

Abstract
The extracytoplasmic assembly of the Dot/Icm type IVb secretion system (T4SS) of Legionella pneumophila is dependent on correct disulfide bond (DSB) formation catalyzed by a novel and essential disulfide bond oxidoreductase DsbA2 and not by DsbA1, a second nonessential DSB oxidoreductase. DsbA2, which is widely distributed in the microbial world, is phylogenetically distinct from the canonical DsbA oxidase and the DsbC protein disulfide isomerase (PDI)/reductase of Escherichia coli. Here we show that the extended N-terminal amino acid sequence of DsbA2 (relative to DsbA proteins) contains a highly conserved 27-amino-acid dimerization domain enabling the protein to form a homodimer. Complementation tests with E. coli mutants established that L. pneumophila dsbA1, but not the dsbA2 strain, restored motility to a dsbA mutant. In a protein-folding PDI detector assay, the dsbA2 strain, but not the dsbA1 strain, complemented a dsbC mutant of E. coli. Deletion of the dimerization domain sequences from DsbA2 produced the monomer (DsbA2N), which no longer exhibited PDI activity but complemented the E. coli dsbA mutant. PDI activity was demonstrated in vitro for DsbA2 but not DsbA1 in a nitrocefin-based mutant TEM β-lactamase folding assay. In an insulin reduction assay, DsbA2N activity was intermediate between those of DsbA2 and DsbA1. In L. pneumophila, DsbA2 was maintained as a mixture of thiol and disulfide forms, while in E. coli, DsbA2 was present as the reduced thiol. Our studies suggest that DsbA2 is a naturally occurring bifunctional disulfide bond oxidoreductase that may be uniquely suited to the majority of intracellular bacterial pathogens expressing T4SSs as well as in many slow-growing soil and aquatic bacteria.
INTRODUCTION
Legionella pneumophila and related species reside in aquatic environments as obligate intracellular parasites of amoebic hosts (1, 2). When transmitted to susceptible humans via aerosols, these bacteria can cause an acute pneumonia known as Legionnaires' disease (LD) (3, 4). Since human-to-human transmission does not occur with LD, there is little opportunity for evolution of virulence or for development of antibiotic resistance. Like many aquatic microorganisms, L. pneumophila displays a dimorphic life cycle, alternating between vegetative replicative intracellular bacteria and planktonic terminally differentiated cysts that are highly infectious and resilient to environmental stresses (5, 6, 7, 8). Both ultrastructural and proteomic analyses of the transition to cyst forms indicate substantial remodeling of the cell envelope, and the heightened infectivity suggests that the type IVb Dot/Icm secretion system (T4SS), the major virulence determinant, has become fully functional (7, 9, 10, 11). Since much of the cell remodeling occurs extracytoplasmically, we have been investigating proteins that participate in these processes. These studies led to identification of disulfide bond oxidoreductase DsbA2, which, in addition to essential functions, seemed to be required for proper assembly or function of the Dot/Icm T4SS (10). Phylogenetic analysis revealed that DsbA2 formed a distinct clade from DsbA and was broadly distributed in the microbial world from environmental species to nearly all microbial pathogens expressing T4SSs (10).
The DSB oxidoreductase family is part of the thioredoxin (TRX) superfamily of proteins, which are defined by the presence of one or more thioredoxin folds (12). These enzymes participate in disulfide bond formation through a conserved Cys-X-X-Cys (CXXC) active site motif, and they can either reduce or oxidize target substrates. DsbA is maintained in the oxidized (disulfide) form by a membrane-spanning DsbB partner which delivers reducing equivalents from DsbA into the electron transport chain via quinone reduction (13, 14). Similarly, DsbC and DsbG are maintained in the reduced (thiol) form by DsbD, which transfers reducing equivalents from the cytoplasmic thioredoxin/thioredoxin reductase system across the cytoplasmic membrane to these periplasmic partners (15, 16, 17). Separation of the two systems is considered important to avoid futile cycling of reducing equivalents, and this is accomplished through the high affinity of DsbD for homodimeric DsbC and DsbG, which are poor substrates of DsbB (15, 18, 19). While DsbA catalyzes the formation of consecutive disulfide bonds in nascent polypeptides entering the periplasm, DsbC, a protein disulfide isomerase (PDI), is thought to repair inappropriate disulfides or introduce nonconsecutive disulfide bonds through a process of reduction and re-formation of disulfide bonds to aid correct protein folding (15, 19, 20). It is generally believed that once correct disulfide bonds are formed, the rapid folding of proteins and their interactions with periplasmic chaperones or other assembly systems protect these bonds from further redox action (21).
As noted previously, the genes encoding DsbC and DsbG are absent from the genomes of DsbA2-expressing species, and some species, like Coxiella burnetii, also lack DsbA (10). In contrast to dsbA mutants in other bacteria, including pathogens (17, 22), we showed that dsbA1 mutants of L. pneumophila were indistinguishable from wild-type parental strains for infectivity (amoeba and HeLa cell models) and motility and were without correlating phenotypes, suggesting that DsbA2 likely plays a greater role in managing disulfide bonding and protein folding (10). We also showed that expression of a mutant DsbA2 (P198T) protein in L. pneumophila produced a dominant negative effect on DsbA2 function, resulting in loss of motility and infectivity; the latter was traced to a functional defect in the Dot/Icm secretion apparatus (10). Based on DsbA2 existing as a mixture of oxidized and reduced forms in the periplasm of L. pneumophila and together with the ability of DsbA2P198T mutant protein to form stable disulfide bonds with substrate proteins, including the cysteine-containing components (DotG and DotC) of the core Dot/Icm structure, we concluded that DsbA2 was functionally similar to DsbA in delivering disulfide bonds to nascent polypeptides (10).
Here we show that the extended N-terminal amino acid sequence of DsbA2 (compared to that of DsbA) contains a highly conserved dimerization domain enabling DsbA2 to exist as a homodimer in the periplasm of L. pneumophila. Through complementation tests, we show that expression of dsbA2 in an Escherichia coli dsbC mutant restored protein disulfide isomerase activity to nearly wild-type levels. While DsbC-DsbA chimeras have been shown to exist as a mixture of reduced thiol and disulfide moieties that enable oxidase and PDI activities in E. coli (19), DsbA2 appears to be an example of a naturally occurring bifunctional DSB oxidoreductase that is widely distributed in the microbial world and especially in those intracellular parasites also expressing T4SSs.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Legionella pneumophila strains and plasmids are listed in Table 1. Strains were grown aerobically at 37°C on buffered charcoal yeast extract agar (BCYE) (23) or in buffered yeast extract (BYE) broth and supplemented with α-ketoglutaric acid (1 mg/ml), ferric pyrophosphate (250 μg/ml), l-cysteine (40 μg/ml), thymidine (100 μg/ml), and antibiotics where required. Starter cultures were prepared as previously described (24) and used to inoculate prewarmed BYE to an optical density at 620 nm (OD620) of 0.2. For growth curve determinations, samples were taken every 2 h (triplicate) and optical density was determined at 620 nm. E. coli strains used in these studies are listed in Table 1. These strains were grown at 37°C on Luria-Bertani (LB) agar or in LB broth supplemented with the appropriate antibiotics. Antibiotics (Sigma-Aldrich Ltd.) were added to media at the following concentrations, when appropriate: streptomycin (100 μg/ml), kanamycin (40 μg/ml), gentamicin (10 μg/ml), chloramphenicol (20 μg/ml), and ampicillin (100 μg/ml). All strains were stored at −85°C in nutrient broth containing 10% dimethyl sulfoxide.
