

Abstract
Escherichia coli possesses two type 1A topoisomerases, Topo I (topA) and Topo III (topB). Topo I relaxes excess negative supercoiling, and topA mutants can grow only in the presence of compensatory mechanisms, such as gyrase mutations. topB mutants grow as well as wild-type cells. In vitro, Topo III, but not Topo I, can efficiently decatenate DNA during replication. However, in vivo, a chromosome segregation defect is seen only when both type 1A topoisomerases are absent. Here we present experimental evidence for an interplay between gyrase and type 1A topoisomerases in chromosome segregation. We found that both the growth defect and the Par− phenotypes of a gyrB(Ts) mutant at nonpermissive temperatures were significantly corrected by deleting topA, but only when topB was present. Overproducing Topo IV, the major cellular decatenase, could not substitute for topB. We also show that overproducing Topo III at a very high level could suppress the Par− phenotype. We previously found that the growth and chromosome segregation defects of a triple topA rnhA gyrB(Ts) mutant in which gyrase supercoiling activity was strongly inhibited could be corrected by overproducing Topo III (V. Usongo, F. Nolent, P. Sanscartier, C. Tanguay, S. Broccoli, I. Baaklini, K. Drlica, and M. Drolet, Mol. Microbiol. 69:968-981, 2008). We show here that this overproduction could be bypassed by substituting the gyrB(Ts) allele for a gyrB+ one or by growing cells in a minimal medium, conditions that reduced both topA- and rnhA-dependent unregulated replication. Altogether, our data point to a role for Topo III in chromosome segregation when gyrase is inefficient and suggest that Topo I plays an indirect role via supercoiling regulation.
INTRODUCTION
DNA topoisomerases are ubiquitous enzymes found in eubacteria, archaebacteria and eukaryotes that solve the topological problem associated with replication, transcription, and recombination (1). Type 1A topoisomerases cleave one DNA strand at a time to change the DNA linking number in single steps, and they all require, to various extents depending on the enzyme, an exposed single-stranded region within the DNA substrate. Escherichia coli possesses two type 1A enzymes, namely, topoisomerase I (Topo I) (topA) and Topo III (topB). Topo I binds to single-stranded DNA regions close to double-stranded ones (2), and this explains its major role in the relaxation of excess negative supercoiling, which is mostly generated during transcription (3–5). Consistent with this function is the finding that many topA mutants can grow owing to the presence of compensatory mutations in gyrA or gyrB that reduce the supercoiling activity of gyrase (6, 7). One major consequence of excess negative supercoiling is R-loop formation and RNA degradation (5, 8, 9).
Topo III is a very-low-abundance protein (10), and its cellular role is not well defined. This is largely due to the fact that, as opposed to topA-null mutants, topB-null mutants display no obvious phenotype (they grow as well as wild-type strains, at least under standard laboratory conditions [10]). Topo III requires stable single-stranded DNA regions for activity and does not efficiently relax negatively supercoiled DNA (11). In fact, Topo III plays no role in supercoiling regulation in vivo (12, 13). In vitro, Topo III is a potent decatenase provided that single-stranded regions are present on the DNA (11).
DNA gyrase, the enzyme responsible for the introduction of negative supercoiling in DNA, plays major roles in replication. First, via negative supercoiling, gyrase facilitates DNA melting at oriC in order for replication initiation to take place (14). Second, gyrase removes the left-handed positive supercoiling generated in front of moving replication forks (15). In fact, it is believed that most of the intertwining generated by replication is normally eliminated by gyrase (16). The positive supercoiling generated by replication can also migrate behind the replication fork, provided that the replication complex is free to rotate, which leads to the formation of precatenanes (intertwining of the pair of replicated chromosome segments) (17–19) that can be removed by Topo IV, the major cellular decatenase. Once the chromosome is fully replicated, the remaining precatenanes becomes catenanes that are also eliminated by topoisomerase IV, thus allowing chromosome segregation to take place. In vitro, Topo III alone can support replication presumably by acting on single-stranded DNA at the replication fork to remove precatenanes (20–22). This mechanism has been proposed to explain the suppression of the chromosome segregation defect of Topo IV mutants by Topo III overproduction at very high levels (22).
Interestingly, one of the first mutations isolated that caused a chromosomal segregation defect mapped to a subunit of gyrase (23). The observed phenotype was named Par−, and it is characterized by anucleate cells, guillotined cells, and long filaments with abnormal nucleoid structures. As Topo IV, not gyrase, was later shown to be the major cellular decatenase in vivo (24, 25), this result suggested that the gyrase mutation somehow reduced the efficiency of decatenation by Topo IV. This has been explained based on the observation that negative supercoiling strongly favors the decatenation reaction of Topo IV over its catenation reaction (26–29). Moreover, defective gyrase would favor the accumulation of precatenanes. Such precatenanes are good substrates for Topo IV only when their density is low so that their crossing angle is optimal for enzyme activity (16). Thus, defective gyrase (and supercoiling) could lead to chromosome segregation defects by rendering Topo IV inefficient for two reasons: by promoting the catenation reaction and by causing the accumulation of precatenanes.
Despite biochemical and genetic evidence for the involvement of Topo III but not Topo I in chromosome segregation, a defect in this process could be observed in vivo only when both type 1A topoisomerases were absent (30). Since only topA mutants, not topB ones, display severe growth defects, it is possible that the topA topB phenotype reflects an absolute requirement for Topo III in chromosome segregation when topA is absent. To test this hypothesis, we initiated a study to understand how the various compensatory mechanisms for the absence of topA, such as gyrase mutations, RNase HI overproduction and others to be presented elsewhere (V. Usongo, C. Tanguay, and M. Drolet, unpublished data), can modulate this topA topB phenotype. While we were testing the effect of modulating gyrase activity by using a gyrB(Ts) allele that is known to compensate for the absence of topA at 37°C (31), we found that deleting topA was able to compensate for the strong gyrase inhibition at 39 to 40°C and above (up to 42°C). This temperature also coincided with the appearance of the strong Par− phenotype. This complementation, achieved by deleting topA, was found to be totally dependent on the presence of an active topB gene. Here we present these results and others that allow us to conclude that Topo III can play an important role in chromosome segregation in vivo and that topA likely influences this process indirectly by regulating replication via supercoiling.
MATERIALS AND METHODS
E. coli strains.
Strains used in this work are described in Table 1. Strains carrying the gyrB(Ts) allele carry in fact a gyrB gene with two mutations, one (gyrB221) conferring coumermycin resistance and one (gyrB203) conferring temperature sensitivity. These two mutations were obtained simultaneously following mutagenesis of a wild-type strain with N-methyl-N′-nitro-N-nitrosoguanidine (NTG). They have always been used together in our studies (4, 8, 9, 31–34) and in studies from other groups (35), and DNA supercoiling is fully restored to a wild-type level when strains carrying these mutations are exposed to permissive temperatures (30°C and below) (31). Strains were constructed by transduction with phage P1vir as previously described (36). When needed, tetracycline (10 μg/ml), or kanamycin (50 μg/ml) was added to the medium. The gyrB+ allele was transduced into gyrB(Ts) recipients by selecting first for a nearby Tn10 marker and then for thermoresistant growth (42°C). The presence of the wild-type gyrB allele was confirmed by sequencing.
Table 1.
