

Abstract
Azotobacter vinelandii is a bacterium which undergoes a differentiation process leading to the formation of metabolically dormant cysts. During the encystment process, A. vinelandii produces alkylresorcinol lipids (ARs) that replace the membrane phospholipids and are also components of the layers covering the cyst. The synthesis of ARs in A. vinelandii has been shown to occur by the activity of enzymes encoded by the arsABCD operon, which is expressed only during the differentiation process. Also, the production of ARs has been shown to be dependent on the stationary-phase sigma factor RpoS, which is also implicated in the control of the synthesis of other cyst components (i.e., alginate and poly-β-hydroxybutyrate). In this study, we identified ArpR, a LysR-type transcriptional regulator expressed only during encystment that positively regulates arsABCD transcription. We show that this activation is dependent on acetoacetyl-coenzyme A (acetoacetyl-CoA), which might provide a metabolic signal for encystment. We also show that RpoS regulates arsABCD expression through the control of arpR transcription.
INTRODUCTION
Azotobacter vinelandii is a soil bacterium that undergoes differentiation to form cysts resistant to desiccation. A mature cyst consists of a contracted cell, known as the central body, surrounded by a capsule made up of a thin laminated outer layer, called the exine, and a thicker inner layer, the intine (1). When growing on glucose or sucrose, cysts are formed at late stationary phase, from <0.01% of the cells (2). However, encystment can be induced in a higher percentage of cells (10% or more of the cells) by removing glucose or sucrose from exponentially growing cultures and replacing it with n-butanol or β-hydroxybutyrate as the sole carbon source (2, 3).
Alkylresorcinols (ARs) are phenolic lipids that are synthesized by plants but are rare in animals, fungi, and bacteria (4). Together with alkylpyrones (APs), ARs are important lipid components of the A. vinelandii cyst. These lipids are synthesized only during the differentiation process, where they replace the phospholipids of the membranes and also participate in the formation of the exine (5). ARs play a structural role in the cysts, and mutant strains unable to synthesize these lipids produce cysts with a defective exine showing an agglutination phenotype, although they are still resistant to desiccation (2).
Little is known about the control of encystment in A. vinelandii at the genetic level. A role in this process has been established for the transcriptional regulator AlgR (6) and the alternative sigma factor AlgU (sigma E) (7, 8). The sigma factor RpoS (σs) is also involved in the control of this process. It is required for full expression of the genes involved in the synthesis of two important cyst components: the polysaccharide alginate, an essential component of the cyst capsule (9); and polyhydroxybutyrate (PHB), a reserve polymer present in the cysts (10). Additionally, it was recently established that RpoS is somehow involved in the control of AR synthesis, because an rpoS mutant was shown to be unable to synthesize these lipids (11). However, the mechanism of regulation exerted by this sigma factor was not established.
The enzymes responsible for the synthesis of ARs and APs are encoded by the arsABCD operon (12). Expression of these genes is very low in exponentially growing vegetative cells and increases slightly (14-fold) during stationary phase, when a low percentage of encystment occurs. However, with encystment induction, their expression is induced 200-fold (2).
Because arsABCD are the only genes known to be induced specifically during encystment, we considered it important to better understand the mechanisms involved in the regulation of their expression. In this work, we identified ArpR, a LysR-type transcriptional regulator (LTTR), as the regulatory protein involved in the activation of transcription of these genes during encystment. We established that this activation is dependent on other signals, such as the presence of acetoacetyl-coenzyme A (acetoacetyl-CoA). We also showed that RpoS regulates the synthesis of ARs by controlling the expression of the arpR gene.
MATERIALS AND METHODS
Bacterial strains, plasmids, media, and growth conditions.