Table 1.
Strains and plasmids
| Strain or plasmid | Description | Reference or source |
|---|---|---|
| L. pneumophila strains | ||
| Philadelphia-1 | Spontaneous Strr mutant of Philadelphia-1 | 40 |
| 130b (AA100) | ATCC BAA-74 | |
| AA100ΔdsbA1 | Deletion of dsbA1 | 10 |
| E. coli strains | ||
| DH5α | F− endA1 hsdR17 (rK− mK−) supE44 thi-1 recA1 gyrA (Nalr) relA1 (lacIZYA argF)U169 deoR (80dlac(lacZ)M15) | Clontech |
| BL21(DE3) | CodonPlus RIL B F− ompT hsdS(rB− mB−) dcm Tetr gal (DE3) endA hte argU ileY leuW Camr | Stratagene |
| JCB570 | ER1821 | J. C. Bardwell |
| JCB571 | dsbA | J. C. Bardwell |
| RGP209 | ER1821 dsbC | 20 |
| RGP663 | ER1821 + pPDI detector plasmid | 20 |
| RGP665 | RGP209 + pPDI detector plasmid | 20 |
| Plasmids | ||
| pPDI detector | pBR322 Bla S81C, T108C | 20 |
| pET15b | Expression vector, His tag | Novagen |
| pBC | pBSK with chloramphenicol cassette | Stratagene |
| pMMB206 | Derivative of pRSF1010; Ptac promoter and IPTG-inducible lacIQ system; Cmr | 42 |
| pET15bdsbA1 | dsbA1 His tag | 10 |
| pET15bdsbA2 | dsbA2 His tag | 10 |
| pET15bdsbA2N | dsbA2 His minus dimerization domain | This study |
| pET15bdsbA2LDN | dsbA2 His minus leader and dimerization sequences | This study |
| pBCdsbA1 | dsbA1 in pBC cloning vector | This study |
| pBCdsbA2 | dsbA2 in pBC cloning vector | This study |
| pBCdsbA2N | dsbA2 minus dimerization domain in pBC | This study |
| pBCdsbA2LDN | dsbA2 minus leader and dimerization sequences | This study |
Protein purification and estimation of mass.
An N-terminal hexa-His (H6)-tagged DsbA2 and DsbA1 were constructed in pET15b expression vector and cloned into E. coli BL21(DE3) CodonPlus RIL (Stratagene) as previously described (10). N-terminal H6-tagged DsbA2LDN, minus the leader sequence and dimerization domain, was created using primers DsbA2LDNNdeI (5′-GGAATTCCATATGGCCGCAATTCAGGAAAAT) and DsbA2expBamHI (5′-TTAGATGGATCCTTAATTGCCAGCCGCC). The amplicon was cloned into pET15b and expressed in the BL21 strain. All genetic constructs were verified by DNA sequencing, and protein expression was determined by SDS-PAGE and immunoblotting with anti-DsbA2 serum (10). DsbA2LDN was purified by nickel affinity chromatography (Novagen, Madison, WI) as previously described (10). The molecular masses of DsbA2 and DsbA1 (ca. 3 mg/ml protein) were estimated by gel filtration in 50 mM Tris-HCl, 100 mM NaCl at pH 8 on a calibrated HiLoad 16/60 Superdex-200 column (GE Healthcare).
Isolation of native DsbA2 by osmotic shock.
Stationary-phase bacteria (strain AA100) were collected by centrifugation at 13,000 × g for 15 min at 4°C, and the pellet (1 g [wet weight]) was suspended in 40 ml of sucrose buffer (0.5 M sucrose, 30 mM Tris-HCl plus 1 mM EDTA at pH 8) and gently shaken for 10 min at 25°C. Following centrifugation, the pellet was suspended in 20 ml of ice-cold 30 mM Tris-HCl plus 1 mM EDTA buffer, gently shaken for 10 min at 4°C, and centrifuged. The shockate was decanted and concentrated to 5 ml and loaded onto the calibrated HiLoad 16/60 Superdex-200 column, and fractions corresponding to the homodimer and monomer were subjected to SDS-PAGE. The proteins were transferred to nitrocellulose and developed with DsbA2-specific antibody diluted 1:10,000 in phosphate-buffered saline (PBS) with 0.1% Tween 20 as previously described (10, 25).
Complementation studies.
Wild-type (WT) E. coli (JCB570) and a dsbA mutant (JCB571) were kindly provided by J. C. Bardwell. The plasmid pBC containing the coding sequence of either dsbA1 or dsbA2 or empty vector was transformed into the E. coli strains and selected by chloramphenicol resistance. To construct an in-frame deletion of the dimerization domain of DsbA2 (DsbA2N), a vector-free strategy was employed using overlapping primers (DsbA2NF, 5′-TGGCAGCTGATCAGGAAAATGCTGAACAAG; DsbA2NR, 5′-GCATTTTCCTGATCAGCTGCCATTATTGCA) to join the 5′ leader sequence with sequences downstream of the dimerization domain and using flanking primers Com1FSD (5′-GGGAATTCTAAGGGGAATTACGTGAAATTTAC) and DsbA2BamHIR (5′-TAAGGATCCTTAATTGCCAGCCGCC) to amplify the joined construct (24). The resulting amplicon was cloned into pBC and pMMB206 vectors. Soft agar LB plates (0.4% agar) supplemented with 1 mM IPTG were prepared, and 2 μl of cell suspension was inserted into the middle of the plate. The LB plates were incubated overnight at 30°C, and motility was assessed by measuring the diameter of spreading bacterial growth and reported as a percentage of the WT control. Motility assays were performed in triplicate, and the results from representative plates are presented.
PDI detector assay.
The PDI detector assay utilizes the TEM1 β-lactamase of pBR322 with an engineered nonconsecutive disulfide bond (cysteine residues added, S81C and T108C) that requires disulfide bond isomerase activity to be properly folded in the E. coli periplasm (20). Strains kindly provided by J. C. Bardwell and listed in Table 1 include RGP209 (dsbC mutant control), RGP663 plus pPDI detector plasmid (wild-type positive control), and RGP665 (RGP209 plus pPDI detector plasmid). Ampicillin resistance was evaluated at 0, 1, 2, or 3 g/liter. For complementation studies, dsbA1, dsbA2, dsbA2LDN, and dsbA2N were cloned into pBC with selection for chloramphenicol. Bacterial cells were grown overnight in the appropriate antibiotic, diluted 1:20, and grown for 2 to 3 h to an OD600 of 0.7 with 1 mM IPTG induction. Cells were then serially diluted and plated in triplicate on LB plates with 0, 1, 2, or 3 g/liter Amp, and CFU counts were recorded at 24 h. One set of representative plates depicting typical results is presented.
Insulin reduction assay.
