

Background: XPC-RAD23B and Rad4-Rad23 proteins are primary damage recognition factors in nucleotide excision repair in human and yeast cells, respectively.
Results: XPC-RAD23B and Rad4-Rad23 have contacts with damaged DNA in the same positions.
Conclusion: Both proteins reveal similar topography in the complex with damaged DNA in solution.
Significance: This study fills the gap between biochemical results for XPC-RAD23B and x-ray data for Rad4-Rad23.
Keywords: DNA Binding Protein, DNA Damage, DNA Nucleotide Excision Repair, DNA-Protein Interaction, Photoaffinity Labeling, Rad4-Rad23, XPC-RAD23B
Abstract
The human XPC-RAD23B complex and its yeast ortholog, Rad4-Rad23, are the primary initiators of global genome nucleotide excision repair. The interaction of these proteins with damaged DNA was analyzed using model DNA duplexes containing a single fluorescein-substituted dUMP analog as a lesion. An electrophoretic mobility shift assay revealed similarity between human and yeast proteins in DNA binding. Quantitative analyses of XPC/Rad4 binding to the model DNA structures were performed by fluorescent depolarization measurements. XPC-RAD23B and Rad4-Rad23 proteins demonstrate approximately equal binding affinity to the damaged DNA duplex (KD ∼ (0.5 ± 0.1) and (0.6 ± 0.3) nm, respectively). Using photoreactive DNA containing 5-iodo-dUMP in defined positions, XPC/Rad4 location on damaged DNA was shown. Under conditions of equimolar binding to DNA both proteins exhibited the highest level of cross-links to 5I-dUMP located exactly opposite the damaged nucleotide. The positioning of the XPC and Rad4 proteins on damaged DNA by photocross-linking footprinting is consistent with x-ray analysis of the Rad4-DNA crystal complex. The identity of the XPC and Rad4 location illustrates the common principles of structure organization of DNA damage-scanning proteins from different Eukarya organisms.
Introduction
The genetic stability of organisms is achieved by a broad spectrum of repair mechanisms (1–4), among which nucleotide excision repair (NER)2 is of high significance. This process removes a wide range of lesions distorting the double helix such as pyrimidine dimers arising as a result of UV irradiation and bulky chemical adducts resulting from environmental factors or chemotherapeutic agents. In eukaryotes, two distinct NER subpathways are operative, namely transcription-coupled repair and global genome repair (GG-NER), which employ a partly different subset of NER proteins. Among the products of xeroderma pigmentosum (XP) genes, the XP-C complementing protein (XPC) is specifically required for GG-NER. The human XPC, RAD23B, and CEN2 (centrin 2) proteins form the damage recognition complex that is the first to the site of damage in the GG-NER pathway. After recognizing and binding to the damaged DNA the XPC-RAD23B-CEN2 complex, downstream proteins are recruited sequentially, and GG-NER progresses through double helix unwinding, damaged fragment excision, and DNA resynthesis steps due to the involvement of >30 proteins (5). Replication protein A (RPA) and XPA have also been shown to possess damage recognition ability depending on the type of damage in both GG-NER and transcription-coupled repair (6). The helicase activities of the XPB and XPD subunits of the transcription factor II (TFIIH) are responsible for unwinding the damaged DNA producing a partially open region of DNA duplex (7). This is followed by 5′- and 3′-incisions performed by XPF-ERCC1 and XPG endonucleases, respectively (8). Finally, PCNA, RPA, DNA polymerases δ, ϵ, and κ, and DNA ligases (I or IIIα) participate in gap filling and ligation of the newly synthesized DNA (9, 10).
The coordination and spatial organization of the protein complexes of the NER machinery on damaged DNA remain undetermined. In particular, protein-DNA complex topography during the NER damage recognition step is still under discussion. The XPC protein is a 940-amino acid polypeptide that is the primary damage sensor, and its correct location on the damaged site is necessary for recruitment of the next NER factor (11). Based on binding experiments, the XPC-RAD23B complex exhibits high affinity for a branched DNA structure containing a junction between the double and single-stranded regions that is a common feature of most NER substrates (12). Based on this binding property, it was assumed that XPC-RAD23B recognizes this kind of DNA structure and contacts with a DNA junction. Footprinting assay indicated that XPC-RAD23B forms contacts with a symmetrical area around the damaged site (bubbled with or without the pyrimidine(6-4) pyrimidone adducts) (13). The presence of the acetylaminofluorene adducts or of the loop in the DNA structure caused asymmetrical contacts of XPC-RAD23B with the 5′-side of the looped or damaged DNA (12, 13). Despite the interaction of XPC with nucleotides surrounding the lesion, photocross-linking experiments were able to detect contacts with the damaged base only in the case of a lesion containing a photoreactive group attached by a long linker (14, 15). Further truncation analyses revealed that different XPC structure domains contribute to recognition of different segments of damaged DNA. The main part of the protein (1–740 amino acids) binds to the undamaged double-stranded segment and damage recognition ability is mediated by the small part (∼740–831 amino acids) that interacts selectively with the single-stranded undamaged region opposite the lesion (16). This finding has been strongly supported by a structural analysis of the Sacchromyces cerevisiae XPC ortholog, Rad4, which was bound to damaged DNA (17). However, there was no direct evidence for a relationship of the structural data obtained with the yeast protein and the human protein. Rad4p and XPC share only 23% homology in amino acid sequences that mainly belongs to domains responsible for DNA binding (17). In the present study, we performed detailed comparative analysis of XPC and Rad4 interactions with damaged DNA in solution using electrophoretic mobility shift assay, fluorescent depolarization measurements, and photoaffinity labeling techniques.
EXPERIMENTAL PROCEDURES
Materials
[γ-32P]ATP (3000 Ci/mmol) was produced in the Laboratory of Radiochemistry of the Institute of Chemical Biology and Fundamental Medicine; phage T4 polynucleotide kinase was purchased from Biosan (Novosibirsk, Russia); stained molecular mass markers were from Bio-Rad, and reagents for electrophoresis and buffer components were from Sigma or made in Russia (extra pure grade). Oligonucleotides bearing a 5I-dUMP or fluorescein dUMP derivative or both analogs were synthesized by Dr. V. Silnikov (Nanotech-C, Novosibirsk, Russia). Schematic view of the DNA duplexes and structures of the modified nucleotides are presented in Fig. 1, and the sequences of the oligonucleotides are presented in Table 1.
