

Abstract
Rex factors are bacterial transcription factors thought to respond to the cellular NAD+/NADH ratio in order to modulate gene expression by differentially binding DNA. To date, Rex factors have been implicated in regulating genes of central metabolism, oxidative stress response, and biofilm formation. The genome of Enterococcus faecalis, a low-GC Gram-positive opportunistic pathogen, encodes EF2638, a putative Rex factor. To study the role of E. faecalis Rex, we purified EF2638 and evaluated its DNA binding activity in vitro. EF2638 was able to bind putative promoter segments of several E. faecalis genes in an NADH-responsive manner, indicating that it represents an authentic Rex factor. Transcriptome analysis of a ΔEF2638 mutant revealed that genes likely to be involved in anaerobic metabolism were upregulated during aerobic growth, and the mutant exhibited an altered NAD+/NADH ratio. The ΔEF2638 mutant also exhibited a growth defect when grown with aeration on several carbon sources, suggesting an impaired ability to cope with oxidative stress. Inclusion of catalase in the medium alleviated the growth defect. H2O2 measurements revealed that the mutant accumulates significantly more H2O2 than wild-type E. faecalis. In summary, EF2638 represents an authentic Rex factor in E. faecalis that influences the production or detoxification of H2O2 in addition to its more familiar role as a regulator of anaerobic gene expression.
INTRODUCTION
Ongoing bacterial metabolism and growth require maintenance of the redox balance in the cell. To achieve this balance, bacteria have evolved mechanisms to monitor their redox state and convert redox signals into adaptive regulatory outputs. For example, changes in availability of oxygen or metabolic activity can influence the relative levels of the dinucleotides NAD+ and NADH in the cell (1), and changes in this ratio are detected by the Rex family of transcription factors that are widespread in the genomes of Gram-positive bacteria. Rex factors have been extensively studied in Streptomyces coelicolor, Thermus aquaticus, Thermus thermophilus, Bacillus subtilis, and Staphylococcus aureus (2–8). Rex factors respond to the cellular NAD+/NADH ratio to modulate expression of genes involved in anaerobiosis, fermentative metabolism, biofilm formation, and oxidative stress (2–4, 7, 9, 10). Structural studies of Rex factors have identified dinucleotide binding pockets in the C-terminal Rossmann fold domain of the protein. NADH binding in this region leads to a conformational change in a Rex homodimer and a subsequent displacement of Rex from its recognition sites on DNA, leading to derepression of the downstream genes (2–4).
Enterococcus faecalis, a low-GC Gram-positive bacterium and an opportunistic pathogen, is a facultative anaerobe and a commensal member of the gastrointestinal microbiota in insects and animals, including humans (11, 12). E. faecalis is a hardy organism that exhibits substantial resistance to diverse environmental stresses, including antibiotics, bile detergents, and oxidative stress. E. faecalis can produce large quantities of reactive oxygen species, such as superoxide and hydrogen peroxide (13, 14). To cope with the stress this imposes, E. faecalis expresses numerous gene products responsible for detoxification of reactive oxygen species (15), including an NADH peroxidase (16, 17), a heme-dependent catalase that is functional only when heme is available in the medium (18, 19), a manganese-dependent superoxide dismutase (20), and a glutathione reductase (21), among others. As a facultative anaerobic member of the gastrointestinal (GI) tract, E. faecalis must adapt to fluctuations in the availability of oxygen and nutrients so that it can proliferate and compete with other community members. However, the mechanisms used by E. faecalis to monitor environmental conditions, such as the level of oxygen available, in order to mount an adaptive response are not well understood.
Although Rex factors are widespread in the genomes of Gram-positive bacteria, most genomes encode only one Rex factor homolog. The genome of E. faecalis, however, encodes two putative Rex factors (EF2638 and EF2933) that exhibit the characteristic Rex family bipartite domain architecture consisting of an N-terminal DNA-binding domain coupled to a C-terminal domain bearing a dinucleotide-binding Rossmann fold. The mechanisms by which multiple Rex factors in a single organism coordinate with each other to modulate gene expression in response to the cellular redox state are unknown. To our knowledge, the role of the putative Rex factors in E. faecalis has not been studied.
In this work, we demonstrate that EF2638 is an authentic Rex factor capable of interacting with DNA in an NADH-responsive manner. Our data indicate that EF2638 has the ability to regulate genes through its DNA binding activity, that EF2638 contributes to NAD+/NADH homeostasis, and that elevated levels of H2O2 impose oxidative stress on the ΔEF2638 mutant to account for its growth defect under certain environmental conditions. In addition, our results establish that the two Rex factors encoded in the E. faecalis genome are not functionally redundant.
MATERIALS AND METHODS
Bacterial strains, growth media, oligonucleotides, and chemicals.
The strains and plasmids used in this study are listed in Table 1. Oligonucleotides used for plasmid construction were synthesized by Integrated DNA Technologies, Inc. Escherichia coli strains were grown either in LB (Difco) or brain heart infusion (BHI) (Difco) at 37°C with shaking at 225 rpm. E. faecalis strains were cultured in Mueller-Hinton (MH) broth prepared according to the manufacturer's instructions (Difco), or MM9YE, a previously described semidefined medium (25) supplemented with 0.3% glucose or 0.3% glycerol (ACROS). Where indicated, MM9YE-based medium was supplemented with bovine catalase (∼500 U/ml) or porcine hematin (8 μM). When required, antibiotics were added at the following concentrations: for E. coli, 50 μg/ml kanamycin (Kn), 100 μg/ml erythromycin (Em); for E. faecalis, 10 μg/ml erythromycin, 10 μg/ml chloramphenicol (Cm). All chemicals and antibiotics were purchased from Sigma unless otherwise indicated.
Table 1.
Plasmids and strains used in this study
| Strain or plasmid | Relevant genotype or description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| TOP10 | E. coli routine cloning host | Invitrogen |
| Electro10 | E. coli cloning host for pCJK218-based plasmids | Agilent |
| BL21/DE3 | E. coli protein expression host | Lab stock |
| E. faecalis | ||
| OG1 | Wild-type reference strain | 22 |
| DV87-4 | OG1 ΔEF2638-2 | This work |
| DV106 | DV87-4 (EF1116-EF1117)2::EF2638-3 | This work |
| DV122-1 | OG1 ΔEF2933-2 | This work |
| Plasmids | ||
| pET28b | E. coli expression vector (Knr) | Novagen |
| pCJK3 | pTRKL2 derivative with pJMA61-derived transcriptional terminator | This work |
| pCJK4 | Plasmid carrying promoterless lacZ | This work |
| pJRG32 | pCJK47 derivative with a synthetic P-pheS* cassette (Emr) | 23 |
| pVE6007 | Temperature-sensitive derivative of pWV01 (Cmr) | 24 |
| pCJK47 | E. faecalis allelic exchange vector with pheS* counterselectable marker (Emr) | 25 |
| pCJK218 | pLT06 derivative with a synthetic P-pheS* cassette (Cmr) | This work |
| pCJK141 | pCJK47 derivative enabling ectopic integration of genes at the EF1116-7 locus | This work |
| pDV75-2 | pCJK218 derivative enabling ectopic integration of genes at the EF1116-7 locus | This work |
| pDV41-1 | EF2638 cloned in pET28b | This work |
| pDV42-4 | ΔEF2638-2 (ΔP7-L211) in pJRG32 | This work |
| pDV92 | ΔEF2933-2 (ΔP6-N211) in pCJK218 | This work |
| pDV59-3 | pCJK4::EF2638p (424 bp upstream of start codon)-lacZ | This work |
| pCJK221 | pCJK4::EF1929p (399 bp upstream of start codon)-lacZ | This work |
| pDV80 | pDV75-2::EF2638-3 with C-terminal Strep-tag | This work |
Plasmid construction.
A plasmid (pDV41-1) to overexpress C-terminally tagged EF2638-His6 for purification from E. coli was constructed by amplifying full-length EF2638 from E. faecalis OG1RF genomic DNA and introducing it into pET28b with primer-encoded NcoI/XhoI restriction sites.