Reductase activity was assessed by an insulin precipitation assay with minor modifications (10, 13). Bovine insulin was dissolved in Tris-HCl to 10 mg/ml (1.67 mM) and titrated to pH 7.5, creating a clear solution. Reactions (triplicate) were carried out in 200 μl of 100 mM sodium phosphate buffer, pH 7.0, 150 μM insulin, 0.33 mM dithiothreitol (DTT), and 2 mM EDTA; reaction mixtures were incubated in a 96-well plate format at room temperature in a VersaMax (Molecular Devices) plate reader, and absorbance was measured at 650 nm. The insulin reduction assay was initiated by adding 5 μM DsbA2, DsbA2LDN, and DsbA1 protein purified as H6-tagged proteins following induction in E. coli strain BL21. Both time to start of insulin reduction and specific activity were determined in triplicate using the enzyme kinetics program as previously described (10). The results from a typical experiment are presented.
Isomerization assay.
TEM1 β-lactamase (WT and PDI detector mutant enzymes Bla and MBla) was obtained from spent culture supernatants of strains RGP663 and RGP665 following overnight growth in LB medium. Supernatants were concentrated by spin columns (12 ml concentrated to 0.5 ml) (Amicon Ultracel 10k). Bla activity was tested by spotting 5 μl of concentrated supernatant onto nitrocefin-impregnated paper disks (Becton, Dickinson). The relative concentration of Bla in each fraction was determined by SDS-PAGE following staining with Coomassie brilliant blue. For the PDI assay, either purified leaderless H6-tagged DsbA1 (6.9 mg/ml) or H6-DsbA2 (3 mg/ml) was added to concentrated culture supernatant from strain RGP665 in a ratio of 1:5 and incubated at 37°C for 30 min. Nitrocefin (Calbiochem, La Jolla, CA) was prepared according to manufacturer's instructions for a spectrophotometric assay, and a working dilution of 500 μg/ml was prepared in 0.1 M sodium phosphate buffer at pH 7.0. The refolding assay was configured in a 96-well microplate (Costar, Corning, NY) assay (100 μl/well) containing sodium phosphate buffer and 15 μl of MBla plus DsbA1 or MBla plus DsbA2, and the assay was started by the addition of 10 μl of nitrocefin stock. Controls contained no DsbA2 or DsbA1 protein or contained WT Bla as a positive control. The rates of hydrolysis of nitrocefin at 486 nm were obtained over the linear range of the reaction (30 min) at 30°C in a Molecular Dynamics plate reader; all reactions were run in triplicate, and a representative recording of reaction kinetics is presented.
Periplasmic redox status of DsbA2.
The in vivo redox status of DsbA2 was determined by alkylation of free thiol groups by 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) (Molecular Probes, Eugene, OR) essentially as described previously (10, 26). Briefly, L. pneumophila was grown to stationary phase (∼24 h), and half of the culture was collected by centrifugation and washed once in sterile water and then suspended to an OD660 of 0.5 in sterile filtered tap water. The water-suspended bacteria were kept in the dark for 48 h at 22°C, a period of time previously shown to promote differentiation into cyst-like dormant forms (7). The stationary-phase bacteria were collected by centrifugation and were divided into aliquots, one of which was first treated with 10 mM DTT and then trichloroacetic acid (TCA) precipitated and alkylated with 100 mM AMS; one was TCA precipitated and alkylated with AMS, and one served as an untreated control. The water-treated bacteria were TCA precipitated and treated with 100 mM AMS. MalPEG5000 (N-ethylmaleimide covalently bound to polyethylene glycol) was used to alkylate free thiols of DsbA2 and DsbA2N expressed in E. coli. Aliquots were treated as described for AMS except that 2 mM DTT was used to reduce DsbA2 prior to alkylation with 5 mM MalPEG5000. In both treatments, samples were analyzed by SDS-PAGE and immunoblotting with anti-DsbA2 serum (10). AMS increases protein mass by 490 Da and MalPEG by 5,000 Da, observed as band shifts.
RNA isolation.
Total L. pneumophila RNA was extracted from cells at an OD620 of 0.9 by the RiboZol RNA extraction reagent (Amresco, OH). Total RNA was treated with DNase I, and RNA concentrations were determined using a NanoDrop ND-1000 spectrophotometer (Nanodrop Technologies). First-strand cDNA was generated using the SuperScript first-strand synthesis system for reverse transcriptase PCR (RT-PCR) (Invitrogen, CA). When the RT reaction was completed, samples were adjusted to 200 μl with molecular biology water.
Real-time PCR analysis of gene expression.
The expression of a dsbA1 and dsbA2 was measured in early exponential and stationery phases of growth. Real-time PCR assays were performed using the CFX96 real-time PCR detection system (Bio-Rad). The final reaction volume (25 μl) contained 12.5 μl SYBR green PCR master mix (Applied Biosystems), 1 μl 10 μM forward primer, 1 μl 10 μM reverse primer, 5 μl cDNA template, and 5.5 μl molecular biology water. The real-time PCR assays were carried out using a standard program: 95°C for 10 min followed by 35 cycles at 95°C for 15 s, 55°C for 15 s, and 60°C for 30 s. Gene expression was analyzed using the comparative threshold cycle (2−ΔΔCT) method with rplJ expression as an endogenous control (24). All quantitative PCRs (qPCRs) were carried out in triplicate and the mean and standard deviation computed.
Phylogenetic analysis.
DsbA1 and DsbA2 amino acid sequences used in phylogenetic analysis were obtained from the LegioList web server (http://genolist.pasteur.fr/LegioList/genome.cgi) and orthologous genes from GenBank by BLASTP search using DsbA1 and DsbA2 of L. pneumophila Philadelphia-1 and DsbA, DsbC, and DsbG from E. coli. Phylogenetic trees were generated by multiple sequence alignment of unedited DsbA2 sequences using CLUSTALW. Analyses were further refined by BLASTP search using the highly conserved dimerization domain sequence (SLSDAQKKEIEKVIHDYLINNPEVLLEASQA).
RESULTS
Previous phylogenetic analysis of the DsbA2 clade indicated that this group has diverged from the DsbA lineage of disulfide bond oxidoreductases (10). Comparison of the structures of DsbA2, DsbA, and DsbC shows that DsbA2 contains an additional 56 amino acids (86 total) from the N terminus to the CXXC motif compared with 31 for DsbA and 107 for DsbC. A refined BLASTP search using the dimerization domain region depicted in Fig. 1A revealed a 27-amino-acid (aa) sequence (bold in Fig. 1A) present in all the bacterial species expressing DsbA2 listed in Table 2. Moreover, this sequence is more discriminating in searches than the whole protein or the C-terminal DsbA-related sequences used previously (10). Table 2 includes representative species based on a similarity cutoff of ∼e−34 and is not meant to be comprehensive. We noticed that below this cutoff, the DsbA2 clade begins to merge into the DsbA clade. The DsbA2 clade includes a group of intracellular parasites that commonly cause disease in humans, animals, and plants and share a T4SS. Other genera that do not express T4SSs include the nitrogen-fixing endosymbionts associated with leguminous plants, including Bradyrhizobium and related genera (not listed), and many soil and aquatic free-living genera, including Azospirillum Rhodopseudomonas, Rhodospirillum, and Caulobacter. One exception is Bordetella pertussis, which has a T4SS (pertussis toxin [PtL] transporter) and expresses a DsbA/DsbC system similar to the E. coli system.
Fig 1.