E. coli strains used in this study
| Strain | Genotype | Reference or source |
|---|---|---|
| RFM443 | rpsL galK2 Δlac74 | 31 |
| RFM445 | rpsL galK2 gyrB221(Cour) gyrB203(Ts) Δlac74 | 31 |
| RFM475 | rpsL galK2 gyrB221(Cour) gyrB203(Ts) Δ(topA cysB)204 Δlac74 | 31 |
| NF88 | RFM475 gyrB+ | This work |
| MD897 | DM4100 ΔtopB::kan | 30 |
| CT170 | RFM475 ΔtopB::kan | This work |
| RFM430 | rpsL galK2 ΔtrpE | 32 |
| PH379 | RFM430 rnhA::cam | 32 |
| SB383 | RFM475 rnhA::cam pPH1243 | 32 |
| NF98 | SB383 gyrB+ | This work |
Plasmids.
pPH1243 is a pTrc99a derivative carrying the topB gene under the control of the IPTG (isopropyl-β-d-thiogalactopyranoside)-inducible Ptrc promoter (33). pET11-parEC produces a ParEC fusion protein that is active as a Topo IV (37).
Plasmid extraction for supercoiling analysis.
pPH1243 DNA extraction for supercoiling analysis was performed as described previously (32). Chloroquine gel electrophoresis and in situ hybridization of the dried gels were done as previously reported (32).
Western blot analysis.
Western blot analysis was performed as described previously (32).
Microscopy.
Cells were grown overnight on LB plates supplemented, when required, with cysteine (50 μg/ml) and appropriate antibiotics. When needed, IPTG (1 mM) was added to the plates. The plates were incubated at 37°C. After overnight growth, cells were resuspended in prewarmed (37°C) liquid LB medium (supplemented as required) to obtain a starting optical density at 600 nm (OD600) of about 0.01. Cells were grown at 37°C to an OD600 of 0.8. The cells were prepared for microscopy as described before (32). Pictures were randomly taken and randomly selected to calculate the number of cells in each category.
Flow cytometry.
Overnight cultures were prepared and diluted, and cells were grown in either LB or M9 glucose medium supplemented as appropriate. When the OD600 reached 0.3, the cells were either recovered (Fig. 1) or treated with rifampin (300 μg/ml) to prevent the initiation of new rounds of replication and then incubated for an additional 2 h, to allow the ongoing replication rounds to terminate (run-out experiments [see Fig. 8]). Cells were washed twice with TE buffer (Tris [10 mM], pH 8.0; EDTA [1 mM]) before being fixed with ice-cold ethanol (77%). After one wash with TE buffer, the cells were stained with Syto 16 (Molecular Probes). RNase A (200 μg/ml) was also added during the staining (30 min). Flow cytometry was performed on a Becton, Dickinson FACSCalibur. DNA/mass ratios were obtained by dividing the average Syto 16 green fluorescence by the average forward light scatter (FSC), which is roughly equivalent to cell mass.
Fig 1.

Deleting topA complements the growth and Par− phenotype of the gyrB(Ts) strain. (a) Cells of strains RFM445 [gyrB(Ts)] and RFM475 [topA gyrB(Ts)] were grown at 37°C to an OD600 of 0.7 and streaked on LB plates. The plates were incubated at 42°C for 24 h. (b, c, and d) Flow cytometry analysis of RFM443, RFM445, and RFM475 cells grown in LB medium. Cell mass (left panels) and DNA content (right panels) parameters are shown. M1 and M2 correspond to anucleate cells (the percentages are indicated in each panel) and to cells with DNA, respectively. (e) DNA/mass ratios were obtained from the data shown in panels b to d (37°C, 40°C, and 42°C, respectively) except for strains RFM445 and RFM475 grown at 42°C, where the data of two additional experiments were used to obtain an average DNA/mass ratio.
Fig 8.

Effects of the growth medium on unregulated replication in topA and rnhA mutants. Rifampin run-out experiments for flow cytometry analyses were performed as described in Materials and Methods. (a) RFM443 (wild-type), RFM445 [gyrB(Ts)], and RFM475 [gyrB(Ts) topA] cells grown in LB or M9 glucose medium; (b) RFM430 (wild-type) and PH379 (rnhA) cells grown in LB or M9 glucose medium.
RESULTS
Deleting topA partially corrects the growth defect and the Par− phenotypes of the gyrB(Ts) mutant at nonpermissive temperatures.
We found that deleting topA (strain RFM475) allowed a gyrB(Ts) mutant (strain RFM445) to grow at 42°C on LB plates (Fig. 1a). The growth inhibition of a strain carrying the same gyrB(Ts) allele was previously shown to correlate with the inhibition of replication initiation at nonpermissive temperatures (38, 39). Moreover, a strong Par− phenotype for this allele at a nonpermissive temperature has been described (35). Therefore, this result suggested that the absence of Topo I sufficiently increased the negative supercoiling level in the gyrB(Ts) mutant to allow replication initiation and chromosome segregation to take place in strain RFM475.
We used flow cytometry to measure cell mass and DNA content in strains RFM445, RFM475, and RFM443 as a wild-type control. Figure 1b shows that the average cell mass at 37°C was similar for the three strains, with that of RFM475 being slightly lower. DNA content had a somewhat wider distribution for RFM445 compared to RFM443 and that of RMF475 was significantly wider, with the average DNA content being higher in this strain. As a result, strain RFM475 clearly had a higher DNA/mass ratio (Fig. 1e). This could be due, at least in part, to unregulated replication in strain RFM475 (see below).
At 40 and 42°C, strain RFM445 did not form visible colonies on plates but grew sufficiently in liquid (five generations) to obtain cells for flow cytometry analysis. Figure 1c shows a bimodal distribution of the average cell mass for strain RFM445 at 40°C. This also coincided with the appearance of a high peak on the left side of the DNA histogram which corresponds to nonspecific binding of the dye. This indicates the accumulation of small anucleate cells (55% of the cells were anucleate), a typical manifestation of the Par− phenotype. Moreover, DNA labeling shifted toward the right side of the histogram for strain RFM445, reflecting the presence of longer cells, another manifestation of the Par− phenotype. Based on these criteria, it can be deduced that deleting topA (strain RFM475) significantly corrected the Par− phenotype of the gyrB(Ts) strain (9.4% anucleate cells [Fig. 1c]). This was later confirmed by fluorescence microscopy.
Figure 1d shows that the Par− phenotype was exacerbated in both RFM445 and RFM475 at 42°C (79.4 and 32.8% anucleate cells, respectively) compared to 40°C but that deleting topA (strain RFM475) still significantly corrected this phenotype. In fact, the Par− phenotype was even stronger in strain RFM445 cultivated at 40°C than in strain RFM475 grown at 42°C (55.06% versus 32.8% anucleate cells, respectively). The DNA/mass ratio clearly dropped for strains RFM445 and RFM475 at 40°C compared to 37°C (Fig. 1e). This probably indicates reduced replication initiation due to the lack of negative supercoiling at oriC (14). We also noted that the drop at 42°C was reproducibly more important for strain RFM445 compared to RFM475 (Fig. 1e). We believe that this indicates a partial correction of the replication initiation defect in strain RFM475 conferred by the absence of topA. Thus, deleting topA significantly corrected the Par− phenotype of the gyrB(Ts) strain at nonpermissive temperatures plus all other supercoiling-dependent processes, e.g., replication initiation, that are required for growth.
The correction of the growth and the Par− phenotypes of the gyrB(Ts) mutant by deleting topA depends upon the presence of the topB gene.