To improve upon and extend previously described versions of the markerless allelic exchange technology used for genetic manipulation of E. faecalis, modified plasmids for markerless exchange were developed (see Fig. S1 in the supplemental material). First, we exchanged the pheS* counterselectable marker found in the temperature-sensitive allelic exchange vector pLT06 (26) with a previously described synthetic pheS* allele (23) bearing synonymous substitutions at the wobble position in many of the codons, to prevent recombination of the plasmid-borne pheS* with the chromosomal allele, thereby creating pCJK218. Second, we developed a system to stably introduce cloned genes into the chromosome of E. faecalis at an ectopic locus. Such a system enables cloned genes to be present in a single copy, and without the burden of maintaining an independent plasmid, for the purposes of complementation analysis. Our system is analogous to a previously described strategy (27), although in our case integration into the E. faecalis chromosome occurs at a distinct site from that utilized in the previous report. The system we developed enables genes to be integrated in the intergenic region between the convergently transcribed genes EF1116 and EF1117. This intergenic region encodes a stem-loop transcriptional terminator (28), and in our constructs this terminator is duplicated such that a copy of the terminator flanks both sides of the ectopically integrated sequence to prevent any transcriptional readthrough from (or out into) adjacent genes. The plasmid enabling integration at the EF1116-7 locus (pDV75-2) was developed in stages: first, segments of the E. faecalis OG1RF chromosome to serve as the substrates directing homologous recombination were amplified by PCR. The segments that encode portions of the EF1116 and EF1117 genes (∼820 bp and ∼760 bp, respectively) were assembled together by restriction digest and PCR into an intact fragment in which the two segments are separated by a short multiple-cloning site (the NotI-NcoI fragment from pCJK47). This intact fragment was then introduced into pCJK47 using primer-encoded XbaI/SphI restriction sites, creating pCJK141. Subsequently, the XbaI/SphI fragment from pCJK141 was transferred to pCJK218 by restriction digest of pCJK141 with XbaI to open the plasmid, T4 polymerase treatment to create a blunt end from the XbaI sticky end, and finally an SphI restriction digest to release the now blunt-end/SphI fragment of interest. This fragment was subsequently ligated into SmaI/SphI-digested pCJK218, creating pDV75-2. A derivative of pDV75-2 containing an epitope-tagged version of EF2638 and its putative promoter (pDV80) was created by first amplifying full-length EF2638, including a 424-bp region (containing the promoter) upstream of the start site, and then introducing the fragment into PstI/EcoRI-digested pDV75-2 using primer-encoded PstI/MfeI sites.
A plasmid to construct operon fusions with lacZ (pCJK4) was developed by first amplifying a short segment encoding a transcriptional terminator from pJMA61 and introducing it into pTRKL2 (29) using primer-encoded EcoRV/BglII restriction sites, creating pCJK3. Subsequently, a promoterless lacZ gene derived from pTRK390 (30) was amplified and introduced into pCJK3 using primer-encoded SmaI/StuI restriction sites. The final plasmid therefore encodes a transcriptional terminator to prevent readthrough into lacZ and 3 unique restriction sites (PstI, BamHI, XmaI) to enable cloned promoters to be introduced upstream of lacZ. Plasmids pDV59-3 and pCJK221 were constructed by amplifying putative promoter segments for EF2638 (424 bp upstream of the EF2638 start codon) and EF1929 (399 bp upstream of the EF1929 start codon) and introducing them into pCJK4 using primer-encoded PstI/BamHI (EF2638) or PstI/SmaI (EF1929) restriction sites.
Construction of the ΔEF2638 mutant.
Modification of the EF2638 locus in the E. faecalis OG1 chromosome was performed using markerless exchange as described previously (25). A derivative of pJRG32 carrying an in-frame deletion allele of EF2638 (pDV42-4) was constructed using a BsaI-based cloning scheme to seamlessly fuse two PCR amplicons flanking EF2638 to generate the in-frame deletion. The deletion allele was designed such that the first and last 6 codons remained (94% of the open reading frame [ORF] was deleted). This deletion allele was transferred to the native EF2638 locus in the OG1 chromosome using pVE6007 as a helper plasmid as previously described (31). Deletion mutants were isolated by plating on counterselection medium containing p-Cl-Phe (25) at 30°C, for 2 to 3 days. Two such ΔEF2638 mutants isolated completely independently from each other were analyzed and found to exhibit identical phenotypes.
Complementation of the ΔEF2638 mutant.
For complementation analysis, a fragment encoding an epitope-tagged EF2638 and its putative promoter (same promoter used for lacZ fusion) was inserted in a single copy into an ectopic locus in the E. faecalis chromosome. This fragment was amplified from E. faecalis OG1RF genomic DNA and introduced into pDV75-2 using primer-encoded PstI/MfeI restriction sites, creating pDV80. The cloned EF2638 allele encoded a C-terminal Strep tag epitope tag (WSHPQFEK). A procedure similar to that described by Thurlow and coworkers (26) was used to obtain recombinants in which the cloned fragment had been transferred to the chromosome by recombination. Briefly, pDV80 was introduced into the E. faecalis ΔEF2638 mutant (DV87-4) by electroporation, with selection on BHI supplemented with Cm and X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) (100 μg/ml) at 30°C. Subsequently, transformants were used to inoculate cultures, grown to optical density at 600 nm (OD600) of ∼0.2 and shifted to 42°C for ∼3 h. Dilutions were spread on BHI supplemented with Cm and X-Gal (100 μg/ml) at 42°C. Blue colonies were screened for the integration event by PCR, and the integrant colonies were used to inoculate cultures in BHI (30°C, 225 rpm, overnight). The mutants with EF2638-Strep integrated ectopically in the E. faecalis chromosome were isolated by plating on counterselection plates as described for the construction of the ΔEF2638 mutant above and confirmed by PCR. Two such complemented mutants isolated completely independently from each other were analyzed and found to exhibit identical phenotypes. In addition, we found that ectopic introduction of wild-type EF2638 (lacking the epitope tag) in the identical fashion also complemented the ΔEF2638 mutation (not shown).
Construction of the ΔEF2933 mutant.
A mutant lacking EF2933 was constructed using markerless exchange as described above for the ΔEF2638 complementation strain. A derivative of pCJK218 carrying an in-frame deletion allele of EF2933 (pDV92) was constructed using a BsaI-based cloning scheme as described above. The deletion allele was designed such that the first and last 5 codons remained (95% of the ORF was deleted). Deletion mutants were isolated by plating on counterselection medium containing p-Cl-Phe (25) at 30°C, for 2 to 3 days. Two such ΔEF2933 mutants isolated completely independently from each other were analyzed and found to exhibit identical phenotypes.
Overexpression and purification of E. faecalis EF2638.
Overnight cultures of E. coli BL21 [DE3] (pDV41-1) were grown in LB supplemented with 50 μg/ml kanamycin at 37°C, diluted 50-fold into 200 ml of the same medium, incubated 3 h at 37°C at 250 rpm, and then induced with 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) (Gold Biotechnology) for 1 h at 30°C. Bacteria were collected by centrifugation (10,816 × g, 8 min, 4°C) and cell pellets resuspended in 7 ml binding buffer (50 mM Tris, 300 mM NaCl, 5 mM imidazole, pH 8). Cell suspensions were treated with lysozyme (1 mg/ml) in lysozyme buffer (10 mM Tris, 50 mM NaCl, 10 mM EDTA, pH 8) for 20 min at 37°C. Cells were disrupted by sonication, and debris was removed by centrifugation (26,892 × g, 15 min, 4°C) followed by filtration of supernatant through a 0.22-μm-pore-size filter. The filtered supernatant was loaded onto an Ni column (Profinity IMAC Ni-charged resin; Bio-Rad) equilibrated with binding buffer. Columns were subsequently washed with 5 column volumes of binding buffer, followed by 5 column volumes of wash buffer (50 mM Tris, 300 mM NaCl, 20 mM imidazole, pH 8) and eluted in elution buffer (50 mM Tris, 300 mM NaCl, 500 mM imidazole, pH 8). Eluted fractions were analyzed by 10% SDS-PAGE, and fractions containing the protein were dialyzed against dialysis buffer (100 mM MOPS [pH 7.5], 0.8 M LiCl, 1 mM EDTA). Dialyzed protein was stored at −80°C.
