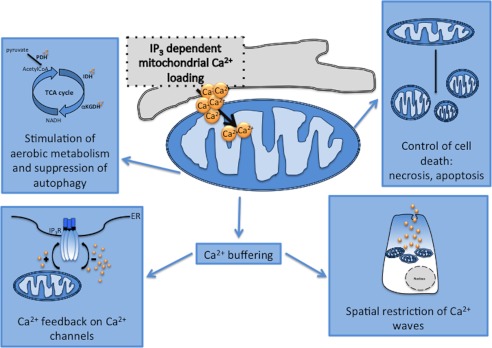
Pleiotropic roles of mitochondrial Ca2+ homeostasis. PDH, pyruvate dehydrogenase; IDH, isocitrate dehydrogenase; αKGDH, α-ketoglutarate dehydrogenase.
Increased electron feeding into the respiratory chain allows an increase in ATP production, as demonstrated by the direct measurement of ATP levels in the mitochondria and cytosol. Interestingly, this Ca2+-mediated activation of aerobic metabolism appears to be impaired in some mitochondrial genetic disorders, such as respiratory chain defects, in which the partial reduction of ΔΨ greatly impairs the transmission of Ca2+ signals to mitochondria and hence ATP production. Accordingly, in cell lines harboring these mutations (such as the tRNA defects), restoration of Ca2+ signals (e.g. using inhibitors of mitochondrial Ca2+ efflux) markedly increases ATP production (41).
Importantly, mitochondrial Ca2+ is also involved in the regulation of cell death pathways. Mitochondrial Ca2+ overload has long been known to be associated with the process of necrosis, such as that due to ischemia-reperfusion of the heart and excitotoxicity of neurons (for a review, see Ref. 42). Cellular (and hence mitochondrial) Ca2+ overload, in conjunction with accumulation of reactive oxygen species, favors the sustained opening of the high conductance cyclosporin A-sensitive permeability transition pore (PTP), which causes the rapid collapse of ΔΨm and the swelling of mitochondria, with consequent loss of pyridine nucleotides and cytochrome c. The ensuing bioenergetic crisis and ATP depletion send the cells to necrotic cell death (43, 44).
In more recent years, however, Ca2+ dysregulation and, more specifically, [Ca2+]m increases have been shown to play a regulatory role in the more controlled process of apoptotic cell death. The initial evidence was provided by the demonstration that the anti-apoptotic oncogene bcl-2 affects intracellular Ca2+ homeostasis by regulating the Ca2+ leak from the ER and/or the release kinetics upon cell stimulation. The most logical target for this signaling alteration was mitochondria, which, in the initiation step of the intrinsic pathway of apoptosis, release essential components of the apoptosome (such as cytochrome c, SMAC/DIABLO, and apoptosis-inducing factor). In this release, a key role is played by organelle fragmentation and swelling following PTP opening (45), a process triggered by increases in matrix [Ca2+], in conjunction with a variety of pathological challenges (e.g. oxidative stress or production of C2-ceramide). Thus, in a stressed cell, cytosolic Ca2+ waves, such as those evoked by a physiological stimulation, rather than stimulating aerobic metabolism, trigger an intracellular wave of PTP opening that ultimately leads to cell death. Interestingly, in the ER, Bcl-2 reduces mitochondrial Ca2+ loading (46–49), whereas the pro-apoptotic members of the protein family (such as Bax) exert the opposite effect (50). Furthermore, Bcl-2 localizes also to mitochondrial membranes, where it has been demonstrated to protect cells against mitochondrial Ca2+ overload (51).
Mitochondrial Ca2+ signals have recently been shown to play a major role in the regulation of autophagy. Indeed, a recent seminal paper by Foskett and co-workers (52) clearly demonstrated that [Ca2+]m exerts an inhibitory effect on AMP-activated protein kinase activity. Indeed, the authors showed that in DT40 IP3R1/2/3 knock-out cells (in which IP3R-dependent cellular Ca2+ signals were abrogated), autophagy was maximally activated also in the presence of nutrients (52). The effect could be ascribed to ablation of mitochondrial Ca2+ loading, as MCU blockers had the same effect as IP3R inhibition. This study led to the interesting conclusion that apoptosis and autophagy, which share molecular regulators (such as Beclin), also utilize a common second messenger (Ca2+ and its loading into mitochondria) to drive cell fate in opposite directions (high mitochondrial [Ca2+], pro-cell death; low [Ca2+], autophagic rescue).