As gyrase supercoiling activity was strongly inhibited at 40°C and above in strains carrying the gyrB(Ts) allele, it was possible that Topo IV could not efficiently perform DNA decatenation. In this situation, Topo III activity could be required if it can actually play a role in chromosome segregation in vivo. The correction of the growth defect of the gyrB(Ts) strain by deleting topA gave us the opportunity to test this possibility. We used P1vir transduction to introduce a topB-null allele into the topA gyrB(Ts) strain, RFM475. As a control for transduction efficiency, we also introduced the topB-null allele within isogenic wild-type (RM443) and gyrB(Ts) (RFM445) strains. For these strains, topB-null transductants were obtained after 18 h of incubation at 37°C, whereas 48 h was required to obtain transductants of strain RFM475 at the same temperature. The disruption of the topB gene was confirmed by PCR for 8 transductants of each group (data not shown).
Upon restreaking on LB plates, transductants of strains RFM443 and RFM445 formed medium-sized colonies after 18 h of incubation, whereas it took 48 h for RFM475 transductants to form colonies of a similar size (not shown). The vast majority of the colonies obtained after 48 h were homogeneous in size. We obtained similar results for strain DM800, a widely used topA-null mutant that also contains the naturally acquired gyrB225 compensatory mutation (6) (data not shown). Similar results in terms of the number of transductants and their growth rate were also reported by another group for strain DM750, a topA-null mutant carrying the naturally acquired gyrA224 compensatory mutation (40). Thus, E. coli cells lacking both type 1A topoisomerases are viable but grow slowly.
Next, we tested the ability of topA gyrB(Ts) cells lacking the topB gene to form colonies on LB plates at 35 and 42°C. Figure 2a shows that whereas RFM475 cells grew better at 42°C than at 35°C, cells of strain CT170, a topB-null transductant of strain RFM475, formed colonies at 35°C but did not grow at 42°C. The growth behavior of strain RFM475 was previously reported and was shown to be due to the reactivation of gyrase activity as the temperature was decreased, so that the gyrB(Ts) allele could no longer compensate for the absence of Topo I (31). The opposite behavior of strain CT170 shows that topB is required for the growth of the topA gyrB(Ts) strain at higher temperatures.
Fig 2.

The topB gene is required for the growth of the topA gyrB(Ts) strain at nonpermissive temperatures. (a) Cells of strains RFM475 [topA gyrB(Ts)] and CT170 (RFM475 topB) were grown at 37°C to an OD600 of 0.7 and streaked on LB plates. The plates were incubated for 24 h at 35 or 42°C. (b) Cell growth of strains RFM475 and CT170 was monitored in liquid LB at 35, 37, 39, and 40°C.
The growth of the two isogenic strains (RFM475 and CT170) was also monitored in liquid medium at different temperatures, and in some cases cell samples were recovered for DAPI (4′,6′-diamidino-2-phenylindole) staining and prepared for fluorescence microscopy to examine cell morphology and DNA content. The growth rates at 35°C were very similar for both strains, whereas when the temperature was raised to 40°C, the growth rate increased for strain RFM475 but did not significantly change for strain CT170 (Fig. 2b). The temperature of 37°C was found to be the optimal temperature for the growth of the topA topB mutants (Fig. 2b) (Usongo et al., unpublished). Major differences between RFM475 and CT170 strains were observed from 39°C. At 40°C, the growth of strain CT170 stopped during log phase at an OD600 of 0.6 (Fig. 2).
One typical phenotype of topA topB-null mutants that was previously reported is the formation of very long filaments with unsegregated nucleoids having abnormal structures (30). Such filamentous cells were also observed for our topA topB-null mutant (strain CT170; >14 chromosomes/cell) (Fig. 3; also, see Fig. S3 and S4 in the supplemental material). Importantly, the proportion of such cells dramatically increased as the incubation temperature was raised to 39°C, with a maximum being reached at 40°C (7% versus 24%, respectively, for 37 and 40°C; this roughly corresponds, respectively, to 30% and 70% of the total cell mass [Fig. 3]). This dramatic increase correlated with temperatures at which the gyrB(Ts) Par− phenotype started to appear.
Fig 3.

The topB gene is required for chromosome segregation in the topA gyrB(Ts) strain at nonpermissive temperatures. Cells were grown and prepared for microscopy as described in Materials and Methods. Superimposed pictures of phase contrast and fluorescence (Adobe Photoshop) of DAPI-stained cells from strains RFM475 [topA gyrB(Ts)] and CT170 (RFM475 topB) were used to calculate the number of cells in the different categories. The total is the number of cells that were examined to calculate the percentages of cells in each category. The low-DNA-content category likely reflects guillotined cells, a manifestation of the Par− phenotype (23). N/A, cells in this category were not counted. chr, chromosomes.
As expected because of the Par− phenotype, the proportion of anucleate cells also considerably increased at high temperatures (4% versus 48%, respectively, for 37 and 40°C [Fig. 3]). Although the isogenic RFM475 strain [topA gyrB(Ts)] did not exhibit significant formation of very long filaments at any temperature tested (less than 1% at 40°C [Fig. 3; also, see Fig. S1 and S2 in the supplemental material), it also produced a large proportion of anucleate cells at higher temperatures (less than 0.1% versus 40%, respectively, for 37 and 40°C [Fig. 3]). As predicted from the results shown in Fig. 1, anucleate cells accumulated at a higher frequency in strain RFM445 (0.1% versus 67%, respectively, for 37 and 40°C [see Fig. S5 in the supplemental material]). However, RFM445 produced considerably fewer very long filaments than CT170 (4% versus 24%, respectively, for RFM445 and CT170 at 40°C). A ΔtopB::kan derivative of RFM445 [gyrB(Ts) topB] behaved like RFM445 except that it produced a higher number of longer cells (data not shown). Thus, in the topA deletion strain, the topB gene is required to alleviate the Par− phenotype related to defective gyrase activity. When topB is deleted, the Par− phenotype is mostly manifested as very long filaments with unsegregated nucleoids. This could be due to the fact that the absence of topA (increased negative supercoiling) confers to the cells the ability to grow for a while, whereas growth is much more restricted in the single gyrB(Ts) mutant. We can conclude that Topo III at its wild-type level can perform DNA decatenation to allow chromosome segregation to take place when gyrase is defective.
Overproducing Topo IV cannot bypass the requirement for Topo III activity for the growth of the topA gyrB(Ts) strain.
Our data indicated that Topo IV could not fully support chromosome segregation in the gyrB(Ts) mutant at nonpermissive temperatures, despite the increase in negative supercoiling conferred by deleting topA. This could indicate either that there was not enough Topo IV or that it could not substitute for Topo III. To address this issue, we introduced the plasmid pET11-parEC (37) into strains RFM475 [topA gyrB(Ts)] and CT170 [topA topB gyrB(Ts)]. This plasmid produces a ParEC fusion protein that is fully active as a Topo IV protein in vitro and that was shown to complement the thermosensitive growth of parE(Ts) and parC(Ts) strains (37). We found that this plasmid was able to stimulate the growth of strain RFM475 (Fig. 4a). This result is in agreement with the previous finding showing that Topo IV overproduction can compensate for the absence of topA (41) and also confirmed that an active Topo IV could be produced from this plasmid. The results of Western blot experiments showed that Topo IV as a fusion protein was indeed produced in both RFM475 and CT170 strains carrying pET11-parEC (Fig. 4b).
Fig 4.
