

Background: SK2 channels modulate NMDA-dependent neuronal excitability and provide neuroprotection against excitotoxicity.
Results: We identify mitoSK2/KCa2.2 channels in neuronal mitochondria and demonstrate their protective function in cells lacking NMDAR.
Conclusion: SK2 channels prevent mitochondrial dysfunction and completely restore cell viability independently of NMDAR modulation.
Significance: Understanding how mitochondrial SK2 channels operate is crucial to develop novel therapeutic strategies for diseases caused by mitochondrial demise.
Keywords: Calcium, Mitochondrial Apoptosis, Neuroprotection, Patch Clamp Electrophysiology, Potassium Channels, CyPPA, SK2 Channels
Abstract
Small conductance calcium-activated potassium (SK2/KCa2.2) channels are known to be located in the neuronal plasma membrane where they provide feedback control of NMDA receptor activity. Here, we provide evidence that SK2 channels are also located in the inner mitochondrial membrane of neuronal mitochondria. Patch clamp recordings in isolated mitoplasts suggest insertion into the inner mitochondrial membrane with the C and N termini facing the intermembrane space. Activation of SK channels increased mitochondrial K+ currents, whereas channel inhibition attenuated these currents. In a model of glutamate toxicity, activation of SK2 channels attenuated the loss of the mitochondrial transmembrane potential, blocked mitochondrial fission, prevented the release of proapoptotic mitochondrial proteins, and reduced cell death. Neuroprotection was blocked by specific SK2 inhibitory peptides and siRNA targeting SK2 channels. Activation of mitochondrial SK2 channels may therefore represent promising targets for neuroprotective strategies in conditions of mitochondrial dysfunction.
Introduction
Potassium channels are highly diverse transmembrane proteins with multiple functions in the physiology of excitable cells. Respective dysfunctions have been linked to degeneration of neurons in various neurological diseases (1); e.g. ATP-sensitive potassium (KATP)3 channels, large conductance Ca2+-regulated (BKCa) channels, and voltage-dependent potassium (Kv1.3) channels have been suggested to be involved in Alzheimer and Parkinson diseases (2–4). Despite the detailed knowledge about these members of the potassium channel family, relatively little is known about the pathophysiology correlated with another family member, i.e. small conductance calcium-activated potassium (KCNN/SK/KCa2) channels. In neurons, SK channels are closely associated with synaptic NMDA receptors (NMDARs) in the plasma membrane of dendritic spines where they control excitability by reducing the amplitude of evoked synaptic potentials after NMDAR stimulation (5, 6). At the sites of increased NMDAR-mediated Ca2+ influx, SK channels mediate afterhyperpolarization (7), thereby providing feedback control of NMDA receptor activity (5) and excitotoxic increases in [Ca2+]i (8). Recent findings demonstrated potential protective effects of SK channel activation in models of NMDAR-mediated glutamate toxicity in cultured neurons and in a mouse model of cerebral ischemia (8, 9); however, their specific mode of action remains unclear.
In addition to their well documented expression at the plasma membrane, several members of the potassium channel family have also been found at the inner mitochondrial membrane, including mitoKv1.3 channels, mitoKATP channels, mitoBKCa channels, intermediate conductance Ca2+-regulated (mitoIKCa) channels, and two-pore domain (mitoTASK-3) channels (10–13). In particular, mitoKATP, mitoBKCa, and mitoKv1.3 channels have been proposed as potential targets of neuroprotective approaches (14, 15). Under pathological conditions, enhanced cytosolic Ca2+ concentrations result in loss of mitochondrial membrane potential (ΔΨm), decreased ATP levels, formation of reactive oxygen species (ROS), and failure of the mitochondrial Ca2+ retention capacity. Loss of ΔΨm was also accompanied by opening of the permeability transition pore, release of mitochondrial proapoptotic proteins, and ultimately activation of cellular death pathways (16). Our recent studies demonstrated a promising potential of SK channel activators in paradigms of neuronal death induced by excitotoxic stimuli in vitro and in a model of cerebral ischemia in vivo (8, 9). In these model systems, neuronal death occurs through intrinsic pathways of programmed cell death mediated by glutathione depletion, activation of 12-lipoxygenase, accumulation of intracellular peroxides, loss of ΔΨm, mitochondrial fragmentation, and release of apoptosis inducing-factor (AIF) to the nucleus (17–21). Here, we sought to investigate whether protective effects of SK channel activation are mediated at the level of mitochondria independently of effects on NMDAR-mediated Ca2+ influx at the plasma membrane. This study provides insights into the expression and the physiological function of SK.2 channels in neuronal mitochondria and their potential role as therapeutic targets in neurological diseases where mitochondrial damage and associated intrinsic pathways of neuronal cell death play an important role in the underlying pathology.
EXPERIMENTAL PROCEDURES
Neuronal HT-22 Cells
HT-22 hippocampus-derived cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing calcium (Invitrogen) or in DMEM without calcium. Both culture media were supplemented with 10% fetal calf serum, 100 units ml−1 penicillin, 100 μg ml−1 streptomycin, and 2 mm glutamine. The compounds EDTA, iberiotoxin, apamin, and cell-permeable N-cyclohexyl-N-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-4-pyrimidinamine (CyPPA), NS8593, diazoxide, 5-hydroxydecanoate, and glibenclamide were obtained from Sigma.
Mitoplast Preparation
Neuronal HT-22 cells collected from at least three T 75-cm2 flasks in the isolation buffer (220 mm mannitol, 70 mm sucrose, and 2 mm HEPES adjusted with Tris to pH 7.4 and containing 0.5 mg/ml bovine serum albumin) and homogenized with a Dounce glass homogenizer. The mitochondrial isolation was performed as described previously (22, 23). In short, the homogenate was centrifuged twice at 420 × g for 1 min, and the supernatants were layered on top of 5 ml of 0.5 m sucrose and centrifuged for 10 min at 410 × g. The top layer, which contained smaller mitochondria and other membranes, was discarded, and the pellet was gently resuspended into the 0.5 m sucrose (lower) layer. The sample was diluted to 0.3 m sucrose with glass-distilled water and pelleted at 750 × g for 5 min. The pellet was resuspended in 5 ml of isolation buffer, layered on 0.5 m sucrose, and centrifuged at 240 × g for 3 min. The top layer was collected, and the giant mitochondria were pelleted at 720 × g for 5 min in 0.25 m sucrose. The mitoplast preparation was performed as described previously (24).
Patch Clamp Recordings of Mitoplasts
Patch clamp recordings were performed using the bath and pipette solutions described previously (22). Isolated mitoplasts were placed in 35-mm dishes (Corning) filled with a bath solution containing 150 mm KCl, 0.1 mm CaCl2 (or 1 μm CaCl2), and 20 mm HEPES, pH 7.2 with KOH. Pipettes had a tip resistance of 4.0–7.0 megaohms when filled with the pipette solution containing 150 mm KCl, 0.1 mm CaCl2 (or 1 μm CaCl2), and 20 mm HEPES, pH 7.2 with KOH. After 15 min of settling, patch clamp recordings were performed at room temperature (21–22 °C) using an EPC-10 (HEKA) amplifier. For data acquisition, Patchmaster software (HEKA) was used, and data were analyzed with Fitmaster (HEKA). Electrophysiological data are reported as mean ± S.E. (n = number of cells; n = 4). Statistical differences were evaluated using paired Student's t tests. Significance (indicated by asterisks) was assumed for p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***). Drugs were freshly prepared from DMSO stocks for each measurement, and the final DMSO concentration did not exceed 0.1%.
xCELLigence Impedance-based System
HT-22 cells were seeded at a density of 8,000–10,000 cells/well in a 96-well E-plate (Roche Diagnostics GmbH). The impedance, depicted as cell index, was used to monitor the real time kinetics of cellular growth and alteration of cell morphology. Twenty-four hours after seeding, the cells were treated with different modulators of SK channels in the presence or absence of toxic concentrations of glutamate. The optimal glutamate concentration was chosen based on our previous results established by the xCELLigence system (25). Quantification of cell viability was performed by MTT reduction assay (at 0.5 mg ml−1) performed after 14–16-h treatment. The absorbance of each well was determined with an automated FLUOstar Optima reader (BMG Labtechnologies GmbH, Inc., Offenburg, Germany).