Dimerization domain and secondary structure comparison. (A) DsbA2 amino acid sequence depicting the leader sequence and the detached dimerization domain sequence deleted to create DsbA2N. The underlined sequences (27 amino acids) of the dimerization domain are conserved, and larger letters represent amino acids that are highly conserved among DsbA2 members listed in Table 2. The CXXC and cis-proline regions are in boldface. (B) Secondary structure predictions for L. pneumophila DsbA1, which is similar to DsbA of E. coli, E. coli DsbC, and L. pneumophila DsbA2 are depicted. The CXXC region and the cis-proline motifs are boxed. Note that the cis-P region of DsbA2 is similar to that of DsbC.
Table 2.
DSB composition of genera expressing DsbA2
| Bacterium | Presence ofa: |
||||||
|---|---|---|---|---|---|---|---|
| DsbA1 | DsbA2 | DsbB | DsbD | CcmG | DsbC/G | T4SDS | |
| Legionella | + | + | + | + | + | − | + |
| Rickettsia | + | + | + | + | + | − | + |
| Francisella | + | + | + | + | − | − | ? |
| Brucella | + | + | + | + | + | − | + |
| Anaplasma | + | + | + | + | + | − | + |
| Agrobacterium | + | + | + | + | + | − | + |
| Coxiella | − | + | + | + | − | − | + |
| Ehrlichia | + | + | + | + | + | − | + |
| Afipia | + | + | + | + | + | − | + |
| Rhodopseudomonas | + | + | + | + | + | − | − |
| Bradyrhizobium | + | + | + | + | + | − | − |
| Azospirillum | + | + | + | + | + | − | − |
| Bartonella | + | + | + | + | + | − | + |
| Caulobacter | + | + | + | + | + | − | − |
| Rhodospirillum | + | + | + | + | + | − | − |
| Aeromonas | + | + | + | + | + | − | − |
| Bordetella | + | − | + | + | + | + | + |
| Escherichia | + | − | + | + | + | + | − |
This is a partial list from more than 60 genomes identified by BLASTP with DsbA2 and the 27-amino-acid dimerization domain sequences depicted in Fig. 1A. For DsbA1, DsbB, DsbD, DsbC, DsbG, and CcmG, the E. coli protein sequences were used for the searches against each genus. The presence or absence of a type IV secretion system (T4SS) was determined from the literature. The question mark for Francisella tularensis indicates that the system may be incomplete. CcmG is a thioredoxin-like periplasmic enzyme associated with cytochrome c maturation.
In addition, the spacing between the CXXC motif and the resolving cis-proline motif is conserved between DsbA2 and DsbA compared with DsbC. Finally, as highlighted in Fig. 1B, the first amino acid at the N terminus of the cis-proline in DsbA2 (threonine) is similar to that of DsbC and not that of DsbA, which substitutes valine. Further genomic analysis using E. coli DsbC and DsbG amino acid sequences in the BLASTP searches revealed that the genera expressing DsbA2 listed in Table 2 also lacked orthologues of DsbC and DsbG. Our previous studies showed that L. pneumophila ΔdsbA1 was indistinguishable from the wild-type strain for motility and infectivity for amoebae and HeLa cells, and in the close relative Coxiella burnetii, dsbA1 is completely absent from sequenced genomes (10). While there is plenty of precedent for interchangeability of DsbA and DsbC functions in E. coli (18, 19), we investigated whether DsbA2 represents a functional equivalent of DsbC or if equilibrium of monomers and homodimers is responsible for the apparent bifunctional phenotype and thus a variation of the E. coli DsbA/DsbC paradigm.
DsbA2 exists as a homodimer in E. coli and L. pneumophila.
To test whether DsbA2 exists as a mixture of monomers and dimers, size exclusion chromatography was employed. For this study, H6-tagged DsbA1 and DsbA2 were treated with iodoacetamide to limit formation of spurious disulfides prior to nickel interaction chromatography. Each protein was applied to a calibrated gel filtration column, and, as seen in Fig. 2A, DsbA1 eluted at 95 min, with an apparent mass of 27 kDa, consistent with a monomer. In Fig. 2B, DsbA2 eluted as a single peak at 75 min, with an apparent mass of 60 kDa, consistent with the size of the homodimer. Subsequent purifications of DsbA2 indicated that inclusion of iodoacetamide was unnecessary (data not presented). To ensure that the homodimeric form was not an artifact generated in E. coli, we applied osmotic shockates from L. pneumophila similarly treated with iodoacetamide over the same calibrated column, and DsbA2 eluted as the dimer (Fig. 2C). The elution of DsbA2 as a single peak suggests that if the monomer form is present in L. pneumophila, it is below the limit of detection in our assay.
Fig 2.

Size exclusion of native proteins from L. pneumophila. (A) DsbA1 elutes as a single peak at 95 min, consistent with a monomer and an estimated mass of 25 kDa. (B) DsbA2 elutes at 75 min, consistent with the homodimer and estimated mass of 60 kDa. The HiLoad 16/60 Superdex-200 column was calibrated with RNase A (13.7 kDa), chymotrypsinogen A (25 kDa), ovalbumin (43 kDa), albumin (67 kDa), and blue dextran 2000 (void volume). (C) Immunoblot of DsbA2 containing fractions prepared by osmotic shock from L. pneumophila and collected from the calibrated column. The DsbA2 protein from L. pneumophila eluted at 75 min, consistent with the homodimer. The immunoblot was developed with antibody specific for DsbA2.
Legionella pneumophila dsbA1 and dsbA2N, but not dsbA2, restore motility to a dsbA mutant of E. coli.
In E. coli, DsbA is required for disulfide bond formation in the flagellar P-ring protein (FlgI), which is necessary for motility in soft agar (27). As seen in Fig. 3A, wild-type E. coli strain JCB570 is motile in 0.4% soft agar, whereas ΔdsbA mutant strain JCB571 is not. We introduced L. pneumophila DsbA1 and DsbA2 expressed from pBC plasmids into JCB571 to test whether expression of these proteins could restore motility. As seen in Fig. 3A, dsbA1 complemented motility to ∼60% that of wild type (diameter of spreading growth), whereas dsbA2 did not, consistent with the general view that dimeric proteins, such as DsbC, are poor substrates of the E. coli DsbB (18, 19, 28). We next removed the dimerization domain of DsbA2 (DsbA2N) by an in-frame deletion that retained the signal sequence as depicted in Fig. 1A to create the monomer. The DsbA2N monomer was confirmed by gel filtration. Expression of dsbA2N in JCB571 restored motility to nearly the wild-type level (>70%) of the control (Fig. 3B). While growth rate differences might account for the partial complementation in our studies, these were not apparent; more likely, these differences reflect different efficiencies of DsbA1 and DsbA2N oxidation by E. coli DsbB compared with DsbA. These results indicate that it is the dimerization domain of DsbA2 that prevents interaction of the homodimer with DsbB.
Fig 3.

Complementation of motility defect in soft agar. Spreading motility by E. coli in soft agar is dependent on functional flagella and chemotaxis. (A) Wild-type E. coli (JCB570) is motile in soft agar at 30°C. Strain JCB571(dsbA) has a dsbA mutation and is nonmotile in this medium. Strain JCB571(pBCLpdsbA2) did not complement the motility defect, whereas JCB571(pBCLpdsbA1) complemented (∼60%) this strain for motility. (B) Wild-type E. coli strain JCB570 is motile in soft agar, and the dsbA mutant strain JCB571 is not. Expression of pBCdsbA2N (pdsbA2N), which contains an in-frame deletion of the dimerization domain depicted in Fig. 1A, restored motility to ∼70% of wild-type strain JCB570 activity. The results presented in both panels are representative of typical results obtained from triplicate platings.
