

Background: An epigenetic switch regulates biofilm formation.
Results: Sequence-specific DNA binding by SinR has been visualized, and the macromolecular interactions in the epigenetic switch have been analyzed.
Conclusion: DNA binding by SinR requires precise protein-DNA contacts and SinR-induced DNA deformation.
Significance: The macromolecular interactions at the center of biofilm formation have been analyzed.
Keywords: Bacillus, Biofilm, Protein-Nucleic Acid Interaction, Protein-Protein Interactions, Thermodynamics, X-ray Crystallography
Abstract
Bacterial biofilms are complex communities of cells that are attached to a surface by an extracellular matrix. Biofilms are an increasing environmental and healthcare issue, causing problems ranging from the biofouling of ocean-going vessels, to dental plaque, infections of the urinary tract, and contamination of medical instruments such as catheters. A complete understanding of biofilm formation therefore requires knowledge of the regulatory pathways underpinning its formation so that effective intervention strategies can be determined. The master regulator that determines whether the Gram-positive model organism Bacillus subtilis switches from a free-living, planktonic lifestyle to form a biofilm is called SinR. The activity of SinR, a transcriptional regulator, is controlled by its antagonists, SinI, SlrA, and SlrR. The interaction of these four proteins forms a switch, which determines whether or not SinR can inhibit biofilm formation by its repression of a number of extracellular matrix-associated operons. To determine the thermodynamic and kinetic parameters governing the protein-protein and protein-DNA interactions at the heart of this epigenetic switch, we have analyzed the protein-protein and protein-DNA interactions by isothermal titration calorimetry and surface plasmon resonance. We also present the crystal structure of SinR in complex with DNA, revealing the molecular basis of base-specific DNA recognition by SinR and suggesting that the most effective means of transcriptional control occurs by the looping of promoter DNA. The structural analysis also enables predictions about how SinR activity is controlled by its interaction with its antagonists.
Introduction
The Gram-positive model organism Bacillus subtilis has evolved a number of adaptive responses to meet the various challenges posed by changes in its environment. In addition to the free-living, motile form most commonly found in liquid media, B. subtilis can differentiate under conditions of nutrient starvation to form spores, a dormant cell type highly resistant to extremes of heat, pH, and salt. Alternatively, B. subtilis can form a biofilm, a lifestyle more commonly associated with microorganisms in their natural environments (1, 2). A biofilm is an architecturally complex community of microorganisms that is attached to surfaces by an extracellular matrix of exopolysaccharide, protein, and DNA (3, 4). Biofilms are found in an array of environmental niches, including the hulls of ocean-going vessels, medical instruments, catheters, teeth, and the urinary tract. Biofilms are thus a societal problem of significant magnitude.
The propensity for wild isolates of B. subtilis to form biofilms was discovered only quite recently (5). The B. subtilis biofilm is a complex community of bacteria with multiple, different specializations. For instance, some members of the biofilm are dedicated to excreting the extracellular matrix that holds the biofilm together, whereas others are fated to sporulate and are typically located at the end of tongue like protrusions, similar to the fruiting bodies of the myxobacteria or fungi (5).
Although multiple gene products are necessary for biofilm development in B. subtilis, the decision to switch from a free-living, planktonic state to a sessile, biofilm forming state is governed by SinR, the master regulator of biofilm formation (6–9). SinR is a transcriptional repressor of the operons for exopolysaccharide production (epsA-O (5)) and the production of the secreted, amyloid-like protein component of the matrix, TasA (yqxM-sipW-tasA (7, 8, 10)), thus blocking biofilm formation. The consensus DNA binding sequence for SinR comprises a 7-bp pyrimidine-rich sequence (5′-GTTCTYT-3′, with Y representing an unspecified pyrimidine base), which can be found in various orientations and permutations at SinR operator sites (8, 11), although SinR appears to have a preference for binding sites containing inverted repeats (11). The binding of SinR to repress DNA transcription is dependent upon the stoichiometry of the repressor and its antagonist, SinI (7, 12). When sufficient SinI is present, an SinR-SinI complex is formed that inhibits the SinR-DNA interaction, causing derepression (13, 14), brought about not by the occlusion of the DNA binding sites in SinR by SinI, but by the dissociation of the SinR tetramer into a SinR-SinI heterodimer (14).
The presence of three different promoters in the sinIR locus results in a complex transcription profile. For instance, SinR is constitutively expressed at low levels throughout growth, whereas SinI is expressed at low levels during vegetative growth and at higher levels during sporulation because it is under the control of the sporulation response regulator, Spo0A (15, 16). However, a subset of cells within the population expresses SinI at much higher levels (17), leading to biofilm formation. It has been proposed that these high SinI-expressing cells go on to become specialized for the production of the biofilm matrix for the entire community (17).
The SinR-SinI switch is made yet more complex by SlrR (18), a transcriptional regulator that contains regions of homology to both SinR and SinI. slrR is located immediately adjacent to epsA on the B. subtilis chromosome, and thus its expression is under direct negative control by SinR. The repression is relieved when SinR is inhibited by SinI (18). SlrR binds to SinR, and this complex represses genes involved in flagellar biosynthesis and cell separation (19, 20). SlrR expression is controlled by SinR; the SinR-SlrR complex no longer represses SlrR expression, thus creating a self-reinforcing double-negative feedback loop. Finally, SlrA is an additional SinR antagonist that is homologous to SinI (19). Pulldown assays have indicated that a tight complex could be formed between SinR and SlrA that antagonized the DNA binding properties of SinR (9), but no quantitative data are available for this complex. Although SlrA was initially described as an activator of SlrR (19), it would seem that SlrR antagonizes SlrA, forming part of a negative feedback loop that alleviates the effects of SlrA (9).
The complex interplay between these transcription factors and their antagonists, as well as the feedback loops created by these interactions, explains how B. subtilis adapts to challenging environmental conditions (Fig. 1). Cells either develop motility, with the aim of moving to a better environment, or become sessile and cooperate with other cells to form a biofilm.
FIGURE 1.

A schematic view of the control network of SinR. SinR is a tetrameric repressor of two major biofilm operons, epsA-O and yqxM-sipW-tasA. The repression is relieved by the action of SinI and SlrA, which bind to and inactivate SinR. SlrR, itself under control by SinR, repurposes SinR to become a repressor of motility genes.
To understand the nature of the SinR-SinI complex at the heart of this epigenetic switch, the crystal structure of the SinR-SinI complex was solved previously (14). The N-terminal domain of SinR (SinR1–64) resembles a Cro-like helix-turn-helix (HTH)2 DNA binding domain, whereas the C-terminal domain of SinR (SinR75–111) forms a pair of α-helices called a helical hook. The helical hook structure is replicated in SinI and is used by both proteins to drive heterodimerization (14). The structures of the two isolated domains of SinR have also been solved, confirming that SinR homodimerization occurs using the same helical hook interactions as seen in the SinR-SinI heterodimer (11). The interaction between the two proteins has also been studied by analytical ultracentrifugation (21) and size exclusion chromatography coupled with multiangle laser light scattering (11). However, these studies have provided neither the molecular basis of the tetramerization of SinR nor an explanation of the sequence specificity of SinR binding to DNA. Equilibrium and rate constants of the various protein-protein and protein-DNA interactions are also lacking, which are necessary to understand the regulation of the switch on a systems level. Therefore, to understand the hierarchy of the protein-protein and protein-DNA interactions in this molecular switch, we have obtained the missing quantitative data and have determined the crystal structure of SinR in complex with DNA, enabling the interactions that provide specificity in DNA recognition to be visualized. Finally, an alternative model of the SinR tetramer, as well as its implications for the looping of DNA at SinR-regulated promoters, is discussed.