Cellular Compartment Fractionation
HT-22 cells were lysed in 250 mm sucrose, 20 mm HEPES, 3 mm EDTA, Complete Mini protease inhibitor mixture tablet, and phosphatase inhibitor mixtures 1 and 2 (Sigma-Aldrich). Cell lysates, crude mitochondria, and mitoplasts were prepared from cultured HT-22 cells as described previously (26, 27). Plasma membrane fractionation was performed as described previously (28). Immunoblotting was performed with the following commercially available antibodies: anti-Hsp60 (Abcam ab3080), anti-cytochrome c (MitoSciences MSA06), anti-ATP5A (MitoSciences MS507), anti-porin (MitoSciences MSA03), and anti-actin (Abcam ab8227). Alkaline carbonate extraction from crude mitochondria and proteinase K digestion of mitoplasts were performed as described previously (25, 26).
Measurements of Lipid Peroxidation with BODIPY Assay
For detection of cellular lipid peroxidation, cells were loaded with 2 μm BODIPY 581/591 C11 (Invitrogen). Flow cytometry was performed using a FACScan FACSCalibur flow cytometer (BD Biosciences). Data were collected from at least 10,000 cells (n = 3).
Measurement of ROS Production
Total cellular ROS generation was determined using the cell-permeable dye CM-H2DCFDA (Invitrogen). Treatment with 3 mm glutamate and 25 μm CyPPA was performed simultaneously at the indicated time points. After 6 or 8 h, cells were incubated with 2 μm CM-H2DCFDA. Fluorescence was detected at 520 nm using a FACSCalibur flow cytometer (BD Biosciences). Data from three independent experiments were collected (10,000 cells/sample).
Measurement of Mitochondrial Superoxide Production
MitoSOX (Invitrogen) was applied to neuronal cells according to the manufacturer's protocol. MitoSOX fluorescence was analyzed using a FACS-Guava easyCyteTM HT System (easyCyte 6-2L, Merck Millipore, Merck KGaA). Data were collected from at least 10,000 cells (n = 3). Measurements were performed in triplicates and are representative of at least three independent experiments.
Measurement of Mitochondrial Calcium
Mitochondrial calcium was determined using the cell-permeable dye dihydrorhodamine-2 AM (Invitrogen). Treatment with 3 mm glutamate and 25 μm CyPPA was performed simultaneously at the indicated time points. After 13 h, cells were then preloaded with 2 μm dihydrorhodamine-2 AM (prepared by reduction of rhodamine-2 AM following the manufacturer's protocol). Fluorescence was detected using a FACS-Guava easyCyte HT System (easyCyte 6-2L, Merck Millipore, Merck KGaA). Data from three independent experiments were collected (10,000 cells/sample).
Inhibitory Peptide and siRNA Transfection
Lipofectamine 2000 (Invitrogen) at a concentration of 1.5 μl ml−1 and mitoGFP (1–2 μg) were incubated separately in Opti-MEM I (Invitrogen) for 10 min. Afterward, they were gently mixed and incubated for 20 min at room temperature. The mitoGFP vector was kindly provided by Andreas Reichert (Frankfurt, Germany). Cellular loading of peptides was performed with the cationic lipid mixture Pro-JectTM Protein Transfection Reagent kit according to the manufacturer's instructions (Pierce 89850). The peptide sequence is NH2-ELQAQQEELEARLAALESR-acid (for SK1 channels), NH2-DLNERSEDFEKRIVTLETK-acid (for SK2 channels), and NH2-ELNDRSEDLEKQIGSLESK-acid (for SK3 channels). Experiments using siRNA specific for SK2 channels (NCBI RefSeq accession number NM_080465) were performed with SMARTpool ON-TARGETplus Kcnn2 siRNA according to the manufacturer's instructions (Dharmacon, Thermo Fisher Scientific, Inc., Waltham, MA). In brief, HT-22 cells were transfected with 50 nm SK2 channel siRNA or with two different concentrations of peptides (29) (50 and 75 μm) in 24-well plates (60,000 cells/well) for 24 h. Afterward, cells were seeded in 96-well plates, and after another 24 h, HT-22 cells were treated with glutamate (3 mm). The evaluation of the impact of these peptides was carried out by MTT assays and using the xCELLigence technique. Prediction of coiled coil domains of the C terminus of SK channels was determined using the Coils Version 2.2 program. Characterization of peptide specificity for SK channels were performed according to Tuteja et al. (29).
Analysis of Mitochondrial Morphology
Cells were transfected with mitoGFP (as described above) or labeled with MitoTracker DeepRed according to the manufacturer's protocol (Invitrogen). Cells were treated with glutamate in the presence or absence of CyPPA and EDTA for 14–15 h followed by fixation with 4% paraformaldehyde and DAPI counterstaining for nuclei. We distinguished three categories of mitochondria based on their morphology: category I, long, tubular mitochondria; category II, short tubules or large round organelles; category III, small fragmented mitochondria (17). Images were collected with a DMI6000B fluorescence microscope equipped with a DCF360FX camera (Leica, Wetzlar, Germany).
Mitochondrial Transmembrane Potential Measurements (TMRE Assay)
Mitochondrial membrane potential was determined by TMRE fluorescent dye. Cells were stained with 200 nm TMRE (Invitrogen) according to the manufacturer's protocol. As a positive control for mitochondrial membrane potential loss, cells were treated for 20 min with m-chlorophenylhydrazone. TMRE fluorescence was analyzed using a FACS-Guava easyCyte HT System (easyCyte 6-2L, Merck Millipore, Merck KGaA). Data were collected from at least 10,000 cells (n = 3). Measurements were performed in triplicates and are representative of at least three independent experiments.
RT-PCR Analysis
Total RNA was extracted using the NucleoSpin RNA II kit (Macherey-Nagel GmbH and Co. KG, Düren, Germany) following the manufacturer's instructions. RT reactions were conducted using SuperScript® III One-Step RT-PCR System (Invitrogen) in a SensoQuest Labcycler (SensoQuest Biomedizinische Elektronik GmbH, Göttingen, Germany). The following primers synthesized by Eurofins MWG Operon (Ebersberg, Germany) were used: KCNN1/SK1/KCa2.1 (240 bp), 5′-CTGTGGGAAGGGCGTGTGTCTG-3′ and 5′-CCGAACCCGGCTTTGGTCTGG-3′; KCNN2/SK2/KCa2.2 (220 bp), 5′-GTGCTCTTGGTTTTAGTATCTCG-3′ and 5′-CAACCTGCACCCATTATTCC-3′; KCNN3/SK3/KCa2.3 (400 bp), 5′-GCCAACAAGCGGAAAAACCAAAAC-3′ and 5′-CCAGGCGTGCCGTCCAGAAGAAC-3′. cDNAse digestion and RT-PCR without reverse transcriptase in the reaction mixture were used as negative controls for genomic DNA contamination. The following amplifications of cDNA by PCR using specific primers were carried at the following steps: 1) denaturing at 95 °C for 4 min, 2) 94 °C for 30 s, 3) Tm (annealing temperature) for 30 s depending on the SK isoform of interest, and 4) extension at 72 °C for 30 s with 30 cycles from steps 2 to 4. The final extension step was set to 72 °C for 5 min. The Tm was 63 °C for SK1, 57.3 °C for SK2, and 61 °C for SK3.