FIGURE 1.

Schematic view of the DNA duplexes and structures of the nucleotide analogs used for the experiments (see Table 1 for oligonucleotide sequences).
TABLE 1.
Sequences of the 48-mer oligonucleotides
| Name | Sequencesa |
|---|---|
| 1 | 5′-ctatggcgaggcgaItaagttgggcaacgtcagggtcttccgaacgac-3′ |
| 2 | 5′-ctatggcgaggcgattaagItgggcaacgtcagggtcttccgaacgac-3′ |
| 3 | 5′-ctatggcgaggcgattaagttgggIaacgtcagggtcttccgaacgac-3′ |
| 4 | 5′-ctatggcgaggcgattaagttgggcIacgtcagggtcttccgaacgac-3′ |
| 5 | 5′-ctatggcgaggcgattaagttgggcaacgIcagggtcttccgaacgac-3′ |
| 6 | 5′-ctatggcgaggcgattaagttgggcaacgtcagggIcttccgaacgac-3′ |
| FI1 | 5′-gtcgttcggaagaccctgacgFtacccaacttaaIcgcctcgccatag-3′ |
| FI2 | 5′-gtcgttcggaagaccctgacgFtacccaacItaatcgcctcgccatag-3′ |
| FI3 | 5′-gtcgttcggaagaccctgacgFIacccaacttaatcgcctcgccatag-3′ |
| FI4 | 5′-gtcgttcggaagacccIgacgFtacccaacttaatcgcctcgccatag-3′ |
| Fa | 5′-gtcgttcggaagaccctgacgtFacccaacttaatcgcctcgccatag-3′ |
| Fg | 5′-gtcgttcggaagaccctgacgtFgcccaacttaatcgcctcgccatag-3′ |
| N | 5′-ctatggcgaggcgattaagttgggtaacgtcagggtcttccgaacgac-3′ |
| Nm | 5′-ctatggcgaggcgattaagttgggcaacgtcagggtcttccgaacgac-3′ |
| 48c | 5′-gtcgttcggaagaccctgacgttgcccaacttaatcgcctcgccatag-3′ |
a Modifications are indicated as follows: underlined I indicates 5I-dUMP and underlined F indicates Flu-dUMP.
Protein Purification
The recombinant human FLAG-XPC and His-tagged RAD23B (RAD23B-His) proteins were purified, and the XPC-RAD23B heterodimer was reconstituted in vitro as described previously (18). Recombinant human RPA was isolated from Escherichia coli as indicated (19). The plasmid containing the cDNA of human RPA was a kind gift of Dr. K. Weisshart (Leibniz Institute for Age Research, Fritz Lipmann Institute, Jena, Germany). Recombinant hXPA bearing the N-terminal polyhistidine fragment was expressed in the E. coli BL21(DE3)LysS strain, using the pETI5b-XPA recombinant plasmid kindly provided by Dr. O. Schärer (State University of New York, Stony Brook, NY). Protein isolation was performed according to Ref. 20 with one modification: EDTA was not added during purification. The truncated N-terminal His6-tagged Rad4 protein (residues 101–632) was expressed together with a Rad23 fragment in which the ubiquitin association 1 domain (residues 135–229) was replaced by a thrombin protease site and purified as described (17).
Preparation of 5′-32P-labeled DNA Duplexes
A radioactive label was inserted into the 5′-end of 5I-dUMP-containing oligonucleotides using phage T4 polynucleotide kinase as described (21). Labeled oligonucleotides were purified by electrophoresis under denaturing conditions followed by passive elution with 3 m LiClO4 and acetone precipitation or using MicroSpinTM G-25 columns (Amersham Biosciences). DNA duplexes were prepared by annealing 5′-32P-labeled oligonucleotides with complementary oligonucleotides in a 1:1 ratio. Oligonucleotides were incubated for 5 min at 95 °C, cooled slowly to 75 °C, kept for 15 min at this temperature, and slowly cooled to room temperature. The hybridization degree was monitored by electrophoresis in 10% polyacrylamide gel (acrylamide/bis-acrylamide = 40:1). TBE buffer (50 mm Tris, 50 mm H3BO3, and 1 mm EDTA, pH 8.3) was used as the electrode buffer.
Electrophoretic Mobility Shift Assay
Protein-DNA complexes were analyzed by gel retardation. The reaction mixture (10 μl) contained 50 mm Tris-HCl, pH 7.5, 100 mm KCl, 1 mm dithiothreitol, and 0.6 mg/ml BSA (buffer A), 10 nm 5′-32P-labeled DNA and either XPC-RAD23B or Rad4-Rad23 at various concentrations. Protein complexes with DNA were formed for 20 min at 37 °C. Loading buffer (1:5 v/v) containing 20% glycerol, 0.015% bromphenol blue in buffer A was then added to the sample. Temperature of loading buffer was 37 °C. Protein-nucleic acid complexes were separated in native 5% polyacrylamide gel (acrylamide/bis-acrylamide = 60:1). Electrophoresis was carried out in TBE as running buffer for 3 h at 17 V/cm and 4 °C. Positions of radioactively labeled oligonucleotide and protein-DNA complexes were visualized by phosphorimaging using a Molecular Imager FX Pro+ (Bio-Rad).
Fluorescence Depolarization Measurements
Fluorescein-containing DNA structures were also used for fluorescence anisotropy analysis. Binding of such DNA to XPC-RAD23B increases fluorescence anisotropy of the fluorescein group and can be used as an indicator of complex formation. The fluorescence intensity of free and protein-bound DNA probe was almost the same. All experiments were performed at 37 °C on a POLARstar Optima multidetection microplate reader (BMG Labtech, Offenburg, Germany). The excitation wavelength was set at 495 nm, and emission wavelength was set at 520 nm. After sample equilibration, 50 scanning data points were collected for each titration point, and the average was used as a value. For better accuracy, we obtained each value 10 times and then took the average. All titrations were performed in a 96-well assay “V” bottom black plate (Axigen), and thus, one well contained one titration point. These experiments were carried out in buffer A. The volume of the reaction mixture was 200 μl. Measurements were performed at four DNA concentrations (0.5, 1, 3, and 5 nm) for the KD determination. During the competition experiments a constant concentration of labeled DNA (3 nm) was used and various concentrations of unlabeled DNA competitors. All reactions were mixed at room temperature in 0.6 ml tubes (Axigen) and were then loaded onto the plate. The plate was incubated at 37 °C in the reader. Each experiment was repeated at least three times.