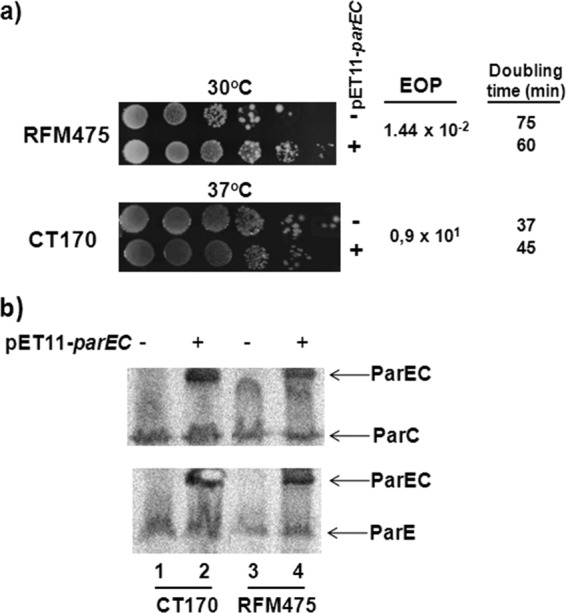
Effect of overproducing a functional ParEC fusion protein on the growth of RFM475 and CT170 strains. (a) The effect of pET11-parEC on the growth of RFM475 and CT170 cells was monitored by spotting 10 μl of serial 10-fold dilutions of cells grown in LB to an OD600 of 0.6 (at 30 and 37°C, respectively, for RFM475 and CT170) from 100 to 10−5 (from left to right) on LB plates that were incubated at 30°C for 48 h (RFM475) or 37°C for 24 h (CT170). Cells grown in liquid were also used to calculate the efficiency of plating (EOP; number of viable cells [colonies] without plasmid divided by the number of viable cells carrying pET11-parEC) and the doubling time. The results shown here are representative of three independent experiments. (b) CT170 and RFM475 cells with or without pET11-parEC were grown on LB plates at 37°C for 24 h. Aliquots of cells were recovered for Western blotting with anti-ParC (top) or anti-ParE (bottom) antibodies as described in Materials and Methods.
We found that overproducing Topo IV did not improve the growth of strain CT170. There was even a slight but reproducible negative effect (CT170; one log drop in the efficiency of plating for cells carrying pET11-parEC that also had a slightly higher doubling time than CT170 cells without pET11-parEC [Fig. 4a]). The reason for this effect is currently unknown. Nevertheless, it clearly indicates that Topo IV cannot substitute for Topo III in chromosome segregation when gyrase supercoiling activity is significantly impaired. This result is also in line with a previous report showing that overproducing Topo IV could not complement the growth defect of DM750 and DM800 topA cells in which the topB gene had been inactivated (30).
Topo III overproduction at a very high level can correct the Par− phenotype of the gyrB(Ts) mutant at a nonpermissive temperature.
The fact that the gyrB(Ts) strain RFM445 displays a strong Par− phenotype despite the presence of the topB gene may indicate that there is not enough Topo III to fully decatenate chromosomal DNA to allow segregation. To test this hypothesis, we introduced the plasmid pC18pBAD33 (a kind gift from R. J. DiGate, University of the Sciences, Philadelphia, PA) into strain RFM445 to overproduce Topo III at a very high level. This plasmid has the topB gene placed under the control of the strong arabinose-inducible promoter PBAD and a strong Shine-Dalgarno box. The same amount of RFM445/pC18pBAD33 cells from a concentrated glycerol stock were streaked on three LB plates containing ampicillin and either arabinose (0.05%), glucose (0.2%) or no sugar. The plates were incubated for 3 days at 42°C. We found that growth was essentially restricted to the beginning of the streaks, as very few isolated colonies were obtained (not shown). This was expected, as negative supercoiling is not restored by overproducing Topo III. Cells from the beginning of the streaks were recovered for DAPI staining and prepared for fluorescence microscopy to examine cell morphology and DNA content.
Figure 5a shows that when Topo III was not overproduced (0.2% glucose), a large number of anucleate cells accumulated. Most strikingly, lemon-shaped cells with a huge mass of unsegregated DNA at the center were produced. However, very few anucleate cells and no lemon-shaped cells were produced in the absence of sugar or with 0.05% arabinose (Fig. 5b and c, respectively). Western blotting (Fig. 5e) showed that a large quantity of Topo III was produced only when cells were grown on plates with arabinose or no sugar.
Fig 5.

Overproducing Topo III at a very high level corrects the Par− phenotype of a gyrB(Ts) strain at 42°C. RFM445 strain [gyrB(Ts)] carrying pC18pBAD33 was grown on LB plates with glucose 0.2% (a), no added sugar (b), or arabinose 0.05% (c) at 42°C for 72 h. In all cases, growth was essentially restricted to the beginning of the streaks, as was expected, since the chromosomal DNA is extensively relaxed. Cells that were able to grow were shown not to be revertants, as they kept their thermosensitive growth phenotype. Aliquots of cells were recovered for fluorescence microscopy to examine cell morphology and DNA content. The images are pictures of phase contrast and fluorescence that were superimposed (using Adobe Photoshop). (d) An aliquot of cells from the LB plate with no sugar was streaked on an LB plate with glucose and incubated for 72 h at 42°C. Growth was restricted to the beginning of the streaks. Cells were recovered for fluorescence microscopy as described above. Data in panels a to d are representative of three independent experiments. (e and f) Western blots showing the level of Topo III overproduction from cells carrying either pC18pBAD33 (e) or pPH1243 (f). The bottom image in panel e is an overexposure of the gel to reveal the Topo III band in the 0.2% glucose sample.
Since the cells were recovered after 3 days of incubation, we were aware of the possibility that suppressor mutations leading to the loss of the Par− phenotype could have accumulated. To test this possibility, we took cells from the LB plate with no sugar that had been incubated for 3 days and streaked them on an LB plate with glucose. After 3 days of incubation, cells from the beginning of the streaks were recovered for DAPI staining and prepared for fluorescence microscopy. The fact that a large number of anucleate cells and lemon-shaped cells accumulated indicated that the correction of the Par− phenotype was due to Topo III overproduction and not to the accumulation of suppressor mutations. Moreover, in an independent experiment, we found that RFM445 strain without plasmid produced similar amount of anucleate cells and lemon-shaped cells whether it was grown on LB plates with no sugar or with glucose or arabinose (data not shown).
In another experiment, we used the plasmid pPH1243, in which the topB gene with its poor Shine-Dalgarno sequence is under the control of the IPTG-inducible Ptrc promoter. However, the level of Topo III overproduction achieved with this plasmid after the addition of IPTG was similar to the level obtained from pC18pBAD33 in the presence of glucose (Fig. 5f, compare lanes 2 and 3). This would explain our failure to observe an effect of pPH1243 on chromosome segregation in strain RFM445 at 42°C (data not shown). Thus, overproduction of Topo III at a very high level can substantially correct the Par− phenotype of the gyrB(Ts) strain.
Increasing gyrase activity in a topA rnhA gyrB(Ts) mutant allows chromosome segregation and growth independent of Topo III overproduction.
We previously described a topA gyrB(Ts) mutant in which the depletion of RNase HI (rnhA) activity triggered the inhibition of the supercoiling activity of gyrase and lead to chromosome segregation defects and growth inhibition (32). Similarly, this extensive supercoiling inhibition was observed in a triple topA rnhA gyrB(Ts) mutant (strain SB383) in which Topo III overproduction from an IPTG-inducible topB gene on a plasmid (pPH1243), corrected both the growth and chromosome segregation defects.