Protein Analysis
Neuronal cells were lysed in 20 mm Tris, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Triton X-100, pH 7.4, Complete Mini protease inhibitor mixture tablet, and phosphatase inhibitor mixtures 1 and 2 (Sigma-Aldrich). The membranes were incubated overnight with primary antibodies at 1:500 dilution for rabbit anti-SK1 channel (Sigma-Aldrich P9372) and 1:3,000 dilution for rabbit anti-SK2 channel (molecular mass, 64 kDa) (30), Hsp60 (molecular mass, 60 kDa), ATP5A (inner mitochondrial membrane) (molecular mass, 60 kDa), cytochrome c (mitochondrial intermembrane space) (molecular mass, 15 kDa), actin (cytosol) (molecular mass, 43 kDa), and porin (integral inner membrane protein) (molecular mass, 31 kDa) at 4 °C and afterward with peroxidase-conjugated secondary antibodies at a 1:2,500 dilution.
Immunocytochemistry
Cells were fixed using 4% paraformaldehyde and permeabilized using 0.04% Triton X-100. Incubation with primary antibody against SK2 channels (30) at a concentration of 1:100 was conducted overnight at 4 °C followed by secondary anti-rabbit antibodies coupled to Alexa Fluor® 488 (Invitrogen). Images were acquired using a confocal laser-scanning microscope (Axiovert 200, Carl Zeiss, Jena, Germany). Light was collected through a 63× 1.4 numerical aperture or 100× 1.3 numerical aperture oil immersion objectives.
Statistical Analysis
All data are given as means ± S.D. For statistical comparisons between two groups, Student's t test was used; multiple comparisons were performed by ANOVA followed by Scheffé's post hoc test. Calculations were made with the Winstat standard statistical software package (Robert Fitch Software, Bad Krozingen, Germany). A statistically significant difference was assumed at p < 0.05 (*), p < 0.01 (**), or p < 0.001 (***).
RESULTS
Localization of SK2 Channels in the Inner Mitochondrial Membrane
Two independent computational methods (TargetP 1.1 (31) and Mitoprot II) support a mitochondrial targeting sequence only in a subgroup of SK channels, namely SK2 (NCBI RefSeq accession number NP_536713).
Immunofluorescence staining specific for SK2 channels showed extensive overlap between SK2 channel and mitochondrial staining in neuronal HT-22 cells (Fig. 1A). Isolation of subcellular compartments revealed an enrichment of SK2 channels from whole-cell extracts to crude mitochondria and mitoplasts, suggesting that these channels reside in the inner mitochondrial membrane (Fig. 1B). Furthermore, alkaline carbonate extraction of crude mitochondria (Fig. 1C) and proteinase K treatment (Fig. 1D) of mitoplasts suggested that SK2 channels span the mitochondrial inner membrane. The mitochondrial SK2 (mitoSK2) channels were still detected after protease digestion of membrane preparations, whereas they were digested by proteinase K after elution from the membrane by detergents (Fig. 1D). Together with the computational prediction of a mitochondrial targeting sequence, these data strongly point toward the localization of SK2 channels at the inner mitochondrial membrane.
FIGURE 1.
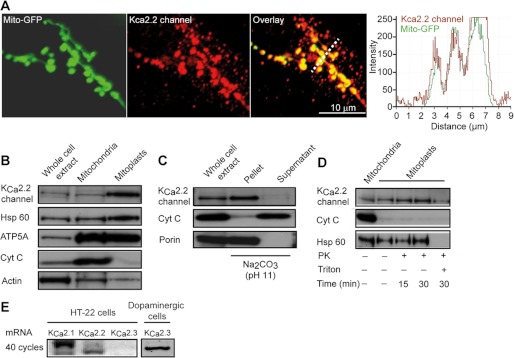
SK2 channels are located at the inner mitochondrial membrane of neuronal HT-22 cells. A, immunofluorescence staining of SK2 channels in HT-22 cells expressing mitochondrially targeted GFP. B, immunoblot analysis of whole-cell extract, cytosol supernatant, or crude mitochondrial pellets using antibodies against SK2 and control antibodies for protein location (Hsp60, mitochondrial matrix; ATP5A, inner mitochondrial membrane; cytochrome c (Cyt C), mitochondrial intermembrane space; and actin, cytosol). C, immunoblot analysis of soluble (supernatant) and insoluble (pellet) fractions following alkaline carbonate extraction of mitochondrial fractions with antibodies against porin (integral inner membrane protein) and cytochrome c (soluble intermembrane space protein). D, immunoblot analysis of crude mitochondria and mitoplasts and after proteinase K (PK) treatment. E, mRNA analysis of SK channel subtypes. As a positive control, we show the expression of SK3/KCa2.3 channel mRNA in dopaminergic cells.
Functional Characterization of SK2 Channels in the Inner Mitochondrial Membrane
To ultimately prove the localization of functional SK2 channels in the inner mitochondrial membrane, we performed patch clamp experiments of mitoplasts according to previously described protocols (22, 24). To isolate SK2 channel currents, we used the specific SK2/SK3 channel activator CyPPA (32) and the SK channel antagonists NS8593 (33) and apamin (34). These drugs can be used to characterize SK2 channel functions in mitoplasts because (i) SK1 and SK3 channels were not detected in HT-22 mitochondria (Fig. 1E), and (ii) BK and SK4/IK channels expressed in mitochondria are not affected by the pharmacological modulators in the applied concentration range (32, 33, 35). Whole-cell recordings were performed applying a voltage ramp protocol (Fig. 2A). The mean capacity of the mitoplasts was 4.9 ± 1.3 pF (n = 8). Whole-cell measurements yielded outward rectifying currents (Fig. 2B) despite the symmetrical potassium distribution as described previously for measurements of mitoplast potassium currents (22). Application of 25 μm CyPPA, however, did not result in a significant increase of inward or outward currents (Fig. 2, B and C). This finding is in contrast to the expected increase in SK currents by CyPPA, which should significantly increase the Ca2+ sensitivity of the channels (32). However, because the recordings were performed in the presence of high Ca2+ concentrations (0.1 mm) similar to those described previously (23), the SK2 channels might have been already fully activated, preventing an additional current increase by CyPPA. In fact, the half-maximal Ca2+ sensitivity of SK2 is about 0.5 μm (7, 35, 36); therefore, we next used solutions with only 1 μm Ca2+. Under these conditions, when SK2 channels were not fully activated, 10 μm CyPPA caused a significant current increase (Fig. 2, D–G). Fig. 2D illustrates the time course of current augmentation by CyPPA. Note that the current-voltage relationship recorded with a reduced Ca2+ concentration was less outwardly rectifying (Fig. 2, B and E), which indicates a reduction of the outward current component when Ca2+ is decreased. Consistently, CyPPA primarily enhanced outward currents (Fig. 2, E–G). Outward conductance was increased 1.63 ± 0.08-fold, whereas the inward conductance increased only 1.19 ± 0.14-fold (n = 4) (Fig. 2G). The mean current densities before and after application of 10 μm CyPPA (n = 4) are illustrated in Fig. 2E. The difference current (before and after drug application) illustrates the outwardly rectifying CyPPA-induced current of mitoplasts (Fig. 2F). The CyPPA-induced current had an amplitude of 26.7 pA/pF at +100 mV and an inward amplitude of −7.7 pA/pF at −100 mV (n = 4) (Fig. 2F). In symmetrical potassium concentration, macroscopic SK2 channel currents have a strong inward rectification (33, 37). Thus, according to our patch clamp recordings, SK2 channels should be orientated in the inner mitochondrial membrane with the outer vestibule of the channel facing the inner matrix. This orientation would explain the outward rectification in the mitoplast whole-cell recordings. A similar orientation is known for mitoKATP channels (38). To verify the orientation of the channel with N and C termini of the SK2 channel facing the intermembrane compartment, we applied apamin, which blocks SK2 by binding to the outer vestibule (34). At high concentrations, apamin did not result in a rapid block of currents (Fig. 2H, upper panel), suggesting that the outer vestibule of the channel is facing the inner matrix and thus is not rapidly accessible to the toxin. In contrast, the SK channel blocker NS8593 (33), which binds to the C terminus facing the inter membrane compartment and here the bath solution, caused a rapid block of currents (Fig. 2H, lower panel). Thus, our data indicate that the outer vestibule of SK2 channels is facing into the inner matrix of the mitochondria. We further investigated the size of SK2 currents present without activation by CyPPA using the SK channel blocker NS8593. Similarly to CyPPA, NS8593 primarily affected the outward currents of mitoplasts (Fig. 2, I–K). 10 μm NS8593 decreased outward currents by a factor of 0.48 ± 0.10 (n = 4), and inward currents were decreased by a factor of 0.77 ± 0.09 (n = 4) (Fig. 2K). The mean current densities before and after application of 10 μm NS8593 (n = 4) are illustrated in Fig. 2I. The difference current (before and after drug application) illustrates the outwardly rectifying NS8593-sensitive SK2 current present in mitoplast (Fig. 2J). The NS8593-sensitive current had an amplitude of 36.1 ± 9.5 pA/pF at +100 mV and an inward amplitude of −5.4 ± 2.5 pA/pF at −100 mV (n = 4) (Fig. 2J). Accordingly, our data indicate that SK2 channels are located and functionally active in the inner mitochondrial membrane. The patch clamp recordings further suggested an “inverse” orientation of the mitoSK2 channels with the N and C termini directed into the mitochondrial intermembrane space, thereby mediating K+ flux into the mitochondrial matrix upon activation by CyPPA under physiological conditions, i.e. at a ΔΨm of about −150 mV.
