

Abstract
Glutamate is usually synthesized from acetyl coenzyme A (acetyl-CoA) via citrate, isocitrate, and 2-oxoglutarate. Genome analysis revealed that in Syntrophus aciditrophicus, the gene for Si-citrate synthase is lacking. An alternative pathway starting from the catabolic intermediate glutaconyl-CoA via 2-hydroxyglutarate could be excluded by genomic analysis. On the other hand, a putative gene (SYN_02536; NCBI gene accession no. CP000252.1) annotated as coding for isopropylmalate/citramalate/homocitrate synthase has been shown to share 49% deduced amino acid sequence identity with the gene encoding Re-citrate synthase of Clostridium kluyveri. We cloned and overexpressed this gene in Escherichia coli together with the genes encoding the chaperone GroEL. The recombinant homotetrameric enzyme with a C-terminal Strep-tag (4 × 72,892 Da) was separated from GroEL on a Strep-Tactin column by incubation with ATP, K+, and Mg2+. The pure Re-citrate synthase used only acetyl-CoA and oxaloacetate as the substrates. As isolated, the enzyme contained stoichiometric amounts of Ca2+ (0.9 Ca/73 kDa) but achieved higher specific activities in the presence of Mn2+ (1.2 U/mg) or Co2+ (2.0 U/mg). To determine the stereospecificity of the enzyme, [14C]citrate was enzymatically synthesized from oxaloacetate and [1-14C]acetyl-CoA; the subsequent cleavage by Si-citrate lyase yielded unlabeled acetate and labeled oxaloacetate, demonstrating that the enzyme is a Re-citrate synthase. The production of Re-citrate synthase by S. aciditrophicus grown axenically on crotonate was revealed by synthesis of [14C]citrate in a cell extract followed by stereochemical analysis. This result was supported by detection of transcripts of the Re-citrate synthase gene in axenic as well as in syntrophic cultures using quantitative reverse transcriptase PCR (qRT-PCR).
INTRODUCTION
Syntrophic bacteria participate in the anaerobic part of the carbon cycle by oxidizing short-chain fatty acids and aromatic compounds to acetate, CO2, and H2 or formate. This process is thermodynamically possible only in syntrophic coculture with methanogens or sulfate reducers to keep the steady-state concentration of H2 or formate at very low values (1–3). Without syntrophic bacteria, fatty acids and aromatic compounds would accumulate, decreasing the pH and causing a bottleneck in the anaerobic food chain.
Syntrophus aciditrophicus strain SBT (ATCC 700169), a strictly anaerobic, Gram-negative deltaproteobacterium, degrades benzoate and certain fatty acids in syntrophic association with hydrogen- or formate-using microorganisms (4–6). The strain also thrives axenically on crotonate, which is oxidized to acetate and reduced to cyclohexane carboxylate and some benzoate (7–9). Studies with 13C-labeled substrates suggested that degradation and synthesis of benzoate use the same pathway, in which glutaconyl coenzyme A (glutaconyl-CoA) serves as the central intermediate (10). The genome of S. aciditrophicus has been sequenced (5), and the metabolism has been partially reconstructed, although biochemical and metabolic approaches are required to fully understand the carbon and energy flow of the organism.
Glutamate, one of the main cellular building blocks, is generally synthesized from acetyl-CoA and oxaloacetate via citrate, isocitrate, and 2-oxoglutarate (Fig. 1). Genome analysis revealed that in S. aciditrophicus, no gene for the common Si-citrate synthase had been detected. An alternative pathway from oxaloacetate to 2-oxoglutarate via fumarate could be excluded because fumarate reductase/succinate dehydrogenase is absent in the genome of S. aciditrophicus. Therefore, we speculated that glutaconyl-CoA could be the precursor of 2-oxoglutarate for glutamate biosynthesis. Several bacteria living in anoxic environments within humans and animals are able to ferment glutamate via 2-oxoglutarate, (R)-2-hydroxyglutarate, (R)-2-hydroxyglutaryl-CoA, and glutaconyl-CoA (11–13).
Fig 1.

Proposed pathway of crotonate fermentation and three hypothetic routes to glutamate in S. aciditrophicus starting either from labeled acetate (red) or from labeled CO2 (blue). Bold compound names connected by bold arrows indicate the fermentation of crotonate, oxidatively to acetate and reductively to cyclohexane carboxylate and some benzoate, assuming a rapid equilibration between acetate, acetyl-CoA, and crotonyl-CoA. Names in regular text connected by thin arrows illustrate possible intermediates of glutamate biosynthesis, including the isotope patterns. Note that the isotope pattern of glutamate readily distinguishes between the pathways via Re- and Si-citrate synthase, whereas the pathways via the hydration of glutaconyl-CoA to (R)-2-hydroxyglutaryl-CoA and that via Re-citrate synthase would yield identical patterns.
The recent discovery of a gene encoding Re-citrate synthase in Clostridium kluyveri and the detection of a homologue in S. aciditrophicus SYN_02536 (49% sequence identity between the deduced proteins), annotated as isopropylmalate/homocitrate/citramalate synthase genes (14, 15), prompted us to consider an alternative pathway that could proceed via citrate. Moreover, there are an increasing number of anaerobic bacteria containing Re-citrate synthase (16–20). As shown in Fig. 1, Re-citrate synthase catalyzes the addition of acetyl-CoA to oxaloacetate from the Re-side (from the front in Fig. 1) and Si-citrate synthase from the Si-side (from the back in Fig. 1). Therefore, both enzymes most likely have different active sites and are phylogenetically unrelated. The work of Li et al. (15) demonstrated that Si-citrate synthases form a family different from that of the Re-citrate synthases, which belong to the family of homocitrate and isopropylmalate synthases. Figure 1 illustrates that isotope labeling cannot distinguish between the pathway via Re-citrate synthase and that via glutaconyl-CoA. Therefore, we used a biochemical approach and expressed the SYN_02536 gene of S. aciditrophicus in Escherichia coli and characterized the purified protein as Re-citrate synthase. Bioinformatics helped to exclude the pathway via hydration of glutaconyl-CoA to (R)-2-hydroxyglutaryl-CoA.
MATERIALS AND METHODS
Bacterial strain and chemicals.
Citrate lyase of Klebsiella pneumoniae (Si-face specific), Si-citrate synthase of porcine heart, malate dehydrogenase, acetyl-CoA synthetase, and CoA were purchased from Roche (Mannheim, Germany) and Sigma-Aldrich (Munich, Germany). S. aciditrophicus SBT (ATCC 700169T) is available from the McInerney laboratory. Acetyl-CoA was synthesized from CoA and acetic anhydride by an improvement of the method of Simon and Shemin (21, 22).
Heterologous overexpression of the putative gene for Re-citrate synthase.