Legionella pneumophila DsbA2, but not DsbA1 or DsbA2N, exhibits PDI activity.
In E. coli, DsbA catalyzes disulfide bonding between consecutive cysteine residues as the nascent polypeptide enters the periplasm (26). For those proteins for which nonconsecutive disulfide bonding is required for proper folding, consecutive disulfide bonds must be reduced and then re-formed between nonconsecutive cysteine residues. In E. coli, the protein disulfide isomerase DsbC catalyzes this reaction (20). In our previous studies, we noted that some of the proteins captured by DsbA2 contained an odd number of cysteine residues, and prediction software analyses suggested that some of these might be nonconsecutive (10). Since DsbA2 exists as a homodimer, we tested the possibility that DsbA2 might complement a dsbC mutant of E. coli by restoring PDI activity. To test this hypothesis, dsbA2, dsbA2N, and dsbA1 were cloned into an E. coli strain carrying a dsbC mutation and containing a pPDI detector system (20). In this system, a TEM β-lactamase (Bla) which naturally contains two cysteine residues at positions 52 and 98 that are not required for biological activity was engineered to contain an additional two cysteine residues (81 and 108) positioned so that DsbA will introduce consecutive disulfide bonds and produce an inactive enzyme (20). In the presence of DsbC, biological activity is restored by PDI activity (20). In this PDI detector assay, only dsbA2 expression restored ampicillin resistance to nearly wild-type DsbC control levels at 2 or 3 g/liter of drug (Fig. 4A and B). In contrast, DsbA1 (depicted in Fig. 4A and B), DsbA2LDN (Fig. 4A), and DsbA2N (Fig. 4B) failed to complement in the PDI detector assay. The potent PDI activity displayed by DsbA2 in E. coli also suggests that DsbA2 is an efficient substrate of the DsbD reductase system, perhaps tipping the balance in favor of PDI/reductase activity over oxidase activity. To confirm the redox status of DsbA2 and DsbA2N in E. coli, we used the thiol alkylating agent MalPEG5000 to alkylate free thiols, and those thiols reactive with MalPEG resulted in an increased mass of 5,000 Da per cysteine residue. As seen in Fig. 5, whole E. coli bacteria expressing DsbA2 were first treated with DTT before alkylation with MalPEG (lane 1) or in the absence of prior DTT treatment (lane 3) and in both cases showed shifted high-molecular-weight bands resulting from complete alkylation by MalPEG relative to untreated control (lane 2). In contrast, when DsbA2N was treated with reducing agent and then MalPEG, multiple shifted bands were observed (Fig. 5, lane 6), whereas treatment with MalPEG without reducing agent (lane 8) produced no shifted bands and showed no differences from that of the untreated control (lane 7), indicating that DsbA2N is maintained as the disulfide in E. coli.
Fig 4.

Complementation tests in PDI detector system. The PDI detector system uses a β-lactamase engineered to form consecutive disulfide bonds which require DsbC PDI activity to correct to nonconsecutive to facilitate protein folding (20). Growth on ampicillin indicates that β-lactamase is properly folded. Ampicillin concentrations are 0, 1, 2, or 3 g/liter. Bacterial dilutions are plated from top to bottom as detailed in the text. (A) +, wild-type E. coli strain RGP663 plus pPDI detector plasmid; −, strain RGP665dsbC plus pPDI detector plasmid; 1, strain RGP665 plus pPDI detector plasmid plus pBCdsbA1; 2, strain RGP665 plus pPDI detector plasmid plus pBCdsbA2; LDN; strain RGP665 plus pPDI detector plasmid plus pBCdsbA2LDN (LDN, deleted leader and dimerization domain sequences). (B) Replicate PDI detector assay that includes RPG665 plus pPDI detector plasmid plus pBCdsbA2N containing the leader sequence and an in-frame deletion of the dimerization domain sequences.
Fig 5.

DsbA2 and DsbA2N redox status in E. coli. (A) Redox status of DsbA2 in E. coli. Lane 1, whole cells reduced with DTT for 30 min and alkylated with MalPEG5000 (reduced control); lane 2, untreated control; lane 3, whole cells treated with MalPEG5000. Lanes 4 and 5 contain the molecular weight standards. (B) Redox status of DsbA2N in E. coli. Lane 6, whole cells treated with DTT for 30 min and then alkylated with MalPEG5000; lane 7, untreated control; lane 8, whole cells treated with MalPEG5000. The molecular weight of DsbA2 is ∼28,000, and that of DsbA2N is ∼26,000. Anti-DsbA2 mouse serum was used to identify the DsbA2 proteins. The multiply shifted bands are indicative of DsbA2 or DsbA2N covalent complexes with MalPEG5000, which adds 5,000 Da per cysteine. In E. coli, DsbA2 is maintained as the free thiol and DsbA2N as the disulfide.
These results indicate that DsbA2 but not DsbA2N is fully reduced to the free thiol in the periplasm of E. coli.
In vitro PDI activity.
To confirm in vivo PDI activity for DsbA2, we developed an assay in which the PDI detector TEM β-lactamase serves as the substrate. In this assay, gain of Bla activity, as measured with the chromogenic substrate nitrocefin, is dependent on PDI enzyme reducing and shuffling disulfide bonds until correct nonconsecutive disulfides are formed (29). As seen in Fig. 6, the Bla activity of MBla in concentrated spent culture supernatants is below the level of detection over the course of the assay. Incubation of DsbA2, but not DsbA1 with MBla efficiently corrected disulfide bonding and catalyzed protein folding, as noted by a gain in enzyme activity with nitrocefin. Compared with the WT Bla similarly concentrated from culture supernatants, near-equivalent activity was obtained. These studies were repeated several times, and results of a representative experiment are presented. These studies confirm the biological assay for PDI activity for DsbA2.
Fig 6.

Nitrocefin PDI folding assay. TEM1 β-lactamase (Bla) and pPDI mutant TEM1 β-lactamase (MBla) were obtained by concentrating culture supernatants from strains RGP663 and RGP665, respectively. MBla contains consecutive disulfide bonds and is inactive (nitrocefin negative). DsbA1 or DsbA2 was added to aliquots of MBla for 30 min at 37°C, and 15-μl aliquots were tested in triplicate for Bla activity in microplates with nitrocefin as described in the text. Cleavage of the β-lactam of nitrocefin is measured as a change in absorbance at 486 nm over a 30-min assay period. DsbA2, but not DsbA1, exhibited PDI activity.
Insulin reduction assay of DsbA1, DsbA2, and DsbA2LDN.
An insulin reduction assay is often used to demonstrate thioredoxin-like activity of DSBs (13, 30). This assay can distinguish between strong oxidases like DsbA and reductases such as thioredoxin by both the time to reduction and the rate of insulin precipitation resulting from the reduction of its disulfide bond (31). In this regard, we previously demonstrated that DsbA2 was similar to thioredoxin in this assay (10). As seen in Fig. 7, DsbA1 of L. pneumophila has a lag time of 20 min compared to DsbA2, which has a lag time of 5 min, more typical of reductases like DsbC and thioredoxin. However, as a monomer, DsbA2LDN is much more active in this assay than DsbA1, though the time to reduction is doubled for the monomer compared with the dimer. These results suggest that monomeric DsbA2LDN, while structurally more similar to DsbA than DsbC, favors the reductase function (10).