FIGURE 2.

Patch clamp recordings of mitoSK2 channels in mitoplast. A, mitochondria were stained with mitochondrial MitoTracker Deep Red. A photomicrograph of a mitoplast during a whole-cell patch clamp recording with remnants of the outer matrix visible at one side of the mitoplast is shown. (The black arrow indicates the inner mitochondrial membrane, and the blue arrow indicates membrane fragments. Scale bar, 5 μm). The illustrated voltage ramp protocol was used for all patch clamp recordings. B, representative whole-cell recording of a mitoplast using symmetrical bath and pipette solutions with high Ca2+ concentrations (100 μm) before and after application of 25 μm CyPPA. C, changes of relative currents analyzed at +100 and −100 mV by 25 μm CyPPA (n = 3). D, wash-in and current activation by 10 μm CyPPA using symmetrical bath and pipette solutions with 1 μm Ca2+. Currents were analyzed at the end of the voltage ramp (+100 mV), which was repeated every 5 s. E, average currents of mitoplasts analyzed before (n = 4) and after application of 10 μm CyPPA (n = 4). The S.E. is illustrated every 2 mV. F, difference current from E illustrates the CyPPA-induced current. G, relative currents from E analyzed at +100 and −100 mV. H, wash-in of 5 μm apamin or 10 μm NS8593 using symmetrical bath and pipette solutions with 1 μm Ca2+. I, average currents of mitoplasts analyzed before (n = 4) and after application of 10 μm NS8593 (n = 4). J, difference current from I illustrates the NS8593-sensitive current. K, relative currents from I analyzed at +100 and −100 mV. n.s., not significant; ***, p < 0.001. Error bars represent S.D.
Protective Role of SK2 Channels in Mitochondria
After characterizing functional SK2 channel expression in mitochondria, we next examined the effects of mitoSK2 channel modulation on mitochondrial integrity. Under standard culture conditions, mitochondria appeared predominantly tubular in control cells, whereas the organelles were increasingly fragmented after glutamate treatment; this was attenuated in cells treated with CyPPA (Fig. 3, A and B). Notably, the proposed CyPPA-mediated combined activation of mitoSK2 channels and SK2 channels at the plasma membrane overall provided stronger protection than inhibition of Ca2+ influx through the plasma membrane by the Ca2+ chelator EDTA, which only partly prevented glutamate-induced mitochondrial fragmentation (Fig. 3B). These findings suggest that opening of SK2 channels mediates effects beyond inhibition of Ca2+ influx from the extracellular space and that the plasma membrane SK2 channels are not the main mediators of the mitochondrial integrity protection.
FIGURE 3.

Activation of SK2 channels preserves mitochondrial integrity and ΔΨm. A, changes of mitochondrial morphology after 14–16 h of glutamate (3 mm) exposure of HT-22 cells visualized using MitoTracker Red. B, mitochondrial morphology was classified into three categories of fragmentation (category I, tubulin-like; category II, large fragmented structures; category III, small fragmented structures; 500 cells/condition, n = 4). C, mitochondrial membrane potential was measured using the fluorescent dye TMRE and FACS analysis. HT-22 cells treated with CyPPA (25 μm) were challenged for 12–13 h with glutamate. m-Chlorophenylhydrazone (CCCP) (50 μm) was used as positive control for loss of ΔΨm (**, p < 0.01 versus control non-treated neurons; ***, p < 0.001 versus control non-treated neurons; ###, p < 0.001 versus glutamate; ANOVA and Scheffé's test; n = 3). D, mitochondrially dependent peroxide formation was measured using the fluorescent dye MitoSOX and FACS analysis up to 13 h after glutamate exposure. E, lipid peroxidation was measured after BODIPY staining by FACS up to 12 h of glutamate exposure (10,000 cells/condition; n = 3; *, p < 0.05 versus non-treated neurons; ***, p < 0.01 versus non-treated neurons; #, p < 0.05 versus glutamate-treated neurons; ANOVA and Scheffé's test). F and G, soluble ROS were determined by CM-H2DCFDA and FACS analysis 6–8 h after onset of glutamate exposure (10,000 cells/sample). Error bars represent S.D. Ctrl, control; DCF, dichlorodihydrofluorescein.
Furthermore, glutamate caused a significant loss of ΔΨm within 10–12 h (17–19), whereas CyPPA prevented such glutamate-induced mitochondrial membrane depolarization (Fig. 3C). Interestingly, activation of SK2 channels alone induced a slight depolarization of ΔΨm. This observation was in line with a K+ influx via mitoSK2 channels at hyperpolarized ΔΨm as evident from the currents recorded from the inner mitochondrial membrane. Despite the orientation of the SK2 channel in the inner mitochondrial membrane and the resulting ability of the channels to primarily conduct K+ currents out of the mitoplasts (Fig. 2B), there is a resulting K+ influx under physiological conditions due to the very hyperpolarized ΔΨm of about −150 mV, creating a strong electrical driving force for a K+ influx. Thus, opening of the mitoSK2 channels by CyPPA under physiological conditions further increased K+ flow into the mitochondrial matrix, thereby decreasing ΔΨm. In contrast, under conditions of oxidative stress when ΔΨm has collapsed to values higher than the K+ Nernst potential, activation of mitoSK2 should result in a K+ efflux from mitochondria, thereby rescuing ΔΨm. This rehyperpolarization, however, will be most likely only incomplete and will not reach a ΔΨm of −150 mV as supported by our ΔΨm measurements (Fig. 3C).
In the present model of oxidative cell death in HT-22 cells, glutamate-induced ROS production occurs in two phases: a first triggering phase that is mainly dependent on glutathione depletion and enhanced lipoxygenase activity and a second phase with a pronounced increase in ROS levels that is dependent on Bid-mediated mitochondrial dysfunction that cannot be reversed by lipoxygenase inhibition (18). In line with these previous observations, we found a significant increase in mitochondrial ROS formation after glutamate treatment (Fig. 3D). Furthermore, CyPPA significantly reduced but did not fully block lipid peroxidation or soluble ROS production in HT-22 cells exposed to glutamate (Fig. 3, E, F, and G). These results support the conclusion that opening of mitochondrial SK2 channels prevents the second phase of ROS production that marks irreversible mitochondrial damage. Furthermore, mitochondrial calcium influx is highly increased in response to toxic concentrations of glutamate (data not shown). To confirm a functional contribution of mitochondrial SK2 channels to mitochondrial calcium homeostasis, we investigated the effect of SK2 channel activators in conditions of glutamate-induced increased mitochondrial calcium uptake. Indeed, SK2 channel opening prevented mitochondrial calcium overload and mitochondrial superoxide formation, suggesting a neuroprotective role of SK2 channel activator in models of mitochondrial dysfunction (data not shown).