To test the hypothesis that Topo III acts by compensating for the weak gyrase activity in strain SB383 [topA rnhA gyrB(Ts)/pPH1243], we first performed P1vir transduction to replace the gyrB(Ts) allele of strain SB383 with a wild-type one. Although the gyrB(Ts) allele present in strain SB383 compensated for the lack of topA, we reasoned that the response leading to gyrase inhibition in the absence of RNase HI would be sufficient to allow a gyrB+ derivative of SB383 to grow. Indeed, gyrB+ transductants of strain SB383 could be obtained. Moreover, hypernegative supercoiling was not observed following the addition of a translation inhibitor (spectinomycin) to a gyrB+ transductant (strain NF98 [topA rnhA gyrB+/pPH1243]) (Fig. 6a, lane 2).
Fig 6.
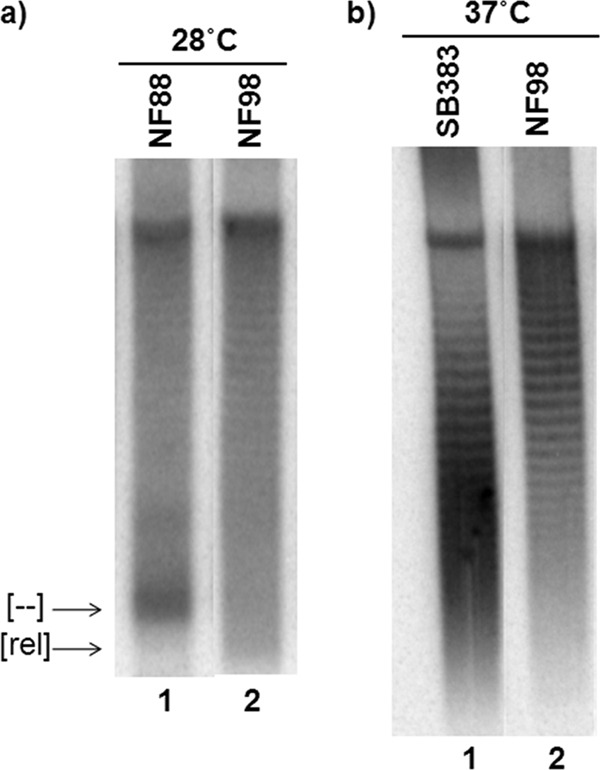
Effects on DNA supercoiling of substituting the gyrB(Ts) allele of strain SB383 [topA rnhA gyrB(Ts)] for a gyrB+ one. (a) NF88 (RFM475 gyrB+) and NF98 (SB383 gyrB+) cells were grown in LB with IPTG (1 mM) at 37°C to an OD600 of 0.6, at which time spectinomycin (250 μg/ml) was added, and 15 min later the cells were transferred to 28°C for 30 min. pPH1243 DNA was extracted, and the topoisomers were resolved following electrophoresis in an agarose gel containing 7.5 μg/ml of chloroquine. At this chloroquine concentration, the relaxed topoisomers migrate more rapidly than the negatively supercoiled ones, except the hypernegatively supercoiled topoisomers, which also migrate rapidly. (b) SB383 and NF98 cells were grown in LB at 37°C to an OD600 of 0.6, at which time pPH1243 DNA was extracted and the topoisomers were resolved as described above. The gels were hybridized with a probe to detect pPH1243 topoisomers. [--] and [rel] indicate hypernegatively supercoiled and extensively relaxed DNA, respectively.
This treatment was previously shown to strongly stimulate hypernegative supercoiling in topA-null mutants (34), but not when rnhA was absent (as in strain SB383) (35). As a control, we showed that hypernegative supercoiling accumulated in strain NF88 (topA gyrB+/pPH1243) after the addition of spectinomycin (Fig. 6a, lane 1). Steady-state supercoiling was significantly higher in strain NF98 than in SB383 (Fig. 6b), which is in agreement with the presence of the gyrB+ allele in strain NF98. Thus, despite the presence of the response leading to gyrase inhibition in the absence of RNase HI, gyrase supercoiling activity was higher in strain NF98 than in strain SB383. In a recent study, deleting the topA gene in an rnhA mutant with a wild-type gyrase was shown to generate nonviable cells (42). Presumably, the excess negative supercoiling together with the absence of RNase HI caused the accumulation of stable R loops that inhibited growth and precluded the expression of the cellular response leading to gyrase inhibition.
As would be predicted if increasing gyrase activity could bypass the need for Topo III overproduction, strain NF98 grown in the absence of IPTG produced many fewer anucleate cells than strain SB383 grown under the same conditions (29 and 2% anucleate cells, respectively, for strains SB383 and NF98 [Fig. 7; also, see Fig. S6a and S7a in the supplemental material]). Cells with a low DNA content and long filaments with unsegregated nucleoids were also produced in small amounts in strain NF98. Noticeably, the suppression was so efficient in this strain that the addition of IPTG to overproduce Topo III did not further reduce the number of cells with chromosome segregation defects (Fig. 7; also, see Fig. S7b). As expected, strain NF98, but not SB383, was able to grow overnight on LB plates in the absence of IPTG (see Fig. S8a in the supplemental material), and both strains produced similar amounts of Topo III protein with and without IPTG (see Fig. S8b). Thus, the need to overproduce Topo III for growth and chromosome segregation could be bypassed by increasing gyrase supercoiling activity in a topA rnhA gyrB(Ts) strain. This result further supports the interplay between gyrase and Topo III in chromosome segregation.
Fig 7.
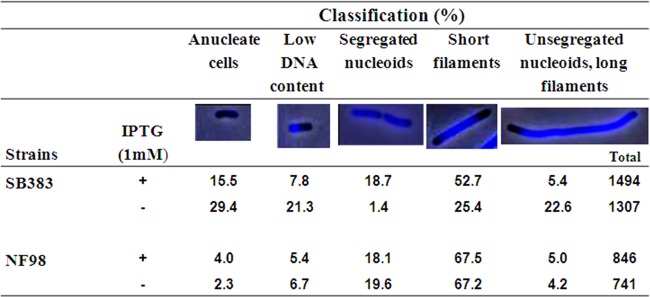
Substituting the gyrB(Ts) allele of strain SB383 [topA rnhA gyrB(Ts)] for a gyrB+ one substantially corrects the chromosomal segregation defect. Cells were grown and prepared for microscopy as described in Materials and Methods. Phase contrast and fluorescence images of DAPI-stained cells from strain SB383 [topA rnhA gyrB(Ts)/pPH1243] and its gyrB+ derivative (NF98) were superimposed (by using Adobe Photoshop) and were used to calculate the number of cells in the different categories. The total is the number of cells that were examined to calculate the percentages of cells in each category. The low-DNA-content category likely reflects guillotined cells, a manifestation of the Par− phenotype (23). Cells in the short-filaments category have nucleoids that are not fully segregated. These cells were most likely viable, since they increased in proportion in strains that grew better (e.g., SB383 with IPTG).
The absence of topA and rnhA causes unregulated replication.