Electrophoretic mobility shift assay (EMSA) for DNA binding.
Putative promoter segments of EF0255 (EF0255p), EF2933 (EF2933p), EF2638 (EF2638p), and EF1929-27 (EF1929p) were amplified from E. faecalis OG1RF genomic DNA. These segments included the upstream region as well as a few nucleotides into the actual reading frame; if the sequence was much larger than 300 bp, the segments were split into two smaller fragments containing a 30- to 50-bp overlap. The segments corresponded to 260 bp for EF0255p (includes both predicted Rex binding sites), 219 bp for EF2933p, 457 bp for EF2638p, and 304 bp for EF1929p. Double-stranded fragments were labeled at the 3′ end with digoxigenin (DIG) via the DIG gel shift kit, 2nd generation (Roche Applied Science), according to the manufacturer's instructions. DIG-labeled probes (∼5 fmol) and purified EF2638-His6 (1 μM) were incubated in binding buffer (0.1 M MOPS [pH 7.5], 0.8 M LiCl, 1 mM EDTA, 1 mM MgCl2, 5% glycerol) supplemented with 1 μg poly(dI-dC) and 1 μg poly l-lysine, for 30 min at room temperature (RT). The effect of NAD+, NADH, and NADPH on electrophoretic mobility was tested by the addition of the dinucleotides (10 mM final concentration) to the reaction following an initial incubation in the absence of dinucleotides (15 min, RT). Reactions were then incubated an additional 15 min, RT. Loading dye was added and samples were subjected to electrophoresis at 50 V for 3 h at 4°C on a 6% polyacrylamide gel in 0.5× Tris-borate-EDTA that had been prerun for 15 min at 120 V. After electrophoresis, DNA was transferred onto positively charged nylon membranes (Roche), subjected to UV cross-linking, probed using anti-digoxigenin-alkaline phosphatase-conjugated antibody (Roche), and detected with 3-(4-methoxyspiro {1,2-dioxetane-3,2′-(5′-chloro)tricyclo [3.3.1.13,7]decan}-4-yl)phenyl phosphate (CSPD) (Roche) by following the manufacturer's directions.
β-Galactosidase activity measurements.
β-Galactosidase activity was measured using o-nitrophenyl-β-d-galactopyranoside (ONPG) as the substrate. Overnight cultures of plasmid-bearing strains were diluted to an OD600 of 0.01 in MH broth supplemented with erythromycin (10 μg/ml) and cultured until an OD600 of 0.2 at 37°C and 225 rpm. Cultures were then chilled on ice and pelleted (1,503 × g, 10 min, 4°C). Cells were resuspended in Z buffer (60 mM Na2HPO4 · 7H2O, 40 mM Na2HPO4 · H2O, 10 mM KCl, 1 mM MgSO4 · 7H2O, 50 mM β-ME). A portion (200 μl) of each sample was treated with 25 μl 0.1% sodium dodecyl sulfate (SDS) and 50 μl chloroform for 10 min at RT. ONPG (200 μl, 4 mg/ml) was added to samples and incubated for 10 min at RT, and reactions were stopped by the addition of 500 μl 1 M Na2CO3. Cellular debris was removed by centrifugation (21,130 × g, 5 min, RT), and absorbance was measured at 420 and 550 nm; samples were normalized for OD600. Samples were analyzed in triplicate, and experiments were conducted at least two times. Statistical analysis was performed using a two-tailed Student t test.
NAD+/NADH extraction and cycling assay.
Dinucleotides were extracted and assayed according to a previously described method (32), with modifications. Cultures growing exponentially were collected by centrifugation (10,816 × g, 8 min, 4°C). Cell pellets were washed once with water, collected by centrifugation (16,100 × g, 2 min, 4°C), resuspended in 1,100 μl water, and then split into two 500-μl samples and centrifuged again (16,100 × g, 2 min, 4°C). Cell pellets were frozen in a dry ice-ethanol bath. NAD+ and NADH were extracted by the addition of 100 μl 0.2 M HCl and 100 μl 0.2 M NaOH, respectively. Samples were boiled for 10 min and neutralized, and cell debris was collected by centrifugation at 5,000 × g for 5 min at 4°C. Control experiments using pure NAD+ or NADH that had been subjected to acid or base treatment established the specificity of the extraction procedure for the respective nucleotide. Supernatants were transferred to new tubes and kept on ice. Extracts were used immediately in the cycling assay. The reaction mixture for the cycling assay was comprised of 20 μl 1 M bicine (pH 8), 20 μl 16.6 mM PES, 20 μl 4.2 mM MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide], 20 μl 100% EtOH, 8 μl H2O, and 2 μl yeast alcohol dehydrogenase (yADH). A total of 40 μl of each nucleotide extract was added to the wells of a 96-well plate, samples were treated with 80 μl of the reaction mixture, and absorbance was measured kinetically at 570 nm for 15 min. The concentrations of NAD+ and NADH were determined using an NAD+ standard curve, and results are expressed as the NAD+/NADH ratio. The experiment was done a minimum of two times, and the results represent the means ± standard errors. Statistical analysis was performed using a two-tailed Student t test.
Quantitation of hydrogen peroxide.
H2O2 present in culture supernatants was quantified using Amplex Red (Invitrogen), according to the manufacturer's instructions. Overnight cultures were grown under static conditions in MM9YE supplemented with 0.3% glucose, diluted to an OD600 of 0.01 in 75 ml of the same medium in 250-ml flasks. Cultures were incubated either statically or with aeration at 37°C and collected at an OD600 of 0.4. After the bacteria were collected by centrifugation (10,826 × g, 8 min, 4°C), culture supernatants were filtered through 0.22-μm-pore-size filters prior to analysis with Amplex Red. Results are expressed as H2O2 concentration determined using a standard curve generated as per the manufacturer's directions. Standards were prepared in equivalent culture medium. Control experiments in which aliquots of the culture supernatants were treated with bovine catalase (∼500 U/ml, 30 min, RT) before Amplex Red analysis established that the Amplex Red signal was indeed due to H2O2. The experiments were performed a minimum of two times. Data are presented as means ± standard errors. Statistical analysis was performed using a two-tailed Student t test.
Microarray analysis of gene expression.
Custom 12- by 135K microarrays (NimbleGen) were designed to interrogate expression of each ORF encoded in the E. faecalis OG1RF genome sequence. Although an annotated version of the complete OG1RF genome is now available (NCBI accession no. CP002621), at the time of array design, this annotation was not complete. Consequently, the arrays were designed on the basis of an OG1RF annotation generated with the Rapid Annotation Using Subsystem Technology (RAST) server (33), which has been previously described (34). This annotation contains 2,562 ORFs, of which unique probe sets were successfully designed for 2,477 ORFs. The probe set for most ORFs contains 18 probes per ORF (43,638 probes for all ORFs), each of which is present in triplicate on the array for a total of 130,914 probes per array.
For microarray analysis of gene expression, E. faecalis cultures were grown in MH broth at 37°C with aeration to exponential phase (OD600 = 0.16). Cultures were rapidly chilled in an ice water bath, and bacteria were subsequently collected by centrifugation. Cell pellets were immediately resuspended in RNAProtect reagent (Qiagen) to kill cells and stabilize the RNA. RNA was recovered from cells using the RNeasy minikit (Qiagen) according to the manufacturer's instructions, with slight modifications. The frozen cell pellets were thawed and resuspended in lysis buffer (10 mM Tris, 1 mM EDTA, pH 8) containing 250 U/ml mutanolysin and 15 mg/ml lysozyme with incubation at 37°C for 10 min. Buffer RLT (Qiagen RNeasy minikit) was then added, and the samples were processed according to the manufacturer's instructions. Purified RNA was subjected to cDNA synthesis, labeling, hybridization to the microarrays, scanning, and normalization via RMA at the Carver Center for Genomics at the University of Iowa. Intensity data from biological replicate cultures, prepared ∼1 year apart, were averaged, and genes for which the log2 of the expression was greater than 2-fold different in the ΔEF2638 mutant compared to the wild type are included in Table 2. Statistical significance of the differences was assessed via t test, and in all cases the P value was <0.001.