Activation of SK2 Channels Protects HT-22 Cells against Glutamate Toxicity
Mitochondrial calcium overload has been implicated in the irreversible activation of cell death mechanisms. Accordingly, we investigated whether mitoKCa2 channel activation affected glutamate-induced cell death. As shown in Fig. 4, CyPPA provided robust and long lasting protection against glutamate toxicity as shown by MTT assays and live cell measurements of cellular impedance (Fig. 4, A, B, and C). Although the MTT assay measured the reductive capacity of viable cells, the impedance measurements indicated detachment of dying cells in the model of glutamate toxicity as validated earlier (25). The neuroprotective effect obtained with CyPPA was apparently attributed to the modulation of SK2 channels because SK3 channels were not expressed in HT-22 cells (Fig. 1E). Furthermore, only inhibitory peptides (29) (Fig. 4, D and E) or siRNA (Fig. 4F) targeting SK2 channels significantly attenuated the protective effect of CyPPA, whereas specific inhibition of SK1 or SK3 channels did not affect CyPPA-mediated protection (Fig. 4, G and H). These inhibitory peptides bind the coiled coil domain of SK channels, inhibiting the pore formation and thus the function of SK channels. The design of the inhibitory peptides was performed using the Coils Version 2.2 program, and the specificity for each SK channel subtype was validated previously in patch clamp experiments by Tuteja et al. (29). These results provide strong evidence for a cytoprotective role of SK2 channels at the pharmacological level and by specific gene silencing.
FIGURE 4.

Cytoprotective effects by SK2 channel activation in neuronal HT-22 cells. A, cell impedance was detected after incubation of HT-22 cells with CyPPA (1–50 μm) and glutamate (3 mm at 0 h; n = 8). B, xCELLigence real time impedance-based measurements of cells treated with CyPPA at different time points following glutamate application. The time point of treatment is marked as “0 h” in the graph (n = 8). C, MTT analysis of cells treated with CyPPA (25 μm) 0, 1, 3, 5, and 7 h after the application of glutamate. (*, p < 0.05; ***, p < 0.001 versus glutamate-treated neurons; ANOVA and Scheffé's test; n = 6). D and E, HT-22 cells were transfected with a 50 μm concentration of specific inhibitory peptides for SK2/KCa2.2 channels before exposure to CyPPA (5–10 μm) and glutamate (3 mm; n = 8). Cell impedance (D) and MTT assays (measured at 14 h after glutamate application) (E) show that only SK2/KCa2.2 inhibitory peptides reduce the protective effect of CyPPA (***, p < 0.001 versus CyPPA and glutamate treatment in non-transfected cells; ANOVA and Scheffé's test; n = 6). F, non-transfected and siRNA-transfected HT-22 cells targeting SK2/KCa2.2 channels were treated with CyPPA (5 μm) and challenged with glutamate (3 mm). Morphological alterations were detected by a real time impedance-based system. G, HT-22 cells were transfected with specific inhibitory peptides for SK1/KCa2.1 (G) and SK3/KCa2.3 (H) channels (50 μm). Afterward, transfected cells were treated with 5 μm CyPPA for SK1/KCa2.1 channels (G) and 10 μm CyPPA for SK3/KCa2.3 channels (H). Glutamate was applied together with CyPPA, and the cellular index was measured using the xCELLigence system (n = 8). Error bars represent S.D. inh, inhibitory; Ctrl, control.
We next attempted to differentiate between potential effects of SK2 channel activation on inhibition of Ca2+ influx at the level of the cell membrane and protective effects at the mitochondrial level. To this end, we evaluated the effects of CyPPA in the absence of extracellular Ca2+ or in the presence of millimolar concentrations of the Ca2+ chelator EDTA. The results from these experiments indicated that SK channel activation at the level of mitochondria significantly exceeded protection achieved by inhibition of Ca2+ influx from the extracellular space (Fig. 5, A and B). In fact, EDTA alone only partially rescued HT-22 cells from glutamate toxicity, whereas the cells were protected by 80% when CyPPA was added in addition to EDTA (Fig. 5). Moreover, glutamate was still able to promote cellular demise independently of extracellular calcium (Fig. 5B). Notably, activation of SK2 channels by CyPPA protected the cells from glutamate toxicity under both conditions in the presence and in the absence of extracellular calcium (Fig. 5B). These results support a mechanism of cytoprotection through mitoSK2 channel activation in neuronal cells that resulted in more pronounced protective effects than inhibition of calcium influx from the extracellular space alone.
FIGURE 5.
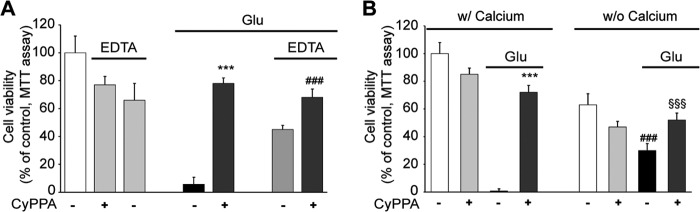
Activation of SK2 channels mediates neuroprotection in the absence of extracellular calcium. A, cells were treated with CyPPA and glutamate in the presence or absence of EDTA (1 mm) up to 15 h before MTT analysis (***, p < 0.001 versus glutamate-treated neurons; ###, p < 0.01 versus neurons treated with EDTA and glutamate; ANOVA and Scheffé's test; n = 6). B, cells were treated with CyPPA (25 μm) and glutamate (3 mm) in the presence (w/) or absence (w/o) of extracellular calcium for 16 h before MTT analysis (***, p < 0.001 versus glutamate-treated neurons; ###, p < 0.001 versus glutamate-treated neurons in calcium-containing medium; §§§, p < 0.001 versus glutamate-treated neurons in medium without calcium; ANOVA and Scheffé's test; n = 6). Error bars represent S.D.
SK2 Channel-mediated Neuroprotection Is Independent of KATP Channel Modulation
Diazoxide can activate potassium channels in both mitochondria and plasma membranes (10, 11). The cell viability MTT assay showed a dose-dependent neuroprotection in HT-22 cells treated with diazoxide with a maximal rescue against glutamate-induced cell demise at a concentration of 1 mm (Fig. 6A). However, diazoxide offered only a transient cell protection as measured by the real time impedance-based system (Fig. 6B). To further evaluate the protective potential of KATP channels, we performed real time impedance measurements of HT-22 cells exposed to glutamate and different concentrations of glibenclamide. Although this blocker of KATP channels did not promote cellular death when it was applied alone, it further increased the glutamate-induced cell death (Fig. 6C). Furthermore, the transient neuroprotective effect elicited by diazoxide was partially blocked by glibenclamide treatment, confirming that diazoxide-induced cell survival requires activation of KATP channels (Fig. 6D). In addition, glibenclamide was not able to abrogate the neuroprotective effect offered by CyPPA, further supporting a specific role for SK2 potassium channels in the neuroprotective effects of CyPPA (Fig. 6E).
FIGURE 6.

Activation of SK2 channels inhibits oxytosis independently of KATP channel pathways. Cells were treated with different concentrations of diazoxide (250 μm, 500 μm, and 1 mm) and stimulated with glutamate (3 mm). Diazoxide mediated neuroprotection in a dose-dependent manner as analyzed by an MTT assay (14 h following glutamate application) (A) and xCELLigence system (B). (*, p < 0.05; ***, p < 0.001 versus glutamate-treated neurons were considered to be significant; ANOVA and Scheffé's test; n = 8). C, xCELLigence analysis of HT-22 cells treated with glibenclamide (5–50 μm) in the presence or absence of glutamate (3 mm). The time point of treatment is marked as 0 h in the graph (n = 8). D, xCELLigence analysis of HT-22 cells treated with diazoxide (1 mm) with or without glibenclamide (20 μm) and challenged with glutamate (3 mm). E, xCELLigence analysis of HT-22 cells treated with CyPPA (25 μm) with or without glibenclamide (20 μm) and challenged with glutamate (3 mm). F, MTT assay of cells treated with CyPPA (25 μm) and 5-hydroxydecanoate (5HD) (500 μm) or glibenclamide (20 μm) and stimulated with glutamate (3 mm) (***, p < 0.001 versus glutamate-treated neurons were considered to be significant; ANOVA and Scheffé's test; n = 8). Error bars represent S.D. Ctrl, control.