Data Fitting and Processing
Anisotropy mean values were plotted against the protein concentration and fitted using a simple one-site or n identical binding site models. The fluorescence anisotropy change is proportional to the concentration of protein-DNA complex and can be defined as shown in Equation 1,
 |
where
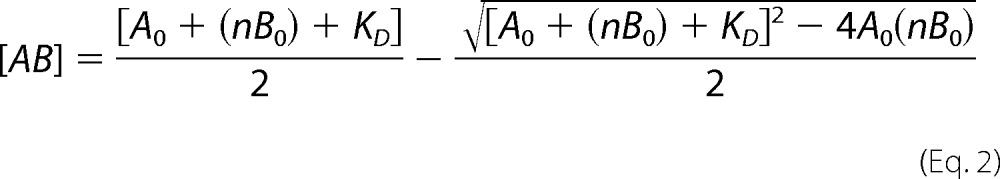 |
with rAB being the anisotropy of the protein-DNA complex, A0 and B0 representing the total concentrations of protein and DNA, respectively, Δr representing relative anisotropy, and KD representing the dissociation constant of protein-DNA complex. The experimentally determined relative anisotropy change is Δr = ((r − rB)/rB), where rB indicates the anisotropy of free DNA. These relative anisotropy values were plotted against A0. The KD values for fluorescein-containing DNA were calculated from the resulting binding curves. Data sets were least-square fitted with the program OriginPro (version 8, Microcal, Northampton, MA) using the Levenberg-Marquardt algorithm.
Two DNA concentrations (B1 and B2) served to determine stoichiometry of DNA-protein complex under titration conditions. The degree of binding can be described as shown in Equation 3,
 |
where A1 and A2 are the protein concentrations that have the same value of Δr (22, 23).
The KD values for DNA structures without fluorescein were determined from competition experiments. The reaction mixtures contained increasing amounts of DNA competitor. Anisotropy values were plotted against the DNA concentration ratio. Constants of dissociation values were calculated from the simple equation,
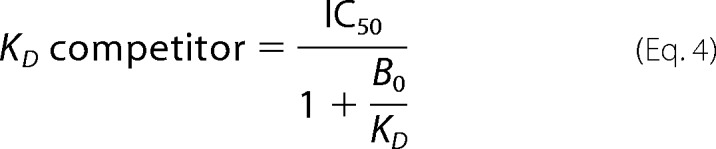 |
where IC50 is the concentration of competitor necessary to displace half of the fluorescein-containing DNA. The IC50 value was determined as the concentration of competitor in the point of half-maximum anisotropy.
Photoaffinity Labeling
Protein modification by photoreactive DNA structures was performed in a reaction mixture (10 μl) containing buffer A, 10 nm 5′-32P-labeled photoreactive DNA, and the corresponding protein(s). Reaction mixtures were incubated for 20 min at 37 °C and then UV-irradiated for 1 h in an ice bath using a Bio-Link BLX-312 cross-linker (Vilber Lourmant), a wavelength of 312 nm, and a light intensity of 5 mJ/(cm2·s). The reaction was terminated by 1:5 (v/v) dilution of the sample with stop buffer (5% SDS, 5% 2-mercaptoethanol, 0.3 m Tris-HCl, pH 7.8, 50% glycerol, and 0.005% bromophenol blue). Samples were heated at 97 °C for 5 min and loaded on the gel. DNA-protein adducts were separated by electrophoresis in the Laemmli system (10% polyacrylamide gel, acrylamide/bis-acrylamide, 60:1) (24). Gels were dried and exposed to autoradiography.
RESULTS
XPC-RAD23B and Rad4-Rad23 Binding to Damaged DNA
Although the Rad4-Rad23 crystal structure has been solved, biochemical study of this protein is very limited in contrast to XPC-RAD23B. We analyzed binding of both proteins with DNA in the same conditions using two different types of DNA binding assays. EMSA experiments demonstrate preference in the binding of both proteins to damaged DNA (Fig. 2). Despite the total similarity in interaction with DNA, there are some differences between the DNA binding modes of these proteins. XPC-RAD23B displays one band with DNA (Fig. 2A) corresponding to the binding of single protein molecule. A DNA complex with two XPC-RAD23B molecules can be clearly detected only with large excess of XPC-RAD23B over DNA (lanes 6 and 12 in Fig. 2A). Rad4-Rad23 displays at least two DNA-protein complexes of different mobility in the presence of unbound DNA (for example lanes 3 or 8 in Fig. 2B) and also in the presence of DNA competitor (lanes 14 and 15, and lanes 19 and 20 in Fig. 2B). This could be caused by the smaller size of the DNA binding site for Rad4 in comparison with XPC. According to structural data, an 11-mer duplex segment will be sufficient for the binding of the Rad4 transglutaminase homology domain/BHD1 domains (17); therefore, a 48-mer duplex can bind four Rad4 molecules. These complexes were displayed in the presence of large protein excess (Fig. 2C).
FIGURE 2.

Binding of XPC-RAD23B and Rad4-Rad23 to DNA analyzed by EMSA. The reaction mixtures (10 μl) contained buffer A, 10 nm 5′-32P-labeled DNA and the indicated concentrations of XPC-RAD23B (A) or Rad4-Rad23 (B). A schematic view of the DNA structures is presented at the top: triangle indicates the position of the bulky lesion. 5-Fold excess of nonradioactive undamaged 48-mer duplex was added in the reaction mixtures to compete with binding of the DNA studied. The right panels show the quantitative analysis of the data presented in the left panels. Error bars indicate the S.E. of five independent experiments. C, Rad4-Rad23-DNA complexes with different gel-shift mobility formed in the presence of protein excess.