The bacterial strains used are described in the figures and text. Plasmids used in this study are listed in Table 1. A. vinelandii cells were grown on Burk's medium (16) supplemented with 2% sucrose as a carbon source (BS) for vegetative growth or with 0.2% n-butanol (BBOH) for encystment induction. Liquid cultures were carried out in 125-ml flasks containing 25 ml of medium in a rotatory shaker at 250 rpm and 30°C. Inocula for all experiments were grown on BS, washed twice with Burk's medium without a carbon source (Burk's buffer), and transferred to the indicated medium. Escherichia coli strains were grown at 37°C on Luria-Bertani or M9 medium. For chloramphenicol acetyltransferase (CAT) activity and Western blot assays, bacterial samples were collected from shaken cultures grown at 37°C in 50 ml of M9 medium supplemented with sucrose. Triparental and biparental matings were carried out as previously reported (16). A. vinelandii transformation was carried out as described by Bali et al. (17). When antibiotics were necessary, they were used at the following concentrations (in μg per ml) for A. vinelandii and E. coli, respectively: rifampin (Rif), 15 and 0; nalidixic acid (Nal), 30 and 0; spectinomycin (Sp), 100 and 50; and kanamycin (Km), 1 and 30.
Table 1.
Plasmids used in this study
| Plasmid | Description | Reference or source |
|---|---|---|
| pACYC177 | Cloning vector containing the p15A replication origin | New England Biolabs |
| pACYC/PKK | Derivative of pACYC177 and pKK232-8 | This study |
| pACYC/PKK-ArsA | pACYC/PKK containing an arsA-cat transcriptional fusion from nucleotides −250 to +191 | This study |
| pBAD-ArpR | pBAD/His A derivative expressing ArpR-His from the ara promoter | This study |
| pBAD/His A | Expression vector for constructing N-terminal His fusions, with ara promoter; Apr | Invitrogen |
| pBBR1MCS-2 | Vector for cloning PCR products with a Km resistance cassette | 13 |
| pBBR-ArpR | pBBR1MCS-2 derivative expressing ArpR-His gene under the control of the Km promoter | This study |
| pBluescript KS(+) | Plasmid for subcloning DNA, unable to replicate in A. vinelandii | Stratagene |
| pKK232-8 | pBR322 derivative containing a promoterless chloramphenicol acetyltransferase gene (cat); Apr | 14 |
| pMAL-c2X | Vector for constructing MBP fusions; Apr | New England Biolabs |
| pMAL-ArpR | pMAL-c2X derivative expressing MBP-ArpR | This study |
| pMOSBlue | Plasmid for cloning PCR products | Amersham |
| pMOSgA2 | pMOSBlue derivative carrying gusA::Sp cassette with its own SD | This study |
| pMOSR-His | pMOSBlue derivative expressing ArpR tagged with a six-His tag at the N terminus | This study |
| pMP01 | pBSL97 derivative with an Sp-gusA cassette | 15 |
| pOV7 | pBluescript KS(+) carrying 6-kb PstI fragment containing Tn5SSgusA40 insertion of OV7 | |
| pYRR40 | pMOSBlue derivative carrying 2.6-kb fragment containing arsA | This study |
| pYRR41 | pMOSBlue derivative carrying 1.8-kb fragment containing arpR | This study |
| pYRR50 | pMOSBlue derivative carrying an arsA::gusA transcriptional fusion | This study |
| pYRR52 | pMOSBlue derivative carrying an arpR::gusA transcriptional fusion | This study |
β-Glucuronidase and chloramphenicol acetyltransferase (cat) activity determinations.
β-Glucuronidase activity was measured as reported by Miller; 1 U corresponds to 1 nmol of p-nitrophenyl-β-d-glucuronide hydrolyzed per minute per mg of protein. The amount of protein was determined by the method of Lowry et al. (18). All measurements were done in triplicate. For CAT activity assay, samples from shaken cultures in LB medium were collected at 3, 4, 6, and 12 h of growth for the arsA-cat gene fusion and at 12 h for the arpR-cat gene fusion. CAT assays were performed spectrophotometrically as described previously (19). One unit of β-glucuronidase activity corresponds to 1 nmol of substrate (5-bromo-4-chloro-3-indolyl-β-d-glucuronic acid [X-Gluc]) hydrolyzed min−1 mg protein−1. The CAT specific activity was quantified as μmol of substrate (chloramphenicol) hydrolyzed min−1 mg of protein−1.