The putative gene for Re-citrate synthase (SYN_02536; gene accession no. CP000252.1) was amplified from chromosomal DNA of S. aciditrophicus by PCR. The PCR fragments were cloned into the entry vector pE_blue using site-specific overhangs created by digestion of both the PCR fragment and the cloning vector with LguI and ligation with T4 ligase in a one-step reaction. The cloned gene verified by DNA sequencing was subcloned into the pASK-IBA3plus vector (IBA GmbH, Göttingen, Germany), which has a C-terminal-fused Strep-tag II peptide for purification of the gene product. For overproduction of the recombinant protein, the plasmid was transformed into E. coli BL21 CodonPlus (DE3)-GroEL, harboring an extra plasmid encoding the chaperone GroEL. The cells were grown in Tryptone-phosphate medium (2% Bacto tryptone, 0.2% Na2HPO4, 0.1% KH2PO4, 0.8% NaCl, 1.5% yeast extract, 0.2% glucose) to limit inclusion body formation (23) at 37°C under oxic conditions until the mid-exponential phase (optical density at 600 nm [OD600] of 0.5 to 0.7). The expression of the gene was induced with 0.4 μM anhydrotetracycline, and the chaperone gene was induced with 0.1 mM isopropyl-β-thiogalactopyranoside (IPTG). After overnight growth, the cells were harvested and washed in 50 mM potassium phosphate (pH 7.0) and reinoculated in fresh medium containing 60 μM chloramphenicol to inhibit new protein synthesis and incubated for 2 h at room temperature for the chaperone to fold the protein correctly (24).
Purification of recombinant Re-citrate synthase.
Recombinant Re-citrate synthase was purified oxically using a Strep-Tactin column (IBA GmbH, Göttingen, Germany) at ca. 8°C. The harvested E. coli cells were suspended in equilibration buffer (50 mM potassium phosphate [pH 7.4], 75 mM NaCl) and disrupted by sonication. Cell debris and membranes were removed by ultracentrifugation at 100,000 × g for 1 h. The supernatant was loaded on the affinity Strep-Tactin column. The column was washed with at least 10 column volumes of equilibration buffer. To release the contaminant chaperone from the target protein, the column resin was incubated with dissociation buffer (20 mM HEPES-NaOH [pH 7.0], 10 mM MgCl2, 10 mM ATP, 150 mM KCl) for 2 h at 8°C followed by washing with two column volumes of dissociation buffer (25). The protein was eluted with a mixture of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 2.5 mM d-desthiobiotin. Re-citrate synthase was separated from traces of copurified chaperone by gel filtration (HiLoad26/60 Superdex200; GE Healthcare, München, Germany) in 50 mM Tris-HCl (pH 8.0)-150 mM NaCl. The purified protein was concentrated by ultrafiltration and stored at −80°C. Purification steps were analyzed by SDS-PAGE. The protein concentration was determined by the Bradford method (26). To determine the molecular mass of the purified recombinant Re-citrate synthase, a sample was loaded on the Superdex column (see above) with a flow rate of 2 ml/min with 50 mM Tris-HCl (pH 8.0)–150 mM NaCl. Albumin (66 kDa), alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), and ferritin (440 kDa) were used as molecular mass standards.
Determination of citrate synthase activity.
Citrate synthase activity was measured at room temperature under air or anoxically in a Coy anaerobic chamber in an atmosphere consisting of 95% N2 and 5% H2. The assay mixture contained 100 mM Tris-HCl (pH 8.0), 0.2 mM 5,5′-dithiobis(2-nitrobenzoate) (DTNB), 0.2 mM oxaloacetate, 0.2 mM MnCl2, and 0.2 mM acetyl-CoA. The reaction was started by addition of the enzyme and monitored spectrophotometrically by the formation of the anion of thionitrobenzoate from DTNB and CoA at 412 nm (Δε412, 14.2 mM−1 cm−1) (27). The reaction was also measured in the absence of DTNB by the decreasing absorbance of the thioester bond of acetyl-CoA and the enolate of oxaloacetate at 232 nm (Δε232, 5.4 mM−1 cm−1) (28, 29).
Determination of the stereospecificity of citrate synthase.
The synthesis of [14C]citrate was performed under oxic conditions following a previously described procedure (15). The reaction mixture (1 ml) contained 50 mM Tris-HCl (pH 8.0), [1-14C]acetate (4.8 × 104 dpm for Si-citrate synthase, 1.08 × 106 dpm for the recombinant Re-citrate synthase), 1 mM ATP, 0.1 mM CoA, 2 mM (S)-malate, 1 mM NAD+, 20 mM MgCl2, 100 mM KCl, 0.2 mM MnCl2, 3 U acetyl-CoA synthetase, 1 U malate dehydrogenase, and 1 U Si-citrate of pig heart or 1 U recombinant Re-citrate synthase. The reaction was followed by monitoring the formation of NADH spectrophotometrically at 340 nm. After overnight incubation at room temperature, the reaction was stopped by heating the mixture to 95°C for 10 min. Proteins were removed by centrifugation, and the supernatant was applied to an ion-exchange column (Dowex 1×8 formate, 200-to-400 mesh; Sigma-Aldrich, München, Germany). After loading the sample, the column was washed with 10 volumes of 1 M formic acid to elute unreacted [14C]acetate. Citrate was eluted with 4 M formic acid. A 50-μl sample of each fraction was added to 5 ml Quicksave A scintillation fluid (Zinsser Analytic; Frankfurt am Main, Germany), and 14C radioactivity was measured by scintillation counting. The radioactive fractions were combined and brought to dryness by evaporation under vacuum, and the residue was dissolved in 500 μl of 50 mM Tris-HCl (pH 8.0).
For the cleavage of citrate to acetate and malate, the reaction mixture (total volume, 1.0 ml) contained 50 mM Tris-HCl (pH 8.0), 0.3 mM NADH, 0.2 mM MgCl2, 18 U malate dehydrogenase, 0.25 U Si-citrate lyase (K. pneumoniae), and the [14C]citrate that was synthesized and purified as described above. The reaction was observed by monitoring the oxidation of NADH at 340 nm spectrophotometrically. After incubation at room temperature for 30 min, the reaction was stopped by heating of the mixture to 95°C for 5 min. The supernatant was applied to a column (Dowex 1×8 formate, 200-to-400 mesh). Acetate was eluted with 0.2 M formic acid, and malate was eluted with 1 M formic acid (15). The radioactivity of each fraction was determined by scintillation counting as described above.
Metal analysis.
An inductively coupled plasma-optical emission spectrometer (ICP-OES) was used for analyzing metal ions in the purified recombinant Re-citrate synthase. The enzyme was inactivated by adding 100 μM EDTA (pH 8.0) and incubated at room temperature for 10 min. To remove EDTA, the sample was applied to a PD-10 desalting column (GE Healthcare, Munich, Germany). Enzyme with and without EDTA treatment as well as buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl) was sent to The Chemical Analysis Laboratory—Center for Applied Isotope Studies (University of Georgia, Athens) for ICP-OES analysis.
Stereospecificity of native Re-citrate synthase.
S. aciditrophicus cells grown on crotonate (3 g) were suspended in 50 mM Tris-HCl (pH 8.0) and disrupted by sonication. After ultracentrifugation at 100,000 × g for 1 h, 0.2 ml of the cell-free supernatant (25 mg protein/ml) was used for synthesis of [14C]citrate by the same method described above but in the presence of additional 1 mM CoA, 0.2 mM CoCl2, and 0.2 mM phenanthroline (total volume, 1 ml). Isolation and stereochemical determination of [14C]citrate were performed as shown above. To ascertain the identities of the isolated [14C]citrate and [14C]malate, both compounds were analyzed by thin-layer chromatography (TLC). The solvent system was isobutanol-formic acid-H2O (30/5/7.5). The radioactive spots on the TLC plate (TLC silica gel 60 F254 aluminum sheet; Merck, Germany) were detected by a Strom 860 molecular imager (Molecular Dynamics, Sunnyvale, CA). [14C]citrate made by the recombinant Re-citrate synthase was used as a control.