To determine in further detail whether the mechanism whereby CyPPA reduces glutamate toxicity involves the mitoKATP as well as mitochondrial SK2 channels, we applied 5-hydroxydecanoate, an agent that specifically blocks mitoKATP channels but not plasma membrane KATP channels (39). Adding 5-hydroxydecanoate did not reduce neuroprotection against glutamate toxicity mediated by CyPPA in HT-22 cells (Fig. 6F). Overall, these results suggest that the activation of SK2 channels alone is sufficient for protection from glutamate toxicity, and these neuroprotective effects were not affected by inhibitors of KATP channels at the plasma membrane or in mitochondria.
To further investigate whether CyPPA neuroprotection is indeed mediated via SK2 channels and is not dependent on blocking BK channels as reported previously (32), we monitored cell death in the presence of a specific BK channel blocker, iberiotoxin. Impedance measurements revealed no protection against glutamate damage when different concentrations of iberiotoxin (40) (15–500 nm) were applied (data not shown). These experiments suggest that CyPPA neuroprotective properties are not associated with BK channel inhibition.
To further support the cytoprotective role of SK2 channels at the level of mitochondria, we induced mitochondrial damage by expression of tBid, which mimics the major step toward mitochondrial demise in the glutamate-induced death program in HT-22 cells (17, 18). Here, CyPPA partially prevented tBid-induced cell death (Fig. 7A), supporting the conclusion that the SK channel activator exerted protection at the level of the mitochondria. In contrast, EDTA failed to revert tBid-promoted cellular death, confirming that inhibition of calcium influx from the extracellular space was not sufficient to prevent mitochondrial demise and cell death triggered by tBid (Fig. 7A).
FIGURE 7.

Activation of SK2 channels prevents tBid toxicity and mitochondrial AIF release. A, HT-22 cells transfected with tBID for 24 h were treated with CyPPA (25 μm). MTT analysis was performed 13–15 h after glutamate application. The results shown represent mean ± S.D. (**, p < 0.01 versus tBid-treated neurons; ANOVA and Scheffé's test). B, AIF immunostainings show that upon glutamate damage mitochondrial AIF initially accumulates around the nucleus followed by its nuclear translocation. HT-22-cells were treated with CyPPA in the presence or absence of glutamate. C, cells were fixed and immunostained 12 h after the treatment, and nuclei were stained with DAPI. CyPPA treatment completely prevented translocation of AIF to the nucleus (scale bars, 20 μm). D, quantification of AIF translocation upon glutamate challenge in the presence and absence of CyPPA. Results are presented as percentage of total counted cells from three independent experiments using at least 300 counted cells/condition without the knowledge of treatment history. Error bars represent S.D. Ctrl, control.
Finally, we investigated the effects of CyPPA on mitochondrial translocation of the proapoptotic protein AIF to the nucleus (Fig. 7, B and C). Such AIF translocation is a result of the detrimental mitochondrial membrane permeabilization, and it is essential for cellular death in the present model system of glutamate-induced oxytosis (19) (Fig. 7B). Quantification of AIF translocation revealed that activation of SK2 channels prevented AIF translocation after exposure to glutamate, suggesting that SK2 channels located in mitochondria were critical for the regulation of intrinsic pathways of apoptosis that involve loss of ΔΨm, mitochondrial fragmentation, and release of mitochondrial AIF to the nucleus (Fig. 7, C and D).
DISCUSSION
The data of the present study suggest that SK2 channels mediate neuroprotective effects at the level of mitochondria, thereby extending the previously suggested mechanism of action of these channels at the plasma membrane. This is further supported by the fact that in the present model system using immortalized HT-22 neurons cell membrane NMDA receptors are not expressed (41, 42), and SK2 channel activation is neuroprotective even in the absence of extracellular Ca2+.
Recent findings on the role of SK channel activation by CyPPA in spinocerebellar ataxia type 2 point to an emerging new therapeutic strategy for neurodegenerative diseases (43–45). However, the molecular mechanisms underlying the pathology of neurodegeneration are not well understood.
Our study provides several mechanisms of action that could explain the proposed cytoprotective effects mediated by SK2 channel activation. These may include, for example, preconditioning effects mediated by a decrease of ΔΨm and associated increases in ROS production, which have been also suggested, as a possible mechanism of mitoKATP channel-mediated cytoprotection (10, 11). Here, pharmacological activation of SK2 channels induced slight ΔΨm depolarization and a small increase in mitochondrial ROS production, suggesting a direct regulatory function of SK2 channels in mitochondrial membrane potential that may be linked to a protective effect similar to KATP channel activities. However, in our model system, KATP channel activation provides only transient protection that is not comparable with the pronounced effect of CyPPA. Therefore, activation of mitoSK2 channels may mediate protective effects exceeding those of mitoKATP channel activation.
Other reports suggested that mitochondrial K+ channel activation might inhibit permeability transition pore formation as a potential neuroprotective mechanism (16). Formation of the permeability transition pore has been acknowledged as a possible common trigger for the release of proapoptotic factors and associated cell death pathways in response to cellular stress. For example, formation and mitochondrial translocation of proapoptotic tBid initiated permeability transition pore formation in mitochondria and coincided with a collapse of mitochondrial membrane potential and mitochondrial integrity (17–21). Here, we found that activation of SK2 channels prevented tBid-induced cell death by interfering with tBid toxicity at the level of mitochondria. Moreover, SK2 channel activation preserved ΔΨm, mitigated ROS production, and prevented mitochondrial fragmentation under conditions of cellular stress. Overall, these findings imply that mitoSK2 channel activation prevented mitochondrial membrane permeabilization, breakdown of ΔΨm, and subsequent release of mitochondrial proteins such as proapoptotic AIF.
Notably, breakdown of ΔΨm is a critical step in these mechanisms of intrinsic death pathways, and mitoSK2 channel activation can apparently stabilize ΔΨm particularly under conditions of cellular stress. The mitoSK2 channels might because of their rectification pattern exert a very large capacity to conduct outward currents, which may become particularly activated when ΔΨm decreases under conditions of oxidative stress to values more depolarized than the K+ Nernst potential. Under these conditions, the direction of physiological K+ flow could be reverted, increasing the K+ efflux from the mitochondrial matrix and thereby partially rescuing ΔΨm. Consequently, this may also prevent mitochondrial superoxide production, calcium overload, and osmotic swelling of the mitochondrial matrix, thereby blocking intrinsic pathways of mitochondrial demise and cell death (46) as demonstrated here in models of glutamate toxicity and tBid expression. The findings in the applied model systems of intrinsic programmed cell death exposed a protective role of SK2 channels at the level of mitochondria that exceeded the therapeutic potential of the established KATP channels.
The current findings are also highly relevant for protective effects of SK channel activators in primary neuronal cultures in vitro and in models of neuronal death in vivo. We have shown recently that activation of SK channels attenuated excitotoxic neuronal death in vitro (9), reduced brain damage in animal models of ischemic stroke (8), and prevented microglial activation (47, 48). Furthermore, it was demonstrated by others that activation of SK channels diminished CA1 hippocampal neuronal damage following global cerebral ischemia induced by cardiac arrest (49). A very recent study also linked SK channels to mitochondrial proteins, showing that activation of SK channels attenuated hyperexcitability in Phosphatase and tensin homolog-induced kinase 1 (PINK1)-deficient mice and in HtrA2/Omi-deficient mice (50). However, the protective effects in these model systems were attributed to SK channel activity at the level of the plasma membrane. Our data now show functional expression of SK2 channels at the inner mitochondrial membrane of neuronal cells, providing protective roles under conditions of cellular stress. In particular, we found that inhibition of Ca2+ influx through the plasma membrane only provided partial protection in models of glutamate-induced oxytosis and tBid toxicity (41, 42, 51) (Figs. 4, 5, and 6). Thus, activation of SK channels at the plasma membrane and the resultant inhibition of Ca2+ influx as mimicked by EDTA or Ca2+ withdrawal were not as sufficient as the preservation of mitochondrial integrity and function by CyPPA. In fact, pharmacological SK2 activation exceeded the effects of EDTA and Ca2+ withdrawal alone, supporting the conclusion that the protective effect was mediated at the level of mitochondria.