DNA manipulations.
DNA manipulations were performed according to standard protocols as described by Sambrook et al. (20). Restriction enzymes were obtained from Invitrogen and used according to the manufacturer's instructions. Standard procedures for isolation of total genomic DNA, restriction endonuclease digestion, agarose gel electrophoresis, purification of DNA from agarose, DNA ligations, and transformation of E. coli were carried out as described by Sambrook et al. (20). DNA sequences were determined by the dideoxy chain termination method (21), using a PerkinElmer/Applied Biosystems DNA sequencer. The radioactive probes were prepared by random priming using a Rediprime DNA labeling system (GE Healthcare). The oligonucleotides used in this work were synthesized at the oligonucleotide synthesis facility at the Instituto de Biotecnología/UNAM, and their sequences are available upon request.
Transposon mutagenesis and identification of mutants affected in AR synthesis.
Mutagenesis of A. vinelandii AEIV was carried out using E. coli S17-1 λ-pir containing the promoter-probe minitransposon mTn5SSgusA40 as described by Wilson et al. (22). The mTn5 mutant library obtained was stained for visualization of ARs as described by Segura et al. (2). Briefly, A. vinelandii mutants were grown for 5 days on Burk's medium containing 0.2% n-butanol to induce encystment. The petri dishes were then sprayed with a solution of 0.5% Fast Blue B in 5% acetic acid. AR-producing colonies turned dark red after a few minutes of reaction with the staining solution.
Determination of alkylresorcinol production.
The production of ARs was measured as previously described (23). Briefly, the lipids were extracted with acetone for 20 min at room temperature. The acetone extract was removed, and a second extraction was done for 12 h at room temperature. The resulting extracts were mixed and used for spectrophotometric determination of alkylresorcinols by the use of Fast Blue B as previously described (24). Orcinol was used as a standard. The protein content of the cells used for AR determination was quantified by the method of Lowry et al. (18).
qRT-PCR.
Expression levels of arsA and arpR were measured by quantitative reverse transcription-PCR (qRT-PCR) as previously reported (25). Total RNA extraction from A. vinelandii SW136 and its isogenic mutants was performed as reported by Barry et al. (26). To eliminate genomic DNA, RNA was treated with DNase I (Fermentas Inc.) according to the manufacturer's instructions, and its concentration and purity were measured by the ratio of the absorbances at 260 and 280 nm. The cDNA was synthesized using 500 ng of each DNase-treated RNA, a cDNA synthesis kit (Revert Aid H First Strand kit; Fermentas Inc.), and 5 pmol of the specific reverse primer. The cDNA obtained was used as the template for real-time PCR (qRT-PCR) assays. The primers used (sequences are available from the authors) were as follows: arsA-RT-F and arsA-RT-R for arsA, arpR-RT-F and arpR-RT-R for arpR, and fw-gyrA and rev-gyrA for gyrA. The gyrA gene was used as an internal control in the same samples to normalize the results obtained. All qRT-PCRs were performed in duplicate for each gene of each strain. The quantification technique used to analyze the data was the 2−ΔΔCT method reported by Livak and Schmittgen (27). The reproducibility of the whole procedure was determined by performing cDNA synthesis and qPCR experiments with two separate RNAs extracted from each strain. Similar results were obtained for the transcription of all measured genes in the repetitions and with the internal control (gyrA) used for normalization.
Construction of arpR::mTn5 and rpoS::Sp mutant strains.
An arpR::mTn5SSgusA40 insertion was obtained after transposon mutagenesis of A. vinelandii strain AEIV, a nonsequenced wild-type strain kindly provided by Svein Valla (Norwegian University of Science and Technology). The strain containing this transposon insertion was named OV7. A 6-kb PstI DNA fragment from OV7, containing the mTn5 insertion, was cloned into pBluescript KS (Stratagene). The resultant plasmid (pOV7), which was unable to replicate in A. vinelandii, was used to transfer the arpR::mTn5SSgusA40 insertion to strain SW136 (2) by transformation and selection of a spectinomycin-resistant transformant (strain SW7). The disruption of arpR by double recombination was confirmed by PCR analysis of strain SW7.