Finally, the consequences of mitochondrial accumulation are not limited to events occurring within the organelle. Indeed, the fact that mitochondria can take up large Ca2+ loads (albeit with a low affinity mechanism) places them, at least in principle, among the buffers that should be taken into account when estimating the factors that reduce the amplitude of the cytosolic [Ca2+] increases evoked by channel opening and/or that slow the diffusion of Ca2+ waves. Although we refer to more exhaustive reviews for a detailed coverage of this topic, we will briefly sketch two conceptually different mechanisms through which mitochondrial buffering plays a functional role (53).
The first relates to the possibility that strategic positioning of mitochondria endows cells with a highly polarized complex morphology with a high capacity sink draining a large amount of Ca2+ and preventing (or delaying) Ca2+ diffusion to distant sites. In pancreatic acinar cells, a mitochondrial belt maintains Ca2+ rises in the apical region (where they are generated), limiting its effect to granule secretion. When the buffering capacity of mitochondria is overwhelmed, as in supramaximal stimulation or by impairing mitochondrial uptake capacity, the Ca2+ wave diffuses to the whole cell, thus reaching also the nucleus, where it promotes gene transcription and long-lasting changes (54). In neurons, mitochondria located in proximity to the plasma membrane accumulate large amounts of Ca2+ (up to >10 mmol/kg of dry weight), most likely stored as calcium phosphate precipitates (55). By this means, they modulate synaptic [Ca2+] increases, strongly affecting neurotransmitter release. Interestingly, genetic ablation of a low affinity Ca2+-binding protein highly expressed in neurons (parvalbumin) induces mitochondrial biogenesis and redistribution as a compensatory mechanism, thus indicating that the cell regulates these organelles as key components of its Ca2+-buffering repertoire (56).
The second, conceptual mechanism is the generation of signaling microdomains with the ER/SR or the plasma membrane, in which mitochondrial Ca2+ uptake controls the microenvironment of the resident channels, thus modulating the positive or negative feedback of the cation on the channel itself. A revealing example was provided in the late 1990s by Lechleiter and co-workers (57), who demonstrated that Ca2+ uptake by energized mitochondria controls the kinetics of IP3R Ca2+ release and hence coordinates IP3-induced Ca2+ waves. Interestingly, this concept is not restricted to ER channels but plays a role also in the control of plasma membrane channels. A large body of work has been carried out in T lymphocytes, which depend on store-operated channel activity to sustain the prolonged [Ca2+] increases needed for cell activation (58). In this system, mitochondria buffer the local [Ca2+] increases in the proximity of the channel, thus preventing rapid inactivation. Interestingly, in mast cells, mitochondria are more distant from the plasma membrane but still participate in shaping the [Ca2+] increase and hence the functional response by buffering the global cytosolic rise, thus influencing both slow inactivation of ORAI channels and ER Ca2+ loading (30).
The Essential Components of the Ca2+ Transport Machinery
Outer Mitochondrial Membrane
Ca2+ accumulation in the mitochondrial matrix requires the crossing of two membranes, the outer (OMM) and inner (IMM) mitochondrial membranes. Attention has been placed mostly on the transport systems of the ion-impermeable IMM, as the OMM is permeable to solutes of <5 kDa and thus also to Ca2+. However, the dependence of rapid Ca2+ uptake into the matrix on high [Ca2+] microdomains generated at the mouths of open ER/SR (or plasma membrane) channels implied that also passive equilibration across the OMM could represent a rate-limiting step. The high permeability of the OMM is due mostly to its most abundant protein, the voltage-dependent anion channel, which allows the shuttling of all energy-related metabolites, including succinate, pyruvate, malate, NADH, ATP, ADP, and phosphate, from the cytosol to the mitochondria. Voltage-dependent anion channel overexpression augments agonist-dependent Ca2+ rises, implying that the repertoire of OMM channels is a kinetic bottleneck in the transfer of the Ca2+ microdomain to the IMM (59, 60).
Inner Mitochondrial Membrane
Thermodynamic Principles
A much more substantial problem for Ca2+ accumulation in the matrix is represented by the ion-impermeable inner membrane (Fig. 3). Indeed, the electron transport chain translocates H+ ions across the IMM, generating an electrochemical proton gradient (ΔμΗ+), which is mostly in the form of a membrane potential difference (ΔΨ). ΔμΗ+ drives the flow of H+ through the ATP synthase in a reaction coupled to the generation of ATP from ADP and inorganic phosphate (61) or through leak pathways, such as the uncoupling proteins (which cause H+ cycling and increased respiration and heat production) (62). ΔΨ also represents a very large driving force for Ca2+ accumulation. Based on the Nernst equation (Equation 1, where cat is cation),
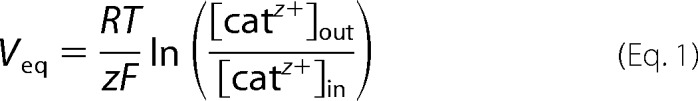 |
equilibrium is reached at an inside [Ca2+] 106-fold greater than that outside the IMM.