A quantitative analysis of binding of XPC-RAD23B and Rad4-Rad23 proteins to damaged DNA was also performed using fluorescence anisotropy measurements. In this study, we also used fluorescein group mimicking DNA damage as a fluorescent probe. Typical titration curve for XPC-RAD23B binding to fluorescein containing DNA is presented in Fig. 3A. Significant changes in fluorescence intensity of the fluorescein moiety upon formation of protein-DNA complexes were not observed. The amount of active protein in the protein preparations was estimated by the titration of reaction mixtures containing a damaged DNA duplex with increasing amounts of XPC-RAD23B (Fig. 3B) and Rad4-Rad23 (Fig. 3C) under conditions of stoichiometric protein-DNA binding. The DNA concentration in these experiments was 10 nm. The relative anisotropy values were plotted against the protein concentrations. The linear parts of the binding curve were fitted and extrapolated separately. From the intersection of the lines the XPC-RAD23B and Rad4-Rad23 concentrations needed to completely bind the DNA were determined (∼56 and ∼31 nm, respectively). Thus, the active protein percentage in the XPC-RAD23B preparation is ∼18% and in the Rad4-Rad23 is ∼32%. For all anisotropy measurements, the protein concentrations given have been corrected for the DNA binding activity. Affinity of XPC/Rad4 for undamaged DNA (without the fluorescein moiety) was estimated from competition experiments (Fig. 3D).
FIGURE 3.

Fluorescence anisotropy measurements of XPC-RAD23B binding to DNA. A, typical titration curve for XPC-RAD23B binding to fluorescein containing DNA (1 nm). Fluorescein group mimicking DNA damage was also used as a fluorescent probe. B and C, analysis of DNA binding activity of the XPC-RAD23B and Rad4-Rad23 protein preparations. The reaction mixtures containing 10 nm damaged DNA duplex were titrated by the increasing amounts of XPC-RAD23B (B) or Rad4-Rad23 (C). D, competition of the binding of the XPC-RAD23B complex to the fluorescent damaged DNA duplex (3 nm) by addition of undamaged DNA duplex. The reaction mixture contained increasing amounts of DNA competitor. Anisotropy values were plotted against the DNA concentration ratio. E, analysis of the stoichiometry of DNA-protein complexes under titration conditions. The left panel shows binding isotherms for 3 and 10 nm DNA. The right panels show quantitative analysis of binding isotherms presented in the left panels.
The stoichiometry of DNA-protein complexes was determined using two DNA concentrations (3 and 10 nm). Binding isotherms for these DNA concentrations are presented in the left panel of Fig. 3E. The right panels show quantitative analysis of binding isotherms presented in the left panels. An extrapolation of resulted linear plot Δr versus Σϴ to the maximum value of relative anisotropy (Δrmax ∼5.2) shows that at saturation, 1.2 molecule of XPC-RAD23B binds to damaged DNA duplex. Some differences from 1:1 stoichiometry can be explained by nonspecific binding to the DNA part 5′-side from a lesion. Nonspecific binding usually presents in binding experiments if indirect methods are used to monitor binding. Nevertheless, under experimental conditions, the binding curves fitted satisfactorily using a simple 1:1 model (of one protein molecule bound per DNA molecule). The same experiments were performed for Rad4-Rad23 using 1 and 3 nm DNA (data not shown). Values of KD for XPC-RAD23B and Rad4-Rad23 complexes with damaged DNA were 0.5 ± 0.1 and 0.6 ± 0.3 nm, respectively. XPC-RAD23B affinity to undamaged DNA was one order lower (KD ∼ 5 ± 2 nm). In the case of Rad4-Rad23, the difference between the affinities to damaged and undamaged DNA was less (KD for complex with undamaged DNA duplex is 2.5 ± 1.6 nm).
Location of XPC and Rad4 on Damaged DNA
To further examine XPC positioning around the lesion, we used photoreactive DNA duplexes bearing a fluorescein residue as the damage recognized by the NER system and a 5I-dUMP residue as cross-linking group. 5I-dUMP was introduced in a defined position of the undamaged or damaged strand of the DNA duplex (Fig. 1 and Table 1). The iodine atom in 5I-dUMP chromophore has a van der Waals radius very close to that of the methyl group of thymidine. Due to this similarity with the natural nucleotide, this analog causes minimal changes in DNA helix and therefore in DNA-protein interactions. It is suggested that a photoreactive group with zero length linker should react only with amino acid residues of the appropriate specificity in tight contact with DNA (25). The efficiency of XPC cross-linking with photoreactive group depended on the position of the 5I-dUMP and protein concentration (Fig. 4A). Under conditions corresponding to binding of a single XPC-RAD23B molecule to the DNA (Fig. 4B, lane 2), the highest level of cross-links was observed for 5I-dUMP located exactly opposite the damaged nucleotide (“zero” position) (Fig. 4A, lane 7, and 4C). Maximum cross-linking shifted in direction of the 5′-end of the undamaged strand (Fig. 4A, lane 4, and 4C) when the XPC concentration was increased ∼2.5-fold. In these conditions, two complexes of XPC-RAD23B with DNA were observed (Fig. 4B, lane 5). The complex with the lower mobility can be attributed to binding of the second XPC-RAD23B molecule to the XPC-RAD23B-DNA complex (Fig. 4D).
FIGURE 4.

Topography of the XPC-RAD23B location on damaged DNA revealed by photocross-linking footprinting. The damaged DNA model substrates carry photoreactive 5I-dUMP substitutions at the indicated positions of radioactively 5′-end-labeled damaged or undamaged strands. A, the reaction mixtures (10 μl) contained buffer A, 10 nm 5′-32P-labeled photoreactive DNA and XPC-RAD23B at the indicated concentrations. Shown is a schematic view of the DNA structures presented at the top: the triangle indicates the position of the bulky lesion; dashes and numbers indicate the position of 5I-dUMP relative to the lesion. B, analysis of the XPC-RAD23B binding products to damaged DNA. The reaction mixtures (10 μl) contained buffer A, 10 nm 5′-32P-labeled photoreactive DNA, and the indicated concentrations of the XPC-RAD23B. C, quantitative analysis of the data from A for 90 and 250 nm protein concentrations. The relative intensities of the protein-DNA adducts were plotted against 5I-dUMP positions in the DNA. Averages and experimental errors were taken from at least five experiments. D, localization of XPC-RAD23B on damaged DNA duplex based on maximum photocross-linking intensity.