EXPERIMENTAL PROCEDURES
Cloning and Overexpression
For all constructs, coding sequences were amplified by the PCR with Phusion high fidelity DNA polymerase (Thermo Scientific) using oligodeoxynucleotide primers (Eurofins MWG Operon) and B. subtilis 168 genomic DNA as the template. The CloneJET PCR cloning kit (Thermo Scientific) was used for sinI, sinR, and slrR PCR products. The fragments were then restricted with NdeI and EcoRI and ligated into pET24a. The ligation product was transformed into Escherichia coli DH5α-competent cells where the presence of the insert was verified by colony PCR with primers for T7 promoter and terminator regions. For slrA, the amplified fragment was digested with NcoI and SalI restriction enzymes and ligated into pET28a. The ligation product was transformed into E. coli DH5α-competent cells where the presence of the insert was verified by colony PCR with primers for T7 promoter and terminator regions. All the constructs were used for overexpression of full-length, untagged proteins. All restriction enzymes, T4 DNA ligase, CloneJET PCR cloning kit, and DNA polymerase were used as recommended by their manufacturers, and all clones generated were verified by DNA sequencing.
The final constructs were all transformed into E. coli BL21 (DE3) for protein overexpression. Cultures were grown in Lennox L broth at 37 °C until the optical density at 600 nm reached 0.6–0.8, at which point overexpression was induced with the addition of isopropyl-1-thio-β-d-galactopyranoside to a final concentration of 0.1 mm. Cells were harvested by centrifugation 4 h after induction, with the exception of SlrR, which was incubated at 18 °C overnight to maximize the production of soluble protein.
Protein Purification
All proteins were expressed and purified as full-length, untagged proteins. For SlrA, cell pellets were resuspended in a buffer containing 20 mm Tris·HCl, pH 8.0, 250 mm NaCl, disrupted by sonication (4 × 25 s), and clarified by centrifugation (46,000 × g for 25 min), and the supernatant was loaded onto an SP Sepharose cation exchange column (GE Healthcare). Proteins were eluted with a linear gradient of 250–750 mm NaCl in 20 mm Tris·HCl, pH 8.0. Fractions containing SlrA were pooled and concentrated by ultrafiltration to ∼1 ml and loaded directly onto a Superdex 75 16/60 (GE Healthcare) gel filtration column, pre-equilibrated with 50 mm HEPES·NaOH, pH 7.0, 500 mm NaCl. Fractions containing SlrA were pooled and concentrated for further analysis by ITC and SPR.
For SinI, cells were resuspended in a buffer containing 50 mm Tris·HCl, pH 8.0, prior to sonication. Cell debris was pelleted by centrifugation, and the supernatant was loaded onto an ANX Sepharose anion exchange column (GE Healthcare). Bound proteins were eluted with a linear gradient of 0–500 mm NaCl in 50 mm Tris·HCl, pH 8.0. Fractions containing SinI were pooled and concentrated by ultrafiltration before further purification by size exclusion chromatography as described above for SlrA.
To purify SinR, cells were resuspended in a buffer containing 50 mm Tris·HCl, pH 8.0, and disrupted by sonication before the addition of concentrated NaCl to a final concentration of 1 m. Cell debris was pelleted by centrifugation. The supernatant was diluted 4-fold before loading onto a heparin-Sepharose column (GE Healthcare), and bound proteins were eluted using a linear gradient of 250 mm to 1 m NaCl. Fractions containing SinR were poled and concentrated by ultrafiltration before final purification by size exclusion chromatography as above.
To purify SlrR, cells were resuspended in a buffer containing 50 mm Tris·HCl, pH 8.0, 5 mm MgCl2, 5 μg/ml DNase I (Sigma-Aldrich) and sonicated. NaCl was added to 1 m, and the cell lysate was clarified by centrifugation as described for SinR. The supernatant was diluted 2-fold before purification by heparin-Sepharose with the application of a linear gradient of 500 mm to 1 m NaCl. Further attempts to concentrate SlrR beyond 0.5 mg/ml for purification by size exclusion chromatography were unsuccessful.
The SinR-SlrR complex was purified by mixing cell pellets in the approximate ratio of 2 parts SlrR to 1 part SinR before disruption and purification by heparin-Sepharose chromatography as described for SlrR. Fractions containing the SinR-SlrR complex were pooled, concentrated by ultrafiltration, and further purified by size exclusion chromatography as described for SlrA.
All proteins were estimated by SDS-PAGE to be greater than 90% pure. Protein concentrations were monitored during purification by the Bradford method using lysozyme as the standard. Final protein concentrations were determined by measuring the absorbance at 280 nm.
Preparation of Oligodeoxynucleotides
All oligodeoxynucleotides used for ITC and crystallization were synthesized on the 1.0-μmol scale and provided by the manufacturer (Eurofins MWG Operon) in the high purity, salt-free form. The oligodeoxynucleotides were used without further purification. The inverted repeat duplex for crystallography was prepared by heating the single-stranded oligodeoxynucleotide sequences 5′-ATTGTTCTCTAAAGAGAACTT-3′ and 5′-AAAGTTCTCTTTAGAGAACAA-3′ to 95 °C before allowing the duplex to cool to room temperature over 15 min. Double-stranded DNA was prepared for ITC by resuspending single-stranded oligodeoxynucleotides in ITC buffer (20 mm HEPES·NaOH, pH 7.0, 500 mm NaCl). The two oligodeoxynucleotides were then mixed and heated to 90 °C for 10 min, and the duplex was then left to cool to room temperature.
Surface Plasmon Resonance
SPR experiments were carried out on a Biacore X1000 (GE Healthcare) using CM5 sensor chips. Proteins were immobilized onto the sensor surface by amine coupling. The surface was activated by injection of 35 μl of a 1:1 mixture of 100 mm N-hydroxysuccinimide and 400 mm N-ethyl (dimethylaminopropyl) carbodiimide hydrochloride, at a flow rate of 5 μl/min. Proteins were immobilized onto the chip using the optimal preconcentration conditions as determined by trial experiments, to an immobilized level of 400 response units for SinR and SinI and 1000 response units for SlrA on flow cell 2. Flow cell 1 was used as an activated blank control for in-line subtraction. Unoccupied groups on the flow cells were blocked by injecting 35 μl of 1 m ethanolamine·HCl, pH 8.5. Interaction experiments were carried out in a running buffer of 20 mm HEPES·NaOH, pH 7.0, 500 mm NaCl and at a flow rate of 30 μl/min. The high salt concentration was necessary to prevent precipitation of SinR and to suppress artifacts caused by protein aggregation. The interaction between SinR and SlrR was monitored using multicycle kinetics, and the sensor surface was regenerated between runs by injecting 50 μl of 10 mm NaOH. All other interaction experiments were performed using the single cycle mode in which interactions were monitored without the need for regeneration of the sensor surface. Binding curves were corrected, aligned, and fitted to a 1:1 Langmuir binding model using the Biacore x1000 evaluation program (GE Healthcare).