Replication initiation that takes place at oriC is tightly regulated so that it occurs once and only once per cell cycle (43). This process is synchronized with the initiation mass. Unregulated replication initiation could be especially harmful when gyrase activity is suboptimal for chromosome segregation. Interestingly, flow cytometry studies have revealed asynchronous replication in the topA-null mutant DM800 (44). We used flow cytometry in rifampin run-out experiments with cells grown in LB medium to investigate the regulation of replication initiation in a set of isogenic strains that included RFM443 (wild-type), RFM445 [gyrB(Ts)], and RFM475 [topA gyrB(Ts)]. As shown in Fig. 8a, both RFM443 and RFM445 cells contained 2n chromosomes, thus showing that replication initiation was well regulated in these strains. However, the removal of the topA gene from the gyrB(Ts) strain almost completely eliminated the 2n chromosomal pattern (strain RFM475 [Fig. 8a]). Thus, the absence of Topo I leads to the appearance of unregulated replication in strain RFM475.
In the absence of RNase HI, replication can initiate from R loops at sites other than oriC (45). Several of these sites (oriKs) are located close to the terminal (Ter) region. Replication in rnhA mutants was named cSDR (constitutive stable DNA replication) because it could continue for several hours following the addition of protein synthesis inhibitors. cSDR is not synchronized with the cell cycle but is sensitive to rifampin. Our rifampin run-out experiments of cells grown in LB medium indicated that replication in rnhA cells was highly unregulated, with the loss of discrete chromosomal peaks being observed (strain PH379 [Fig. 8b]). Thus, extensive unregulated replication is expected to occur in cells lacking both rnhA and topA as is the case for strain SB383 [topA rnhA gyrB(Ts)/pPH1243]. This might be particularly harmful for chromosomal segregation in this strain in which gyrase activity is inhibited.
If unregulated replication is problematic for the growth (and chromosome segregation) of strain SB383, culture conditions that reduce such replication should alleviate the problem. We found that strain SB383 could grow in minimal medium without the need to overproduce Topo III (data not shown). Under these conditions, unregulated replication was clearly reduced in both topA- and rnhA-null mutants. Indeed, flow cytometry in rifampin run-out experiments revealed a near perfect 2n chromosomal pattern with only one small additional peak, reflecting some asynchrony, for the topA-null mutant (strain RFM475 [Fig. 8a]). A nearly perfect 2n chromosomal pattern was also obtained in run-out experiments for rnhA-null cells grown in minimal medium (Fig. 8b). Thus, our results support the hypothesis that unregulated replication due to the absence of topA and rnhA can contribute to growth and chromosomal segregation problems of cells with a defective gyrase. Therefore, the topA gene would indirectly affect chromosomal segregation by regulating replication.
DISCUSSION
Interplay between Topo III and gyrase in chromosome segregation.
In this paper, we present experimental evidence for an interplay between gyrase and Topo III in chromosomal segregation in vivo. This interplay could not be revealed until topA was found to correct the growth defect of the gyrB(Ts) mutant at nonpermissive temperatures. Growth inhibition due to defective gyrase has several causes, including inhibition of replication initiation, chromosome segregation failure (Par−), inhibition of rRNA synthesis, and others (46), that all have at least in part a common denominator, namely, the lack of negative supercoiling. Thus, deleting topA would bring negative supercoiling to a level that would allow these key cellular activities to be sufficiently completed for growth to occur. However, the topB gene would be required due to the Par− phenotype at nonpermissive temperatures.
topA topB-null mutants were previously shown to suffer from severe RecA-dependent chromosomal segregation defects (30). Therefore, it could be argued that the major chromosomal segregation phenotype of the topA topB gyrB(Ts) strain described here was the result of two independent effects: a RecA-dependent effect that was due to the absence of both type 1A topoisomerases plus a Par−-related effect due to defective gyrase activity. However, for several reasons, we do not believe this to be the case. First, we recently found that compensatory mechanisms (including a recA deletion) that significantly correct the chromosomal segregation defects of our topA topB gyrB(Ts) strain at lower temperatures had no effects at higher temperatures (Usongo et al., unpublished). Second, here we present strong evidence for an interplay between gyrase and Topo III in two other instances: overproduction of Topo III at a very high level substantially corrects the Par− phenotype of the gyrB(Ts) strain, and replacement of the gyrB(Ts) allele by a gyrB+ one bypasses the need for Topo III overproduction for growth and chromosome segregation in our topA rnhA gyrB(Ts) strain. Thus, we believe our results are consistent with an interplay between gyrase and Topo III in chromosomal segregation.
While our manuscript was in preparation, a paper was published that demonstrated an interplay between Topo III and Topo IV in chromosome segregation in E. coli (47). However, no clues were provided as to when one specific topoisomerase would be required to allow chromosome segregation to occur. Furthermore, based on the fact that deleting topB had only a minor effect on chromosomal segregation in a strain carrying the same gyrB(Ts) allele as the one used in the present study, the authors concluded that there were no significant interactions between Topo III and gyrase in segregation. However, as shown in our paper and as stated above, a significant interplay between topB and this gyrB(Ts) allele could be seen only when topA was also deleted. Therefore, while the paper by Perez-Cheeks et al. (47) clearly reveals an interplay between Topo III and Topo IV in chromosomal segregation, our paper shows an interplay between Topo III and gyrase in this process.
The interplay between Topo III and Topo IV in chromosomal segregation can thus be explained, at least in part, in the context of chromosomal supercoiling that is regulated by the opposing enzymatic activities of gyrase and Topo I. When gyrase activity is defective, the supercoiling level is low and Topo IV would not be efficient in decatenation. In this context, the activity of Topo III would be required. This would explain why Topo IV overproduction cannot substitute for Topo III in chromosomal segregation at nonpermissive temperatures, in our topA gyrB(Ts) strain. On the other hand, an increase in negative supercoiling conferred by deleting topA in the gyrB(Ts) strain likely improves the ability of Topo IV to perform decatenation, but the supercoiling level is still too low, thus explaining why Topo III is also required.
Replication-induced positive supercoiling becomes particularly problematic when convergent replication forks are about to meet at the Ter region of the E. coli chromosome. Not only does a very high level of positive supercoiling accumulate, but also, the space on the DNA template may become too small to accommodate binding by gyrase. It has been shown in vitro that Topo III together with RecQ and single-stranded DNA-binding protein (SSB) can act at converging replication forks to topologically unlink them (48). In this reaction, RecQ helicase provides the single-stranded DNA substrate to which Topo III can bind. SSB, by interacting with both proteins, mediates the functional cooperation between RecQ and Topo III. However, we found that deleting recQ in our topA gyrB(Ts) strain does not affect the ability of the topA deletion to complement the gyrB(Ts) mutant at high temperatures, thus indicating that RecQ is not required for Topo III to perform its essential function in this situation (Usongo et al., unpublished).
The location where Topo III actually acts in vivo is still unknown. As ssDNA regions are expected to form only transiently in vivo (this would even be more problematic when gyrase is defective) and as the abundance of Topo III is normally very low, it has been difficult to pinpoint the exact site of action of Topo III in vivo. The ability of Topo III to physically interact with SSB protein, as recently shown, may explain how Topo III can have access to its site of action in vivo (48). Indeed, SSB may efficiently bring Topo III to its site of action that may be situated at the replication forks where SSB also binds. Topo III would therefore be properly located to act on precatenanes, as suggested elsewhere (47).
Interplay between Topo I and gyrase in chromosomal segregation.