Table 2.
Genes upregulated in the ΔEF2638 mutant
| ORF | RAST annotation | log2 of ratioa |
|---|---|---|
| EF0094 | Formate/nitrite transporter family protein | 2.75 |
| EF0255 | l-Lactate dehydrogenase (EC 1.1.1.27) | 1.89 |
| EF0475 | Ferrous iron transport protein A | 2.25 |
| EF0634 | Decarboxylase, putative | 2.75 |
| EF0635 | Amino acid permease family protein | 2.83 |
| EF0900 | Alcohol dehydrogenase (EC 1.1.1.1); acetaldehyde dehydrogenase (EC 1.2.1.10) | 2.11 |
| EF1225 | Hypothetical similar to thiamine biosynthesis lipoprotein ApbE | 4.73 |
| EF1226 | Fumarate reductase, flavoprotein subunit precursor (EC 1.3.99.1) | 5.18 |
| EF1227 | Fumarate reductase, flavoprotein subunit precursor (EC 1.3.99.1) | 3.51 |
| EF1326 | Transcriptional regulator, TetR family | 2.28 |
| EF1327 | BadF/BadG/BcrA/BcrD ATPase family protein | 4.89 |
| EF1491 | Ribonucleotide reduction protein NrdI | 2.26 |
| EF1612 | Pyruvate formate-lyase-activating enzyme (EC 1.97.1.4) | 3.87 |
| EF1613 | Pyruvate formate-lyase (EC 2.3.1.54) | 2.36 |
| EF1825 | Conserved domain protein | 3.21 |
| EF1826 | Alcohol dehydrogenase (EC 1.1.1.1) | 2.63 |
| EF2048 | rRNA large subunit methyltransferase N (EC 2.1.1.-) | 2.33 |
| EF2637 | Abortive infection protein | 3.06 |
| EF2754 | Ribonucleotide reductase of class III (anaerobic), large subunit (EC 1.17.4.2) | 2.22 |
| EF2755 | Ribonucleotide reductase of class III (anaerobic), activating protein (EC 1.97.1.4) | 2.16 |
| EF2933 | Redox-sensitive transcriptional regulator (AT-rich DNA-binding protein) | 2.46 |
| EF3245 | Cell envelope-associated acid phosphatase | 2.77 |
| EF3198b | Lipoprotein, YaeC family | −2.07 |
The value is log2 of the ratio (ΔEF2638 mutant/wild type) of averaged expression values for the two strains.
The single gene with a 2-fold-lower expression in the absence of EF2638.
Microarray data accession number.
The microarray data reported here have been deposited in the Gene Expression Omnibus database, under the accession number GSE43228.
RESULTS
Transcriptome analysis reveals a role for EF2638 in regulation of fermentative metabolism.
To explore the physiological role of the putative E. faecalis Rex factor, EF2638, we constructed an in-frame deletion mutant lacking EF2638 in an otherwise wild-type E. faecalis strain (OG1). Because Rex factors are known to be transcriptional repressors in other bacterial species, we anticipated that genes normally repressed by EF2638 would exhibit elevated expression in the mutant. Microarray analysis was performed to compare the global transcriptome of the ΔEF2638 mutant with that of the corresponding wild type during exponential growth in MH broth. Indeed, we found 21 genes encoded at 14 distinct loci whose levels of expression were enhanced by a factor of at least 2 in the absence of EF2638 (Table 2). Strikingly, most of the derepressed gene products are predicted to function primarily in anaerobic fermentative metabolism, including pyruvate formate-lyase (EF1613) and its activating enzyme (EF1612), alcohol/aldehyde dehydrogenase (EF0900), anaerobic ribonucleotide reductase (EF2754-5), subunits of a putative fumarate reductase (EF1226-7), another putative alcohol dehydrogenase (EF1826), and a gene predicted to be involved in anaerobic degradation of glutamate (EF1327). Rex-mediated control of such genes is consistent with observations made previously in other species of bacteria (2, 35, 36) and recent predictions based on bioinformatics analyses (10). We also note that the major lactate dehydrogenase in E. faecalis (EF0255) was upregulated in the ΔEF2638 mutant but did not quite achieve the 2-fold cutoff (1.89-fold). Thus, EF2638 either directly or indirectly controls the expression of numerous enzymes that consume NADH in E. faecalis, consistent with the predicted role of Rex factors in maintenance of cellular redox homeostasis. We also observed derepression of the second putative Rex factor in E. faecalis (EF2933) in the ΔEF2638 mutant. There was little evidence in our transcriptome analysis for a role of EF2638 as an activator of gene expression, as only a single gene achieved 2-fold-lower expression in the ΔEF2638 mutant under the conditions of our experiment.
EF2638 binds DNA in an NADH-responsive manner to regulate gene expression.
Because EF2638 possesses a putative DNA-binding domain, we tested whether recombinant, purified E. faecalis EF2638-His6 can bind DNA in vitro using electrophoretic mobility shift assays (EMSAs). A series of digoxigenin-labeled probes encompassing several putative promoter regions were prepared. These included 2 probes for genes identified as derepressed in the transcriptome analysis (EF2933, EF0255) and for which putative Rex-binding sequences had been proposed previously (37), a probe for EF2638 itself (as Rex homologs are predicted or known to be autoregulated) (7, 10), and a probe for the putative promoter of the glpKOF operon (EF1929-27), as subsequent growth studies with the ΔEF2638 mutant (see below) suggested that these genes might be regulated by EF2638 as well. Purified EF2638-His6 was capable of binding all 4 of the putative promoter segments tested, as indicated by the appearance of a shifted band in the EMSA (Fig. 1), and the addition of excess unlabeled probe competed with the labeled probe for binding, indicating that the interaction was specific. To determine whether EF2638 DNA-binding activity is regulated by the presence of pyridine dinucleotides (as is known to be the case with Rex factors from S. coelicolor, T. aquaticus, B. subtilis, among others) (4, 7, 8), EMSAs were carried out in the presence of NAD+ and NADH (and NADPH in some cases). Neither NAD+ nor NADPH exhibited any effect on DNA binding by EF2638, whereas inclusion of NADH led to an obvious decrease in EF2638-DNA complex formation (Fig. 1). Thus, EF2638 exhibits the properties of an authentic Rex family transcriptional repressor, consistent with the hypothesis that EF2638 regulates gene expression in an NADH-responsive manner to maintain redox homeostasis in E. faecalis.
Fig 1.
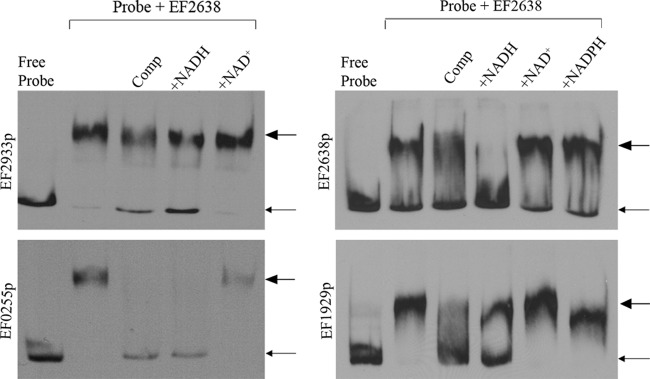
EF2638 binds DNA in an NADH-responsive manner. EMSAs were performed using 3′ DIG-labeled probes containing putative promoter regions of EF2638 (EF2638p), EF1929-27 (EF1929p), EF2933 (EF2933p), or EF0255 (EF0255p). Pure EF2638-His6 was incubated with probes in the presence or absence of pyridine dinucleotide as indicated. Comp, unlabeled specific competitor for each probe: EF2638p, EF2933p, and EF0255p at 150× excess and EF1929p at 250× excess. Small arrows indicate the position of free probe, and large arrows indicate the position of probe-DNA complexes. Representative results from at least three independent experiments are shown.