Isothermal Titration Calorimetry
All ITC experiments were performed on a MicroCal ITC200 (GE Healthcare) at a temperature of 25 °C in a buffer of 20 mm HEPES·NaOH, pH 7.0, 500 mm NaCl. For the SinR-SinI interaction, SinR was used in the cell at a concentration of 38 μm, whereas SinI was injected at an initial concentration of 516 μm for a total of 20 injections; the first three injections were separated by 180 s, and the subsequent 17 injections were separated by 240 s, comprising 0.2 μl of SinI for the first injection and then 2 μl of SinI for the remaining 19 injections. For the SinR-SlrA interaction, SinR was used in the cell at a concentration of 40 μm, whereas SinI was injected at an initial concentration of 400 μm for a total of 20 injections, each separated by 180 s, consisting of 0.2 μl of SinI for the first injection and then 2 μl of SinI for 19 injections. In both cases, the kon measured by SPR was used when fitting the data due to the high c value, the ratio between the analyte concentration and the equilibrium dissociation constant, (a consequence of the high affinity and relatively low ΔH). For the interaction of SinR with inverted repeat DNA, SinR was used in the cell at a concentration of 29 μm with the oligodeoxynucleotide injected at an initial concentration of 197 μm for 1 injection of 0.2 μl and 19 injections of 2 μl, each separated by 180 s. For the single site DNA-SinR interaction, the oligodeoxynucleotide was used in the cell at 142 μm with SinR in the syringe at 1455 μm for 1 injection of 0.2 μl and 19 injections of 2 μl, each separated by 180 s. Finally, for the interaction of SinR with tandem repeat DNA, SinR was used in the cell at 50 μm with the oligodeoxynucleotide in the syringe at a concentration of 500 μm for 1 injection of 0.2 μl and 19 injections of 2 μl, each separated by 180 s. Due to the low c value, the stoichiometry was fixed at 2.0 for fitting of the SinR-DNA tandem repeat data. To provide consistency between all data fits, SinR was considered to be the ligand throughout. All ITC data were fit with Origin 7 using the one-site model.
Crystallization and Structure Determination
SinR, in a buffer of 20 mm HEPES·NaOH, pH 7.0, 500 mm NaCl, was concentrated by ultrafiltration to 10 mg/ml and mixed with DNA (also in 20 mm HEPES·NaOH, pH 7.0, 500 mm NaCl) in the molar ratio 1:1.2 (1.0 mol of SinR dimer to 1.2 mol of DNA duplex). Sitting drop vapor diffusion crystallization trials of the SinR-DNA complex at a concentration of 5 mg/ml were performed with a Mosquito (TTP Labtech) crystallization robot, using commercially available screens (Molecular Dimensions). Crystals of the SinR-DNA complex grew over 1 week against a crystallization buffer containing 0.01 m ZnCl2, 0.1 m sodium acetate, pH 5.0, and 20% (w/v) PEG 6000. Crystals were cryoprotected by soaking for 30 s in the well solution supplemented by the addition of 18% (v/v) ethylene glycol before being loop-mounted and plunged into a pool of liquid nitrogen. The crystals diffracted to 3.0 Å on beamline I02 at the Diamond Synchrotron Light Source, Didcot, Oxfordshire, UK. Diffraction data were processed using XDS (22); the crystals obey primitive orthorhombic symmetry and belong to space group P21212 with unit cell parameters of a = 65.4 Å, b = 79.7 Å, and c = 67.9 Å. There is one copy of the SinR dimer bound to a single DNA duplex in the crystallographic asymmetric unit. The structure of the SinR-DNA complex was solved by molecular replacement using the using residues 1–68 of the B. subtilis SinR-SinI complex (Protein Data Bank (PDB) ID 1B0N (14)) and the DNA from the phage 434 Cro-OR1 complex (PDB ID 3CRO (23)) as search models, after manual correction of the DNA sequence in COOT (24). During the early stages of model building and refinement, it was found that the application of B factor sharpening (using a value of −60 Å2) caused a significant improvement in the quality of the electron density maps. The SinR-DNA model was built in COOT (24) and refined in PHENIX REFINE (25) to a final Rwork of 0.24 and Rfree of 0.26. Although residual electron density remains for SinR75–111, it does not support refinement of these residues, and they have been excluded from the final model.
RESULTS
The SinR-SinI and SinR-SlrA Interactions
Titrating SinR in the cell against SinI or SlrA as injectants in a MicroCal ITC200 produced distinctive binding isotherms with very sharp transitions indicative of tight binding interactions (Fig. 2, A and C). The lack of intermediate points on the binding isotherm means that the equilibrium dissociation constant (Kd) cannot be determined accurately, suggesting that the SinR-SinI and the SinR-SlrA interactions are too tight to be measured directly by ITC (upper limit for Kd ∼10 nm). However, some thermodynamic parameters (Table 1) are directly obtained from the isotherms such as the binding enthalpy, ΔH, and the interaction stoichiometry, n. The fact that SinI and SinR form a very tight complex with 1:1 stoichiometry was not surprising given the previous data on this interaction (11, 14, 21).
FIGURE 2.
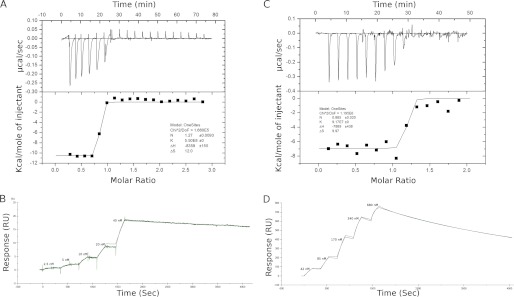
The interactions between SinR-SinI and SinR-SlrA. A, thermodynamics of the SinR-SinI interaction measured by ITC. Sequential injections of SinI (516 μm) were performed into a reaction cell containing SinR (38 μm). Both the raw data (upper panel) and the integrated heats (lower panel) show a sudden sharp transition, indicating that the interaction is too tight to be accurately measured by ITC (Kd ≤10 nm). B, kinetics of the SinR-SinI interaction measured by single cycle SPR. Successive injections of SinR were performed over a CM5 sensor chip immobilized with 400 response units (RU) of SinI followed by a single extended dissociation phase. The experimental data (green line) are overlaid with the best fit to the data (solid black line). C, thermodynamics of the SinR-SlrA interaction measured by ITC. SlrA (400 μm) was injected into a cell containing SinR (40 μm). As with the SinR-SinI interaction, a sharp transition indicates tight binding. D, kinetics of the SinR-SlrA interaction measured by single cycle SPR. SlrA, the ligand, was immobilized on the sensor chip (400 response units), and SinR was the analyte. Five successive injections of SinR were followed by a single dissociation phase.
TABLE 1.