FIGURE 3.

Schematic representation of the essential molecular components of mitochondrial Ca2+ homeostasis: the electron transport chain complex, building up the electrical driving force for accumulation (ΔΨ), MCU, and the mNCX and mHCX exchangers. cyt, cytochrome c; IMS, intermembrane space.
The Transporters
Ca2+ influx occurs via an electrophoretic pathway called the MCU. Ca2+ accumulation by MCU is counteracted predominantly by mNCX (63). This exchanger, first described by Carafoli and co-workers (64), was recently identified by Sekler and co-workers (65), who demonstrated that silencing of NCLX blocks Ca2+ extrusion and that NCLX-mediated mitochondrial Ca2+ transport is inhibited by the NCX blocker CGP-37157.
Both exchangers are ruthenium red (RuR)-insensitive, and indeed, in classical experiments in isolated mitochondria, after challenging mitochondria with a Ca2+ pulse, exchanger-mediated efflux was initiated by RuR addition. mNCX is inhibited by a group of benzothiazepine analogs, such as diltiazem, clonazepam, and CGP-37157 (66). Its Vmax ranges between 0.6 (liver) and 18 nmol (heart) of Ca2+/mg of protein/min (67).
The Discovery of the MCU: Finding the Needle in the Mitochondrial Haystack
Through the years, there has been a wide consensus that the key molecule allowing rapid accumulation of Ca2+ across the ion-impermeable inner channel is the electrophoretic pathway called MCU. Studies in isolated mitochondria demonstrated that it is inhibited by RuR (68) and lanthanides (69), with a Vmax of >1400 nmol of Ca2+/mg of protein/min (70). Electrophysiological recordings of isolated mitoplasts demonstrated that MCU is a highly selective, inwardly rectifying Ca2+ channel (71, 72).
Several candidates have been proposed through the years until the recent discovery. After a few tentative identifications of putative RuR-binding glycoproteins in the 1970s (73), in a series of papers, Sheu and co-workers (74, 75) proposed that mitochondrially sorted RyR1 drives mitochondrial Ca2+ uptake in the heart. Although the tissue distribution and electrophysiological properties seem to exclude RyR1 as the MCU, the possibility that RyR is an additional uptake pathway cooperating with MCU in the heart is still open. More recently, Graier and co-workers (76) proposed that UCP2 and UCP3 are essential components of the MCU machinery. These results, which were questioned on theoretical grounds and based on experiments carried out in mitochondria from UCP2−/− and UCP3−/− mice (77), have recently been ascribed to an indirect effect on ATP production and hence ER Ca2+ loading (78). Finally, in 2009, Clapham and co-workers (79) identified, by siRNA genomic screening in Drosophila, Letm1 as a putative H+/Ca2+ antiporter mediating Ca2+ accumulation in the matrix. Given that the same protein was proposed to be a mitochondrial K+/H+ exchanger (80) and that the proposed stoichiometry and RuR sensitivity differ from those reported for mNCX and mHCX (81, 82), the role of Letm1 awaits further confirmation and, in all cases, does not reflect MCU activity.
The success in MCU identification required an innovative approach. Indeed, (i) the available inhibitors of channel function (e.g. RuR) lacked the protein specificity necessary for direct protein identification; (ii) Saccharomyces cerevisiae appears to have no RuR-sensitive mitochondrial Ca2+ influx (83), thus preventing the use of yeast genetics; and (iii) in silico homology searches with known Ca2+ channels yielded no successful hit. An alternative strategy was made possible by the seminal work of Mootha and co-workers (84), who constructed a mitochondrial gene data set (MitoCarta) by compiling an inventory of gene products with proven mitochondrial localization. By searching the MitoCarta data set, the same group identified a 54-kDa protein, renamed MICU1 (mitochondrial calcium uptake 1). Its silencing in HeLa cells drastically reduced mitochondrial Ca2+ uptake, thus proving that it is an essential component of the Ca2+ uptake machinery (85). However, MICU1 includes two Ca2+-binding EF domains and a single putative transmembrane (TM) domain and thus was unlikely to be the channel itself.