RNA isolation.
S. aciditrophicus was grown axenically on 20 mM crotonate and syntrophically with Methanospirillum hungatei on 20 mM crotonate, 12 mM benzoate, and 10 mM cyclohexane carboxylate. Once in the stationary phase, the cells were rapidly cooled in a dry ice-ethanol bath and collected by centrifugation at 8,000 × g for 15 min, resuspended in RNAlater (Qiagen, Inc., Valencia, CA), and then stored at −80°C. Total RNA was obtained by using the RNeasy minikit (Qiagen). DNA was removed by on-column DNA digestion with the RNase-free DNase set (Qiagen).
qRT-PCR.
RNA was verified to be free of DNA contamination by PCR without reverse transcriptase with the same primer set used for quantitative reverse transcriptase PCR (qRT-PCR). The primers used to detect the Re-citrate synthase gene were SYN_02536F (CCGCTGCAGACGGGCAAACT) and SYN_02536R (GCGATCTGCGTTGCTTCGGC); for the DNA gyrase, the primers were SYN_02049F (TGTGGACGGTTCTCATATCC) and SYN_02049R (TCCTGAATGGTGAAAGAGGG). The PCR was performed using the MyIQ real-time PCR system (Bio-Rad, Hercules, CA) and the iScriptT one-step RT-PCR kit with SYBR green. Each reaction mixture (25-μl total volume) consisted of 12.5 μl IQ SYBR green supermix (Bio-Rad), 8 μl of nuclease-free water, 1.5 μl (10 μM) of each primer, 0.5 μl of reverse transcriptase, and 1.0 μl of RNA. The qPCR generally follows a standard one-step protocol consisting of 10 min at 50°C, 5 min at 95°C, followed by 45 cycles of 95°C for 30 s, 56°C for 30 s, and 72°C for 30 s. No primer dimers were observed in any of the primer sets, as determined by melting curve analysis of qPCR amplicons. Amplification efficiency was determined by testing the primers against decreasing concentrations of DNA. The expression level of SYN_02536 (target gene) was normalized to the expression level of SYN_02049 (DNA gyrase B gene) as the reference gene (30, 31), using the equation NE = (Ereference)CTreference/(Etarget)CTtarget, where NE is normalized expression (E), and CT is the threshold cycle. DNA thermal denaturation curves showed a single peak, indicating that each primer set is specific to one gene. BLAST results showed only a single hit for each primer set, either SYN_02536 or SYN_02049. This DNA gyrase gene was used as the reference gene because there is only a single copy of this gene in the chromosome, it has to be expressed at each replication cycle, and its expression was not significantly different (P > 0.40) under any of the four growth conditions.
RESULTS
Glutaconyl-CoA as precursor of glutamate?
We searched the genome of S. aciditrophicus for genes encoding enzymes that catalyze the hydration of glutaconyl-CoA to (R)-2-hydroxyglutaryl-CoA, liberation of (R)-2-hydroxyglutarate, and its oxidation to 2-oxoglutarate (Fig. 1). A putative glutaconyl-CoA hydratase/2-hydroxglutaryl-CoA dehydratase is encoded by 2 or 3 genes preceded by an activator gene, whose deduced amino acid sequences are related to those of the ATP-dependent benzoyl-CoA reductase. Two of the three genes of S. aciditrophicus annotated as benzoyl-CoA reductase/2-hydroxyglutaryl-CoA dehydratase (SYN_00368, SYN_00369, and SYN_00370) might code for a heterodimeric 2-hydroxyglutaryl-CoA dehydratase similar to those of Clostridium symbiosum or Acidaminococcus fermentans (32). Two genes (SYN_00369 and SYN_00370) are more related to those of the heterodimeric 2-hydroxyisocaproyl-CoA dehydratase of Clostridium difficile, from which the crystal structure is known (33). In both subunits, the three cysteines forming a [4Fe-4S] cluster with one open coordination site are conserved, but the essential glutamate at the active site of the A subunit is replaced by lysine or arginine. In the same transcription unit of these genes, a gene annotated as an activator of 2-hydroxyacyl-CoA dehydratase (SYN_00371) is present. The deduced amino acid sequence of the gene reveals that the 2 cysteines that carry the [4Fe-4S] cluster of the homodimeric activator of 2-hydroxyglutaryl-CoA dehydratase are conserved (34). Although a functional activator appears to be present in S. aciditrophicus, the lack of a conserved active site makes it unlikely that the dehydratase is able to catalyze the hydration of glutaconyl-CoA to 2-hydroxyglutaryl-CoA. Furthermore, this hydration is thermodynamically unfavorable, as has been shown by equilibrium measurements (35).
A gene coding for glutaconate CoA-transferase does not exist in the genome of S. aciditrophicus, but the conversion of 2-hydroxyglutaryl-CoA to 2-hydroxyglutarate could be performed by a hydrolase. The final oxidation to 2-oxoglutarate could be catalyzed by a putative (R)-2-hydroxyglutarate dehydrogenase (SYN-00123 or SYN-01083), in which catalytically important amino acid residues (Arg52, Arg235, Glu264, and His297) of the dehydrogenase of A. fermentans (36) are conserved.
Sequence analysis of the putative gene for Re-citrate synthase.
The gene SYN_02536, annotated as coding for isopropylmalate/homocitrate/citramalate synthase, is composed of 1,905 bp (GC content of 51%, while the GC content is 32% in the gene coding for Re-citrate synthase in C. kluyveri) and codes for 634 amino acids with a calculated molecular mass of 72.4 kDa. The gene with leucine as the start codon was cloned, but expression in E. coli failed. Closer inspection of the nucleotide sequence revealed a Shine-Dalgarno sequence around this leucine codon and suggested a later start by 15 nucleotides with a methionine codon. The shortened gene consists of 1,890 nucleotides and is predicted to encode 629 amino acids with a calculated mass of 71.8 kDa. The deduced amino acid sequence showed 49% identity to that of the Re-citrate synthase of C. kluyveri (15) but only ca. 20% identities to those of the Re-citrate synthases of Syntrophomonas wolfei (37), Dehalococcides sp. strain VS (19), and isopropylmalate synthase of Mycobacterium tuberculosis, the only Re-specific enzyme of this family for which the crystal structure has been reported (38). BLAST results of the S. aciditrophicus genome show that SYN_02536 has 27% identity to SYN_00090 and 22% identity to SYN_02194. Both of these genes are annotated as coding for 2-isopropylmalate synthases.
Interestingly, the C terminus of Re-citrate synthase of S. aciditrophicus is around 150 amino acids longer than those of other Re-citrate synthases. Comparison of the amino acid sequence with sequences in the data bank revealed additional levels of identities (50 to 60%) to several pyruvate carboxyltransferase domains of Deltaproteobacteria and anaerobic Firmicutes. These enzymes catalyze the transfer of CO2 from carboxy-biotin to pyruvate and share with Re-citrate synthase the substrate or product oxaloacetate.
Heterologous overproduction and purification of Re-citrate synthase.