Summary of biophysical constants of protein-protein interactions
N.D. indicates not detected.
| Interaction | ITC |
SPR |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔG | ΔH | −TΔS | Stoichiometry | Kd | kon | koff | Kd | χ2 (U value) | |
| kcal/mol | kcal/mol | kcal/mol | m−1 s−1 | s−1 | nm | ||||
| SinI-SinR | −11.9 | −8.36 ± 0.16 | −3.58a | 1.27 ± 0.01 | ≤10 nm | 2.73 ± 0.01 × 104 | 5 ± 0.06 × 10−5 | 1.83 | 0.12 (5) |
| SlrA-SinR | −10.9 | −7.89 ± 0.44 | −2.97a | 0.89 ± 0.02 | ≤10 nm | 2.16 ± 0.01 × 104 | 2.3 ± 0.006 × 10−4 | 10.6 | 40.8 (1) |
| SlrR-SinR | N.D. | N.D. | N.D. | N.D. | N.D. | 4.85 ± 0.03 × 103 | 2.3 ± 0.015 × 10−4 | 47.5 | 0.10 (NA) |
| SinR-Inverted repeat DNA | −8.77 | 4.33 ± 0.12 | −13.12 | 2.38 ± 0.05 | 360 ± 122 nm | 1 × 105b | 2 × 10−2b | 270 ± 50 | N.D. |
| SinR-Single site DNA | −6.68 | −5.78 ± 0.11 | 0.90 | 1.07 ± 0.01 | 12.5 ± 1.2 μm | N.D. | N.D. | N.D. | N.D. |
| SinR-Tandem site DNA | −6.18 | 1.66 ± 0.13 | −7.84 | ∼2 | 30 ± 7 μm | N.D. | N.D. | N.D. | N.D. |
a Kd measured by SPR was used for fitting ITC data to obtain ΔS.
b Values estimated by simulation software from data of Colledge et al. (11).
The Kd, the association (kon) and dissociation (koff) rate constants for the SinR-SinI and SinR-SlrA interaction were determined by SPR using a Biacore X100 with SinI and SlrA immobilized on SPR chips. Initial experiments determined that the binding was very slow to dissociate and that the dissociation could not be enhanced significantly by any of the commonly used regeneration reagents. Consequently, the interaction was monitored by single cycle kinetics in which successive injections of higher protein concentrations are followed by a single, extended dissociation phase (Fig. 2, B and D). The data were fitted to a simple 1:1 Langmuir binding model to yield the kinetic parameters listed in Table 1.
The SinR-SlrR Interaction
SlrR could be purified to near homogeneity in a single step using heparin-Sepharose pseudo-affinity chromatography; however, it proved impossible to concentrate SlrR, restricting our analysis of its interactions to SPR. The SinR-SlrR interaction was monitored in real time by SPR with SinR immobilized onto the sensor surface to which multiple cycles of association and dissociation were performed with increasing concentrations of SlrR (Fig. 3, upper panel). The response level of these binding curves is significantly lower than expected given the amount of SinR immobilized onto the sensor surface (500 response units). However, this response is proportional to that obtained when either SinI or SlrA was flowed over the same surface (data not shown), indicating that perhaps only a small proportion of SinR is active following immobilization. A slight discrepancy of fit can also be observed, largely, we believe, as a result of noise introduced by double referencing, a procedure that is necessary because of the base-line drift over the time course of the experiment. The data were fitted to a 1:1 Langmuir binding model, and the kinetic parameters are reported in Table 1.
FIGURE 3.

The interaction between SinR and SlrR. Upper panel, kinetics of the SinR-SlrR interaction measured by multicycle SPR. Individual sensorgrams of four different concentrations of SlrR injected over a CM5 sensor surface containing 500 response units (RU) of immobilized SinR are aligned and overlaid with the best fit to the data (solid black lines). Lower panel, a chromatogram showing the elution profile of the SinR-SlrR complex (blue) on a Superdex 75 HR 16/60 size exclusion column overlaid with the elution profile of SinR alone (red). SDS-PAGE analysis of the major peak (inset) shows an approximately equal amount of SinR and SlrR present in the complex. The elution volume of this peak corresponds to an expected molecular mass of 49 kDa based on a calibration curve created using the low molecular mass standards from GE Healthcare.
The apparent molecular mass of the SinR-SlrR complex was estimated by size exclusion chromatography to be 49 kDa, with a stoichiometry of 1:1 (as judged by SDS-PAGE of the peak fractions) (Fig. 3, lower panel). This apparent molecular mass does not correspond either to a SinR1-SlnR1 dimer (expected mass 30.4 kDa) or to a SinR2-SlnR2 tetramer (expected mass 60.8 kDa) given the masses of SinR (12.8 kDa) and SlrR (17.6 kDa). Our best explanation for this discrepancy, given the extended, two-domain architecture of SinR (11, 14), is that the SinR-SlrR complex is probably an elongated dimer rather than a compact tetramer.
The SlrR-SlrA Interaction
To complete the set of protein-protein interactions, we studied the potential for SlrR and SlrA to interact in vitro. Others had used pulldowns with tagged proteins to demonstrate this interaction in vivo (9). However, we were unable to find any evidence for a direct interaction between SlrR and SlrA.
SinR-DNA Interactions
We have extended the analysis to include a thermodynamic characterization of SinR binding to DNA by ITC. Three classes of SinR operator sequences were studied (Fig. 4): tandem, single sites, and inverted repeats of the consensus sequence, the central C of which is found 158 (and 150), 129 and 67 (and 58) bp upstream of the transcription start site of the eps operon, respectively. A clear preference of binding affinity for inverted repeats of the consensus sequence was demonstrated (Fig. 4, left panel, Table 1), in agreement with previous studies (11). SinR binding to sequences containing single copies of the SinR operator sequence was exothermic, with a significantly lower affinity; n was 1.07 ± 0.01 SinR monomers per DNA duplex (Fig. 4, center panel, Table 1), indicating that SinR does not bind to these sites as a tetramer, but probably the SinR dimer binds to single sites on two discrete DNA duplexes. DNA sequences containing tandem repeats of the SinR binding sequence displayed binding affinities similar to that for a single site, although the reaction was slightly endothermic. The stoichiometry was fixed at two SinR molecules to one DNA duplex due to the low c value (Fig. 4, right panel, Table 1).
FIGURE 4.

The interaction between SinR and DNA. Thermodynamics of the SinR-DNA interaction measured by ITC are shown. Three classes of SinR consensus operator sequences (5′-GTTCTYT-3′) were studied containing an inverted repeat sequence (left panel), a single site sequence (center panel), and a tandem repeat sequence (right panel) found 67, 129, and 158 bp upstream from the transcription start site of the eps promoter, respectively. In the case of the tandem repeat DNA sequence, the two mismatches from the consensus sequence were altered so as to match the consensus.