The presence of an MCU regulator further stimulated the search for MCU, and indeed, two groups (our own (87) and Mootha's (88)) found MCU in 2011 through an in silico search of the MitoCarta database. Specifically, we skimmed the list by applying subsequent constraints: 1) a broad expression profile, given the presence of RuR-sensitive mitochondrial Ca2+ uptake in all mammalian tissues; 2) at least two computationally predicted TM α-helixes in the primary sequence (i.e. the expected minimum requirement of all ion channels); 3) the absence in S. cerevisiae, which lacks RuR-sensitive mitochondrial Ca2+ uptake; and 4) the presence in Trypanosomatida, in which RuR-sensitive Ca2+ uptake was reported (86). We ended up with a list of 14 candidates, containing both well known (e.g. subunits of NADH dehydrogenase) and yet unexplored proteins. We finally reasoned that given the key function of ion permeation, the evolutionary pressure was most likely focused on conserving the TM domains and the selectivity filter rather than other traits. We thus carefully aligned the 14 TM domains and intervening loops of candidates across distant species, and it was immediately clear that one of the hits (the product of the CCDC109A gene) met our predictions: two very highly conserved TM domains separated by a small loop identical in sequence from human to kinetoplastids and enriched in acidic residues. CCDC109A (then renamed MCU) became the leading candidate and was validated in studies in intact and permeabilized cells (Fig. 4). Interestingly, Mootha and co-workers (87) also identified the same candidate using a different computational approach. They searched for genes closely related to MICU1 in terms of evolutionary co-occurrence and mRNA/protein coexpression profiles, identifying CCDC109A as the top scoring hit.
FIGURE 4.

Strategy for identifying MCU within the MitoCarta database. TMDs, transmembrane domains.
MCU silencing virtually abolishes mitochondrial Ca2+ uptake in intact and permeabilized cells (87, 88). We cloned the MCU cDNA and demonstrated that its overexpression doubled the [Ca2+]m rise evoked by IP3-coupled agonists while significantly reducing the amplitude of the [Ca2+]c peaks as a consequence of the increased mitochondrial Ca2+ buffering (87). These data demonstrated a crucial role also of this protein in mitochondrial Ca2+ uptake, but direct evidence was necessary to claim that it is MCU, i.e. the bona fide channel. We thus expressed MCU in two different heterologous systems, Escherichia coli and wheat germ cell-free transcription/translation, and inserted the purified protein into the planar lipid bilayer. The electrophysiological properties of MCU matched those previously described in isolated mitochondria, and the current was completely inhibited by RuR and Gd3+. MCU activity was also abolished by the mutation of two negatively charged residues to glutamine (D260Q/E263Q). Furthermore, when overexpressed in HeLa cells, this mutant reduced mitochondrial Ca2+ uptake, acting as a dominant negative, strongly suggesting that MCU forms an active channel as an oligomer (87). Mootha and co-workers (88) demonstrated that a single point mutation of MCU (S259A) retains function but confers resistance to Ru360, indicating that the most potent inhibitors of uniporter activity (RuR and related compounds) act directly on the channel and not with associated regulatory elements.
Conclusions
After a long search in the wrong directions, an unexpected twist rapidly unveiled the real identity of MCU. Now, the last element of the cellular calcium signaling toolkit has been identified, and the direct molecular targeting of mitochondrial Ca2+ homeostasis can be pursued in vivo and in vitro, as well as in organ physiology and disease pathogenesis. To cite only a few examples, the role of mitochondrial Ca2+ signals in aerobic metabolism and in tuning the autophagic response to nutrient deprivation can now be directly investigated and correlated with cell function and trophism, as in the case of muscle adaptation to feeding conditions, aging, or pathological states. In contrast, sensitization to apoptotic and necrotic cell death can be evaluated in important disease models, such as ischemia-reperfusion of the heart and neurodegenerative disorders. Finally, future molecular data should include structural information supporting the discovery of specific inhibitors that may represent a novel important class of pharmacological compounds.
This work was supported, in whole or in part, by National Institutes of Health Grant 1P01 AG025532-01A1. This work was also supported by grants from the Italian Ministries of Health (Ricerca Finalizzata) and of Education, University and Research (PRIN and FIRB), European Union ERC mitoCalcium Project Grant 294777 and FP7 “MyoAGE” Project Grant 223576, the Cariparo Foundation (Padua, Italy), the Italian Association for Cancer Research (AIRC), and Telethon-Italy Grant GPP10005A. This article is part of the Thematic Minireview Series on the Ins and Outs of Calcium Transport.