The microarray results suggested that EF2638 behaves as a transcriptional repressor for nearly all of the genes exhibiting differential expression. As a further test that EF2638 functions as a repressor in vivo, operon fusions of the putative promoters for two of the EMSA-shifted probes (EF2638 and glpKOF) to a promoterless lacZ were constructed. The recombinant constructs were introduced into both wild-type and ΔEF2638 mutant E. faecalis strains, and β-galactosidase activity assays were performed, revealing enhanced β-galactosidase activity from both promoters in the absence of EF2638 (Fig. 2), consistent with the hypothesis that EF2638 functions as a repressor in E. faecalis.
Fig 2.
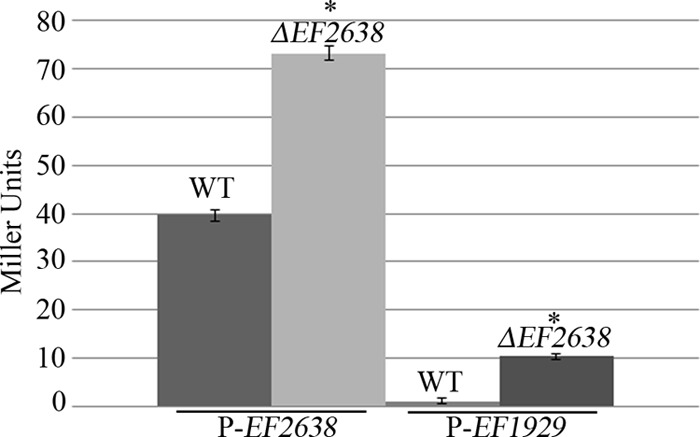
Assay for β-galactosidase activity. Cultures of plasmid-bearing OG1 (wild type) and DV87-4 (ΔEF2638) E. faecalis strains were grown in MH broth supplemented with erythromycin (10 μg/ml). Samples were collected in triplicate, and β-galactosidase activity was assayed as described in Materials and Methods. P-EF2638, putative EF2638 promoter driving the expression of promoterless lacZ; P-EF1929, EF1929-27 putative promoter driving the expression of promoterless lacZ. The data represent the means ± standard errors from two independent experiments. *, P < 0.05 versus wild type.
Redox homeostasis is impaired in the ΔEF2638 mutant.
Rex factors modulate the expression of NADH-consuming enzymes in response to the redox poise to help maintain redox homeostasis in the cell. Because we observed derepression of genes encoding numerous NADH-consuming enzymes in our transcriptome analysis of the ΔEF2638 mutant, we reasoned that the NAD+/NADH balance would be altered in the mutant. To test this, we determined the ratio of NAD+ to NADH in exponentially growing cells using a dinucleotide cycling assay (Fig. 3). As would be expected upon overexpression of NADH-consuming enzymes, the NAD+/NADH ratio was elevated in the ΔEF2638 mutant compared to that in the wild type, supporting the hypothesis that EF2638 helps to maintain NAD+/NADH homeostasis in E. faecalis.
Fig 3.
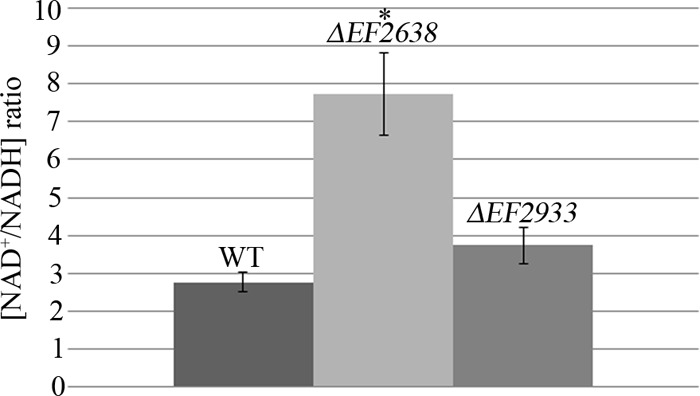
The ΔEF2638 mutant exhibits an elevated NAD+/NADH ratio. Wild-type OG1 (WT), DV87-4 (ΔEF2638), and DV122-1 (ΔEF2933) E. faecalis strains were grown in MH broth to exponential phase. Dinucleotides were extracted and the NAD+/NADH ratio was measured as described in Materials and Methods. Data represent the means ± standard errors from at least two independent experiments. *, P < 0.05 versus wild type.
To evaluate if the second putative E. faecalis Rex factor, EF2933, also plays a role in control of NAD+/NADH homeostasis, we constructed an in-frame deletion mutant lacking EF2933 in an otherwise wild-type E. faecalis strain (OG1). Measurement of the NAD+/NADH ratio revealed that the ΔEF2933 mutant exhibited only a slightly elevated ratio (not statistically significantly different from that of the wild type), suggesting that the contribution of EF2933 to redox homeostasis is relatively minor compared to that of EF2638, at least under the growth conditions we used.
Aerobiosis impairs growth of the ΔEF2638 mutant.
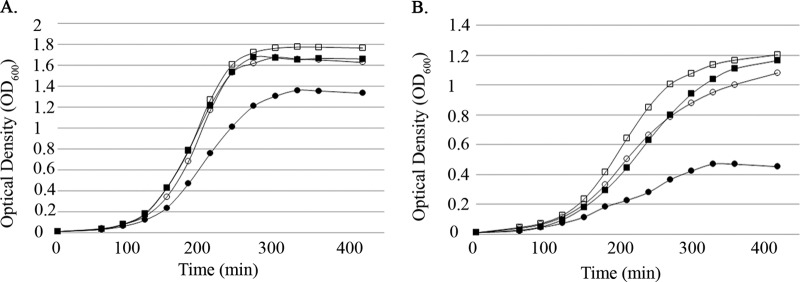
During routine experiments with aerobically incubated (i.e., shaking) liquid cultures, we observed that growth of the ΔEF2638 mutant was noticeably slower than that of the wild type. Careful analysis of the growth kinetics of the mutant (Fig. 4A) revealed that the ΔEF2638 mutant exhibited a growth defect relative to that of the wild type when cultured with aeration. The defect was largely absent if the cultures—which were otherwise identical—were held static during growth (Fig. 4B), suggesting that the ΔEF2638 mutant experiences, or is unable to cope with, elevated levels of oxidative stress that occur as a result of aeration. The growth defect of the mutant in aerated cultures could be complemented by introduction of EF2638 with its native promoter at an ectopic locus in the chromosome of the ΔEF2638 mutant (Fig. 4A), indicating that the defect is indeed due to loss of EF2638 function.
Fig 4.

The ΔEF2638 mutant exhibits a growth defect when grown with aeration. Bacteria were grown in MM9YE supplemented with 0.3% glucose (A and B) or 0.3% glycerol (C and D), and optical density was measured every 30 min at 600 nm. Cultures were grown with aeration (225 rpm, 37°C) (A, C) or statically (B, D). Wild type (OG1), squares; ΔEF2638 (DV87-4) mutant, circles; ΔEF2638 (EF1116-EF1117)2::EF2638-3 (DV106) mutant, triangles. Representative results from at least two independent experiments are shown.
Although the specific source of intracellular reactive oxygen species in the above-described experiment has not been established, certain metabolic pathways in E. faecalis are known to produce reactive oxygen species directly as a by-product. For example, in the presence of O2, E. faecalis can metabolize glycerol via the glpK pathway (38, 39), which includes glpO (glycerol-3-P oxidase) that converts glycerol-3-P to dihydroxyacetone-P with concomitant release of H2O2. Hypothesizing that such oxidative stress would be especially inhibitory for the ΔEF2638 mutant, we evaluated growth in the presence of glycerol. We found that the ΔEF2638 mutant was substantially impaired at growth on glycerol (Fig. 4C and D), even when cultures were incubated statically, consistent with the hypothesis that the ΔEF2638 mutant is impaired at mounting an effective oxidative stress response. The observation that the ΔEF2638 mutant was inhibited in glycerol cultures even under static conditions suggested that the glpKOF operon was being expressed at aberrantly high levels in the ΔEF2638 mutant, leading to efficient H2O2 production even without the use of aeration to promote diffusion of oxygen into the culture.