Crystal Structure of the SinR-DNA Complex
To rationalize base-specific DNA binding by SinR, we solved the crystal structure of the SinR tetramer in complex with duplex DNA. The best crystals diffracted to a resolution of 3.0 Å and were obtained using a 21-bp nonpalindromic sequence, containing a pair of SinR operator inverted repeats with single base adenine overhangs at each 5′ end. The sequence used correlates directly (except for the adenine 5′ overhang) to the inverted SinR binding site of the epsA promoter. The structure of the SinR-DNA complex was solved by molecular replacement using SinR1–64 from the B. subtilis SinR-SinI complex (PDB ID 1B0N (14)) and the DNA, after manual correction of its sequence, from the phage 434 Cro-OR1 complex (PDB ID 3CRO (23) as search models. There are two SinR molecules and one DNA duplex in the asymmetric unit. Overall the electron density for SinR1–64 and for the DNA is of high quality (Fig. 5A). The 5′-adenine overhangs form a noncomplementary adenine-adenine base pair such that the DNA forms a pseudo-continuous B-type helix parallel to the crystallographic c axis. SinR75–111 is, however, almost completely disordered. Residual electron density for one copy of SinR75–111 could be identified, but it was not possible to model this domain with confidence. It is perhaps surprising that SinR75–111 is almost completely disordered given that the isolated domain has been crystallized and its structure has been solved (11). However, the disorder can be explained by the relative lack of crystal contacts and by the connection of this region to the DNA binding domain by a flexible linker that is completely disordered in this structural analysis and in the SinR-SinI complex (14). The final model contains 64 out of 111 residues and 42 DNA bases and has been refined to a crystallographic Rfactor of 0.24 (Rfree = 0.26). A summary of the data collection, refinement, and model quality statistics can be found in Table 2.
FIGURE 5.

Structural basis of SinR-DNA recognition. A, stereo view of the final 2Fobs − Fcalc electron density map colored in gray. Protein and DNA are represented by sticks, with atoms colored by type. The map is displayed at levels of 1.7 σ over the DNA and 1.2 σ over the protein so as not to obscure details in either component. B, stereo view of the SinR DNA binding domain in complex with double-stranded DNA containing a high affinity, inverted repeat SinR binding site taken directly from the eps promoter. The SinR α-helices are labeled, and DNA bases are drawn as colored squares as follows: A (red); T (blue); G (green), and C (yellow). C, expanded stereo view of the specific interactions by which SinR recognizes operator sequences. Contacts between protein side chains and DNA bases are shown as dotted lines with hydrogen bond donor or acceptor status indicated by lowercase lettering. It appears that SinR achieves its sequence specificity through a combination of direct and indirect readout; recognition of the first four bases of the 7-residue motif, 5′-GTTCTYT-3′, and the pyrimidine at position five is sufficient for sequence-specific binding. D, the sequence of the DNA used in structure determination; other than the 5′-A overhang, the sequence shown correlates perfectly to the region 72–53 bp upstream of the transcription start site of the eps promoter. E, schematic of the protein-DNA contacts in the crystal. DNA bases are all represented by hexagons, ribose sugars are represented by pentagons, and phosphates are represented by circles. Each DNA strand is colored separately, and the contacting amino acids are represented as white boxes.
TABLE 2.
Data collection and refinement statistics for the SinR-DNA complex
Values in parentheses are for the highest resolution shell.
| Data collection | |
| Space group | P 21212 |
| Cell dimensions a, b, c (Å) | 65.4, 79.7, 67.9 |
| Wavelength (Å) | 0.977 |
| Resolution (Å) | 47.1–3.0 (3.16–3.0) |
| Rmerge (%)a | 3.2 (66.8) |
| I/σI | 21.3 (2.3) |
| Completeness (%) | 98.4 (98.7) |
| Multiplicity | 3.6 (3.8) |
| Refinement | |
| Resolution (Å) | 40.5–3.0 (3.3–3.0) |
| No. of unique reflections | 13,020 (3204) |
| Rwork/Rfree | 0.242/0.264 (0.332/0.357) |
| No. of atoms | |
| Protein | 978 |
| DNA | 856 |
| B factors | |
| Protein | 96 |
| DNA | 101 |
| Wilson B | 102 |
| r.m.s.b deviations | |
| Bond lengths (Å) | 0.03 |
| Bond angles(°) | 0.8 |
| Ramachandran plot | |
| Favored (%) | 96.7 |
| Allowed (%) | 100 |
a Rmerge = ΣhklΣi|Ii − Im|/ΣhklΣiIi where Ii and Im are the observed intensity and mean intensity of related reflections respectively.
b r.m.s., root mean square.
Both SinR1–64 subunits display the same overall fold (0.4 Å root mean square deviation between chains), similar to previous structures of either SinR1–64 alone (11) or the equivalent domain of full-length SinR in complex with its antagonist SinI (14). SinR1–64 is a compact, five-helix bundle with an HTH DNA binding motif that spans residues 17–36. The two SinR1–64 protomers are arranged such that both HTH motifs insert into adjacent major grooves of the DNA, causing an ∼35° bending of the DNA duplex (Fig. 5B) and a concomitant narrowing of the minor groove between the two consensus sequences. The sugar puckers are predominantly a mixture of C2-endo, C1-exo, and O4-endo, with a mean rise per base pair of 3.2 Å and a mean twist of 34.3° per turn.
Structural Basis for SinR DNA Recognition
The structure of the SinR-DNA complex permits an examination of the mode of DNA binding and the way in which SinR recognizes specific operator sequences. In common with other HTH-type DNA-binding proteins, the recognition helix of the HTH motif (α3) inserts deep into the major groove. There are no significant differences between the two protein chains in the asymmetric unit or in their interactions with the DNA. SinR binds with high affinity to a consensus GTTCTYT DNA sequence, and there are a number of direct contacts that confer sequence specificity to the SinR-DNA interaction (Fig. 5, C–E). The first base in this sequence, guanine, makes two hydrogen bonds; the side chains of Ser-18 and Lys-28 contact the N7 and O6 atoms of guanine, respectively. These interactions require the presence of hydrogen bond acceptors at key points in the base, a requirement that is sufficient to discriminate guanine from all other possibilities. The second base in the sequence, thymine, does not appear to make any polar contacts with the protein. Recognition is achieved by the positioning of the extracyclic, C7 methyl group of the thymine in a largely hydrophobic environment created by the side chains of Leu-17 and the aliphatic portion of Lys-28. The two subsequent base pairs of the motif, T:A at position 3 and C:G at position 4, both lie close to the side chain hydroxyl of Ser-29, which can, for instance, utilize the oxygen lone pair in accepting a hydrogen bond from the N6 of the adenine at position 3. Furthermore, Ser-29 can donate its proton in forming a bifurcated hydrogen bond to both the O6 of the guanine and the O4 of the thymine. These interactions restrict the 16 sequence possibilities for positions 3 and 4 to either TC or GA. The thymines at position 5 and 7 do not appear to make any direct contacts with the protein, and instead contacts between protein and DNA take place predominantly to the DNA backbone. The pyrimidine at position six is discriminated for because of a hydrogen bond between the Nϵ2 of Gln-39 and the N7 of the corresponding guanine on the opposite strand in the duplex. This particular interaction can only occur with purines, explaining neatly the preference for pyrimidine at position six. It is also possible that water-mediated interactions occur between the latter part of the motif and the side chains of Ser-33 and Gln-39, which are not visible at the resolution of the current diffraction data. The importance of Ser-18, Lys-28, and Ser-29 for sequence-specific DNA interactions is underlined by the near invariance of the latter two amino acids in SinR orthologues and by the fact that position 18 is occupied predominantly by serine or threonine. In addition to the sequence-specific protein DNA contacts, sequence-independent contacts to the phosphate DNA backbone are provided by the side chains of Ser-16, at the N terminus of the scaffolding helix, α2; Tyr-30, Ser-32, Arg-36 on α3; Gln-39 on the α3-α4 loop; and Ser-43 and Lys-49 on α4, with further contacts made from the main chain carbonyl oxygens of Leu-17 and Ala-27 (Fig. 5, C–E).