- ER
- endoplasmic reticulum
- SR
- sarcoplasmic reticulum
- MCU
- mitochondrial calcium uniporter
- mNCX
- mitochondrial Na+/Ca2+ exchanger
- mHCX
- mitochondrial H+/Ca2+ exchanger
- IP3
- inositol 1,4,5-trisphosphate
- RyR
- ryanodine receptor
- IP3R
- IP3 receptor
- PTP
- permeability transition pore
- OMM
- outer mitochondrial membrane
- IMM
- inner mitochondrial membrane
- RuR
- ruthenium red
- TM
- transmembrane.
REFERENCES
- 1. Ringer S. (1883) A third contribution regarding the influence of the inorganic constituents of the blood on the ventricular contraction. J. Physiol. 4, 222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 3. Tsien R. Y., Pozzan T., Rink T. J. (1982) T-cell mitogens cause early changes in cytoplasmic free Ca2+ and membrane potential in lymphocytes. Nature 295, 68–71 [DOI] [PubMed] [Google Scholar]
- 4. Miyawaki A., Griesbeck O., Heim R., Tsien R. Y. (1999) Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc. Natl. Acad. Sci. U.S.A. 96, 2135–2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rudolf R., Mongillo M., Rizzuto R., Pozzan T. (2003) Looking forward to seeing calcium. Nat. Rev. Mol. Cell Biol. 4, 579–586 [DOI] [PubMed] [Google Scholar]
- 6. Cobbold P. H., Cuthbertson K. S. (1990) Calcium oscillations: phenomena, mechanisms and significance. Semin. Cell Biol. 1, 311–321 [PubMed] [Google Scholar]
- 7. Hajnóczky G., Robb-Gaspers L. D., Seitz M. B., Thomas A. P. (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 [DOI] [PubMed] [Google Scholar]
- 8. Dolmetsch R. E., Xu K., Lewis R. S. (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936 [DOI] [PubMed] [Google Scholar]
- 9. Rizzuto R., Pozzan T. (2006) Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol. Rev. 86, 369–408 [DOI] [PubMed] [Google Scholar]
- 10. Pozzan T., Rizzuto R., Volpe P., Meldolesi J. (1994) Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 74, 595–636 [DOI] [PubMed] [Google Scholar]
- 11. Pinton P., Pozzan T., Rizzuto R. (1998) The Golgi apparatus is an inositol 1,4,5-trisphosphate-sensitive Ca2+ store, with functional properties distinct from those of the endoplasmic reticulum. EMBO J. 17, 5298–5308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Calcraft P. J., Ruas M., Pan Z., Cheng X., Arredouani A., Hao X., Tang J., Rietdorf K., Teboul L., Chuang K. T., Lin P., Xiao R., Wang C., Zhu Y., Lin Y., Wyatt C. N., Parrington J., Ma J., Evans A. M., Galione A., Zhu M. X. (2009) NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 459, 596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berridge M. J. (2001) The versatility and complexity of calcium signalling. Novartis Found. Symp. 239, 52–64; discussion 64–77, 150–159 [PubMed] [Google Scholar]
- 14. Vasington F. D., Murphy J. V. (1962) Ca ion uptake by rat kidney mitochondria and its dependence on respiration and phosphorylation. J. Biol. Chem. 237, 2670–2677 [PubMed] [Google Scholar]
- 15. Deluca H. F., Engstrom G. W. (1961) Calcium uptake by rat kidney mitochondria. Proc. Natl. Acad. Sci. U.S.A. 47, 1744–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lehninger A. L., Rossi C. S., Greenawalt J. W. (1963) Respiration-dependent accumulation of inorganic phosphate and Ca ions by rat liver mitochondria. Biochem. Biophys. Res. Commun. 10, 444–448 [DOI] [PubMed] [Google Scholar]
- 17. Mitchell P., Moyle J. (1967) Chemiosmotic hypothesis of oxidative phosphorylation. Nature 213, 137–139 [DOI] [PubMed] [Google Scholar]
- 18. Streb H., Irvine R. F., Berridge M. J., Schulz I. (1983) Release of Ca2+ from a nonmitochondrial intracellular store in pancreatic acinar cells by inositol-1,4,5-trisphosphate. Nature 306, 67–69 [DOI] [PubMed] [Google Scholar]
- 19. Becker G. L., Fiskum G., Lehninger A. L. (1980) Regulation of free Ca2+ by liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 255, 9009–9012 [PubMed] [Google Scholar]
- 20. Carafoli E. (1967) In vivo effect of uncoupling agents on the incorporation of calcium and strontium into mitochondria and other subcellular fractions of rat liver. J. Gen. Physiol. 50, 1849–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rizzuto R., Simpson A. W., Brini M., Pozzan T. (1992) Rapid changes of mitochondrial Ca2+ revealed by specifically targeted recombinant aequorin. Nature 358, 325–327 [DOI] [PubMed] [Google Scholar]