Fig 7.

Insulin reduction assay. Reductase activities of equimolar concentrations of DsbA1, DsbA2, and DsbA2N were followed spectrophotometrically at 650 nm in a microplate assay as described in the text. The assays were performed in triplicate, and a representative assay is presented. The time to reduction indicates that DsbA2N is intermediate between DsbA2 and DsbA1 (more reducing than oxidizing).
Growth stage expression of dsbA1 and dsbA2.
To determine at what stage of growth dsbA1 and dsbA2 are expressed, RT-qPCR was performed on cDNA prepared from total RNA obtained from exponential- and stationary-phase-grown L. pneumophila. As seen in Fig. 8, dsbA1 was expressed at a 2-fold-higher rate in stationary phase than exponential phase, whereas dsbA2 expression in stationary phase was increased more than 5-fold. We had previously reported that DsbA2 protein levels were more abundant in differentiated cyst forms than stationary-phase forms (7), and based on transcript levels, both dsb genes appear to be upregulated in stationary phase.
Fig 8.
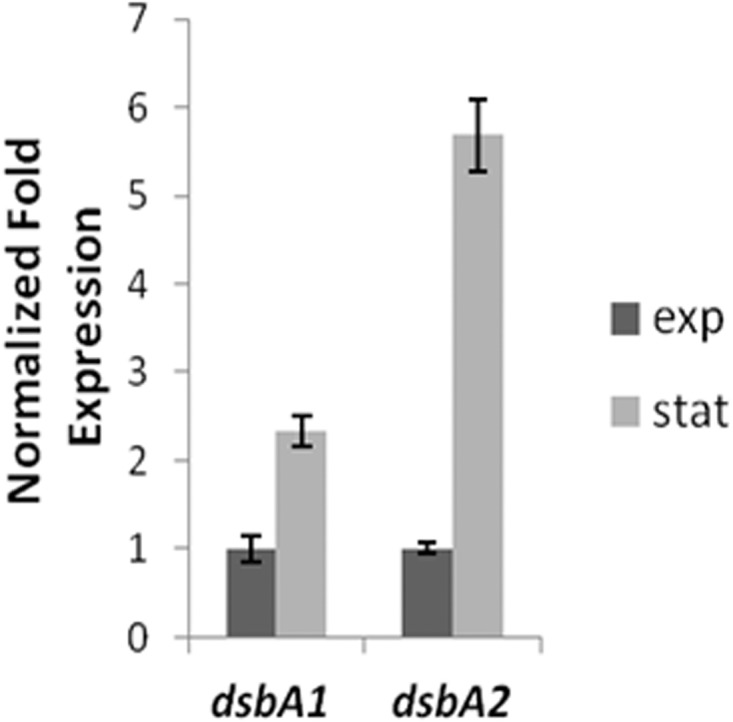
Growth state transcript levels of dsbA1 and dsbA2. L. pneumophila dsbA1 and dsbA2 mRNA transcript levels were determined with total RNA obtained during exponential (E) phase and stationary (S) phase as described in the text. dsbA1 and dsbA2 are upregulated in stationary phase: dsbA1 2-fold and dsbA2 5-fold. (DsbA1 E, 1 ± 0.158; DsbA2 E, 1 ± 0.182; DsbA1 S, 2.33 ± 0.06; DsbA2 S, 5.68 ± 0.415).
Redox status of DsbA2 is unchanged through cyst phase.
In Gram-negative bacteria, DsbA is maintained fully oxidized by DsbB, while DsbC is maintained fully reduced by the DsbD system. We previously reported that DsbA2 existed as a mixture of oxidized and reduced forms in L. pneumophila during stationary phase (10). We examined bacteria harvested from stationary phase as well as from water-differentiated metabolically dormant forms (7). Collected bacteria were TCA precipitated and subjected to AMS alkylation; this adds ∼500 Da to free cysteine thiol residues, which can then be observed as a shift in apparent mass by SDS-PAGE and immunoblotting. As seen in Fig. 9, even after suspension in tap water for several days, DsbA2 exists as a mixture of thiols and disulfides in the cyst-like forms. Since these forms are nearly dormant metabolically and exhibit no measurable respiration rate (7), it is not known how the mixture is maintained under these conditions.
Fig 9.

Redox status of DsbA2 in L. pneumophila. Lane 1, whole cells treated with DTT for 30 min and then subjected to AMS alkylation (reduced control). Lane 2, whole cells untreated with AMS (oxidized control). Lane 3, stationary-phase L. pneumophila treated with AMS. Lane 4, differentiated cyst-like forms after suspension in tap water for 48 h. The cyst-like forms are resistant to antibiotics and are nearly dormant metabolically (7). The doublets depicted in lanes 3 and 4 indicate that DsbA2 is present in a mixture of thiol and disulfide forms.
DISCUSSION
The results of our studies show that DsbA2 of L. pneumophila, which contains a 56-amino-acid N-terminal extension relative to DsbA of E. coli, forms a homodimer in L. pneumophila and displays protein disulfide isomerase activity in both in vivo and in vitro assays. We previously reported that DsbA2 behaved similarly to DsbA by catalyzing the formation of disulfide bonds in secreted proteins of L. pneumophila (10). Consistent with DsbA-like activity, a DsbA2 cis-proline (P198T) mutant protein formed stable disulfide cross-linked complexes with substrate proteins (10). Only the disulfide is capable of capturing substrate proteins (26). Previous studies found that L. pneumophila DsbA1 was dispensable for growth, motility, and infectivity in cell-based assays and that this gene is naturally absent from the genome of C. burnetii, which also expresses a dsbB gene (10). Taken together, these findings suggest that the DsbA2 lineage may be naturally bifunctional and therefore functionally distinct from the DsbA/DsbC paradigm so well characterized in E. coli.
In support of this notion, we determined that DsbA2 was present as a mixture of reduced thiol and disulfide forms in vivo, which contrasts with DsbA and DsbC, which are mostly oxidized and reduced, respectively (28). We determined by gel filtration that the mixture of oxidized and reduced forms was not due to equilibrium of monomer and dimer forms. Our studies do not rule out the possibility that each arm of the DsbA2 dimer might contribute to the mixture, one as the disulfide and the other as the free thiol. Our studies suggest that structural differences within the thioredoxin folds of DsbA2 might render them more accessible to the DsbB oxidases of L. pneumophila. In this regard, L. pneumophila expresses two alleles, DsbB1 and DsbB2, also known as LidJ (32), that show sequence divergence from E. coli DsbB. As previously reported, L. pneumophila also expresses two alleles of DsbD (10). We suggest that these competing oxidases and reductases interact with DsbA2 at differing efficiencies to maintain an equilibrium between oxidized and reduced forms. Interestingly, Segatori et al. showed that a chimera composed of the dimerization domain of DsbC fused to the catalytic domain of DsbA also existed as a mixture in E. coli and displayed bifunctional activity, including partial complementation of a dsbA mutant for motility (19). Since DsbA2 did not restore motility to a dsbA mutant, DsbA2 is either a poor substrate of DsbB or a good substrate of DsbD. Our studies found that DsbA2 is maintained as the free thiol in E. coli by DsbD and efficiently complemented a DsbC mutant in the PDI detector assay. By demonstrating that DsbA2N was able to complement a dsbA mutant of E. coli, we conclude that the lack of interaction between DsbA2 and DsbB is most likely the result of steric hindrance by the homodimer.