To obtain an rpoS mutant of A. vinelandii SW136, this strain was transformed with total DNA extracted from strain CNS59, an rpoS::Sp mutant derived from strain ATCC 9046 (9). A spectinomycin-resistant transformant was selected and named SW9. The presence of the rpoS::Sp mutation in strain SW9 was confirmed by PCR analysis.
Construction of arsA::gusA and arpR::gusA transcriptional fusions.
To construct arsA::gusA and arpR::gusA transcriptional fusions, DNA fragments containing the intergenic regulatory region and part of the coding regions of arsA and arpR were amplified by PCRs using primers UIARS and LIARS and primers PwarsR and ARSRlw, respectively. These PCR products were cloned into vector pMOSBlue, resulting in plasmids pYRR40 and pYRR41, respectively (Table 1). A DNA fragment containing the gusA reporter and spectinomycin resistance (Sp) genes was amplified using oligonucleotides UGA and SPlow and the plasmid pMP01 (15). This fragment was cloned into pMOSBlue, and the resulting plasmid was named pMOSgA2. For construction of the arsA::gusA-Sp transcriptional fusion, the pYRR40 plasmid was digested with the HindIII and EcoRI restriction enzymes and then ligated to a HindIII-EcoRI fragment containing the gusA-Sp reporter and antibiotic resistance genes, excised from pMOSgA2. The resulting plasmid was named pYRR50. To construct the arpR::gusA-Sp transcriptional fusion, we used plasmid pYRR41. The gusA-Sp reporter and spectinomycin resistance genes were inserted into the NdeI-PstI sites of pYRR41, resulting in plasmid pYRR52.
Plasmid pYRR50 was used to transform the A. vinelandii SW136 wild-type strain and its mutant derivatives SW7 and SW9. Spectinomycin-resistant transformants of each strain were selected, resulting in strains YRR30, YRR32, and YRR34, respectively. The integration of the arsA::gusA-Sp transcriptional fusion into the chromosome of each strain by gene replacement was confirmed by PCR analysis.
For the construction of strains YRR50 and YRR52, i.e., derivatives of SW136 and SW9, respectively, carrying the arpR::gusA-Sp transcriptional fusion, the corresponding strains were transformed with plasmid pYRR52, and spectinomycin-resistant transformants were isolated and confirmed by PCR analysis to contain the arpR::gusA-Sp transcriptional fusion in the chromosome.
Construction of plasmids pACYC/PKK-ArsA, pBAD-ArpR, and pBBR-ArpR.
To demonstrate the regulation of arsA expression by ArpR, two plasmids were constructed for heterologous expression in E. coli DH5α. Plasmid pACYC/PKK-ArsA (Table 1) was generated to construct a transcriptional fusion of the arsA regulatory region with the cat reporter gene. This plasmid was derived from pACYC177 and pKK232-8, which were digested with BamHI-PstI and SmaI-NdeI, respectively, filled with Klenow polymerase (Roche), and ligated with T4 DNA ligase (Roche), producing plasmid pACYC/PKK. The arsA promoter region was excised from vector pYRR40 by use of the SacII and BglI enzymes, and this fragment was cloned into the same restriction sites of pACYC/PKK, leaving the cat reporter gene under the control of the arsA promoter in plasmid pACYC/PKK-ArsA. The second plasmid, named pBAD-ArpR (Table 1), was constructed for the inducible expression of the ArpR regulator. To construct this plasmid, the coding region of arpR was amplified from A. vinelandii SW136 total DNA by using oligonucleotides arpRu and arpRw (sequences are available from the authors) and a high-fidelity Taq polymerase from Invitrogen. This PCR product was digested with the SacI and EcoRI restriction enzymes, purified from an agarose gel, and cloned into the pBAD/His A vector (Invitrogen), producing plasmid pBAD-ArpR, allowing the arabinose-induced expression in E. coli of a six-His-tagged ArpR protein.