The severe chromosomal segregation and growth phenotypes seen here when both type 1A topoisomerases were absent may indicate that Topo I can substitute for Topo III in chromosomal segregation. The much lower efficiency of Topo I in this process compared to Topo III could be compensated for by its much higher abundance than Topo III. Although this is possible, an alternative and non-mutually exclusive explanation that may reflect a real function of Topo I would be that it indirectly affects chromosome segregation through supercoiling by limiting firing from oriC. By doing so, Topo I would control the number of replication forks traveling on the chromosome. This would facilitate linkage removal, especially when gyrase is defective, as is often the case in topA mutants.
In fact, in an in vitro oriC-based replication system, Topo III was shown to support replication fork progression and to perform the final decatenation step, whereas inhibition of replication initiation from oriC was the only effect seen for Topo I (20, 21). Moreover, a topA deletion has been shown to suppress the growth defect of a dnaA46(Ts) mutant at the nonpermissive temperature (49) and to cause replication from oriC to be unregulated (44), as also shown here in our topA-null strain. Furthermore, the replication initiation defect of the gyrB(Ts) strain at nonpermissive temperatures was shown here to be corrected by deleting topA. This supports the hypothesis that the lack of Topo I activity promotes replication from oriC by causing negative supercoiling to increase in this region, thus facilitating DNA melting. Thus, in the topA gyrB(Ts) strain, unregulated replication from oriC would make the defective gyrase unable to efficiently support chromosome segregation. The wild-type level of Topo III activity would then be indispensable, but sufficient, for chromosome segregation. Alternatively, we also have to consider the possibility that Topo I, through supercoiling regulation, may affect chromosome segregation indirectly by modulating gene expression.
In the topA rnhA gyrB(Ts) strain SB383, the absence of RNase HI further enhanced the level of unregulated replication already caused by the absence of topA, by allowing replication from stable R loops (cSDR). Moreover, since one major function of Topo I is to inhibit R-loop formation (5, 8), the absence of topA is also expected to stimulate cSDR. Together with the fact that gyrase supercoiling activity is significantly inhibited in the absence of RNase HI (32), this high level of unregulated replication could explain why Topo III needed to be overproduced in strain SB383 to allow growth and chromosomal segregation. In this context, our results suggest that the previously reported effect of rnhA on chromosomal segregation (32) is indirect and likely related to cSDR. Thus, Topo I and RNase HI may facilitate chromosomal segregation by limiting replication.
Interestingly, when the topA rnhA gyrB(Ts) strain was grown in a minimal medium instead of a rich one, chromosomal segregation was significantly improved, and this coincided with a clear reduction in the amount of unregulated replication conferred by deleting topA or rnhA. This correlation strongly supports the link between unregulated replication and chromosomal segregation defects. Furthermore, the results of transposon mutagenesis to isolate suppressors of the growth defect of strain SB383 indicate that, indeed, unregulated replication conferred by the absence of topA and rnhA significantly contributes to the chromosomal segregation defect seen in this strain (Usongo et al., unpublished). Thus, Topo I, via supercoiling regulation can likely affect chromosomal segregation in two ways: by affecting the efficiency of decatenation by Topo IV and by regulating replication initiation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Russell DiGate for strain DM4100 ΔtopB::kan, plasmid pC18pBAD33, and anti-Topo III antibodies, Hiroshi Hiasa for plasmid pET11-parEC, and Kenneth Marians for anti-ParC and anti-ParE antibodies. We also thank Serge Sénéchal for excellent technical assistance with flow cytometry and Patrick Hallenbeck for English editing.
This work was supported by grants from the CIHR and the NSERC to M.D. C.T. and V.U. were supported by a scholarship from la Faculté des études supérieures et postdoctorales from the Université de Montréal.
Footnotes
Published ahead of print 8 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02001-12.
REFERENCES
- 1. Champoux JJ. 2001. DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem. 70:369–413 [DOI] [PubMed] [Google Scholar]
- 2. Kirkegaard K, Wang JC. 1985. Bacterial DNA topoisomerase I can relax positively supercoiled DNA containing a single-stranded loop. J. Mol. Biol. 185:625–637 [DOI] [PubMed] [Google Scholar]
- 3. Drlica K. 1992. Control of bacterial DNA supercoiling. Mol. Microbiol. 6:425–433 [DOI] [PubMed] [Google Scholar]
- 4. Massé E, Drolet M. 1999. Relaxation of transcription-induced negative supercoiling is an essential function of Escherichia coli DNA topoisomerase I. J. Biol. Chem. 274:16654–16658 [DOI] [PubMed] [Google Scholar]
- 5. Drolet M. 2006. Growth inhibition mediated by excess negative supercoiling: the interplay between transcription elongation, R-loop formation and DNA topology. Mol. Microbiol. 59:723–730 [DOI] [PubMed] [Google Scholar]
- 6. DiNardo S, Voelkel KA, Sternglanz R, Reynolds AE, Wright A. 1982. Escherichia coli DNA topoisomerase I mutants have compensatory mutations in DNA gyrase genes. Cell 31:43–51 [DOI] [PubMed] [Google Scholar]
- 7. Pruss GJ, Manes SH, Drlica K. 1982. Escherichia coli DNA topoisomerase I mutants: increased supercoiling is corrected by mutations near gyrase genes. Cell 31:35–42 [DOI] [PubMed] [Google Scholar]
- 8. Massé E, Drolet M. 1999. Escherichia coli DNA topoisomerase I inhibits R-loop formation by relaxing transcription-induced negative supercoiling. J. Biol. Chem. 274:16659–16664 [DOI] [PubMed] [Google Scholar]
- 9. Baaklini I, Usongo V, Sanscartier P, Nolent F, Hraiky C, Drlica K, Drolet M. 2008. Hypernegative supercoiling inhibits growth by causing RNA degradation. J. Bacteriol. 190:7346–7356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. DiGate RJ, Marians KJ. 1989. Molecular cloning and DNA sequence analysis of Escherichia coli topB, the gene encoding topoisomerase III. J. Biol. Chem. 264:17924–17930 [PubMed] [Google Scholar]
- 11. DiGate RJ, Marians KJ. 1988. Identification of a potent decatenating enzyme from Escherichia coli. J. Biol. Chem. 263:13366–13373 [PubMed] [Google Scholar]
- 12. Zechiedrich EL, Khodursky AB, Bachellier S, Schneider R, Chen D, Lilley DM, Cozzarelli NR. 2000. Roles of topoisomerases in maintaining steady-state DNA supercoiling in Escherichia coli. J. Biol. Chem. 275:8103–8113 [DOI] [PubMed] [Google Scholar]
- 13. Lopez CR, Yang S, Deibler RW, Ray SA, Pennington JM, DiGate RJ, Hastings PJ, Rosenberg SM, Zechiedrich EL. 2005. A role for topoisomerase III in a recombination pathway alternative to RuvABC. Mol. Microbiol. 58:80–101 [DOI] [PubMed] [Google Scholar]
- 14. Funnell BE, Baker TA, Kornberg A. 1986. Complete enzymatic replication of plasmids containing the origin of the Escherichia coli chromosome. J. Biol. Chem. 261:5616–5624 [PubMed] [Google Scholar]