The localization of XPC-RAD23B is consistent with the x-ray data of a complex of the yeast ortholog of XPC, Rad4, with a fragment of damaged DNA. Despite performing the same function, the sequence identity of the structural domains of the human and yeast proteins is not extensive. To check the correlation between the x-ray and photocrosslinking data on the localization of Rad4 on damaged DNA, the same Rad4 preparation was used for the cross-linking experiments (Fig. 5A) as for the x-ray analysis. The behavior of Rad4 was similar to that of XPC in these experiments. Under conditions corresponding to binding of one Rad4 molecule to DNA (Fig. 5B, lane 2) the highest yield of Rad4-DNA adducts was obtained for a DNA structure with photoreagent in zero position (Fig. 5A, lane 7, and 5C). Modification of Rad4 was more visible for DNA structures with a 5I-dUMP residue on the 5′-side of the lesion in the undamaged strand (Fig. 5A, lanes 3 and 5, and 5C) and at the position of 3′ adjacent to the lesion in the damaged strand (Fig. 5A, lane 17, and 5C). Photoaffinity modification data on the position of Rad4 on the DNA were in line with the x-ray analysis of the Rad4-DNA crystal complex (17). After binding of a second molecule (Fig. 5B, lane 4, and Fig. 5A, even lanes), changes in modification were similar for both proteins (compare Fig. 4C with 5C).
FIGURE 5.

Topography of the Rad4-Rad23 location on damaged DNA revealed by photocross-linking footprinting. A, the reaction mixtures (10 μl) contained buffer A, 10 nm 5′-32P-labeled photoreactive DNA and Rad4-Rad23 at the concentrations analyzed. DNA structures were the same as in the experiments with XPC-RAD23B. B, analysis of the Rad4-Rad23-DNA binding product formation. The reaction mixtures (10 μl) contained buffer A, 10 nm 5′-32P-labeled photoreactive DNA, and the indicated concentrations of Rad4-Rad23. C, quantitative analysis of the data from A for 10 nm and 50 nm protein concentrations. The relative intensities of the protein-DNA adducts were plotted against the 5I-dUMP positions in the DNA. Averages and experimental errors were taken from at least three experiments. D, localization of Rad4-Rad23 on a damaged DNA duplex according to photocross-linking intensity maximum.
The Influence of RPA and XPA on the Location of XPC on Damaged DNA
XPC binding and cross-linking to DNA in different positions around the lesion were moderated by RPA (Fig. 6). One can see that in the presence of RPA, the position of the 5I-dUMP in the undamaged strand corresponding to maximum XPC cross-linking moved over 10 nucleotides: from −6 (Fig. 6A, lane 5) to +4 (lane 12); furthermore, in the presence of RPA, the levels of XPC cross-linking were significantly increased in certain positions in both undamaged and damaged strands (Fig. 6B). This effect can reflect a conformational rearrangement of XPC in the complex with damaged DNA induced by RPA and resulting in tighter contacts of XPC in some positions around the lesion (Fig. 6C). We have shown previously that RPA influences XPC-RAD23B modification due to protein-protein interactions (15, 20). The level of RPA labeling significantly decreases with the simultaneous appearance of the products of XPC-RAD23B modification. These phenomena could be explained by RPA displacement from the undamaged DNA strand. In fact, RPA stimulates XPC-RAD23B binding to DNA (Fig. 7A, compare lanes 1–5 with lanes 7–11 and 13–17, and Fig. 7B, left panel); at the same time, the RPA binding products with DNA were decreased in the presence of XPC (Fig. 7A, compare lanes 6 and 12 with lanes 7–11 and 13–17). This could be caused by XPC-RAD23B-RPA-DNA complex formation. The triple complex containing one XPC-RAD23B and one RPA molecule should have gel mobility intermediate between XPC-RAD23B-DNA and 2(XPC-RAD23B)-DNA complexes. Some bands observed on lanes 11 and 14–17 may be potentially attributed to such complexes. The second XPC-RAD23B molecule is displaced from the DNA by RPA (Fig. 7B, right panel). This second XPC-RAD23B molecule most likely binds to the dsDNA segment nonspecifically, and its location is identified by photocross-links (Fig. 4D).
FIGURE 6.

The XPC-RAD23B location on damaged DNA duplex in the presence of RPA. A, influence of RPA on the level of the XPC-RAD23B modification depends on the positions of the 5I-dUMP in the DNA. B, diagram showing the quantitative analysis of the level of XPC-RAD23B photocross-linking in the presence and absence of RPA. The relative intensities of the protein-DNA adducts were plotted against the positions of the 5I-dUMP in the DNA. Averages and experimental errors were taken from at least five experiments. C, schematic view of the rearrangement of the localization of XPC-RAD23B on DNA in the presence of RPA (based on maximum photocross-linking intensity).
FIGURE 7.

Binding of the XPC-RAD23B to damaged DNA duplex in the presence of different RPA concentrations. A, the damaged DNA duplex was incubated at a concentration 10 nm with the indicated concentrations of XPC-RAD23B without/with various RPA concentrations: 2 or 10 nm. At the concentrations analyzed, the XPC-RAD23B formed two protein-DNA complexes with different electrophoretic mobility. B, the quantitative analysis of the protein-DNA complexes (from A). Percentages of bound DNA were plotted against XPC-RAD23B concentrations.
Similar stimulating effects on XPC cross-linking with a damaged DNA duplex were observed in the presence of XPA except that the position of the maximum level of XPC modification was not changed by addition of XPA. At the same time, the level of XPA cross-linking was markedly diminished in the presence of XPC-RAD23B (data not shown).
DISCUSSION
The XPC complex is now considered as a structure-specific DNA binding factor (26). Extensive biochemical analyses finally revealed that the XPC complex does not directly recognize the damaged DNA structures per se. For example, this protein prefers some specific DNA secondary structures containing a junction between the double-stranded and single-stranded DNA parts (12). The proposed model for XPC-DNA interaction, which was deduced from biochemical data, has been corroborated by the x-ray crystal structure of the complex involving the S. cerevisiae XPC homolog Rad4 and a DNA duplex with a site-specific lesion (a UV-induced cyclobutane pyrimidine dimer within a three-base bubble structure) (17). However, there remained an unsolved feature between the structural data resolved for the yeast protein and data observed in biochemical experiments with the human ortolog. There was a great need to compare the interaction of both proteins with damaged DNA in the same conditions because the structural data regarding XPC are not yet published. Here, we analyzed binding with DNA and the position of the XPC and Rad4 proteins on damaged DNA using the same DNA structures.