To study the biochemical properties of Re-citrate synthase, we cloned the revised Re-citrate synthase gene and overproduced the recombinant protein carrying a C-terminal Strep-tag in E. coli. This protein was always found in inclusion bodies, in spite of efforts to use different overproduction conditions. Therefore, coexpression with the gene of GroEL, a molecular chaperone in E. coli, was used to improve solubility of the target protein. The recombinant plasmid was transformed into an E. coli strain, which contains a plasmid for the production of GroEL. Coexpression of both genes resulted in over 50% soluble Re-citrate synthase, as estimated from the intensities of the Coomassie-stained band in SDS-PAGE. The cell extract was loaded on a Strep-Tactin column and washed with column buffer followed by incubation with MgCl2-ATP-KCl to remove the copurifying chaperone. Finally, Re-citrate synthase was eluted with a buffer containing 2.5 mM dethiobiotin. The purified Re-citrate synthase showed a molecular mass of approximately 65 kDa on SDS-PAGE (Fig. 2), which did not agree well with the predicted molecular mass of 72,891.16 Da calculated from the deduced amino acid sequence together with the 1-kDa Strep-tag II peptide in the absence of the N-terminal methionine. Electrospray-time of flight mass spectrometry (ESI-TOF MS) revealed the same mass (72,891.78 Da) as that predicted from the amino acid sequence. The apparent molecular mass of active Re-citrate synthase was determined by size exclusion chromatography as 271 kDa, indicating a homotetrameric quaternary structure (4 × 72,892 Da = 292 kDa).
Fig 2.

SDS-PAGE of recombinant Re-citrate synthase. Lanes: 1, cell lysate; 2, cell extract after induction with anhydrotetracycline (AHT) (200 μg/liter) and 0.1 mM IPTG; 3, pellet; 4, flowthrough of the Strep-Tactin column; 5, washing; 6, 2nd washing after dissociation; 7, elution; 8, gel filtration; M, molecular mass markers.
Substrate specificity and catalytic properties.
The Re-citrate synthase gene was originally annotated as coding for isopropylmalate/homocitrate/citramalate synthase. The sequence-related enzymes are all Re-face stereospecific with respect to their substrates, 2-oxo-3-methylbutanoate, 2-oxoglutarate, and pyruvate. Therefore, these substrates and oxaloacetate, in case of citrate synthase, were examined using the DTNB assay. Table 1 shows that the purified gene product catalyzed the formation of CoA from acetyl-CoA (Km, 130 μM) only in the presence of oxaloacetate (Km, 85 μM). The other tested 2-oxoacids together with acetyl-CoA gave neither activity nor the combination of propionyl-CoA and oxaloacetate. The activity of Re-citrate synthase was also measured by the absorbance change of the thioester bond of acetyl-CoA and the enolate of oxaloacetate at 232 nm (Δε, 5.4 mM−1 cm−1) (29), yielding around 30% higher values than by the DTNB assay, indicating that DTNB slightly inhibits the enzyme. Other weak inhibitors were 10 mM ATP (30%), 10 mM citrate (40%), or 10 mM 2-oxoglutarate (40%), but not 5 mM NADH. p-Hydroxymercuribenzoate (0.1 mM) completely inactivated the enzyme.
Table 1.
Determination of substrate specificity of Re-citrate synthase
| 2-Oxoacid substrate + acetyl-CoA | Corresponding enzyme | MnCl2 (0.2 mM) | Sp act (U/mg) |
|---|---|---|---|
| Pyruvate | Citramalate synthase | +/− | NDa |
| 2-Oxoglutarate | Homocitrate synthase | +/− | ND |
| 2-Oxo-3-methylbutanoate | Isopropylmalate synthase | +/− | ND |
| Oxaloacetate | Re-Citrate synthase | + | 1.2 |
| Oxaloacetate | Re-Citrate synthase | − | 0.72 |
| Oxaloacetate + propionyl-CoAb | Methylcitrate synthase | + | ND |
ND, not detected.
Acetyl-CoA was replaced by propionyl-CoA.
All of the assays were performed either in the presence or absence of 0.2 mM MnCl2. A lag phase was detected when 0.2 mM MnCl2 was absent in the assay mixture. Preincubation of the enzyme with 0.2 mM metal ion such as Mn2+ or Co2+ helped to eliminate the lag phase. Co2+ even increased the activity by about 70% compared with that obtained with Mn2+ (Fig. 3). The lag was probably not due to a slow activation by metal ions from impurities of the chemicals used in the assay mixture, because the use of ultrapure Tris (Applichem, Germany) or preincubation of Re-citrate synthase in the absence of metal ions in the buffer for 10 min did not change the activity. The highest specific activity of 2.0 U/mg was obtained in the presence of 0.2 mM CoCl2. The enzyme was completely inactivated by 0.2 mM EDTA. The EDTA-inactivated enzyme regained activity by addition of Ca2+, Zn2+, Mn2+, or Co2+ ions at final concentrations of 0.2 mM. Co2+ was the most effective ion in restoring activity, whereas 0.2 mM Zn2+ and 1 mM Ca2+ resulted in lag phases similar to that of the enzyme as isolated. The EDTA-inactivated enzyme regained ca. 20% activity by incubation with 1 mM Ca2+, whereas Mg2+ had no effect at all. ESI-TOF mass spectrometry (see above) revealed that metal ions are not tightly incorporated into the enzyme, because the observed mass matched exactly that calculated from the amino acid sequence. To clarify the metal content, the recombinant enzyme treated with and without EDTA was analyzed by inductively coupled plasma optical emission spectrometry (ICP-OES) (Table 2). The high magnesium content (6.8 atoms/mol subunit) stems from the removal of the chaperone from Re-citrate synthase using 10 mM ATP and 10 mM MgCl2. Since Mg2+ was not able to restore the activity of the EDTA-treated enzyme, the presence of Ca2+ ion in Re-citrate synthase (0.88 atom/mol subunit) could be in part responsible for the activity without addition of Mn2+ or Co2+.
Fig 3.
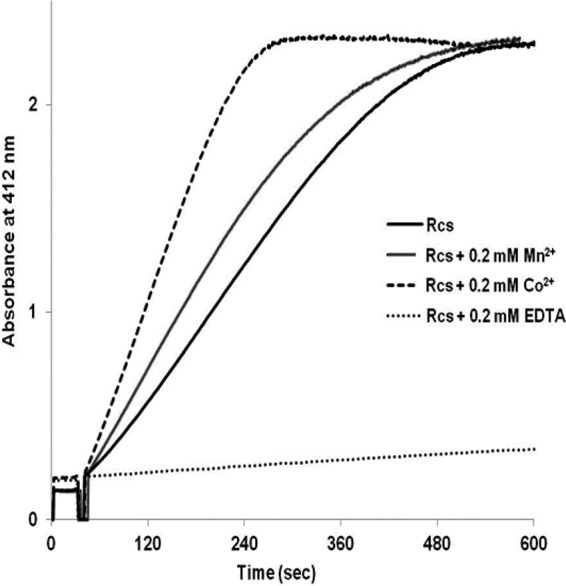
Effect of metal ions and EDTA on product formation. Recombinant Re-citrate synthase (Rcs) as isolated from E. coli was preincubated in the DTNB assay mixture together with 0.2 mM metal ions or 0.2 mM EDTA at room temperature for 30 min. Afterwards, the reaction was started by the addition of oxaloacetate.
Table 2.