- 15. Drlica K, Zhao X. 1997. DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol. Mol. Biol. Rev. 61:377–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang X, Reyes-Lamothe R, Sherratt DJ. 2008. Modulation of Escherichia coli sister chromosome cohesion by topoisomerase IV. Genes Dev. 22:2426–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Champoux JJ, Been MD. 1980. Topoisomerases and the swivel problem, p 809–815 In Alberts B. (ed), Mechanistic studies of DNA replication and recombination: ICN-UCLA symposia on molecular and cellular biology. Academic Press, Inc., New York, NY [Google Scholar]
- 18. Hiasa H, Marians KJ. 1996. Two distinct modes of strand unlinking during theta-type DNA replication. J. Biol. Chem. 271:21529–21535 [DOI] [PubMed] [Google Scholar]
- 19. Ullsperger CJ, Vologodskii AA, Cozzarelli NR. 1995. Unlinking of DNA by topoisomerases during DNA replication, p 115–142 In Lilley DMJ, Eckstein F. (ed), Nucleic acids and molecular biology. Springer-Verlag, Berlin, Germany [Google Scholar]
- 20. Hiasa H, Marians KJ. 1994. Topoisomerase III, but not topoisomerase I, can support nascent chain elongation during theta-type DNA replication. J. Biol. Chem. 269:32655–32659 [PubMed] [Google Scholar]
- 21. Hiasa H, DiGate RJ, Marians KJ. 1994. Decatenating activity of Escherichia coli DNA gyrase and topoisomerases I and III during oriC and pBR322 DNA replication in vitro. J. Biol. Chem. 269:2093–2099 [PubMed] [Google Scholar]
- 22. Nurse P, Levine C, Hassing H, Marians KJ. 2003. Topoisomerase III can serve as the cellular decatenase in Escherichia coli. J. Biol. Chem. 278:8653–8660 [DOI] [PubMed] [Google Scholar]
- 23. Kato J, Nishimura Y, Suzuki H. 1989. Escherichia coli parA is an allele of the gyrB gene. Mol. Gen. Genet. 217:178–181 [DOI] [PubMed] [Google Scholar]
- 24. Adams DE, Shekhtman EM, Zechiedrich EL, Schmid MB, Cozzarelli NR. 1992. The role of topoisomerase IV in partitioning bacterial replicons and the structure of catenated intermediates in DNA replication. Cell 71:277–288 [DOI] [PubMed] [Google Scholar]
- 25. Peng H, Marians KJ. 1993. Decatenation activity of topoisomerase IV during oriC and pBR322 DNA replication in vitro. Proc. Natl. Acad. Sci. U. S. A. 90:8571–8575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ullsperger C, Cozzarelli NR. 1996. Contrasting enzymatic activities of topoisomerase IV and DNA gyrase from Escherichia coli. J. Biol. Chem. 271:31549–31555 [DOI] [PubMed] [Google Scholar]
- 27. Zechiedrich EL, Khodursky AB, Cozzarelli NR. 1997. Topoisomerase IV, not gyrase, decatenates products of site-specific recombination in Escherichia coli. Genes Dev. 19:2580–2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holmes VF, Cozzarelli NR. 2000. Closing the ring: links between SMC proteins and chromosome partitioning, condensation, and supercoiling. Proc. Natl. Acad. Sci. U. S. A. 97:1322–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Witz G, Stasiak A. 2010. DNA supercoiling and its role in DNA decatenation and unknotting. Nucleic Acids Res. 38:2119–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu Q, Pongpech P, DiGate RJ. 2001. Type I topoisomerase activity is required for proper chromosomal segregation in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 98:9766–9771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Drolet M, Phoenix P, Menzel R, Massé E, Liu LF, Crouch RJ. 1995. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: R-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. U. S. A. 92:3526–3530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Usongo V, Nolent F, Sanscartier P, Tanguay C, Broccoli S, Baaklini I, Drlica K, Drolet M. 2008. Depletion of RNase HI activity in Escherichia coli lacking DNA topoisomerase I leads to defects in DNA supercoiling and segregation. Mol. Microbiol. 69:968–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Broccoli S, Phoenix P, Drolet M. 2000. Isolation of the topB gene encoding DNA topoisomerase III as a multicopy suppressor of topA null mutations in Escherichia coli. Mol. Microbiol. 35:58–68 [DOI] [PubMed] [Google Scholar]
- 34. Broccoli S, Rallu F, Sanscartier P, Cerritelli SM, Crouch RJ, Drolet M. 2004. Effects of RNA polymerase modifications on transcription-induced supercoiling and associated R-loop formation. Mol. Microbiol. 52:1769–1779 [DOI] [PubMed] [Google Scholar]
- 35. Steck TR, Drlica K. 1984. Bacterial chromosome segregation: evidence for DNA gyrase involvement in decatenation. Cell 36:1081–1088. [DOI] [PubMed] [Google Scholar]
- 36. Miller JH. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 37. Lavasani LS, Hiasa H. 2001. A ParE-ParC fusion protein is a functional topoisomerase. Biochemistry 40:8438–8443 [DOI] [PubMed] [Google Scholar]
- 38. Filutowicz M, Jonczyk P. 1981. Essential role of the gyrB gene product in the transcriptional event coupled to dnaA-dependent initiation of Escherichia coli chromosome replication. Mol. Gen. Genet. 183:134–138 [DOI] [PubMed] [Google Scholar]
- 39. Filutowicz M, Jonczyk P. 1983. The gyrB gene product functions in both initiation and chain polymerization of Escherichia coli chromosome replication: suppression of the initiation deficiency in gyrB-ts mutants by a class of rpoB mutations. Mol. Gen. Genet. 191:282–287 [DOI] [PubMed] [Google Scholar]
- 40. Li Z, Hiasa H, DiGate R. 2006. Characterization of a unique type IA topoisomerase in Bacillus cereus. Mol. Microbiol. 60:140–151 [DOI] [PubMed] [Google Scholar]
- 41. Kato J, Nishimura Y, Imamura R, Niki Hiraga HS, Suzuki H. 1990. New topoisomerase essential for chromosome segregation in E. coli. Cell 63:393–404 [DOI] [PubMed] [Google Scholar]
- 42. Stockum A, Lloyd RG, Rudolph CJ. 2012. On the viability of Escherichia coli cells lacking DNA topoisomerase I. BMC Microbiol. 12:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leonard AC, Grimwade JE. 5 January 2010, posting date. Chapter 4.4.1, Initiation of DNA replication. In Böck A, Curtiss R, III, Kaper JB, Karp PD, Neidhardt FC, Nyström T, Slauch JM, Squires CL, Ussery D. (ed), EcoSal—Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, DC: doi:10.1128/ecosal.4.4.1. [Google Scholar]
- 44. Olsson JA, Nordström K, Hjort K, Dasgupta S. 2003. Eclipse-synchrony relationship in Escherichia coli strains with mutations affecting sequestration, initiation of replication and superhelicity of the bacterial chromosome. J. Mol. Biol. 334:919–931 [DOI] [PubMed] [Google Scholar]
- 45. Kogoma T. 1997. Stable DNA replication: interplay between DNA replication homologous recombination, and transcription. Microbiol. Mol. Biol. Rev. 61:212–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Drlica K. 1984. Biology of bacterial deoxyribonucleic acid topoisomerases. Microbiol. Rev. 48:273–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Perez-Cheeks BA, Lee C, Hayama R, Marians KJ. 16 October 2012, posting date. A role for topoisomerase III in Escherichia coli chromosome segregation. Mol. Microbiol doi:10.1111/mmi.12039. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Suski C, Marians KJ. 2008. Resolution of converging replication forks by RecQ and topoisomerase III. Cell 30:779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Louarn J, Bouché JP, Patte J, Louarn JM. 1984. Genetic inactivation of topoisomerase I suppresses a defect in initiation of chromosome replication in Escherichia coli. Mol. Gen. Genet. 195:170–174 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.