- 22. Nagai T., Sawano A., Park E. S., Miyawaki A. (2001) Circularly permuted green fluorescent proteins engineered to sense Ca2+. Proc. Natl. Acad. Sci. U.S.A. 98, 3197–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rizzuto R., Brini M., Murgia M., Pozzan T. (1993) Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 262, 744–747 [DOI] [PubMed] [Google Scholar]
- 24. Szabadkai G., Simoni A. M., Rizzuto R. (2003) Mitochondrial Ca2+ uptake requires sustained Ca2+ release from the endoplasmic reticulum. J. Biol. Chem. 278, 15153–15161 [DOI] [PubMed] [Google Scholar]
- 25. Montero M., Alonso M. T., Carnicero E., Cuchillo-Ibáñez I., Albillos A., García A. G., García-Sancho J., Alvarez J. (2000) Chromaffin-cell stimulation triggers fast millimolar mitochondrial Ca2+ transients that modulate secretion. Nat. Cell Biol. 2, 57–61 [DOI] [PubMed] [Google Scholar]
- 26. Rizzuto R., Pinton P., Carrington W., Fay F. S., Fogarty K. E., Lifshitz L. M., Tuft R. A., Pozzan T. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280, 1763–1766 [DOI] [PubMed] [Google Scholar]
- 27. Csordás G., Thomas A. P., Hajnóczky G. (1999) Quasi-synaptic calcium signal transmission between endoplasmic reticulum and mitochondria. EMBO J. 18, 96–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szalai G., Csordás G., Hantash B. M., Thomas A. P., Hajnóczky G. (2000) Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 275, 15305–15313 [DOI] [PubMed] [Google Scholar]
- 29. Mannella C. A., Buttle K., Rath B. K., Marko M. (1998) Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors 8, 225–228 [DOI] [PubMed] [Google Scholar]
- 30. Parekh A. B., Putney J. W., Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810 [DOI] [PubMed] [Google Scholar]
- 31. Glitsch M. D., Bakowski D., Parekh A. B. (2002) Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 21, 6744–6754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. David G., Barrett J. N., Barrett E. F. (1998) Evidence that mitochondria buffer physiological Ca2+ loads in lizard motor nerve terminals. J. Physiol. 509, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Young K. W., Bampton E. T., Pinòn L., Bano D., Nicotera P. (2008) Mitochondrial Ca2+ signalling in hippocampal neurons. Cell Calcium 43, 296–306 [DOI] [PubMed] [Google Scholar]
- 34. Giacomello M., Drago I., Bortolozzi M., Scorzeto M., Gianelle A., Pizzo P., Pozzan T. (2010) Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 38, 280–290 [DOI] [PubMed] [Google Scholar]
- 35. Csordás G., Várnai P., Golenár T., Roy S., Purkins G., Schneider T. G., Balla T., Hajnóczky G. (2010) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hayashi T., Rizzuto R., Hajnóczky G., Su T. P. (2009) MAM: more than just a housekeeper. Trends Cell Biol. 19, 81–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Szabadkai G., Simoni A. M., Chami M., Wieckowski M. R., Youle R. J., Rizzuto R. (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell 16, 59–68 [DOI] [PubMed] [Google Scholar]
- 38. de Brito O. M., Scorrano L. (2008) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456, 605–610 [DOI] [PubMed] [Google Scholar]
- 39. McCormack J. G., Halestrap A. P., Denton R. M. (1990) Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiol. Rev. 70, 391–425 [DOI] [PubMed] [Google Scholar]
- 40. Hansford R. G. (1994) Physiological role of mitochondrial Ca2+ transport. J. Bioenerg. Biomembr. 26, 495–508 [DOI] [PubMed] [Google Scholar]
- 41. Brini M., Pinton P., King M. P., Davidson M., Schon E. A., Rizzuto R. (1999) A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat. Med. 5, 951–954 [DOI] [PubMed] [Google Scholar]
- 42. Orrenius S., Zhivotovsky B., Nicotera P. (2003) Regulation of cell death: the calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 4, 552–565 [DOI] [PubMed] [Google Scholar]
- 43. O'Rourke B. (2000) Pathophysiological and protective roles of mitochondrial ion channels. J. Physiol. 529, 23–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Di Lisa F., Bernardi P. (2009) A CaPful of mechanisms regulating the mitochondrial permeability transition. J. Mol. Cell. Cardiol. 46, 775–780 [DOI] [PubMed] [Google Scholar]
- 45. Rasola A., Bernardi P. (2011) Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50, 222–233 [DOI] [PubMed] [Google Scholar]