H2O2 is responsible for the growth defect of the ΔEF2638 mutant.
The growth analyses described above suggested that H2O2, specifically, could be the proximal cause of the growth defect exhibited by the ΔEF2638 mutant. To test this hypothesis, we supplemented the growth medium with bovine catalase to detoxify H2O2. Bovine catalase substantially improved the growth of the ΔEF2638 mutant in aerated cultures of both glucose- and glycerol-containing media to levels comparable to those of the wild type (Fig. 5A and B), indicating that H2O2 is a significant cause of oxidative stress experienced by the ΔEF2638 mutant. Of note, addition of bovine catalase also somewhat improved the growth of wild-type E. faecalis on glycerol, implying that H2O2 production during culture on glycerol is able to saturate the oxidative stress response of E. faecalis under the conditions used here. Supplementation of the growth medium with exogenous hematin (which is required for E. faecalis to produce an active, endogenously encoded catalase) also improved the growth of aerobically grown cells of the ΔEF2638 mutant (see Fig. S2 in the supplemental material), consistent with the hypothesis that H2O2 is responsible for the growth defect.
Fig 5.
Catalase rescues the growth defect of the ΔEF2638 mutant. Bacteria were grown with aeration (37°C, 225 rpm) in MM9YE containing 0.3% glucose (A) or 0.3% glycerol (B) with or without ∼500 U/ml bovine catalase (open symbols, + catalase; closed symbols, − catalase), and optical density was measured every 30 min at 600 nm. Wild type (OG1), squares; ΔEF2638 (DV87-4) mutant, circles. Representative results from at least two independent experiments are shown.
Analogous growth studies of the ΔEF2933 mutant also revealed an aeration-dependent growth defect (Fig. 6A and B), suggesting that the ΔEF2933 mutant is also impaired at mounting an effective oxidative stress response. In contrast to the ΔEF2638 mutant, however, the addition of catalase to the growth medium did not ameliorate the growth defect, indicating that the underlying cause of growth inhibition is likely to be different in the two Rex mutants.
Fig 6.
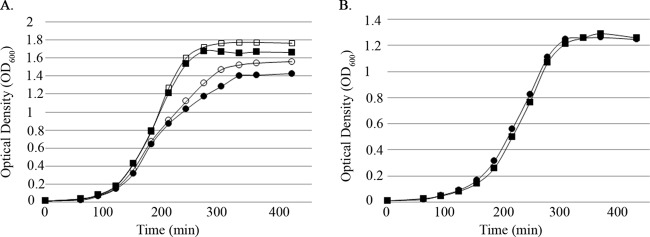
The ΔEF2933 mutant exhibits an aeration-dependent growth defect that is not rescued by catalase. Bacteria were grown in MM9YE containing 0.3% glucose either with aeration (37°C, 225 rpm) (A) or statically (B). Aerated cultures were supplemented with catalase in some cases (open symbols, + catalase; closed symbols, − catalase). Optical density was measured every 30 min at 600 nm. Wild type (OG1), squares; ΔEF2933 (DV122-1) mutant, circles. Representative results from at least two independent experiments are shown.
Given that H2O2 appeared to be a major cause of the growth defect exhibited by the ΔEF2638 mutant, we hypothesized that cultures of the ΔEF2638 mutant would accumulate higher levels of H2O2. Because H2O2 rapidly equilibrates across the cytoplasmic membrane (40), we analyzed H2O2 levels in cell-free culture supernatants using an Amplex Red-based assay. Measurements of the levels of H2O2 in the supernatants of wild-type and ΔEF2638 E. faecalis strains revealed that H2O2 is indeed present at substantially elevated levels in cultures of the ΔEF2638 mutant that have been subjected to aeration (Fig. 7; see also Fig. S3 in the supplemental material). In contrast, when cultures were incubated under static conditions (conditions in which there is no growth defect for the mutant; see Fig. 4), little difference was observed in culture supernatant H2O2 levels, consistent with the hypothesis that H2O2-derived oxidative stress is responsible for the growth defect of the ΔEF2638 mutant. The ΔEF2933 mutant did not exhibit a detectable increase in H2O2 accumulation compared to the wild type, consistent with the results of the growth studies (Fig. 6) in which catalase supplementation failed to improve growth of the ΔEF2933 mutant.
Fig 7.
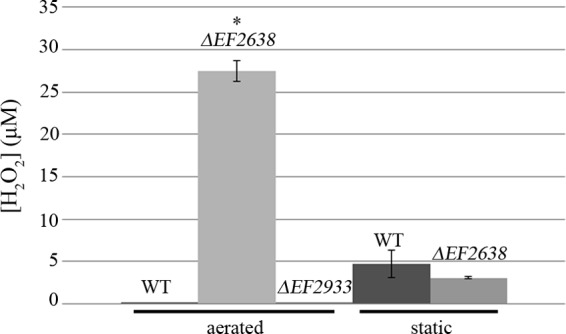
Accumulation of H2O2 by the ΔEF2638 mutant. Wild-type OG1 (WT), DV87-4 (ΔEF2638 mutant), and DV122-1 (ΔEF2933 mutant) E. faecalis strains were grown to mid-log phase at 37°C with aeration (225 rpm) or statically in MM9YE supplemented with 0.3% glucose. Culture supernatants were collected and assayed for hydrogen peroxide using Amplex Red. Data are means ± standard errors from at least two independent experiments. *, P < 0.05 versus wild type.
DISCUSSION
As a natural inhabitant of the GI tract, E. faecalis must adapt to fluctuations in the levels of available oxygen and nutrients to maximize its competitive fitness. The mechanisms used by E. faecalis to achieve this are poorly understood. Changes in oxygen availability and nutrient metabolism can manifest themselves as changes in the redox state of the cell; for example, in E. coli, the steady-state NADH/NAD+ ratio depends on the availability of suitable electron acceptors (1). The ratio is highest under anaerobic conditions and decreases when oxygen is introduced. In Gram-positive bacteria, the Rex transcription factor binds pyridine dinucleotides to monitor the redox state of the cell, thereby enabling Rex to modulate transcription of genes that are important for cellular redox homeostasis, including genes for alternative metabolic pathways, NADH reoxidation, and oxidative stress responses. The genome of E. faecalis encodes 2 putative Rex factors, EF2638 and EF2933, whose functions have not been described.