In summary, using pharmacological approaches and siRNA-mediated gene silencing, the present study demonstrates a protective role for mitochondrial SK2 channels on ΔΨm, mitochondrial integrity, and cell viability in neuronal cells. In particular, these mitoSK2 channels apparently exert increased activity under conditions of cellular stress and may therefore serve as therapeutic targets in neurological diseases where mitochondrial damage is prominent.
Acknowledgments
We thank Alexander Seiler (Roche Diagnostics GmbH) for providing support with the xCELLigence system, Hans Guenther-Knaus for kindly providing SK2 channel antibodies, Sassan Rafizadeh and Nipavan Chiamvimonvat for the specific inhibitory peptides of SK channels, and Emma Jane Esser for editing of the manuscript. We also thank Jürgen Daut for helpful comments on the study.
This work was supported in part by a grant from Alzheimer Forschung Initiative e.V.
- KATP
- ATP-sensitive potassium
- KCNN/SK/KCa2
- small conductance calcium-activated potassium
- BKCa
- large conductance Ca2+-regulated
- Kv1.3
- voltage-dependent potassium
- ΔΨm
- mitochondrial membrane potential
- ROS
- reactive oxygen species
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- AIF
- apoptosis-inducing-factor
- NF-κB
- nuclear factor-κB
- NMDAR
- NMDA receptor
- CyPPA
- N-cyclohexyl-N-[2-(3,5-dimethyl-pyrazol-1-yl)-6-methyl-4-pyrimidinamine
- CM-H2DCFDA
- 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester
- TMRE
- tetramethylrhodamine, ethyl ester
- ANOVA
- analysis of variance
- pF
- picofarad(s)
- tBid
- truncated Bid.
REFERENCES
- 1. Jentsch T. J. (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat. Rev. Neurosci. 1, 21–30 [DOI] [PubMed] [Google Scholar]
- 2. Liu D., Pitta M., Lee J. H., Ray B., Lahiri D. K., Furukawa K., Mughal M., Jiang H., Villarreal J., Cutler R. G., Greig N. H., Mattson M. P. (2010) The KATP channel activator diazoxide ameliorates amyloid-β and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer's disease. J. Alzheimers Dis. 22, 443–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Y., Yang P. L., Tang J. F., Lin J. F., Cai X. H., Wang X. T., Zheng G. Q. (2008) Potassium channels: possible new therapeutic targets in Parkinson's disease. Med. Hypotheses 71, 546–550 [DOI] [PubMed] [Google Scholar]
- 4. Poulopoulou C., Markakis I., Davaki P., Tsaltas E., Rombos A., Hatzimanolis A., Vassilopoulos D. (2010) Aberrant modulation of a delayed rectifier potassium channel by glutamate in Alzheimer's disease. Neurobiol. Dis. 37, 339–348 [DOI] [PubMed] [Google Scholar]
- 5. Ngo-Anh T. J., Bloodgood B. L., Lin M., Sabatini B. L., Maylie J., Adelman J. P. (2005) SK channels and NMDA receptors form a Ca2+-mediated feedback loop in dendritic spines. Nat. Neurosci. 8, 642–649 [DOI] [PubMed] [Google Scholar]
- 6. Faber E. S., Delaney A. J., Sah P. (2005) SK channels regulate excitatory synaptic transmission and plasticity in the lateral amygdala. Nat. Neurosci. 8, 635–641 [DOI] [PubMed] [Google Scholar]
- 7. Stocker M. (2004) Ca2+-activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5, 758–770 [DOI] [PubMed] [Google Scholar]
- 8. Dolga A. M., Terpolilli N., Kepura F., Nijholt I. M., Knaus H. G., D'Orsi B., Prehn J. H., Eisel U. L., Plant T., Plesnila N., Culmsee C. (2011) KCa2 channels activation prevents [Ca2+]i deregulation and reduces neuronal death following glutamate toxicity and cerebral ischemia. Cell Death Dis. 2, e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dolga A. M., Granic I., Blank T., Knaus H. G., Spiess J., Luiten P. G., Eisel U. L., Nijholt I. M. (2008) TNF-α-mediates neuroprotection against glutamate-induced excitotoxicity via NF-κB-dependent up-regulation of K2.2 channels. J. Neurochem. 107, 1158–1167 [DOI] [PubMed] [Google Scholar]
- 10. Bednarczyk P. (2009) Potassium channels in brain mitochondria. Acta Biochim. Pol. 56, 385–392 [PubMed] [Google Scholar]
- 11. Szewczyk A., Kajma A., Malinska D., Wrzosek A., Bednarczyk P., Zabłocka B., Dołowy K. (2010) Pharmacology of mitochondrial potassium channels: dark side of the field. FEBS Lett. 584, 2063–2069 [DOI] [PubMed] [Google Scholar]
- 12. Szabò I., Bock J., Jekle A., Soddemann M., Adams C., Lang F., Zoratti M., Gulbins E. (2005) A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 280, 12790–12798 [DOI] [PubMed] [Google Scholar]
- 13. Szabò I., Zoratti M., Gulbins E. (2010) Contribution of voltage-gated potassium channels to the regulation of apoptosis. FEBS Lett. 584, 2049–2056 [DOI] [PubMed] [Google Scholar]
- 14. Peixoto P. M., Ryu S. Y., Kinnally K. W. (2010) Mitochondrial ion channels as therapeutic targets. FEBS Lett. 584, 2142–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malinska D., Mirandola S. R., Kunz W. S. (2010) Mitochondrial potassium channels and reactive oxygen species. FEBS Lett. 584, 2043–2048 [DOI] [PubMed] [Google Scholar]
- 16. Cheng Y., Debska-Vielhaber G., Siemen D. (2010) Interaction of mitochondrial potassium channels with the permeability transition pore. FEBS Lett. 584, 2005–2012 [DOI] [PubMed] [Google Scholar]
- 17. Grohm J., Kim S. W., Mamrak U., Tobaben S., Cassidy-Stone A., Nunnari J., Plesnila N., Culmsee C. (2012) Inhibition of Drp1 provides neuroprotection in vitro and in vivo. Cell Death Differ. 19, 1446–1458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tobaben S., Grohm J., Seiler A., Conrad M., Plesnila N., Culmsee C. (2011) Bid-mediated mitochondrial damage is a key mechanism in glutamate-induced oxidative stress and AIF-dependent cell death in immortalized HT-22 hippocampal neurons. Cell Death Differ. 18, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Landshamer S., Hoehn M., Barth N., Duvezin-Caubet S., Schwake G., Tobaben S., Kazhdan I., Becattini B., Zahler S., Vollmar A., Pellecchia M., Reichert A., Plesnila N., Wagner E., Culmsee C. (2008) Bid-induced release of AIF from mitochondria causes immediate neuronal cell death. Cell Death Differ. 15, 1553–15563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Culmsee C., Zhu C., Landshamer S., Becattini B., Wagner E., Pellecchia M., Blomgren K., Plesnila N. (2005) Apoptosis-inducing factor triggered by poly(ADP-ribose) polymerase and Bid mediates neuronal cell death after oxygen-glucose deprivation and focal cerebral ischemia. J. Neurosci. 25, 10262–10272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oxler E. M., Dolga A., Culmsee C. (2012) AIF depletion provides neuroprotection through a preconditioning effect. Apoptosis 17, 1027–1038 [DOI] [PubMed] [Google Scholar]
- 22. Sorgato M. C., Keller B. U., Stühmer W. (1987) Patch-clamping of the inner mitochondrial membrane reveals a voltage-dependent ion channel. Nature 330, 498–500 [DOI] [PubMed] [Google Scholar]
- 23. Hochman J., Ferguson-Miller S., Schindler M. (1985) Mobility in the mitochondrial electron transport chain. Biochemistry 24, 2509–2516 [DOI] [PubMed] [Google Scholar]