Preferential binding of the Rad4-Rad23 complex to UV-irradiated as well as N-acetoxy-2-(acetylamino)fluorene-treated DNA was shown previously (27, 28). Here, we demonstrate that the Rad4 complex, as also XPC, can efficiently bind to DNA duplexes containing a single bulky lesion (Fig. 2). In summary, human and yeast orthologs demonstrate a similar behavior in DNA binding. A quantitative analysis of the XPC-RAD23B and Rad4-Rad23 binding to model DNA structures was performed using fluorescence depolarization measurements. This method was used earlier to measure the affinity of the XPC-RAD23B complex for model oligonucleotides containing different DNA lesions. The oligonucleotides carried a terminal fluorescein modification located distantly from the damaged site that served as an indicator of complex formation with the XPC-RAD23B (29). In our study, a fluorescein group mimicking bulky DNA damage was used as fluorescent probe.
The data obtained are in agreement with EMSA experiments. The investigated proteins demonstrate close DNA binding properties. The XPC-RAD23B affinity for undamaged DNA duplex is ∼ 5 ± 2 nm. This value is nearly equal (within a factor of 2) to data obtained in (29), where the affinity of XPC for 36-mer DNA duplexes has been measured. It was shown previously that the affinity of XPC for DNA duplexes depends on the length of the DNA (30). Therefore, we can suggest that the slightly lower affinity to undamaged duplex reported in Ref. 29 is due to smaller DNA length. In another study (31), the authors applied the EMSA method and obtained dissociation constants of the XPC-RAD23B complex with undamaged 49-mer DNA that were the same as we obtained in this work. The duplex bearing fluorescein-substituted dUMP as a bulky damage shows a KD value equal to 0.5 ± 0.2 nm. Similar constant values have been reported by Refs. 29 and 31 for DNA duplexes containing (6-4) photoproducts. The Rad4-Rad23 DNA binding properties have not been studied as extensively as the XPC-RAD23B. Our data show KD values of 0.6 ± 0.3 nm and 2.5 ± 1.6 nm for Rad4-Rad23 complexes with damaged and undamaged DNA, respectively. Earlier KD values for the Rad4-Rad23 complex with undamaged 24-mer DNA and DNA containing cyclobutane pyrimidine dimer in a three-nucleotide bubble were estimated as 500 ± 40 nm and 94 ± 5 nm, respectively (17). We assume that this discrepancy could be explained by differences in DNA lengths and structures as well as by the methods applied.
According to the crystal structure of Rad4, its domain responsible for DNA binding can be divided into four subdomains: transglutaminase homology domain and three consecutive β-hairpin domains designated as BHD1, BHD2, and BHD3, respectively (17). Contacts with DNA shared between these subdomains. The transglutaminase homology domain and BHD1 provide nonspecific binding with the undamaged segment of the duplex on the 5′-side of the lesion; BHD2 and BHD3 fulfill damaged site recognition. BHD2 and BHD3 form a hand-like structure that interacts with the damaged DNA through two central features: the BHD3 β-hairpin inserts through double helix, causing the two base pairs that contain the cyclobutane pyrimidine dimer lesion to entirely flip out of the double helix; the BHD2-BHD3 groove holds the backbone of the undamaged strand and binds to its flipped-out nucleotides. We used these structural data to explain results on the crosslinking of Rad4 and XPC with damaged DNA.
In the case of an equimolar ratio in the XPC-DNA or Rad4-DNA complex, the highest XPC or Rad4 cross-linking level was obtained for DNA with zero position of photoreagent. The undamaged nucleotide opposite the lesion binds in the hydrophobic pocket formed by amino acid residues of the BHD3 domain based on Rad4 crystal structure data (T16u on Fig. 8A). Therefore, we can assume that tight interaction of this nucleotide with the protein leads to the highest level of protein-DNA adducts. Cross-linking products that were obtained for DNA-structures with photoreagent positions from −1 to −13 could result from contacts of the transglutaminase homology domain-BHD1 tandem with the extended dsDNA segment (Fig. 8A). These tandem interactions were produced mainly through polar contacts between the protein and the DNA backbone (17). Protein-DNA contacts with the duplex part on the other side from the lesion were limited, and levels of cross-links to DNA with the photoreagent located in positions +4, +5, and +10 were low. Identical or analogous amino acid residues of BHD2-BHD3 contacting the damaged DNA segment suggest that XPC and Rad4 bind DNA in a similar manner (Fig. 8B). In summary, the photoaffinity modification data reveal that the pattern of XPC-RAD23B and Rad4-Rad23 localization on damaged DNA is the same and that both proteins are located on the undamaged strand of the DNA duplex on the 5′-side from the lesion. The resulting XPC or Rad4 localization mode is in concert with crystal structure data of a complex of Rad4 with a fragment of damaged DNA. The identity of the XPC and Rad4 location revealed by the photocross-linking experiments illustrates the common principles of structure organization for damaged DNA scanning proteins from different eukaryotic organisms. Based on these results, photocross-linking footprinting may be appropriate for the positioning of the DNA-binding proteins on DNA. By using this approach to detect DNA-protein contacts in solution, we found the similarity between mode of XPC and Rad4 interaction with damaged DNA duplex and coincidence with structural data derived earlier for the complex of Rad4 with DNA duplex carrying other kind of bulky lesion.
FIGURE 8.

A, schematic representation of the interactions of the BHD2-BHD3 domains with a 4-bp damaged DNA segment according to the Rad4-Rad23 crystal structure data (17). The BHD2 and BHD3 domains are marked in blue and red, respectively. The flipped-out thymidines of the undamaged strand (T15u and T16u) are in black; the disordered, cyclobutane pyrimidine dimer-linked thymidines (T15d and T16d) and the damaged strand segment are in purple; numbers in the red ovals indicate numbering of nucleotides in the damaged strand relative to the T16d nucleotide; the protein side chain contacts to DNA are marked with orange arrows. B, the BHD2 and BHD3 residues (blue and red, respectively) that contact the damaged DNA segment and the corresponding residues from the XPC structure (17). Analogous and identical residues in these positions suggest that human and yeast orthologs bind to DNA in the similar way.