- 46. Pinton P., Ferrari D., Magalhães P., Schulze-Osthoff K., Di Virgilio F., Pozzan T., Rizzuto R. (2000) Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl-2-overexpressing cells. J. Cell Biol. 148, 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pinton P., Ferrari D., Rapizzi E., Di Virgilio F., Pozzan T., Rizzuto R. (2001) The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20, 2690–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Palmer A. E., Jin C., Reed J. C., Tsien R. Y. (2004) Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc. Natl. Acad. Sci. U.S.A. 101, 17404–17409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Foyouzi-Youssefi R., Arnaudeau S., Borner C., Kelley W. L., Tschopp J., Lew D. P., Demaurex N., Krause K. H. (2000) Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 97, 5723–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Scorrano L., Oakes S. A., Opferman J. T., Cheng E. H., Sorcinelli M. D., Pozzan T., Korsmeyer S. J. (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135–139 [DOI] [PubMed] [Google Scholar]
- 51. Murphy A. N., Bredesen D. E., Cortopassi G., Wang E., Fiskum G. (1996) Bcl-2 potentiates the maximal calcium uptake capacity of neural cell mitochondria. Proc. Natl. Acad. Sci. U.S.A. 93, 9893–9898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cárdenas C., Miller R. A., Smith I., Bui T., Molgó J., Müller M., Vais H., Cheung K. H., Yang J., Parker I., Thompson C. B., Birnbaum M. J., Hallows K. R., Foskett J. K. (2010) Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 142, 270–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Rizzuto R., De Stefani D., Raffaello A., Mammucari C. (2012) Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578 [DOI] [PubMed] [Google Scholar]
- 54. Tinel H., Cancela J. M., Mogami H., Gerasimenko J. V., Gerasimenko O. V., Tepikin A. V., Petersen O. H. (1999) Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO J. 18, 4999–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pivovarova N. B., Hongpaisan J., Andrews S. B., Friel D. D. (1999) Depolarization-induced mitochondrial Ca accumulation in sympathetic neurons: spatial and temporal characteristics. J. Neurosci. 19, 6372–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen G., Racay P., Bichet S., Celio M. R., Eggli P., Schwaller B. (2006) Deficiency in parvalbumin, but not in calbindin D-28k, upregulates mitochondrial volume and decreases smooth endoplasmic reticulum surface selectively in a peripheral, subplasmalemmal region in the soma of Purkinje cells. Neuroscience 142, 97–105 [DOI] [PubMed] [Google Scholar]
- 57. Jouaville L. S., Ichas F., Holmuhamedov E. L., Camacho P., Lechleiter J. D. (1995) Synchronization of calcium waves by mitochondrial substratesin Xenopus laevis oocytes. Nature 377, 438–441 [DOI] [PubMed] [Google Scholar]
- 58. Hoth M., Fanger C. M., Lewis R. S. (1997) Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J. Cell Biol. 137, 633–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rapizzi E., Pinton P., Szabadkai G., Wieckowski M. R., Vandecasteele G., Baird G., Tuft R. A., Fogarty K. E., Rizzuto R. (2002) Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159, 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Madesh M., Hajnóczky G. (2001) VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J. Cell Biol. 155, 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Stock D., Leslie A. G., Walker J. E. (1999) Molecular architecture of the rotary motor in ATP synthase. Science 286, 1700–1705 [DOI] [PubMed] [Google Scholar]
- 62. Cannon B., Nedergaard J. (2004) Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 [DOI] [PubMed] [Google Scholar]
- 63. Rizzuto R., Bernardi P., Favaron M., Azzone G. F. (1987) Pathways for Ca2+ efflux in heart and liver mitochondria. Biochemical J. 246, 271–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Carafoli E., Tiozzo R., Lugli G., Crovetti F., Kratzing C. (1974) The release of calcium from heart mitochondria by sodium. J. Mol. Cell. Cardiol. 6, 361–371 [DOI] [PubMed] [Google Scholar]
- 65. Palty R., Silverman W. F., Hershfinkel M., Caporale T., Sensi S. L., Parnis J., Nolte C., Fishman D., Shoshan-Barmatz V., Herrmann S., Khananshvili D., Sekler I. (2010) NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. U.S.A. 107, 436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cox D. A., Conforti L., Sperelakis N., Matlib M. A. (1993) Selectivity of inhibition of Na+-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J. Cardiovasc. Pharmacol. 21, 595–599 [DOI] [PubMed] [Google Scholar]