In this study, we explored the function of EF2638 in E. faecalis and found that it is indeed an authentic Rex factor. An E. faecalis mutant lacking EF2638 exhibited altered patterns of gene expression, primarily upregulation of genes expected to have critical roles in anaerobic metabolism and reoxidation of NADH, such as pyruvate-formate lyase, a putative fumarate reductase, and alcohol/aldehyde dehydrogenase (Table 2). Regulation of anaerobic gene expression has been described previously for Rex homologs in both S. aureus and B. subtilis (2, 35, 36). A palindromic consensus sequence (TGTGANNNNNNTCACA) for the S. aureus Rex binding site (the “Rex box”) has been described (41), and inspection of the intergenic DNA sequences upstream of many of the genes that are differentially expressed in the ΔEF2638 mutant revealed potential Rex box binding motifs (allowing for 1 or 2 mismatches), including EF0900, EF1613, EF0255, EF1225, and EF2933. We also identified a putative Rex box upstream of EF2638 itself. Mehmeti and coworkers (37) reported the presence of putative Rex boxes upstream of EF1613, EF0255, EF0900, and EF2933 but did not investigate a role for EF2638 in binding to these sites. We found that purified EF2638 was able to bind to several promoter fragments in vitro (Fig. 1), indicating that at least in some cases the effect of EF2638 on transcription is likely mediated by direct interaction with the promoters. Analysis of operon fusions to lacZ revealed that two of these promoters are more active in vivo in the ΔEF2638 mutant (Fig. 2), indicating that increased transcriptional initiation (rather than reduced transcript degradation) is responsible for the elevated transcript abundance in the ΔEF2638 mutant as detected by microarray analysis. Furthermore, DNA binding activity was reduced specifically in the presence of NADH, as has been observed for other Rex factors (4–7), suggesting that EF2638 likely monitors the NAD+/NADH ratio in vivo to control transcription of genes in its regulon. Consistent with this hypothesis, Mehmeti and coworkers (37) also observed that expression of EF0900, EF0255, and EF1613 is upregulated under physiological growth conditions which are expected to result in elevated levels of NADH. In addition, our direct measurements of the NAD+/NADH ratio demonstrated that the redox balance of the cell is substantially perturbed in the absence of EF2638 (Fig. 3), consistent with the hypothesis that a critical physiological function of EF2638 is to maintain redox homeostasis. We also note that the second putative Rex factor encoded in the E. faecalis genome, EF2933, was upregulated in the ΔEF2638 mutant, suggesting an interconnected regulatory network. The details of this regulatory network remain to be elucidated, including whether or not EF2933 functions as an authentic Rex factor in E. faecalis, but our results clearly establish that EF2638 and EF2933 are not entirely functionally redundant. Indeed, the properties of mutants lacking either EF2638 or EF2933 are quite different from each other: while the ΔEF2638 mutant exhibits an altered NAD+/NADH ratio and accumulates aberrantly high levels of H2O2, the ΔEF2933 mutant shares neither of those traits. Furthermore, although both mutants exhibit a growth defect when cultivated with aeration, only the ΔEF2638 mutant can be rescued by the addition of catalase to scavenge H2O2. Further work is therefore required to define the function of EF2933 in E. faecalis.
Unexpectedly, we found that the ΔEF2638 mutant accumulates substantially larger amounts of H2O2 than the otherwise isogenic wild-type E. faecalis when cultured under conditions of aeration (Fig. 7), and this H2O2 accumulates to sufficiently high concentrations to impose a growth defect on the mutant (Fig. 4). Although H2O2 is well known to participate in Fenton chemistry in cells to yield highly reactive and toxic hydroxyl radicals, we suspect that H2O2 itself is the primary cause of the cellular damage leading to growth inhibition of the ΔEF2638 mutant. This suggestion is based on our observation that inclusion of an iron chelator (250 μM 2′2 bipyridyl) in the medium to prevent iron-catalyzed Fenton reactions did not improve growth of the ΔEF2638 mutant under conditions of aeration (not shown), although we cannot unequivocally exclude the possibility that bipyridyl does not penetrate E. faecalis cells efficiently. More work will be required to identify the biological targets that are damaged by H2O2 to manifest the growth defect observed here.
Why does H2O2 accumulate in the ΔEF2638 mutant? It seems possible that the mutant might be generating more H2O2 under conditions of aeration than the wild-type strain. H2O2 production in E. coli can occur as a result of adventitious autoxidation by oxygen of flavin-containing enzymes, including fumarate reductase (42). Our microarray data indicate that genes encoding functions likely relevant under anaerobic conditions, such as a putative fumarate reductase, as well as other NADH-consuming dehydrogenases, are expressed at aberrantly high levels in the ΔEF2638 mutant, when they would normally be repressed. Elevated levels of these enzymes might offer increased opportunity for adventitious autoxidation to occur when the ΔEF2638 mutant is cultured with aeration, thereby increasing the rate of H2O2 production. Alternatively, the H2O2 detoxification systems of the ΔEF2638 mutant might be impaired. E. faecalis encodes an NADH peroxidase (npr) that uses NADH to directly reduce H2O2 to H2O (17). NADH peroxidase is known to be an important defense in E. faecalis against both exogenously added and endogenously produced H2O2 under a variety of growth conditions, and a mutant lacking npr accumulates H2O2 in the growth medium at an enhanced rate (43). The reduced availability of NADH in the ΔEF2638 mutant (Fig. 3) might starve Npr of reducing power needed for the enzyme to function efficiently, leading to accumulation of aberrantly high levels of H2O2. It remains less clear what the relevant source of reducing power in vivo is for the other two known E. faecalis peroxidases (alkyl hydroperoxide reductase and thiol peroxidase), but one can imagine that the redox imbalance present in the ΔEF2638 mutant could indirectly impair the ability of these enzymes to function as well. In any case, the relative importance of the three E. faecalis peroxidases has been reported to vary depending on the growth conditions, such as the available carbon source, and certainly the proximal mechanism(s) responsible for accumulation of H2O2 under a particular set of environmental conditions might be expected to vary in a similar fashion.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant AI081692 from NIAID.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or NIH.
Footnotes
Published ahead of print 15 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02135-12.
REFERENCES
- 1. de Graef MR, Alexeeva S, Snoep JL, Teixeira de Mattos MJ. 1999. The steady-state internal redox state (NADH/NAD) reflects the external redox state and is correlated with catabolic adaptation in Escherichia coli. J. Bacteriol. 181:2351–2357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pagels M, Fuchs S, Pane-Farre J, Kohler C, Menschner L, Hecker M, McNamarra PJ, Bauer MC, von Wachenfeldt C, Liebeke M, Lalk M, Sander G, von Eiff C, Proctor RA, Engelmann S. 2010. Redox sensing by a Rex-family repressor is involved in the regulation of anaerobic gene expression in Staphylococcus aureus. Mol. Microbiol. 76:1142–1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang E, Ikonen TP, Knaapila M, Svergun D, Logan DT, von Wachenfeldt C. 2011. Small-angle X-ray scattering study of a Rex family repressor: conformational response to NADH and NAD+ binding in solution. J. Mol. Biol. 408:670–683 [DOI] [PubMed] [Google Scholar]
- 4. Wang E, Bauer MC, Rogstam A, Linse S, Logan DT, von Wachenfeldt C. 2008. Structure and functional properties of the Bacillus subtilis transcriptional repressor Rex. Mol. Microbiol. 69:466–478 [DOI] [PubMed] [Google Scholar]
- 5. Sickmier EA, Brekasis D, Paranawithana S, Bonanno JB, Paget MS, Burley SK, Kielkopf CL. 2005. X-ray structure of a Rex-family repressor/NADH complex insights into the mechanism of redox sensing. Structure 13:43–54 [DOI] [PubMed] [Google Scholar]
- 6. McLaughlin KJ, Strain-Damerell CM, Xie K, Brekasis D, Soares AS, Paget MS, Kielkopf CL. 2010. Structural basis for NADH/NAD+ redox sensing by a Rex family repressor. Mol. Cell 38:563–575 [DOI] [PubMed] [Google Scholar]