To express the ArpR protein in A. vinelandii, plasmid pBBR-ArpR (Table 1) was constructed. A DNA fragment containing arpR was obtained from pBAD-ArpR by a PCR using primers arpRHIS and arpRp4. The PCR product was cloned into plasmid pMOSBlue, producing plasmid pMOSR-His. The arpR gene, encoding a protein tagged with six His residues at the N terminus, was excised from pMOSR-His by digestion with the HindIII and BamHI enzymes and then cloned into plasmid pBBR1MCS-2 digested with the same enzymes. This produced pBBR-ArpR, a broad-host-range plasmid able to replicate in A. vinelandii, with arpR under the control of the promoter of the kanamycin resistance gene. This plasmid was transferred by conjugation into strains SW136, SW7, and SW9.
Primer extension assays.
The mapping of the transcriptional start sites of arsA was done by primer extension analysis as described by Cocótl-Yañez et al. (11). Total RNA was prepared as reported by Barry et al. (26) from A. vinelandii strains SW136, SW7, and SW9 grown on BBOH medium (encystment induction medium) for 36 h. Primers ArsA-5 (5′-GAGGATCGACGAACGGAC-3′) and ArsA-10 (5′-AGGGGCTTTCGTCGG-3′) were used for arsA. The analysis with both primers was necessary due to the unusually long leader region on the arsA mRNA.
SDS-PAGE and Western blot assays.
E. coli DH5α or A. vinelandii whole-cell extracts were prepared from bacterial samples collected at the indicated time points from cultures in LB or from vegetative or encysting cultures, respectively. Ten micrograms of protein of each extract was subjected to electrophoresis on SDS-12% polyacrylamide gels and then transferred to 0.45-μm-pore-size nitrocellulose membranes (Bio-Rad) by use of a semidry transfer apparatus (Hoefer). The membranes containing the transferred proteins were blocked in 5% nonfat milk for 1 h. Immunoblots were performed with anti-His monoclonal antibody (Roche) at a 1:1,000 dilution. Horseradish peroxidase-conjugated anti-rabbit (Pierce) at a dilution of 1:10,000 was used as the secondary antibody. Bands on the blotted membranes were developed by incubation with the Western Lightning Plus chemiluminescence reagent (Perkin-Elmer).
Construction of plasmid pMAL-ArpR and purification of MBP-ArpR.
To construct a plasmid expressing a recombinant maltose-binding protein–ArpR fusion (MBP-ArpR), a PCR fragment of 1.2 kb containing the coding region of arpR lacking its first and last codons was amplified using primers arpRp3 and arpRp4 and high-fidelity Taq polymerase (Invitrogen). The PCR products were purified from an agarose gel, digested with BamHI and HindIII, and cloned in frame at the 3′ end of the malE gene into the pMAL-p2X vector (New England Biolabs), producing plasmid pMAL-ArpR (Table 1). The correct in-frame cloning of arpR in this plasmid was verified by DNA sequencing. The MBP-ArpR protein was overexpressed from pMAL-ArpR in E. coli BL21 grown in 100 ml of LB with 0.2% glucose when the culture reached an optical density (OD) of 0.5, by adding 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h at 30°C. The cells were harvested by centrifugation at 10,000 × g at 4°C and then washed once with column buffer (20 mM Tris-HCl, pH 7.4, 200 mM NaCl, 1 mM EDTA, 10 mM 2-mercaptoethanol), resuspended in 10 ml of the same buffer, and broken by sonication. Cellular debris was eliminated from the cell extracts by centrifugation. The MBP-ArpR protein was bound to amylose resin (New England Biolabs), washed with 100 ml of column buffer to remove nonspecific bound proteins, and eluted with column buffer containing 10 mM maltose.