The XPC or Rad4 binding mode could be necessary for the formation of a special architecture of the protein-DNA complex in which the DNA site with disrupted hydrogen bonds is adopted for next-step damage verification by downstream damage recognition factors (TFIIH, XPA, and RPA). Subsequently, XPC or Rad4 positioning may guide the formation of an asymmetric preincision complex and help specify binding places for other proteins.
RPA and XPA are very abundant proteins (32, 33), and both have a preference for binding to damaged DNA (34). Our data show that XPA (20) and RPA stimulate XPC binding to the damaged DNA duplex. It was demonstrated previously that XPC physically interacts with XPA (35), and when XPA and XPC are simultaneously present in a reaction mixture, these NER factors mutually stimulate their binding to DNA (20). Photocross-linking experiments reveal that the intensity of XPC cross-linking in the presence of XPA was increased, but the localization of XPC on damaged DNA was not changed. This observation is consistent with our previous results indicating that XPA is located on the damaged strand (36). Therefore, it is possible to assume that XPC and XPA could coexist in the same complex with DNA. Decrease in XPA cross-linking intensity could be explained by XPC-induced perturbations in the XPA structure.
RPA stimulates XPC-RAD23B binding to DNA and influences XPC localization. These effects are most likely provided by protein-protein interactions. This assumption is partially confirmed by our earlier observation that an RPA mutant form depleted of all the domains responsible for protein-protein interactions is unable to stimulate XPC-RAD23B interaction with DNA (20). Moreover, no reports so far existed about a physical interaction between RPA and XPC. Thus, the stimulation by RPA observed could be based on protein-protein interactions mediated by DNA. According to the previously identified RPA location on damaged DNA (36) and results presented here, XPC-RAD23B and RPA are localized on the same region of the damaged area. In contrast to XPA, RPA binding with DNA decreased in the presence of XPC, and this could be caused by protein competition. As Fig. 7 illustrates, the second XPC-RAD23B molecule was displaced from the DNA by RPA. This second XPC-RAD23B molecule most likely binds to the dsDNA segment non-specifically, and its location is identified by photoaffinity modification (Fig. 4D). It was recently shown that RPA plays a regulatory role in the NER incision events (37); therefore, we could not exclude the possibility that XPC-RPA interaction is required for such a role.
Taken together, the data on the influence of RPA and XPA converge on the suggestion that when a lesion was found by RPA or XPA, these proteins stimulate XPC-RAD23B binding with damaged areas, and NER assembly started in the usual order. It is important to note that the obtained data exhibit a significance of protein-protein interactions for correct assembly of NER machinery. Proper orientation of NER factors is provided by the specific DNA-protein and protein-protein interactions and is required for the processing of the NER.
Acknowledgments
We are thankful to Dr. Rashid Anarbaev for the technical assistance in fluorescence anisotropy measurements, Dr. Klaus Weisshart (Leibniz Institute for Age Research, Jena, Germany) for the recombinant plasmid coding the human RPA subunits, to Dr. Orlando Schärer (State University of New York, Stony Brook, NY) for the recombinant plasmid coding hXPA, and to Dr. Anne-Lise Haenni (Jacques Monod Institute, Paris, France) for careful reading of the manuscript and useful comments.
This work was supported by Russian Foundation for Basic Research Grants 10-04-00837, 12-04-31323, 12-04-33162, and 13-04-00538; the Russian Ministry of Education and Science Contract 14.B37.21.0188; and the program of the Presidium of Russian Academy of Sciences “Molecular and Cellular Biology.”
- NER
- nucleotide excision repair
- XP
- xeroderma pigmentosum
- GG
- global genome repair
- RPA
- replication protein A
- 5I-dUMP
- 5-iodo-deoxyuridine monophosphate
- pyrimidine(6-4) pyrimidone adducts
- 6-(1,2)-dihydro-2-oxo-4-pyrimidinyl-5-methyl-2,4-(1H,3H)-pyrimidinediones.
REFERENCES
- 1. Lindahl T., Wood R. D. (1999) Quality control by DNA repair. Science 286, 1897–1905 [DOI] [PubMed] [Google Scholar]
- 2. Hoeijmakers J. H. (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366–374 [DOI] [PubMed] [Google Scholar]
- 3. Schärer O. D. (2003) Chemistry and biology of DNA repair. Angew. Chem. Int. Ed. Engl. 42, 2946–2974 [DOI] [PubMed] [Google Scholar]
- 4. Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 5. Aboussekhra A., Biggerstaff M., Shivji M. K., Vilpo J. A., Moncollin V., Podust V. N., Protić M., Hübscher U., Egly J. M., Wood R. D. (1995) Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80, 859–868 [DOI] [PubMed] [Google Scholar]
- 6. Thoma B. S., Vasquez K. M. (2003) Critical DNA damage recognition functions of XPC-hHR23B and XPA-RPA in nucleotide excision repair. Mol. Carcinog. 38, 1–13 [DOI] [PubMed] [Google Scholar]
- 7. Coin F., Oksenych V., Egly J. M. (2007) Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol. Cell 26, 245–256 [DOI] [PubMed] [Google Scholar]
- 8. Fagbemi A. F., Orelli B., Schärer O. D. (2011) Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair 10, 722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L. H., Fousteri M. I. (2007) Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III α in a cell-cycle-specific manner. Mol. Cell 27, 311–323 [DOI] [PubMed] [Google Scholar]
- 10. Ogi T., Limsirichaikul S., Overmeer R. M., Volker M., Takenaka K., Cloney R., Nakazawa Y., Niimi A., Miki Y., Jaspers N. G., Mullenders L. H., Yamashita S., Fousteri M. I., Lehmann A. R. (2010) Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 37, 714–727 [DOI] [PubMed] [Google Scholar]