Our studies suggest that DsbA2 is not a homodimeric DsbA as demonstrated with the E. coli DsbC-DsbA chimera. We suggest that key amino acid differences in the cis-proline domain (TPA versus VPA in DsbA) similar to DsbC may favor PDI reductase activity. In the insulin reductase assay, DsbA2 and even the DsbA2N monomer exhibited reductase characteristics more similar to those of DsbC than to those of DsbA (Fig. 7). Thus, the DsbA2 lineage appears to be a unique adaptation which combines both oxidase and reductase functions into a single protein in situ. To explore the basis for this adaptation, we have replaced the E. coli dsbB and dsbD genes with alleles from L. pneumophila, and future studies will likely show that structural divergence in the two DsbD alleles may affect reducing efficiency of DsbA2.
A number of refolding assays have been developed to demonstrate in vitro PDI activity (15, 33, 34). These assays often rely on refolding of scrambled enzymes like RNase I that contains consecutive as well as nonconsecutive disulfide bonds and a subsequent assay for gained RNase activity (15, 34). Since most DSB enzymes are technically bifunctional (oxidase and reductase), PDI assays can yield mixed results, such as DsbA refolding RNase I (34). One advantage to the use of the PDI detector MBla for these assays is that the protein does not require denaturation prior to assay and the consecutive disulfide bonds produced are the result of DsbA activity (20). In this folding assay, gain of enzyme activity is readily detected with the chromogenic penicillin substrate nitrocefin. DsbA2 and not DsbA1 efficiently restored the correct nonconsecutive disulfide bond pairing required for protein folding and Bla activity. The adaptation of this assay to a microplate format should enable development of rapid screens of proteins for PDI activity. In our hands, the deep red color of the cleaved β-lactam is easily distinguished by visual observation.
Remarkably, much of the microbial world apparently substitutes DsbA2 for DsbC/DsbG, including many soil and aquatic species such as Caulobacter, Bradyrhizobium, Aeromonas, and Azospirillum and most human, animal, and plant pathogens that express T4SSs as major virulence factors (35). The latter group includes Rickettsia, Legionella, Bartonella, Anaplasma, Brucella, Ehrlichia, Coxiella, Agrobacterium, and Francisella. Most of these genera have doubling times of 2 h or more, which raises the possibility that in fast-growing bacteria, the DsbA/DsbC system is required to accelerate correct disulfide bond formation to enable protein folding to keep pace with cell division. This possibility seems to be supported by growth rate differences noted with E. coli strains carrying single mutations in dsbA and double mutations (dsbA dsbC and dsbA dsbD) (36). In this regard, DsbA accelerates disulfide bond formation between consecutive cysteines as most nascent polypeptides enter the periplasm (15, 37, 38). However, in other proteins, disulfide bonds may form after the protein has entered the periplasm (37) or require correction of mispaired disulfides by DsbC (21). We suspect that in those slow-growing bacteria expressing DsbA2, such as L. pneumophila, disulfide bonds might be introduced after proteins enter the periplasm, which is supported by the efficiency and diversity of proteins captured by the DsbA2P198T mutant protein (10). It follows that the apparent essentiality of dsbA2 is due to both its bifunctional nature and an absence of any backup system to repair misoxidized proteins that would accumulate to toxic levels in the periplasm. We also noted that overexpression of the DsbA2P198T mutant protein rendered L. pneumophila noninfectious by interfering with assembly and function of the T4SSs (10). Since dsbA2 was also essential in strains with mutations in dot/icm structural genes, we concluded that DsbA2 must be associated with proper folding of other proteins whose functions might also be essential, including OmpS porin and periplasmic Hsp60 (10, 39, 40, 41).
In summary, we have shown that the DsbA2 protein of L. pneumophila is a homodimer and is capable of delivering disulfide bonds to native unfolded proteins or through PDI activity to repair inappropriate disulfide bonds. While the DsbA2 lineage represents a subset of the DsbA family, the dimerization domain and threonine substitution for valine in the cis-proline domain are more similar to DsbC and likely contribute to PDI activity. We find it interesting that much of the microbial world relies on DsbA2 instead of the DsbA/DsbC system to manage extracytoplasmic disulfide bonding and protein folding. This genetically diverse group also includes most intracellular parasites that express T4SS, all of which grow very slowly compared to E. coli. Perhaps DsbA2 represents an evolutionary adaptation that is more efficient in orchestrating disulfide bond formation and protein folding that suits the lifestyles of slow-growing bacteria.
ACKNOWLEDGMENTS
We thank J. C. Bardwell and G. Ren for helpful suggestions and providing the bacterial strains and other reagents used in these studies.
This work was supported by NIH Grant R01 AI066058 to P.S.H.
Footnotes
Published ahead of print 22 February 2013
REFERENCES
- 1. Rowbotham TJ. 1980. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33:1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fields BS. 1996. The molecular ecology of Legionellae. Trends Microbiol. 4:286–290 [DOI] [PubMed] [Google Scholar]
- 3. McDade JE, Shepard CC, Fraser DW, Tsai TR, Redus MA, Dowdle WR. 1977. Legionnaires' disease: isolation of a bacterium and demonstration of its role in other respiratory disease. N. Engl. J. Med. 297:1197–1203 [DOI] [PubMed] [Google Scholar]
- 4. Newton HJ, Ang DK, van Driel IR, Hartland EL. 2010. Molecular pathogenesis of infections caused by Legionella pneumophila. Clin. Microbiol. Rev. 23:274–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Faulkner G, Berk SG, Garduno E, Ortiz-Jimenez MA, Garduno RA. 2008. Passage through Tetrahymena tropicalis triggers a rapid morphological differentiation in Legionella pneumophila. J. Bacteriol. 190:7728–7738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Faulkner G, Garduno RA. 2002. Ultrastructural analysis of differentiation in Legionella pneumophila. J. Bacteriol. 184:7025–7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garduno RA, Garduno E, Hiltz M, Hoffman PS. 2002. Intracellular growth of Legionella pneumophila gives rise to a differentiated form dissimilar to stationary-phase forms. Infect. Immun. 70:6273–6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morash MG, Brassinga AK, Warthan M, Gourabathini P, Garduño RA, Goodman SD, Hoffman PS. 2009. Reciprocal expression of integration host factor and HU in the developmental cycle and infectivity of Legionella pneumophila. Appl. Environ. Microbiol. 75:1826–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brand BC, Sadosky AB, Shuman HA. 1994. The Legionella pneumophila icm locus: a set of genes required for intracellular multiplication in human macrophages. Mol. Microbiol. 14:797–808 [DOI] [PubMed] [Google Scholar]