- 7. Brekasis D, Paget MS. 2003. A novel sensor of NADH/NAD+ redox poise in Streptomyces coelicolor A3(2). EMBO J. 22:4856–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gyan S, Shiohira Y, Sato I, Takeuchi M, Sato T. 2006. Regulatory loop between redox sensing of the NADH/NAD(+) ratio by Rex (YdiH) and oxidation of NADH by NADH dehydrogenase Ndh in Bacillus subtilis. J. Bacteriol. 188:7062–7071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bitoun JP, Nguyen AH, Fan Y, Burne RA, Wen ZT. 2011. Transcriptional repressor Rex is involved in regulation of oxidative stress response and biofilm formation by Streptococcus mutans. FEMS Microbiol. Lett. 320:110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ravcheev DA, Li X, Latif H, Zengler K, Leyn SA, Korostelev YD, Kazakov AE, Novichkov PS, Osterman AL, Rodionov DA. 2012. Transcriptional regulation of central carbon and energy metabolism in bacteria by redox-responsive repressor Rex. J. Bacteriol. 194:1145–1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Murray BE. 1990. The life and times of the Enterococcus. Clin. Microbiol. Rev. 3:46–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tannock GW, Cook G. 2002. Enterococci as members of the intestinal microflora of humans, p 101–132 In Gilmore MS, Clewell DB, Courvalin P, Dunny GM, Murray BE, Rice LB. (ed), The enterococci: pathogenesis, molecular biology, and antibiotic resistance. American Society for Microbiology Press, Washington, DC [Google Scholar]
- 13. Huycke MM, Moore DR. 2002. In vivo production of hydroxyl radical by Enterococcus faecalis colonizing the intestinal tract using aromatic hydroxylation. Free Radic. Biol. Med. 33:818–826 [DOI] [PubMed] [Google Scholar]
- 14. Huycke MM, Abrams V, Moore DR. 2002. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23:529–536 [DOI] [PubMed] [Google Scholar]
- 15. Paulsen IT, Banerjei L, Myers GS, Nelson KE, Seshadri R, Read TD, Fouts DE, Eisen JA, Gill SR, Heidelberg JF, Tettelin H, Dodson RJ, Umayam L, Brinkac L, Beanan M, Daugherty S, DeBoy RT, Durkin S, Kolonay J, Madupu R, Nelson W, Vamathevan J, Tran B, Upton J, Hansen T, Shetty J, Khouri H, Utterback T, Radune D, Ketchum KA, Dougherty BA, Fraser CM. 2003. Role of mobile DNA in the evolution of vancomycin-resistant Enterococcus faecalis. Science 299:2071–2074 [DOI] [PubMed] [Google Scholar]
- 16. Ross RP, Claiborne A. 1997. Evidence for regulation of the NADH peroxidase gene (npr) from Enterococcus faecalis by OxyR. FEMS Microbiol. Lett. 151:177–183 [DOI] [PubMed] [Google Scholar]
- 17. Claiborne A, Ross RP, Parsonage D. 1992. Flavin-linked peroxide reductases: protein-sulfenic acids and the oxidative stress response. Trends Biochem. Sci. 17:183–186 [DOI] [PubMed] [Google Scholar]
- 18. Pugh SY, Knowles CJ. 1983. Synthesis of catalase by “Streptococcus faecalis subsp. Zymogenes.” Arch. Microbiol. 136:60–63 [DOI] [PubMed] [Google Scholar]
- 19. Frankenberg L, Brugna M, Hederstedt L. 2002. Enterococcus faecalis heme-dependent catalase. J. Bacteriol. 184:6351–6356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Britton L, Malinowski DP, Fridovich I. 1978. Superoxide dismutase and oxygen metabolism in Streptococcus faecalis and comparisons with other organisms. J. Bacteriol. 134:229–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Patel MP, Marcinkeviciene J, Blanchard JS. 1998. Enterococcus faecalis glutathione reductase: purification, characterization and expression under normal and hyperbaric O2 conditions. FEMS Microbiol. Lett. 166:155–163 [DOI] [PubMed] [Google Scholar]
- 22. Gold OG, Jordan HV, van Houte J. 1975. The prevalence of enterococci in the human mouth and their pathogenicity in animal models. Arch. Oral Biol. 20:473–477 [DOI] [PubMed] [Google Scholar]
- 23. Vesic D, Kristich CJ. 2012. MurAA is required for intrinsic cephalosporin resistance of Enterococcus faecalis. Antimicrob. Agents Chemother. 56:2443–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maguin E, Duwat P, Hege T, Ehrlich D, Gruss A. 1992. New thermosensitive plasmid for Gram-positive bacteria. J. Bacteriol. 174:5633–5638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kristich CJ, Chandler JR, Dunny GM. 2007. Development of a host-genotype-independent counterselectable marker and a high-frequency conjugative delivery system and their use in genetic analysis of Enterococcus faecalis. Plasmid 57:131–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thurlow LR, Thomas VC, Hancock LE. 2009. Capsular polysaccharide production in Enterococcus faecalis and contribution of CpsF to capsule serospecificity. J. Bacteriol. 191:6203–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Debroy S, van der Hoeven R, Singh KV, Gao P, Harvey BR, Murray BE, Garsin DA. 2012. Development of a genomic site for gene integration and expression in Enterococcus faecalis. J. Microbiol. Methods 90:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Le Breton Y, Muller C, Auffray Y, Rince A. 2007. New insights into the Enterococcus faecalis CroRS two-component system obtained using a differential-display random arbitrarily primed PCR approach. Appl. Environ. Microbiol. 73:3738–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. O'Sullivan DJ, Klaenhammer TR. 1993. High- and low-copy-number Lactococcus shuttle cloning vectors with features for clone screening. Gene 137:227–231 [DOI] [PubMed] [Google Scholar]
- 30. O'Sullivan DJ, Walker SA, West SG, Klaenhammer TR. 1996. Development of an expression strategy using a lytic phage to trigger explosive plasmid amplification and gene expression. Biotechnology 14:82–87 [DOI] [PubMed] [Google Scholar]
- 31. Kristich CJ, Manias DA, Dunny GM. 2005. Development of a method for markerless genetic exchange in Enterococcus faecalis and its use in construction of a srtA mutant. Appl. Environ. Microbiol. 71:5837–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Leonardo MR, Dailly Y, Clark DP. 1996. Role of NAD in regulating the adhE gene of Escherichia coli. J. Bacteriol. 178:6013–6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Frank KL, Barnes AM, Grindle SM, Manias DA, Schlievert PM, Dunny GM. 2012. Use of recombinase-based in vivo expression technology to characterize Enterococcus faecalis gene expression during infection identifies in vivo-expressed antisense RNAs and implicates the protease Eep in pathogenesis. Infect. Immun. 80:539–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hecker M, Reder A, Fuchs S, Pagels M, Engelmann S. 2009. Physiological proteomics and stress/starvation responses in Bacillus subtilis and Staphylococcus aureus. Res. Microbiol. 160:245–258 [DOI] [PubMed] [Google Scholar]
- 36. Larsson JT, Rogstam A, von Wachenfeldt C. 2005. Coordinated patterns of cytochrome bd and lactate dehydrogenase expression in Bacillus subtilis. Microbiology 151:3323–3335 [DOI] [PubMed] [Google Scholar]
- 37. Mehmeti I, Jonsson M, Fergestad EM, Mathiesen G, Nes IF, Holo H. 2011. Transcriptome, proteome, and metabolite analyses of a lactate dehydrogenase-negative mutant of Enterococcus faecalis V583. Appl. Environ. Microbiol. 77:2406–2413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bizzini A, Zhao C, Budin-Verneuil A, Sauvageot N, Giard JC, Auffray Y, Hartke A. 2010. Glycerol is metabolized in a complex and strain-dependent manner in Enterococcus faecalis. J. Bacteriol. 192:779–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deutscher J, Bauer B, Sauerwald H. 1993. Regulation of glycerol metabolism in Enterococcus faecalis by phosphoenolpyruvate-dependent phosphorylation of glycerol kinase catalyzed by enzyme I and HPr of the phosphotransferase system. J. Bacteriol. 175:3730–3733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seaver LC, Imlay JA. 2001. Hydrogen peroxide fluxes and compartmentalization inside growing Escherichia coli. J. Bacteriol. 183:7182–7189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuchs S, Pane-Farre J, Kohler C, Hecker M, Engelmann S. 2007. Anaerobic gene expression in Staphylococcus aureus. J. Bacteriol. 189:4275–4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Messner KR, Imlay JA. 2002. Mechanism of superoxide and hydrogen peroxide formation by fumarate reductase, succinate dehydrogenase, and aspartate oxidase. J. Biol. Chem. 277:42563–42571 [DOI] [PubMed] [Google Scholar]
- 43. La Carbona S, Sauvageot N, Giard JC, Benachour A, Posteraro B, Auffray Y, Sanguinetti M, Hartke A. 2007. Comparative study of the physiological roles of three peroxidases (NADH peroxidase, alkyl hydroperoxide reductase and thiol peroxidase) in oxidative stress response, survival inside macrophages and virulence of Enterococcus faecalis. Mol. Microbiol. 66:1148–1163 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.