- 11. Volker M., Moné M. J., Karmakar P., van Hoffen A., Schul W., Vermeulen W., Hoeijmakers J. H., van Driel R., van Zeeland A. A., Mullenders L. H. (2001) Sequential assembly of the nucleotide excision repair factors in vivo. Mol. Cell 8, 213–224 [DOI] [PubMed] [Google Scholar]
- 12. Sugasawa K., Shimizu Y., Iwai S., Hanaoka F. (2002) A molecular mechanism for DNA damage recognition by the xeroderma pigmentosum group C protein complex. DNA Repair 1, 95–107 [DOI] [PubMed] [Google Scholar]
- 13. Sugasawa K., Okamoto T., Shimizu Y., Masutani C., Iwai S., Hanaoka F. (2001) A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 15, 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Reardon J. T., Sancar A. (2002) Molecular anatomy of the human excision nuclease assembled at sites of DNA damage. Mol. Cell. Biol. 22, 5938–5945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Maltseva E. A., Rechkunova N. I., Gillet L. C., Petruseva I. O., Schärer O. D., Lavrik O. I. (2007) Crosslinking of the NER damage recognition proteins XPC-HR23B, XPA and RPA to photoreactive probes that mimic DNA damages. Biochim. Biophys. Acta 1770, 781–789 [DOI] [PubMed] [Google Scholar]
- 16. Camenisch U., Träutlein D., Clement F. C., Fei J., Leitenstorfer A., Ferrando-May E., Naegeli H. (2009) Two-stage dynamic DNA quality check by xeroderma pigmentosum group C protein. EMBO J. 28, 2387–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Min J. H., Pavletich N. P. (2007) Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature 449, 570–575 [DOI] [PubMed] [Google Scholar]
- 18. Nishi R., Okuda Y., Watanabe E., Mori T., Iwai S., Masutani C., Sugasawa K., Hanaoka F. (2005) Centrin 2 stimulates nucleotide excision repair by interacting with xeroderma pigmentosum group C protein. Mol. Cell. Biol. 25, 5664–5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Henricksen L. A., Umbricht C. B., Wold M. S. (1994) Recombinant replication protein A: expression, complex formation, and functional characterization. J. Biol. Chem. 269, 11121–11132 [PubMed] [Google Scholar]
- 20. Krasikova Y. S., Rechkunova N. I., Maltseva E. A., Petruseva I. O., Silnikov V. N., Zatsepin T. S., Oretskaya T. S., Schärer O. D., Lavrik O. I. (2008) Interaction of nucleotide excision repair factors XPC-HR23B, XPA, and RPA with damaged DNA. Biochemistry 73, 886–896 [DOI] [PubMed] [Google Scholar]
- 21. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., pp. 10.2–10.70, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 22. Bujalowski W. (2006) Thermodynamic and kinetic methods of analyses of protein-nucleic acid interactions. From simpler to more complex systems. Chem. Rev. 106, 556–606 [DOI] [PubMed] [Google Scholar]
- 23. Petruseva I. O., Tikhanovich I. S., Chelobanov B. P., Lavrik O. I. (2008) RPA repair recognition of DNA containing pyrimidines bearing bulky adducts. J. Mol. Recognit. 21, 154–162 [DOI] [PubMed] [Google Scholar]
- 24. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 25. Meisenheimer K. M., Koch T. H. (1997) Photocross-linking of nucleic acids to associated proteins. Crit. Rev. Biochem. Mol. Biol. 32, 101–140 [DOI] [PubMed] [Google Scholar]
- 26. Maillard O., Solyom S., Naegeli H. (2007) An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PLoS Biol. 5, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jansen L. E., Verhage R. A., Brouwer J. (1998) Preferential binding of yeast Rad4.Rad23 complex to damaged DNA. J. Biol. Chem. 273, 33111–33114 [DOI] [PubMed] [Google Scholar]
- 28. Guzder S. N., Sung P., Prakash L., Prakash S. (1998) Affinity of yeast nucleotide excision repair factor 2, consisting of the Rad4 and Rad23 proteins, for ultraviolet damaged DNA. J. Biol. Chem. 273, 31541–31546 [DOI] [PubMed] [Google Scholar]
- 29. Hey T., Lipps G., Sugasawa K., Iwai S., Hanaoka F., Krauss G. (2002) The XPC-HR23B complex displays high affinity and specificity for damaged DNA in a true-equilibrium fluorescence assay. Biochemistry 41, 6583–6587 [DOI] [PubMed] [Google Scholar]
- 30. Reardon J. T., Mu D., Sancar A. (1996) Overproduction, purification, and characterization of the XPC subunit of the human DNA repair excision nuclease. J. Biol. Chem. 271, 19451–19456 [DOI] [PubMed] [Google Scholar]
- 31. Wakasugi M., Sancar A. (1999) Order of assembly of human DNA repair excision nuclease. J. Biol. Chem. 274, 18759–18768 [DOI] [PubMed] [Google Scholar]
- 32. Araújo S. J., Nigg E. A., Wood R. D. (2001) Strong functional interactions of TFIIH with XPC and XPG in human DNA nucleotide excision repair, without a preassembled repairosome. Mol. Cell. Biol. 21, 2281–2291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wold M. S. (1997) Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 66, 61–92 [DOI] [PubMed] [Google Scholar]
- 34. Missura M., Buterin T., Hindges R., Hübscher U., Kaspárková J., Brabec V., Naegeli H. (2001) Double-check probing of DNA bending and unwinding by XPA-RPA: an architectural function in DNA repair. EMBO J. 20, 3554–3564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bunick C. G., Miller M. R., Fuller B. E., Fanning E., Chazin W. J. (2006) Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 45, 14965–14979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Krasikova Y. S., Rechkunova N. I., Maltseva E. A., Petruseva I. O., Lavrik O. I. (2010) Localization of xeroderma pigmentosum group A protein and replication protein A on damaged DNA in nucleotide excision repair. Nucleic Acids Res. 38, 8083–8094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Overmeer R. M., Moser J., Volker M., Kool H., Tomkinson A. E., van Zeeland A. A., Mullenders L. H., Fousteri M. (2011) Replication protein A safeguards genome integrity by controlling NER incision events. J. Cell Biol. 192, 401–415 [DOI] [PMC free article] [PubMed] [Google Scholar]