Metal analysis of Re-citrate synthase
| Metal | Buffer (mg/liter) |
Re-Citrate synthase concn (mg/liter) |
Concn of metal/Re-citrate synthase subunit(mol/mol) |
||
|---|---|---|---|---|---|
| −EDTAa | +EDTAb | −EDTAa | +EDTAb | ||
| Co | 0.0083 | 0.006 | 0.0037 | ||
| Mg | 0.20 | 2.9 | 0.23 | 6.8 | 0.039 |
| Mn | 0.001 | 0.0027 | 0.001 | 0.0019 | 0 |
| Ni | 0.014 | 0.026 | 0.018 | 0.012 | 0.002 |
| Zn | 0.051 | 0.097 | 0.054 | 0.043 | 0.002 |
| Ca | 0.67 | 1.23 | 0.98 | 0.88 | 0.27 |
Re-citrate synthase concentration of 1.17 mg/ml.
Re-citrate synthase concentration of 2.13 mg/ml.
Upon exposure to air, the activity of the anoxically purified enzyme supplemented with 0.2 mM Mn2+ or Co2+ at 4°C decreased immediately to 70% and after 24 h to 50% of the anoxically determined activity and thereafter remained constant for 14 days. An explanation of this nonlinear behavior could be the oxidation of cysteine and methionine residues, which partially inactivates the enzymatic activity. Reactivation by incubation with dithothreitol was not attempted. The oxically purified enzyme lost 20% of its activity after each cycle of freezing at −80°C and thawing. This was caused by protein aggregation as revealed by native PAGE.
Stereospecificity of Re-citrate synthase.
Condensation of oxaloacetate with acetyl-CoA leads to the identical product, citrate, regardless of whether Si- and Re-citrate synthase has catalyzed the reaction. Only by use of isotopically labeled substrates stereochemically different citrates are formed. Starting with [1-14C]acetyl-CoA and unlabeled oxaloacetate, Re-citrate synthase gives (R)-[1-14C]citrate, whereas Si-citrate synthase yields (S)-[5-14C]citrate, which can be distinguished by cleavage with the Si-specific citrate lyase of Klebsiella pneumoniae (15). Thus, citrate was enzymatically synthesized from [1-14C]acetate, oxaloacetate, CoA, ATP, and Mg2+ catalyzed by acetyl-CoA synthetase and either Re- or Si-citrate synthase. The labeled citrates were isolated by ion-exchange chromatography and cleaved to oxaloacetate and acetate by Si-specific citrate lyase. In case of Re-citrate synthase, practically no labeled acetate was found and almost the entire radioactivity was detected in oxaloacetate isolated as malate (Fig. 4). In the control with Si-citrate synthase, the entire radioactivity was recovered in the acetate as expected. The result indicates that the gene SYN_02536 of S. aciditrophicus, annotated as an isopropylmalate/homocitrate/citramalate synthase gene, encodes a Re-citrate synthase.
Fig 4.
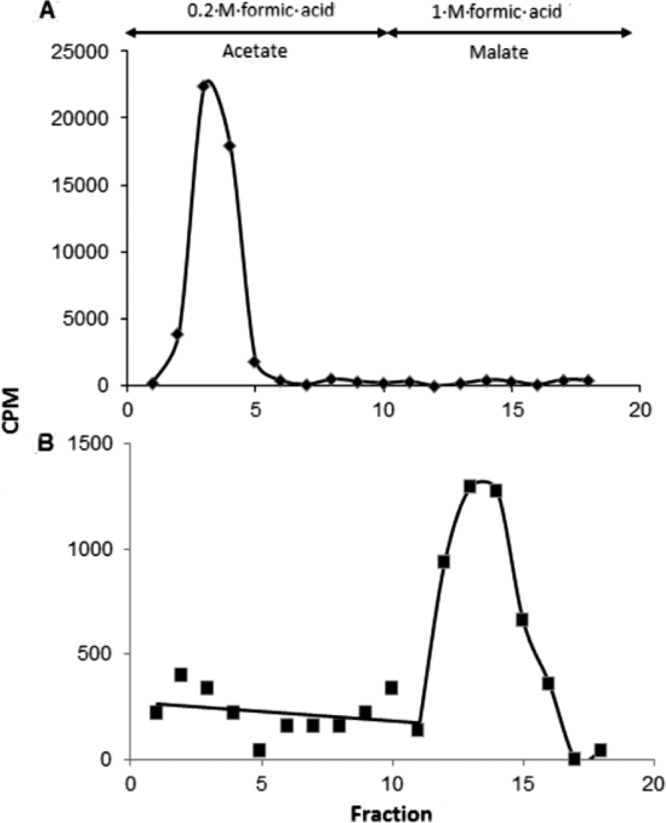
Determination of the stereospecificity of citrate synthase by analysis of [14C]citrate synthesized from [1-14C]acetyl-CoA by Si-citrate synthase of pig heart (A) and the recombinant Re-citrate synthase of S. aciditrophicus (B). The [14C]citrates were converted to acetate and malate in the presence of Si-citrate lyase, malate dehydrogenase, and NADH. Acetate was eluted from the Dowex 1×8 column by 0.2 M formic acid, whereas malate was eluted by 1 M formic acid.
Identification and stereospecificity of Re-citrate synthase in vivo.
To define the active expression of the gene encoding Re-citrate synthase in S. aciditrophicus cells, we tried to raise antibodies against Re-citrate synthase. SDS-PAGE gel fragments with pure recombinant Re-citrate synthase were used as the antigen for antibody production in rabbits. However, antibodies were not produced, which suggested that the gel fragments of Re-citrate synthase were not immunogenic. Attempts to purify the native protein of S. aciditrophicus by using ion-exchange chromatography were performed. These attempts were not successful because other enzymes and components in the purification fractions interfered with the ability to detect Re-citrate synthase activity using the DTNB assay. For instance, there are 2 copies of an acetyl-CoA acetyltransferase gene (SYN_01681 and SYN_02642) in the genome of S. aciditrophicus and the enzymes encoded by those genes react with acetyl-CoA in the assay mixture to yield acetoacetyl-CoA and free CoA that is detected by DTNB. Protein bands on SDS-PAGE with sizes similar to that of Re-citrate synthase were analyzed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry. None of them matched the amino acid sequence of Re-citrate synthase.
To ascertain the existence of Re-citrate synthase in vivo, S. aciditrophicus cells were grown on different substrates and expression of the gene was determined by qRT-PCR as described in Materials and Methods. Expression of SYN_02536, originally annotated as an isopropylmalate/homocitrate/citramalate synthase gene but identified as an Re-citrate synthase gene by our study, was detected under all growth conditions. Compared to the expression of the DNA gyrase subunit gene B (gyrB) (SYN_02049), SYN_02536 expression was found to be highest when the strain was grown axenically on crotonate, with an abundance of 25 times higher than that of gyrB. During syntrophic growth, SYN_02536 was expressed at similar levels under crotonate, benzoate, or cyclohexane carboxylate, always almost 20 times more abundant than gyrB.
The existence and stereospecificity of native Re-citrate synthase were proven by analysis of the [14C]citrate synthesized by cell extracts of S. aciditrophicus. To accumulate [14C]citrate effectively, CoCl2 was added and the iron-sulfur cluster-containing aconitase was suppressed with phenanthroline and oxic conditions. The recombinant Re-citrate synthase was used as a control. Around 3.4% of the added [14C]acetate was converted to [14C]citrate, which was isolated and cleaved to oxaloacetate and acetate. After enzymatic conversion of oxaloacetate to malate, the products were separated by ion-exchange chromatography (see Fig. S1A and B in the supplemental material) and their identities were confirmed by TLC with Rf values of 0.15 for citrate and 0.30 for malate (see Fig. S1C). The entire radioactivity was found in malate, and practically no labeled acetate was detected (Fig. 4). This result demonstrates that the enzyme catalyzing the synthesis of citrate in vivo is an Re-specific citrate synthase.