DNA electrophoretic mobility shift assays (EMSA).
The regulatory region of arsA, contained in a DNA fragment of 291 bp (positions −462 to −171 with respect to the arsA start codon), was amplified by PCR using the primer pair arsAEMSA1 (5′-GCTTGTGTGCTTGATGGT-3) and arsAEMSA3 (5′-GGTCTTCGCCTCTTGTCC-3′), with A. vinelandii SW136 DNA as the template. A 225-bp DNA fragment corresponding to the upstream regulatory region of the rpoS gene was used as a negative control. This fragment was obtained using primers emsaSup and emsaSdwn (11). The PCR products were labeled during the reaction by use of [α-32P]dCTP and were purified using a Geneclean III kit (MP). DNA binding reactions were performed in binding buffer (10 mM Tris-HCl [pH 8], 50 mM KCl, 1 mM dithiothreitol [DTT], 0.5 mM EDTA, 5% glycerol, and 10 mg ml−1 bovine serum albumin [BSA]) by mixing 0.2 pmol of each PCR product and increasing concentrations of purified MBP-ArpR in a total volume of 20 μl. The protein-DNA binding reaction mixtures were incubated at room temperature for 30 min and then electrophoretically separated in 6% nondenaturing polyacrylamide gels in 0.5× Tris-borate-EDTA buffer at room temperature. The gel was dried, and the radioactive signals were detected by autoradiography.
Kd determination.
For estimation of the affinity of ArpR for arsA DNA, the dissociation constant (Kd) was estimated by analysis of gel retardation (DNA binding) assays in both the presence and absence of 5 mM acetoacetyl-CoA. After electrophoresis, the amounts of fragment retarded by different concentrations of MBP-ArpR (0, 25, 50, 100, 200, 500, 1,000, and 3,000 nM protein, using 10 nM labeled DNA) were determined densitometrically from the autoradiographs of dried gels by using a Molecular Dynamics PhosphorImager and ImageQuant software. Plots were constructed and analyzed to estimate the Kd. The conditions used for these experiments were the same as those described for the other EMSAs.
RESULTS
Identification of ArpR, a LysR-type transcriptional regulator whose inactivation impairs the synthesis of ARs.
The expression of the AR biosynthetic operon arsABCD in A. vinelandii is induced during encystment (2). In order to identify regulators involved in this control, we carried out random mTn5 mutagenesis of a wild-type A. vinelandii strain, strain AEIV, and looked for mutants unable to produce these lipids after encystment induction on n-butanol. The production of ARs was visualized by staining the colonies with Fast Blue B, which specifically stains these phenolic lipids, turning them red (2). This mutagenesis produced strain OV7, a mutant which is not stained with Fast Blue B. A 6-kbp PstI DNA fragment from this strain, containing the mTn5 insertion, was cloned into pBluescript KS (Stratagene) to produce plasmid pOV7, which is unable to replicate in A. vinelandii. To corroborate that the observed phenotype was due to the transposon insertion and to transfer this mutation into a strain with a sequenced genome, plasmid pOV7 was transformed into A. vinelandii strain SW136 (2), an encysting strain isogenic to the sequenced strain DJ (28). A spectinomycin-resistant transformant was isolated. The knockout of arpR produced by double recombination was demonstrated in this transformant by PCR analysis (data not shown). This strain, named SW7, showed the same nonstaining phenotype in the presence of Fast Blue B (Fig. 1A).
Fig 1.