In addition to specific polar contacts, there are a number of other ways in which proteins of the wider HTH family achieve sequence specificity (26). The ability of proteins to recognize the propensity of a given DNA sequence to deviate from B-form geometry has been termed “indirect readout” (27), examples of which include alterations to minor and major groove widths and DNA bending and deformability (28). There are significant deviations from the ideal B-form in the SinR-DNA complex, the most striking of which is the narrowing of the minor groove in the region between the two SinR consensus sequences to 8.1 Å when compared with typical values of 11.2 Å. The formation of a narrow minor groove is strongly associated with the presence of “A tracts” (29), AT-rich sequences of 3–4 or more consecutive adenines (or thymines), which are present in the SinR operators controlling the eps and yqxM-sipW-tasA operons. Additional significant deviations from ideal B-form geometry are also observed, such as propeller twisting (Table 3), which may be compensated for by bifurcated, non-Watson-Crick type hydrogen bonding between opposing bases. It is likely that these features constitute a significant aspect of SinR-DNA recognition.
TABLE 3.
Geometrical parameters of DNA base pairs and steps
Parameters are defined, and values were calculated using the program 3DNA (31). N.D. indicates not determined.
| Base pair step parameters |
Groove widths |
Local base pair parameters |
||||||
|---|---|---|---|---|---|---|---|---|
| Step | Roll | Twist | Tilt | Minor | Major | Base | Propeller | Opening |
| ° | Å | ° | ||||||
| N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 1 A-T | −20.7 | 9.0 |
| 1 AA/TT | 10.0 | 24.2 | 2.3 | N.D. | N.D. | 2 A-T | −32.9 | 25.8 |
| 2 AG/CT | 9.4 | 28.9 | 6.0 | N.D. | N.D. | 3 G-C | −11.2 | −2.5 |
| 3 GT/AC | 1.3 | 28.2 | −9.7 | 10.4 | 17.8 | 4 T-A | −19.4 | −2.6 |
| 4 TT/AA | −3.4 | 40.2 | −3.6 | 10.2 | 18.6 | 5 T-A | −27.9 | −5.2 |
| 5 TC/GA | −14.3 | 41.2 | 6.8 | 10.1 | 17.0 | 6 C-G | −27.3 | −2.7 |
| 6 CT/AG | 10.2 | 32.2 | −0.9 | 12.2 | 19.7 | 7 T-A | −15.4 | −0.6 |
| 7 TC/GA | −1.1 | 32.1 | −3.1 | 12.6 | 17.0 | 8 C-G | 4.2 | 1.0 |
| 8 CT/AG | −3.0 | 28.4 | 1.2 | 11.6 | 18.9 | 9 T-A | −24.0 | 1.3 |
| 9 TT/AA | −11.7 | 36.7 | −2.6 | 9.9 | 19.7 | 10 T-A | −17.0 | −2.7 |
| 10 TT/AA | 0.9 | 31.1 | 6.8 | 8.1 | 16.7 | 11 T-A | −50.0 | 22.6 |
| 11 TA/TA | −4.0 | 48.7 | −1.7 | 9.7 | 18.9 | 12 A-T | −48.5 | 10.1 |
| 12 AG/CT | 13.8 | 36.1 | 12.8 | 11.3 | 17.1 | 13 G-C | −32.1 | 8.3 |
| 13 GA/TC | −3.0 | 35.9 | −26.8 | 12.2 | 17.5 | 14 A-T | −13.5 | 6.2 |
| 14 AG/CT | 14.3 | 23.5 | −6.4 | 12.8 | 19.4 | 15 G-C | −20.4 | 3.4 |
| 15 GA/TC | −10.6 | 27.9 | −8.6 | 12.3 | 16.8 | 16 A-T | −29.0 | −3.5 |
| 16 AA/TT | −9.6 | 48.2 | −1.6 | 11.6 | 19.1 | 17 A-T | −37.0 | 8.9 |
| 17 AC/GT | −13.9 | 37.3 | 9.1 | 10.2 | 18.4 | 18 C-G | −30.4 | 15.4 |
| 18 CA/TG | 7.5 | 33.8 | 14.4 | N.D. | N.D. | 19 A-T | −42.7 | 20.0 |
| 19 AA/TT | 17.5 | 38.1 | 5.0 | N.D. | N.D. | 20 A-T | −31.0 | 4.8 |
Quaternary Structure of SinR
It has been shown previously that SinR is predominantly tetrameric in solution (14), which is the form that binds DNA (11, 14). Tetramerization is mostly a function of SinR75–111 (11), whereas SinR1–64 is monomeric in solution (11). The asymmetric unit of the SinR-DNA complex contains two copies of SinR1–64 arranged around a local two-fold axis, in a manner highly reminiscent of the bacteriophage 434 Cro protein when bound to its operator DNA sequence (23). The interface between the two SinR1–64 subunits is relatively small, 520 Å2, and is formed exclusively by the α3-α4 loop and the N-terminal half of α4. The main chain carbonyl oxygen and the side chain Oγ1 of Thr-40 are positioned to form hydrogen bonds with the side chain Nδ2 of Gln-45, and the side chain Oδ1 and Nδ2 of Asn-41 make hydrogen bonds to the main chain amide nitrogen of Gln-45 and to the side chain Oγ of Ser-43, respectively. The interface is completed by a hydrogen bond between the main chain carbonyl oxygen of Pro-42 and the main chain nitrogen of Ile-44. The interface displays only minor differences from perfect symmetry, and the contacts described above are nearly identical between both chains. Interestingly, the same interface between SinR1–64 subunits is observed in the structural analysis of this domain in isolation (11), a monomer in solution under experimental conditions (11).
Although the electron density for SinR75–111 is not sufficient to model this region with any confidence, there are significant residual Fobs − Fcalc difference map features into which the distinctive helical hook can be positioned (Fig. 6A). The density, adjacent to the crystallographic c 2-fold symmetry axis, is consistent with the same four-helical bundle found in the SinR-SinI (14) and the SinR75–111 structures (11) (Fig. 6, A and B). Application of the crystallographic two-fold symmetry axis reveals the likely nature of the SinR tetramer bound to DNA, a relatively compact dimer of dimers linked by associations between opposite rather than adjacent subunits (Fig. 6B). The interface between the two pairs of dimers includes the contributions from the α3-α4 loop detailed above, and, probably, additional contributions provided by the residues 75–111, which cannot be described because of the disorder of this region in the SinR-DNA structure. The arrangement of the DNA binding domains in the tetramer, together with the distinctive bending of DNA when bound to SinR, suggests that it is possible for all four DNA binding domains to engage with DNA simultaneously in the formation of a DNA loop. This activity may explain the observation of multiple species when the promoters from the eps and yqxM-sipW-tasA operons (both of which contain at least one additional SinR consensus sequence ∼60 and 80 bp upstream of the inverted repeat, respectively) were incubated with SinR in electrophoretic mobility shift assays (8). The yqxM-sipW-tasA promoter appears to be a particularly attractive candidate for this activity as an additional inverted repeat, albeit with two mismatches to the consensus, can be found on the same strand ∼80 bp upstream of the first site (Fig. 6C). The mismatch at position 5 of the consensus can be accommodated because SinR makes no direct contacts to this base. The tetrameric arrangement seen in the crystal, a dimer of dimers, is also consistent with the prior observation that SinR exists in equilibrium between dimeric and tetrameric forms with a Kd of 6.7 μm (21).