DISCUSSION
In this work, we characterized the product of the SYN_02536 gene annotated as isopropylmalate/homocitrate/citramalate synthases of S. aciditrophicus as Re-citrate synthase. The enzyme as isolated is active and contains stoichiometric amounts of Ca2+ (0.9/subunit). Mn2+ and Co2+ eliminated the lag phase observed in the progress curve and resulted in linear kinetics and higher rates; Co2+ was most effective in stimulating activity. The Co2+ requirement correlates well with the observation that this metal ion together with 2-oxoglutarate induced maximum acetyl-CoA enolase activity (the exchange of the methyl protons with those of the solvent) of native Re-citrate synthase of Clostridium acidurici (39). The addition of EDTA to the reaction mixture completely inhibited the synthase as well as the enolase activities.
Amino acid sequence comparisons show that Re-citrate synthase belongs to the triosephosphate isomerase (TIM) barrel domain metallolyase superfamily, which includes isopropylmalate synthase (LeuA), homocitrate synthase, citramalate synthase, and pyruvate carboxylase, as well as Re-citrate synthase. These enzymes contain a divalent cation-binding site at the catalytic center in the domain. Furthermore, three invariant residues—aspartate, arginine, and glutamate—are strictly conserved in diverse TIM-barrel metalloenzymes, which catalyze cleavage of carbon-carbon bonds in various organic substrates (40). Re-citrate synthase is structurally related to this superfamily, but only two residues (aspartate and arginine) are conserved. An enzyme, of which the crystal structure is known and is closely related to Re-citrate synthase, is the Zn2+-containing enzyme LeuA that catalyzes the first step of leucine biosynthesis of M. tuberculosis (38). The metal-binding amino acid residues of LeuA are conserved in the amino acid sequences of the Re-citrate synthases not only of S. aciditrophicus but also of C. kluyveri and Dehalococcoides strain CBDB1 (see Fig. S2 in the supplemental material). In LeuA, Zn2+ plays a role in substrate binding and thereby polarizing the carbonyl group of the oxo-substrate, whereas, in Si-citrate synthase, a histidine residue performs these tasks (41). Recently, Ca2+ was assigned as the metal ion instead of the K+ ion in coenzyme B12-dependent diol dehydratase on the basis of quantum mechanical/molecular mechanical calculations (42) and by removal with EDTA followed by reconstitution (43). Even though Ca2+ ion was detected in Re-citrate synthase by ICP-OES, we still do not have a clear picture of its coordination in the enzyme and catalytic function. To date, no crystal structure of a Re-citrate synthase is known and biochemical evidence is not sufficient to explore the catalytic mechanism of the enzyme and the function of the metal ion.
Up to now, Re-citrate synthase has been reported only in some anaerobic microorganisms, while Si-citrate synthase is the predominant enzyme in bacteria and eukaryotes up to mammals. Specific activities similar to that of Re-citrate synthase of S. aciditrophicus (2 U/mg) were also observed for the enzymes of C. kluyveri (15), Dehalococcoides (19), and C. acidurici (44). On the other hand, Si-citrate synthases show much higher specific activities: 20 U/mg (45) for the Si-citrate synthase of G. sulfurreducens, an anaerobic metal-reducing bacterium, and as high as 100 U/mg (46) for the pig heart enzyme. Owing to the higher specific activity of Si-citrate synthases, we speculate that Re-citrate synthases were already present during the emergence of life with a biosynthetic anabolic function. This view is supported by its close relationship to pyruvate carboxylase, an enzyme from the central metabolism involved in the synthesis of oxaloacetate. Later, the more efficient Si-citrate synthase evolved, which by chance catalyzed the attack of acetyl-CoA at the oxo group of oxaloacetate from the opposite site as Re-citrate synthase. Thus, Si-citrate synthase was selected by modern organisms, whereas Re-citrate synthase survived only in a few strict anaerobes. This change in enzyme type was possible because the product citrate remained the same, regardless from which side the oxo group was attacked, and no subsequent enzyme of the metabolic pathway had to be modified. In particular, aconitase, known to be stereospecific for the Re-carboxymethyl group of citrate, required no substitution by a novel enzyme. However, only the Re-types of homocitrate synthase (47), isopropylmalate synthase (48), and citramalate synthase (49) provide the correct substrates with R-configuration for the aconitases (50) mediating the isomerization of the 3-hydroxy acids to the 2-hydroxy acids, the precursors of lysine, leucine, and isoleucine, respectively. Therefore, these three synthases could not readily be exchanged by the more efficient Si-type enzymes and were retained during evolution.
Recently, the existence of Re-citrate synthase has been proven by isotopomer-assisted metabolic analysis from several bacteria by culturing with [1-13C]acetate or 13C-bicarbonate and tracing and analyzing the labeling patterns of metabolites (Fig. 1). Glutamate derived from [1-13C]acetate was mainly labeled on C-1 (α-carboxyl group) in Dehalococcoides strains (19, 51, 52), and this labeling pattern was consistent with that of glutamate in Thermoanaerobacter sp. (16) and Desulfovibrio vulgaris (18). Especially, the pattern of glutamate (C-1 and C-3) in Dehalococcides strains is exactly in accord with the predicted pattern (Fig. 1). It is assumed that in these organisms glutamate is synthesized via Re-citrate synthase.
In our study, the presence of the Re-type citrate synthase and the catalytic properties of the enzyme have been shown. Therefore, we anticipate that glutamate could be synthesized via isocitrate and 2-oxoglutarate in S. aciditrophicus. To assess the pathway of glutamate biosynthesis, labeling patterns of glutamate and other amino acids extracted of S. aciditrophicus cells grown on unlabeled crotonate, together with either [1-14C]acetate or [13C]bicarbonate, are currently in progress. Unfortunately, the hypothetical pathway via glutaconyl-CoA leads to a labeling pattern identical to that obtained via Re-citrate synthase (Fig. 1). We think, however, that our bioinformatic analysis largely excluded this pathway for glutamate biosynthesis. Interestingly, genes related to those of 2-hydroxyacyl-CoA dehydratase with its activator as well as of ATP-dependent benzoyl-CoA reductase are present in almost all anaerobically thriving bacteria or archaea (53). Therefore, these genes may code for enzymes with unknown functions, but certainly not for the benzoyl-CoA reductase present in S. aciditrophicus, which is ATP independent (54). To elucidate these functions would be a promising project for future research.
Supplementary Material
ACKNOWLEDGMENTS
We thank Milko Velarde (Max-Planck-Institut für Biochemie, Martinsried, Germany) for the ESI-TOF measurement, Thorsten Selmer (Fachhochschule Aachen, Germany) for providing the cloning vector pE_blue, and Jörg Kahnt (Max-Planck-Institut für Terrestrische Mikrobiologie, Marburg, Germany) for performing MALDI-TOF mass spectrometry.