- 67. Bernardi P. (1999) Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol. Rev. 79, 1127–1155 [DOI] [PubMed] [Google Scholar]
- 68. Bernardi P., Paradisi V., Pozzan T., Azzone G. F. (1984) Pathway for uncoupler-induced calcium efflux in rat liver mitochondria: inhibition by ruthenium red. Biochemistry 23, 1645–1651 [DOI] [PubMed] [Google Scholar]
- 69. Mela L. (1969) Inhibition and activation of calcium transport in mitochondria. Effect of lanthanides and local anesthetic drugs. Biochemistry 8, 2481–2486 [DOI] [PubMed] [Google Scholar]
- 70. Bragadin M., Pozzan T., Azzone G. F. (1979) Activation energies and enthalpies during Ca2+ transport in rat liver mitochondria. FEBS Lett. 104, 347–351 [DOI] [PubMed] [Google Scholar]
- 71. Kirichok Y., Krapivinsky G., Clapham D. E. (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 [DOI] [PubMed] [Google Scholar]
- 72. Michels G., Khan I. F., Endres-Becker J., Rottlaender D., Herzig S., Ruhparwar A., Wahlers T., Hoppe U. C. (2009) Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation 119, 2435–2443 [DOI] [PubMed] [Google Scholar]
- 73. Panfili E., Sandri G., Sottocasa G. L., Lunazzi G., Liut G., Graziosi G. (1976) Specific inhibition of mitochondrial Ca2+ transport by antibodies directed to the Ca2+-binding glycoprotein. Nature 264, 185–186 [DOI] [PubMed] [Google Scholar]
- 74. Ryu S. Y., Beutner G., Kinnally K. W., Dirksen R. T., Sheu S. S. (2011) Single channel characterization of the mitochondrial ryanodine receptor in heart mitoplasts. J. Biol. Chem. 286, 21324–21329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Beutner G., Sharma V. K., Giovannucci D. R., Yule D. I., Sheu S. S. (2001) Identification of a ryanodine receptor in rat heart mitochondria. J. Biol. Chem. 276, 21482–21488 [DOI] [PubMed] [Google Scholar]
- 76. Trenker M., Malli R., Fertschai I., Levak-Frank S., Graier W. F. (2007) Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat. Cell Biol. 9, 445–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Brookes P. S., Parker N., Buckingham J. A., Vidal-Puig A., Halestrap A. P., Gunter T. E., Nicholls D. G., Bernardi P., Lemasters J. J., Brand M. D. (2008) UCPs–unlikely calcium porters. Nat. Cell Biol. 10, 1235–1237; author reply 1237–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. De Marchi U., Castelbou C., Demaurex N. (2011) Uncoupling protein 3 (UCP3) modulates the activity of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) by decreasing mitochondrial ATP production. J. Biol. Chem. 286, 32533–32541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jiang D., Zhao L., Clapham D. E. (2009) Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science 326, 144–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Dimmer K. S., Navoni F., Casarin A., Trevisson E., Endele S., Winterpacht A., Salviati L., Scorrano L. (2008) LETM1, deleted in Wolf-Hirschhorn syndrome, is required for normal mitochondrial morphology and cellular viability. Hum. Mol. Genet. 17, 201–214 [DOI] [PubMed] [Google Scholar]
- 81. Dash R. K., Beard D. A. (2008) Analysis of cardiac mitochondrial Na+-Ca2+ exchanger kinetics with a biophysical model of mitochondrial Ca2+ handling suggests a 3:1 stoichiometry. J. Physiol. 586, 3267–3285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brand M. D. (1985) The stoichiometry of the exchange catalysed by the mitochondrial calcium/sodium antiporter. Biochem. J. 229, 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Carafoli E., Balcavage W. X., Lehninger A. L., Mattoon J. R. (1970) Ca2+ metabolism in yeast cells and mitochondria. Biochim. Biophys. Acta 205, 18–26 [DOI] [PubMed] [Google Scholar]
- 84. Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., Walford G. A., Sugiana C., Boneh A., Chen W. K., Hill D. E., Vidal M., Evans J. G., Thorburn D. R., Carr S. A., Mootha V. K. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134, 112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Perocchi F., Gohil V. M., Girgis H. S., Bao X. R., McCombs J. E., Palmer A. E., Mootha V. K. (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467, 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Docampo R., Lukeš J. (2012) Trypanosomes and the solution to a 50-year mitochondrial calcium mystery. Trends Parasitol. 28, 31–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L., Koteliansky V., Mootha V. K. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]