- 10. Jameson-Lee M, Garduno RA, Hoffman PS. 2011. DsbA2 (27 kDa Com1-like protein) of Legionella pneumophila catalyzes extracytoplasmic disulfide-bond formation in proteins including Dot/Icm type IV secretion system. Mol. Microbiol. 80:835–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. 2006. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol. Microbiol. 62:1278–1291 [DOI] [PubMed] [Google Scholar]
- 12. Carvalho AP, Fernandes PA, Ramos MJ. 2006. Similarities and differences in the thioredoxin superfamily. Prog. Biophys. Mol. Biol. 91:229–235 [DOI] [PubMed] [Google Scholar]
- 13. Bardwell JC, McGovern K, Beckwith J. 1991. Identification of a protein required for disulfide bond formation in vivo. Cell 67:581–589 [DOI] [PubMed] [Google Scholar]
- 14. Inaba K, Murakami S, Suzuki M, Nakagawa A, Yamashita E, Okada K, Ito K. 2006. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127:789–801 [DOI] [PubMed] [Google Scholar]
- 15. Kadokura H, Beckwith J. 2010. Mechanisms of oxidative protein folding in the bacterial cell envelope. Antioxid. Redox Signal. 13:1231–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Depuydt M, Leonard SE, Vertommen D, Denoncin K, Morsomme P, Wahni K, Messens JK, Carrol S, Collet JF. 2009. A periplasmic reducing system protects single cysteine residues from oxidation. Science 326:1109–1111 [DOI] [PubMed] [Google Scholar]
- 17. Heras B, Shouldice SR, Totsika M, Scanlon MJ, Schembri MA, Martin JL. 2009. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 7:215–225 [DOI] [PubMed] [Google Scholar]
- 18. Bader MW, Hiniker A, Regeimbal J, Goldstone D, Haebel PW, Reimer J, Metcalf P, Bardwell JCA. 2001. Turning a disulfide isomerase into an oxidase: DsbC mutants that imitate DsbA. EMBO J. 20:1555–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Segatori L, Paukstelis PJ, Gilbert HF, Gerorgiu G. 2004. Engineered DsbC chimeras catalyze both protein oxidation and disulfide-bond isomerization in Escherichia coli: reconciling two competing pathways. Proc. Natl. Acad. Sci. U. S. A. 101:10018–10023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ren G, Bardwell JCA. 2011. Engineered pathways for correct disulfide bond oxidation. Antioxid. Redox Signal. 14:2399–2412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Denoncin K, Vertommen D, Paek E, Collet JF. 2010. The protein-disulfide isomerase DsbC cooperates with SurA and DsbA in the assembly of the essential β-barrel protein LptD. J. Biol. Chem. 285:29425–29433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tinsley CR, Voulhoux R, Beretti J, Tommassen J, Nassif X. 2004. Three homologues, including two membrane-bound proteins, of the disulfide oxidoreductase DsbA in Neisseria meningitidis: effects on bacterial growth and biogenesis of functional type IV pili. J. Biol. Chem. 279:27078–27087 [DOI] [PubMed] [Google Scholar]
- 23. Pasculle AW, Feeley JC, Gibson RJ, Cordes LG, Myerowitz RL, Patton CM, Gorman GW, Carmack CL, Ezzell JW, Dowling JN. 1980. Pittsburgh pneumonia agent: direct isolation from human lung tissue. J. Infect. Dis. 141:727–732 [DOI] [PubMed] [Google Scholar]
- 24. LeBlanc JJ, Davidson RJ, Hoffman PS. 2006. Compensatory functions of two alkyl hydroperoxide reductases in the oxidative defense system of Legionella pneumophila. J. Bacteriol. 188:6235–6244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Helsel LO, Bibb WF, Butler CA, Hoffman PS, McKinney RM. 1987. Recognition of a genus-wide antigen of Legionella by a monoclonal antibody. Curr. Microbiol. 16:201–208 [Google Scholar]
- 26. Kadokura H, Tian H, Zander T, Bardwell JCA, Beckwith J. 2004. Snapshots of DsbA in action: detection of proteins in the process of oxidative folding. Science 303:534–537 [DOI] [PubMed] [Google Scholar]
- 27. Dailey FE, Berg HC. 1993. Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 90:1043–1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shouldice SR, Heras B, Walden PM, Totsika M, Schembri MA, Martin JL. 2011. Structure and function of DsbA, a key bacterial oxidative folding catalyst. Antioxid. Redox Signal. 14:1729–1760 [DOI] [PubMed] [Google Scholar]
- 29. Hailu TT, Foit L, Bardwell JCA. 2013. In vivo detection and quantification of chemicals that enhance protein stability. Anal. Biochem. 434:181–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Holmgren A. 1979. Thioredoxin catalyzes the reduction of insulin disulfides by dithiothreitol and dihydrolipoamide. J. Biol. Chem. 254:9627–9632 [PubMed] [Google Scholar]
- 31. Bessette PH, Cotto JJ, Gilbert HF, Georgiou G. 1999. In vivo and in vitro function of the Escherichia coli periplasmic cysteine oxidoreductase DsbG. J. Biol. Chem. 274:7784–7792 [DOI] [PubMed] [Google Scholar]
- 32. Conover GM, Derre I, Vogel JP, Isberg RR. 2003. The Legionella pneumophila LidA protein: a translocated substrate of the Dot/Icm system associated with maintenance of bacterial integrity. Mol. Microbiol. 48:305–321 [DOI] [PubMed] [Google Scholar]
- 33. Berkmen M, Boyd D, Beckwith J. 2005. The nonconsecutive disulfide bond of Escherichia coli phytase (AppA) renders it dependent on the protein-disulfide isomerase, DsbC. J. Biol. Chem. 280:11387–11394 [DOI] [PubMed] [Google Scholar]
- 34. Messens J, Collet JF, Van Belle K, Brosens E, Loris R, Wyns L. 2007. The oxidase DsbA folds a protein with a nonconsecutive disulfide. J. Biol. Chem. 282:31302–31307 [DOI] [PubMed] [Google Scholar]
- 35. Christie PJ, Atmakuri K, Krishnamoorthy V, Jakubowski S, Cascales E. 2005. Biogenesis, architecture, and function of bacterial type IV secretion systems. Annu. Rev. Microbiol. 59:451–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vertommen D, Depuydt M, Pan J, Leverrier P, Knoops L, Szikora JP, Messens J, Bardwell JC, Collet JF. 2008. The disulphide isomerase DsbC cooperates with the oxidase DsbA in a DsbD-independent manner. Mol. Microbiol. 67:336–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kadokura H, Beckwith J. 2009. Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell 138:1164–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zapun A, Bardwell JC, Creighton TE. 1993. The reactive and destabilizing disulfide bond of DsbA, a protein required for protein disulfide bond formation in vivo. Biochemistry 32:5083–5092 [DOI] [PubMed] [Google Scholar]
- 39. Butler CA, Street ED, Hatch TP, Hoffman PS. 1985. Disulfide-bonded outer membrane proteins in the genus Legionella. Infect. Immun. 48:14–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Garduno RA, Garduno E, Hoffman PS. 1998. Surface-associated Hsp60 chaperonin of Legionella pneumophila mediates invasion in a HeLa cell model. Infect. Immun. 66:4602–4610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fernandez R, Logan S, Lee S, Hoffman PS. 1996. Elevated levels of Legionella pneumophila stress protein Hsp60 early in infection of human monocytes and L929 cells correlate with virulence. Infect. Immun. 64:1968–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morales VM, Bäckman A, Bagdasarian M. 1991. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97:39–47 [DOI] [PubMed] [Google Scholar]