This work was supported by Deutscher Akademischer Austauschdienst (DAAD), International Max-Planck-Research School (IMPRS), Deutsche Forschungsgemeinschaft (DFG), and Fonds der Chemischen Industrie to M.K. and W.B. Quantitative PCR analysis was supported by Department of Energy contract no. DE-FG02-96ER20214 to M.J.M. from the Chemical Sciences, Geosciences and Biosciences Division, Office of Basic Energy Sciences.
Footnotes
Published ahead of print 1 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02185-12.
REFERENCES
- 1. McInerney MJ, Struchtemeyer CG, Sieber J, Mouttaki H, Stams AJM, Schink B, Rohlin L, Gunsalus RP. 2008. Physiology, ecology, phylogeny, and genomics of microorganisms capable of syntrophic metabolism. Ann. N. Y. Acad. Sci. 1125:58–72 [DOI] [PubMed] [Google Scholar]
- 2. McInerney MJ, Sieber JR, Gunsalus RP. 2009. Syntrophy in anaerobic global carbon cycles. Curr. Opin. Biotechnol. 20:623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jackson BE, McInerney MJ. 2002. Anaerobic microbial metabolism can proceed close to thermodynamic limits. Nature 415:454–456 [DOI] [PubMed] [Google Scholar]
- 4. Hopkins B, McInerney M, Warikoo V. 1995. Evidence for anaerobic syntrophic benzoate degradation threshold and isolation of the syntrophic benzoate degrader. Appl. Environ. Microbiol. 61:526–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McInerney MJ, Rohlin L, Mouttaki H, Kim U, Krupp RS, Rios-Hernandez L, Sieber J, Struchtemeyer CG, Bhattacharyya A, Campbell JW, Gunsalus RP. 2007. The genome of Syntrophus aciditrophicus: life at the thermodynamic limit of microbial growth. Proc. Natl. Acad. Sci. U. S. A. 104:7600–7605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jackson BE, Bhupathiraju VK, Tanner RS, Woese CR, McInerney MJ. 1999. Syntrophus aciditrophicus sp. nov., a new anaerobic bacterium that degrades fatty acids and benzoate in syntrophic association with hydrogen-using microorganisms. Arch. Microbiol. 171:107–114 [DOI] [PubMed] [Google Scholar]
- 7. Elshahed MS, Bhupathiraju VK, Wofford NQ, Nanny MA, McInerney MJ. 2001. Metabolism of benzoate, cyclohex-1-ene carboxylate, and cyclohexane carboxylate by “Syntrophus aciditrophicus” strain SB in syntrophic association with H2-using microorganisms. Appl. Environ. Microbiol. 67:1728–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Elshahed MS, McInerney MJ. 2001. Benzoate fermentation by the anaerobic bacterium Syntrophus aciditrophicus in the absence of hydrogen-using microorganisms. Appl. Environ. Microbiol. 67:5520–5525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mouttaki H, Nanny MA, McInerney MJ. 2008. Use of benzoate as an electron acceptor by Syntrophus aciditrophicus grown in pure culture with crotonate. Environ. Microbiol. 10:3265–3274 [DOI] [PubMed] [Google Scholar]
- 10. Mouttaki H, Nanny MA, McInerney MJ. 2007. Cyclohexane carboxylate and benzoate formation from crotonate in Syntrophus aciditrophicus. Appl. Environ. Microbiol. 73:930–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Buckel W, Barker HA. 1974. Two pathways of glutamate fermentation by anaerobic bacteria. J. Bacteriol. 117:1248–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Buckel W. 1980. Analysis of the fermentation pathways of clostridia using double labelled glutamate. Arch. Microbiol. 127:167–169 [DOI] [PubMed] [Google Scholar]
- 13. Buckel W. 2001. Unusual enzymes involved in five pathways of glutamate fermentation. Appl. Microbiol. Biotechnol. 57:263–273 [DOI] [PubMed] [Google Scholar]
- 14. Gottschalk G, Barker HA. 1966. Synthesis of glutamate and citrate by Clostridium kluyveri. A new type of citrate synthase. Biochemistry 5:1125–1133 [DOI] [PubMed] [Google Scholar]
- 15. Li F, Hagemeier C, Seedorf H, Gottschalk G, Thauer RK. 2007. Re-Citrate synthase from Clostridium kluyveri is phylogenetically related to homocitrate synthase and isopropylmalate synthase rather than to Si-citrate synthase. J. Bacteriol. 189:4299–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Feng X, Mouttaki H, Lin L, Huang R, Wu B, Hemme CL, He Z, Zhang B, Hicks LM, Xu J, Zhou J, Tang YJ. 2009. Characterization of the central metabolic pathways in Thermoanaerobacter sp. strain X514 via isotopomer-assisted metabolite analysis. Appl. Environ. Microbiol. 75:5001–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gottschalk G. 1969. Partial purification and some properties of the (R)-citrate synthase from Clostridium acidi-urici. Eur. J. Biochem. 7:301–306 [DOI] [PubMed] [Google Scholar]
- 18. Tang Y, Pingitore F, Mukhopadhyay A, Phan R, Hazen TC, Keasling JD. 2007. Pathway confirmation and flux analysis of central metabolic pathways in Desulfovibrio vulgaris Hildenborough using gas chromatography-mass spectrometry and Fourier transform-ion cyclotron resonance mass spectrometry. J. Bacteriol. 189:940–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marco-Urrea E, Paul S, Khodaverdi V, Seifert J, von Bergen M, Kretzschmar U, Adrian L. 2011. Identification and characterization of a Re-citrate synthase in Dehalococcoides strain CBDB1. J. Bacteriol. 193:5171–5178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Crown SB, Indurthi DC, Ahn WS, Choi J, Papoutsakis ET, Antoniewicz MR. 2011. Resolving the TCA cycle and pentose-phosphate pathway of Clostridium acetobutylicum ATCC 824: isotopomer analysis, in vitro activities and expression analysis. Biotechnol. J. 6:300–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simon EJ, Shemin D. 1953. The preparation of S-succinyl coenzyme A. J. Am. Chem. Soc. 75:2520 [Google Scholar]
- 22. Parthasarathy A, Pierik AJ, Kahnt J, Zelder O, Buckel W. 2011. Substrate specificity of 2-hydroxyglutaryl-CoA dehydratase from Clostridium symbiosum: toward a bio-based production of adipic acid. Biochemistry 50:3540–3550 [DOI] [PubMed] [Google Scholar]
- 23. Moore JT, Uppal A, Maley F, Maley GF. 1993. Overcoming inclusion body formation in a high-level expression system. Protein Expr. Purif. 4:160–163 [DOI] [PubMed] [Google Scholar]
- 24. de Marco A. 2007. Protocol for preparing proteins with improved solubility by co-expressing with molecular chaperones in Escherichia coli. Nat. Protoc. 2:2632–2639 [DOI] [PubMed] [Google Scholar]
- 25. Joseph RE, Andreotti AH. 2008. Bacterial expression and purification of interleukin-2 tyrosine kinase: single step separation of the chaperonin impurity. Protein Expr. Purif. 60:194–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254 [DOI] [PubMed] [Google Scholar]