FIGURE 6.

SinR multimerization. A, likely position of the SinR C-terminal domain (residues 75–111). Unmodeled electron density features in the Fobs − Fcalc electron density map, shown in gray and contoured at 2.0 σ, reveal the position of SinR75–111. Positioning a single copy of SinR75–111 on top of this density feature (PDB 2YAL (11)) reveals that the crystallographic two-fold c axis (dashed black line and curved arrows) is coincident with the molecular two-fold axis of the SinR75–111 dimer. B, model of the SinR tetramer bound to two DNA duplexes. The tetramer is composed of dimeric units shown as light and dark shades of green and blue, respectively. The flexibility of the SinR C-terminal domains in the crystal, relative to the DNA binding domains, indicates that there will be dynamism between domains in solution. The connectivity within SinR protomers is indicated by colored dashed lines. C, model for the SinR-induced formation of a DNA loop at the yxqM-sipW-tasA operon, which contains two inverted repeats of the SinR consensus DNA binding sequence as indicated in the text boxes above and below the DNA duplex, with numbering relative to the transcription start site. The most upstream site contains two mismatches to the consensus, shown in red; however, the base at position 5 of the site commencing at −58 does not contribute to base-specific protein interactions with SinR.
DISCUSSION
We have determined biophysical constants for the interactions between the master regulator for biofilm development, SinR, and its three antagonists. Unsurprisingly, given the homology between SinI and SlrA, the characteristics of the interactions of these proteins with SinR are similar, with stoichiometries of 1:1, high affinity, and a mixture of enthalpic and entropic contributions. SlrA probably interacts with SinR by mutual interlocking of helical hooks to form a compact four-helix bundle. The residues forming this interaction motif are maintained in SinI, SlrA, SinR, and SlrR (Fig. 7), suggesting that the formation of the helical bundle drives the interactions involving Sin and Slr proteins. Interestingly, there are two copies of the interaction motif in SlrR; the first copy is separated from the second by an 18-residue sequence that is not present in the other Sin/Slr proteins. Enthalpic contributions are the predominant driving force in the binding of both SinI and SlrA to SinR, an observation that is perhaps surprising given the extensive hydrophobic core of the helical bundle. However, all the proteins exist as homo-oligomers before forming heterodimers with SinR, and thus the entropic and enthalpic terms represent the difference between the primary state, in which the interaction helices are participating in self-self interactions, and the final state upon formation of heteromeric complexes.
FIGURE 7.

Multiple sequence alignments of Sin-Slr proteins. SlrR contains two copies of the interaction domain; the one spanning residues 74–102 is most similar to SinI (42% identities), whereas the second spans residues 121–149 and is most similar to SlrA (50% identities). The core hydrophobic residues, which are shown in the upper panel as a superposition of SinR and SinI, are well conserved across all four proteins.
The limited solubility of SlrR precluded thermodynamic analysis by ITC. We were able to analyze its potential interactions with SinI, SlrA, and SinR by SPR. In contrast to previous studies (9), we were unable to demonstrate interactions between SlrR and SinI and SlrR and SlrA. The high affinity SinR-SlrR interaction (Kd of 47 nm) is still an order of magnitude weaker than the interactions between SinR and SinI or SlrA. The weaker affinity stems from a slower association rate for the SinR-SlrR interaction than the other SinR-containing protein complexes. In general terms, all three protein interactions involving SinR are similar with relatively slow association and disassociation rates, whereas the kinetics of SinR-DNA interactions observed by SPR previously (11) were close to the limits of what can be measured by this technique. The slow kinetics may explain why SinI cannot displace SinR when bound to DNA in a competitive ITC experiment (data not shown). Although it would be thermodynamically favorable for SinR to be displaced from DNA by any of its antagonists, the slow kinetics means it would not happen over a practical timescale. Alternatively, the strength of the self-self interactions in the SinR tetramer may be enhanced in the presence of DNA such that dissociation of the SinR-DNA complex and the formation of four SinR-SinI heterodimers may not be thermodynamically favorable. The differences in kinetics and thermodynamics for the SinR-DNA interactions versus the interactions of SinR with its antagonists may have important implications for the control of SinR in the cell; a rigorous systems approach to the circuitry of the epigenetic switch, now that quantitative data on the various macromolecular interactions are available, may be instructive.
In addition to analyzing the interactions between SinR and its antagonists, we have examined the binding of SinR to DNA. In agreement with others (11), we have found a relatively strong interaction (350 nm) between SinR and DNA sequences containing inverted repeats of the consensus sequence 5′-GTTCTYT-3′ with a 2-bp separation. The affinity of SinR binding to oligodeoxynucleotides containing either single copies or tandem repeats of the consensus sequence are 30- and 100-fold weaker than that of SinR binding to inverted repeats. The observation of the same interface between SinR1–64 subunits in isolation (11) as that seen when SinR is bound to DNA suggests that binding to inverted repeats is favored because of the stabilization of the SinR1–64-SinR1–64 interface. The significant disorder in the structure of the SinR-DNA complex structure suggests that conformational flexibility may also play an important role.
The structure of the SinR-DNA complex explains how SinR recognizes specific DNA sequences. The HTH recognition helix of SinR inserts into the major groove, and SinR is able to make base-specific interactions with a maximum of 5 bases out the 7-bp consensus motif. At three of these positions, the contacts are insufficient to discriminate directly between all 4 possible bases. Two bases that are invariant in the consensus motif do not appear to make direct contact with SinR; thus SinR is likely to recognize its operator sequences by a combination of direct and indirect readout. The region between the inverted repeats, two adenines in both the epsA and the yqxM operons, and the indirect read-out from DNA distorted from the canonical B-form may also contribute to SinR DNA recognition.
Although binding to both single and tandem repeats of the SinR consensus sequence has been demonstrated in vitro, the function of SinR in vivo can be almost entirely explained by its ability to bind with high affinity only to operator sequences close to the yqxM-sipW-tasA and eps operons. This finding is supported by the comparative transcriptomic analysis of ΔsinI and ΔsinR mutants (8), which found that the majority of differentially regulated genes (18 out of 24) were from these operons. Of the other six putative, additional members of the SinR regulon identified, rapG, spoVG, yvfV, yvfW, yvgN, and ywbD (8), only yvfV and yvfW are linked to biofilm formation. Both yvfV and yvfW are part of a three-gene lutABC operon that encodes for lactate utilization genes (30) and that contributes to the formation of complex colonies in the presence of lactate as a carbon source (30). The promoter regions of all these genes contain only single copies of the SinR recognition motif, all with significant deviations from the consensus sequence and that are often distant from the promoter region. Consequently, and with the low binding affinity of SinR for these sequences, it seems likely that the repression of these genes is not as a direct result of SinR DNA binding.