- 24. Kirichok Y., Krapivinsky G., Clapham D. E. (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364 [DOI] [PubMed] [Google Scholar]
- 25. Diemert S., Dolga A. M., Tobaben S., Grohm J., Pfeifer S., Oexler E., Culmsee C. (2012) Impedance measurement for real time detection of neuronal cell death. J. Neurosci. Methods 203, 69–77 [DOI] [PubMed] [Google Scholar]
- 26. Mootha V. K., Lepage P., Miller K., Bunkenborg J., Reich M., Hjerrild M., Delmonte T., Villeneuve A., Sladek R., Xu F., Mitchell G. A., Morin C., Mann M., Hudson T. J., Robinson B., Rioux J. D., Lander E. S. (2003) Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc. Natl. Acad. Sci. U.S.A. 100, 605–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baughman J. M., Perocchi F., Girgis H. S., Plovanich M., Belcher-Timme C. A., Sancak Y., Bao X. R., Strittmatter L., Goldberger O., Bogorad R. L., Koteliansky V., Mootha V. K. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blank T., Nijholt I., Kye M. J., Radulovic J., Spiess J. (2003) Small-conductance, Ca2+-activated K+ channel SK3 generates age-related memory and LTP deficits. Nat. Neurosci. 6, 911–912 [DOI] [PubMed] [Google Scholar]
- 29. Tuteja D., Rafizadeh S., Timofeyev V., Wang S., Zhang Z., Li N., Mateo R. K., Singapuri A., Young J. N., Knowlton A. A., Chiamvimonvat N. (2010) Cardiac small conductance Ca2+-activated K+ channel subunits form heteromultimers via the coiled-coil domains in the C termini of the channels. Circ. Res. 107, 851–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sailer C. A., Kaufmann W. A., Marksteiner J., Knaus H. G. (2004) Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol. Cell. Neurosci. 26, 458–469 [DOI] [PubMed] [Google Scholar]
- 31. Emanuelsson O., Nielsen H., Brunak S., von Heijne G. (2000) Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016 [DOI] [PubMed] [Google Scholar]
- 32. Hougaard C., Eriksen B. L., Jørgensen S., Johansen T. H., Dyhring T., Madsen L. S., Strøbaek D., Christophersen P. (2007) Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br. J. Pharmacol. 151, 655–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strøbaek D., Hougaard C., Johansen T. H., Sørensen U. S., Nielsen E. Ø., Nielsen K. S., Taylor R. D., Pedarzani P., Christophersen P. (2006) Inhibitory gating modulation of small conductance Ca2+-activated K+ channels by the synthetic compound (R)-N-(benzimidazol-2-yl)-1,2,3,4-tetrahydro-1-naphtylamine (NS8593) reduces afterhyperpolarizing current in hippocampal CA1 neurons. Mol. Pharmacol. 70, 1771–1782 [DOI] [PubMed] [Google Scholar]
- 34. Ishii T. M., Maylie J., Adelman J. P. (1997) Determinants of apamin and d-tubocurarine block in SK potassium channels. J. Biol. Chem. 272, 23195–23200 [DOI] [PubMed] [Google Scholar]
- 35. Adelman J. P., Maylie J., Sah P. (2012) Small-conductance Ca2+-activated K+ channels: form and function. Annu. Rev. Physiol. 74, 245–269 [DOI] [PubMed] [Google Scholar]
- 36. Hirschberg B., Maylie J., Adelman J. P., Marrion N. V. (1998) Gating of recombinant small-conductance Ca-activated K+ channels by calcium. J. Gen. Physiol. 111, 565–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Soh H., Park C. S. (2001) Inwardly rectifying current-voltage relationship of small-conductance Ca2+-activated K+ channels rendered by intracellular divalent cation blockade. Biophys. J. 80, 2207–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yarov-Yarovoy V., Paucek P., Jabůrek M., Garlid K. D. (1997) The nucleotide regulatory sites on the mitochondrial KATP channel face the cytosol. Biochim. Biophys. Acta 1321, 128–136 [DOI] [PubMed] [Google Scholar]
- 39. Jabůrek M., Yarov-Yarovoy V., Paucek P., Garlid K. D. (1998) State-dependent inhibition of the mitochondrial KATP channel by glyburide and 5-hydroxydecanoate. J. Biol. Chem. 273, 13578–13582 [PubMed] [Google Scholar]
- 40. Garcia M. L., Galvez A., Garcia-Calvo M., King V. F., Vazquez J., Kaczorowski G. J. (1991) Use of toxins to study potassium channels. J. Bioenerg. Biomembr. 23, 615–646 [DOI] [PubMed] [Google Scholar]
- 41. Maher P., Davis J. B. (1996) The role of monoamine metabolism in oxidative glutamate toxicity. J. Neurosci. 16, 6394–6401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Albrecht P., Lewerenz J., Dittmer S., Noack R., Maher P., Methner A. (2010) Mechanisms of oxidative glutamate toxicity: the glutamate/cystine antiporter system xc− as a neuroprotective drug target. CNS Neurol. Disord. Drug Targets 9, 373–382 [DOI] [PubMed] [Google Scholar]
- 43. Kasumu A. W., Hougaard C., Rode F., Jacobsen T. A., Sabatier J. M., Eriksen B. L., Strøbæk D., Liang X., Egorova P., Vorontsova D., Christophersen P., Rønn L. C., Bezprozvanny I. (2012) Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem. Biol. 19, 1340–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walter J. T., Alviña K., Womack M. D., Chevez C., Khodakhah K. (2006) Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat. Neurosci. 9, 389–397 [DOI] [PubMed] [Google Scholar]
- 45. Alviña K., Khodakhah K. (2010) The therapeutic mode of action of 4-aminopyridine in cerebellar ataxia. J. Neurosci. 30, 7249–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marchi S., Giorgi C., Suski J. M., Agnoletto C., Bononi A., Bonora M., De Marchi E., Missiroli S., Patergnani S., Poletti F., Rimessi A., Duszynski J., Wieckowski M. R., Pinton P. (2012) Mitochondria-ros crosstalk in the control of cell death and aging. J. Signal Transduct. 2012, 329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dolga A. M., Letsche T., Gold M., Doti N., Bacher M., Chiamvimonvat N., Dodel R., Culmsee C. (2012) Activation of KCNN3/SK3/KCa2.3 channels attenuates enhanced calcium influx and inflammatory cytokine production in activated microglia. Glia 60, 2050–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dolga A. M., Culmsee C. (2012) Protective roles for potassium SK/KCa2 channels in microglia and neurons. Front. Pharmacol. 3, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Allen D., Nakayama S., Kuroiwa M., Nakano T., Palmateer J., Kosaka Y., Ballesteros C, Watanabe M., Bond C. T., Luján R., Maylie J., Adelman J. P., Herson P. S. (2011) SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J. Cereb. Blood Flow Metab. 31, 2302–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bishop M. W., Chakraborty S., Matthews G. A., Dougalis A., Wood N. W., Festenstein R., Ungless M. A. (2010) Hyperexcitable substantia nigra dopamine neurons in PINK1- and HtrA2/Omi-deficient mice. J. Neurophysiol. 104, 3009–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eliseev R. A., Salter J. D., Gunter K. K., Gunter T. E. (2003) Bcl-2 and tBid proteins counter-regulate mitochondrial potassium transport. Biochim. Biophys. Acta 1604, 1–5 [DOI] [PubMed] [Google Scholar]