- 27. Riddles PW, Blakeley RL, Zerner B. 1983. Reassessment of Ellman's reagent. Methods Enzymol. 91:49–60 [DOI] [PubMed] [Google Scholar]
- 28. Srere PA, Kosicki GW. 1961. The purification of citrate-condensing enzyme. J. Biol. Chem. 236:2557–2559 [PubMed] [Google Scholar]
- 29. Buckel W, Eggerer H. 1965. On the optical determination of citrate synthase and acetyl-coenzyme A. Biochem. Z. 343:29–43 [PubMed] [Google Scholar]
- 30. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:2002–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guenin S, Mauriat M, Pelloux J, Van Wuytswinkel O, Bellini C, Gutierrez L. 2009. Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental conditions-specific, validation of references. J. Exp. Bot. 60:487–493 [DOI] [PubMed] [Google Scholar]
- 32. Buckel W, Zhang J, Friedrich P, Parthasarathy A, Li H, Djurdjevic I, Dobbek H, Martins BM. 2012. Enzyme catalyzed radical dehydrations of hydroxy acids. Biochim. Biophys. Acta 1824:1278–1290 [DOI] [PubMed] [Google Scholar]
- 33. Knauer SH, Buckel W, Dobbek H. 2011. Structural basis for reductive radical formation and electron recycling in (R)-2-hydroxyisocaproyl-CoA dehydratase. J. Am. Chem. Soc. 133:4342–4347 [DOI] [PubMed] [Google Scholar]
- 34. Locher KP, Hans M, Yeh AP, Schmid B, Buckel W, Rees DC. 2001. Crystal structure of the Acidaminococcus fermentans 2-hydroxyglutaryl-CoA dehydratase component A. J. Mol. Biol. 307:297–308 [DOI] [PubMed] [Google Scholar]
- 35. Parthasarathy A, Buckel W, Smith DM. 2010. On the thermodynamic equilibrium between (R)-2-hydroxyacyl-CoA and 2-enoyl-CoA. FEBS J. 277:1738–1746 [DOI] [PubMed] [Google Scholar]
- 36. Martins BM, Macedo-Ribeiro S, Bresser J, Buckel W, Messerschmidt A. 2005. Structural basis for stereo-specific catalysis in NAD+-dependent (R)-2-hydroxyglutarate dehydrogenase from Acidaminococcus fermentans. FEBS J. 272:269–281 [DOI] [PubMed] [Google Scholar]
- 37. Sieber JR, Sims DR, Han C, Kim E, Lykidis A, Lapidus AL, McDonnald E, Rohlin L, Culley DE, Gunsalus R, McInerney MJ. 2010. The genome of Syntrophomonas wolfei: new insights into syntrophic metabolism and biohydrogen production. Environ. Microbiol. 12:2289–2301 [DOI] [PubMed] [Google Scholar]
- 38. Koon N, Squire CJ, Baker EN. 2004. Crystal structure of LeuA from Mycobacterium tuberculosis, a key enzyme in leucine biosynthesis. Proc. Natl. Acad. Sci. U. S. A. 101:8295–8300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gottschalk G, Dittbrenner S, Lenz H, Eggerer H. 1972. Studies on the re-citrate synthase reaction. Eur. J. Biochem. 26:455–461 [DOI] [PubMed] [Google Scholar]
- 40. Forouhar F, Hussain M, Farid R, Benach J, Abashidze M, Edstrom WC, Vorobiev SM, Xiao R, Acton TB, Fu Z, Kim JJ, Miziorko HM, Montelione GT, Hunt JF. 2006. Crystal structures of two bacterial 3-hydroxy-3-methylglutaryl-CoA lyases suggest a common catalytic mechanism among a family of TIM barrel metalloenzymes cleaving carbon-carbon bonds. J. Biol. Chem. 281:7533–7545 [DOI] [PubMed] [Google Scholar]
- 41. Karpusas M, Branchaud B, Remington SJ. 1990. Proposed mechanism for the condensation reaction of citrate synthase: 1.9-A structure of the ternary complex with oxaloacetate and carboxymethyl coenzyme A. Biochemistry 29:2213–2219 [PubMed] [Google Scholar]
- 42. Kamachi T, Doitomi K, Takahata M, Toraya T, Yoshizawa K. 2011. Catalytic roles of the metal ion in the substrate-binding site of coenzyme B12-dependent diol dehydratase. Inorg. Chem. 50:2944–2952 [DOI] [PubMed] [Google Scholar]
- 43. Toraya T, Honda S, Mori K. 2010. Coenzyme B12-dependent diol dehydratase is a potassium ion-requiring calcium metalloenzyme: evidence that the substrate-coordinated metal ion is calcium. Biochemistry 49:7210–7217 [DOI] [PubMed] [Google Scholar]
- 44. Gottschalk G, Dittbrenner S. 1970. Properties of (R)-citrate synthase from Clostridium acici-urici. Hoppe-Seyler′s Z. Physiol. Chem. 351:1183–1190 [Google Scholar]
- 45. Bond DR, Mester T, Nesbo CL, Izquierdo-Lopez AV, Collart FL, Lovley DR. 2005. Characterization of citrate synthase from Geobacter sulfurreducens and evidence for a family of citrate synthases similar to those of eukaryotes throughout the Geobacteraceae. Appl. Environ. Microbiol. 71:3858–3865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhi W, Srere PA, Evans CT. 1991. Conformational stability of pig citrate synthase and some active-site mutants. Biochemistry 30:9281–9286 [DOI] [PubMed] [Google Scholar]
- 47. Bulfer SL, Scott EM, Couture JF, Pillus L, Trievel RC. 2009. Crystal structure and functional analysis of homocitrate synthase, an essential enzyme in lysine biosynthesis. J. Biol. Chem. 284:35769–35780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. de Carvalho LP, Blanchard JS. 2006. Kinetic and chemical mechanism of α-isopropylmalate synthase from Mycobacterium tuberculosis. Biochemistry 45:8988–8999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ma J, Zhang P, Zhang Z, Zha M, Xu H, Zhao G, Ding J. 2008. Molecular basis of the substrate specificity and the catalytic mechanism of citramalate synthase from Leptospira interrogans. Biochem. J. 415:45–56 [DOI] [PubMed] [Google Scholar]
- 50. Gruer MJ, Artymiuk PJ, Guest JR. 1997. The aconitase family: three structural variations on a common theme. Trends Biochem. Sci. 22:3–6 [DOI] [PubMed] [Google Scholar]
- 51. Tang YJ, Yi S, Zhuang Zinder W-QSH, Keasling JD, Alvarez-Cohen L. 2009. Investigation of carbon metabolism in “Dehalococcoides ethenogenes” strain 195 by use of isotopomer and transcriptomic analyses. J. Bacteriol. 191:5224–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Marco-Urrea E, Seifert J, von Bergen M, Adrian L. 2012. Stable isotope peptide mass spectrometry to decipher amino acid metabolism in Dehalococcoides strain CBDB1. J. Bacteriol. 194:4169–4177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim J, Hetzel M, Boiangiu CD, Buckel W. 2004. Dehydration of (R)-2-hydroxyacyl-CoA to enoyl-CoA in the fermentation of alpha-amino acids by anaerobic bacteria. FEMS Microbiol. Rev. 28:455–468 [DOI] [PubMed] [Google Scholar]
- 54. Kung JW, Löffler C, Dörner K, Heintz D, Gallien S, Van Dorsselaer A, Friedrich T, Boll M. 2009. Identification and characterization of the tungsten-containing class of benzoyl-coenzyme A reductases. Proc. Natl. Acad. Sci. U. S. A. 106:17687–17692 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.