The SinR-DNA structure provides insight into the assembly of the SinR tetramer and the way in which the antagonists SinI and SlrA interfere with DNA binding by SinR. A model for the SinR tetramer, somewhat different from what has been suggested previously (11), is presented in Fig. 6, B and C. Here, the dimeric building blocks of the SinR tetramer, which are linked by the strong association of the C-terminal domains, face opposite sides with respect to a single DNA molecule. The result of this association is that high affinity binding to inverted repeats is a property of the SinR tetramer and not of the dimer. The superposition of four copies of the SinR-SinI complex onto this tetramer explains how derepression is achieved; severe steric clashes between the interaction helices prevent SinR from forming the side by side association required for high affinity DNA binding. The assembly of the SinR tetramer, the nature of the SinR-SlrR complex (and its binding to operator sequences), and the roles that the SlrR-SinI and SlrR-SlrA complexes play in the epigenetic switch will be the focus of further research.
Acknowledgments
We thank Harry J. Gilbert for critical reading of the manuscript, Tony Wilkinson for sharing unpublished observations, the Diamond light source for access to synchrotron radiation, and the Diamond beamline scientists and Arnaud Baslé for help during diffraction data collection.
This work was supported by a Biotechnology and Biological Sciences Research Council grant (to R. J. L.) as part of the BaCell SysMO2 consortium.
This article was selected as a Paper of the Week.
The atomic coordinates and structure factors (code 3zkc) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HTH
- helix-turn-helix
- ITC
- isothermal titration calorimetry
- SPR
- surface plasmon resonance.
REFERENCES
- 1. Davey M. E., O'toole G. A. (2000) Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 64, 847–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Toole G., Kaplan H. B., Kolter R. (2000) Biofilm formation as microbial development. Annu. Rev. Microbiol. 54, 49–79 [DOI] [PubMed] [Google Scholar]
- 3. Sutherland I. W. (2001) The biofilm matrix–an immobilized but dynamic microbial environment. Trends Microbiol. 9, 222–227 [DOI] [PubMed] [Google Scholar]
- 4. Whitchurch C. B., Tolker-Nielsen T., Ragas P. C., Mattick J. S. (2002) Extracellular DNA required for bacterial biofilm formation. Science 295, 1487. [DOI] [PubMed] [Google Scholar]
- 5. Branda S. S., González-Pastor J. E., Ben-Yehuda S., Losick R., Kolter R. (2001) Fruiting body formation by Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 98, 11621–11626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Margot P., Lazarevic V., Karamata D. (1996) Effect of the SinR protein on the expression of the Bacillus subtilis 168 lytABC operon. Microb. Drug Resist. 2, 119–121 [DOI] [PubMed] [Google Scholar]
- 7. Kearns D. B., Chu F., Branda S. S., Kolter R., Losick R. (2005) A master regulator for biofilm formation by Bacillus subtilis. Mol. Microbiol. 55, 739–749 [DOI] [PubMed] [Google Scholar]
- 8. Chu F., Kearns D. B., Branda S. S., Kolter R., Losick R. (2006) Targets of the master regulator of biofilm formation in Bacillus subtilis. Mol. Microbiol. 59, 1216–1228 [DOI] [PubMed] [Google Scholar]
- 9. Chai Y., Kolter R., Losick R. (2009) Paralogous antirepressors acting on the master regulator for biofilm formation in Bacillus subtilis. Mol. Microbiol. 74, 876–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Branda S. S., Chu F., Kearns D. B., Losick R., Kolter R. (2006) A major protein component of the Bacillus subtilis biofilm matrix. Mol. Microbiol. 59, 1229–1238 [DOI] [PubMed] [Google Scholar]
- 11. Colledge V. L., Fogg M. J., Levdikov V. M., Leech A., Dodson E. J., Wilkinson A. J. (2011) Structure and organisation of SinR, the master regulator of biofilm formation in Bacillus subtilis. J. Mol. Biol. 411, 597–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bai U., Mandic-Mulec I., Smith I. (1993) SinI modulates the activity of SinR, a developmental switch protein of Bacillus subtilis, by protein-protein interaction. Genes Dev. 7, 139–148 [DOI] [PubMed] [Google Scholar]
- 13. Lewis R. J., Brannigan J. A., Smith I., Wilkinson A. J. (1996) Crystallisation of the Bacillus subtilis sporulation inhibitor SinR, complexed with its antagonist, SinI. FEBS Lett. 378, 98–100 [DOI] [PubMed] [Google Scholar]
- 14. Lewis R. J., Brannigan J. A., Offen W. A., Smith I., Wilkinson A. J. (1998) An evolutionary link between sporulation and prophage induction in the structure of a repressor:anti-repressor complex. J. Mol. Biol. 283, 907–912 [DOI] [PubMed] [Google Scholar]
- 15. Gaur N. K., Cabane K., Smith I. (1988) Structure and expression of the Bacillus subtilis sin operon. J. Bacteriol. 170, 1046–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamon M. A., Lazazzera B. A. (2001) The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol. Microbiol. 42, 1199–1209 [DOI] [PubMed] [Google Scholar]
- 17. Chai Y., Chu F., Kolter R., Losick R. (2008) Bistability and biofilm formation in Bacillus subtilis. Mol. Microbiol. 67, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chu F., Kearns D. B., McLoon A., Chai Y., Kolter R., Losick R. (2008) A novel regulatory protein governing biofilm formation in Bacillus subtilis. Mol. Microbiol. 68, 1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kobayashi K. (2008) SlrR/SlrA controls the initiation of biofilm formation in Bacillus subtilis. Mol. Microbiol. 69, 1399–1410 [DOI] [PubMed] [Google Scholar]
- 20. Chai Y., Norman T., Kolter R., Losick R. (2010) An epigenetic switch governing daughter cell separation in Bacillus subtilis. Genes Dev. 24, 754–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Scott D. J., Leejeerajumnean S., Brannigan J. A., Lewis R. J., Wilkinson A. J., Hoggett J. G. (1999) Quaternary re-arrangement analysed by spectral enhancement: the interaction of a sporulation repressor with its antagonist. J. Mol. Biol. 293, 997–1004 [DOI] [PubMed] [Google Scholar]
- 22. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mondragón A., Harrison S. C. (1991) The phage 434 Cro/OR1 complex at 2.5 Å resolution. J. Mol. Biol. 219, 321–334 [DOI] [PubMed] [Google Scholar]
- 24. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harrison S. C., Aggarwal A. K. (1990) DNA recognition by proteins with the helix-turn-helix motif. Annu. Rev. Biochem. 59, 933–969 [DOI] [PubMed] [Google Scholar]
- 27. Otwinowski Z., Schevitz R. W., Zhang R. G., Lawson C. L., Joachimiak A., Marmorstein R. Q., Luisi B. F., Sigler P. B. (1988) Crystal structure of trp repressor/operator complex at atomic resolution. Nature 335, 321–329 [DOI] [PubMed] [Google Scholar]
- 28. Rohs R., Jin X., West S. M., Joshi R., Honig B., Mann R. S. (2010) Origins of specificity in protein-DNA recognition. Annu. Rev. Biochem. 79, 233–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rohs R., West S. M., Sosinsky A., Liu P., Mann R. S., Honig B. (2009) The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chai Y., Kolter R., Losick R. (2009) A widely conserved gene cluster required for lactate utilization in Bacillus subtilis and its involvement in biofilm formation. J. Bacteriol. 191, 2423–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lu X.-J., Olson W. K. (2008) 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualisation of three-dimensional nucleic acid structures. Nat. Protoc. 3, 1213–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]