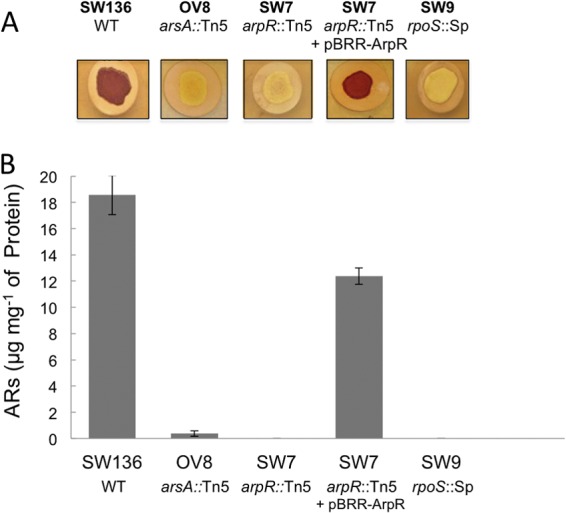
Inactivation of arpR impairs alkylresorcinol production. Staining (A) and quantification (B) are shown for alkylresorcinols produced by A. vinelandii SW136 (WT) and mutant strains OV8 (arsA::Tn5), SW7 (arpR::Tn5), SW9 (rpoS::Sp), and SW7 complemented with a plasmid expressing ArpR. The cells were grown on petri dishes containing encystment induction medium (BBOH). Data for the quantification of alkylresorcinols are averages for two independent experiments.
The production of ARs was quantified for the wild-type SW136 and mutant SW7 strains after 5 days of incubation in BBOH medium. Whereas strain SW136 produced 18 to 19 μg of ARs mg−1 of protein, less than 0.4 μg mg−1 was detected in the SW7 mutant (Fig. 1B), an amount similar to that quantified for strain OV8, an arsA mutant (2), confirming the inability of strain SW7 to produce ARs.
To identify the gene disrupted by the mTn5 insertion, the DNA fragment cloned into pOV7 was used to determine the location of mTn5 by sequencing across the transposon insertion junctions. The transposon was found to lie within nucleotides (nt) 705 to 713 of a gene of 921 bp coding for a putative transcriptional regulator of the LysR family with 306 amino acids and a molecular mass of 33.3 kDa. The deduced protein contains the two conserved domains characteristic of this family of transcriptional regulators: a helix-turn-helix DNA-binding domain in the N terminus and a putative regulatory cofactor-binding domain in the C terminus. The gene was named arpR (alkylresorcinol production regulator) and corresponds to Avin19160 in the A. vinelandii genome annotation (28). The LysR-type transcriptional regulators are often divergently transcribed with respect to the genes they regulate (29). In the case of arpR, apparently no other genes related to the metabolism of ARs are present in this region. Downstream of arpR, and in the opposite direction, there is a gene coding for an ABC transporter, and 1.3 kbp upstream, the gene for an incomplete transposase was found. The arsABCD operon is located elsewhere on the chromosome (28).
Complementation of the arpR mutant.
To further demonstrate that the phenotype of strain SW7 was caused by the inactivation of arpR, we cloned the arpR gene into the broad-host-range plasmid pBBR1MCS (13) under the control of a constitutive promoter. The plasmid thus constructed (pBBR-ArpR) (Table 1) was introduced into mutant SW7 for complementation. The production of ARs was reestablished when the complemented strain was grown on encystment induction medium (Fig. 1A and B), confirming that the lack of synthesis of ARs in strain SW7 is caused by the inactivation of arpR and demonstrating that this gene plays a role in the production of these phenolic lipids.
Inactivation of arpR negatively affects arsA expression from a single promoter.
To investigate the role of ArpR in the regulation of the production of ARs, qRT-PCR was used to determine the effect of the arpR inactivation on the expression of arsA, the gene heading the biosynthetic operon. The level of arsA mRNA was determined for cells harvested 36 h after encystment induction on BBOH medium. The relative expression level of arsA mRNA in the forming cysts of the SW7 mutant with respect to that in the wild-type strain was reduced more than 80% (Fig. 2A). To confirm this result, a transcriptional fusion of the arsA gene with the reporter gene gusA, coding for β-glucuronidase (15), was constructed and introduced into the chromosomes of strains SW136 (wild type) and SW7 (arpR mutant), producing strains YRR30 and YRR32, respectively. A 60% reduction of β-glucuronidase was obtained in encysting YRR32 cells compared to YRR30 cells (Fig. 2B), which shows a transcriptional effect similar to that obtained by qRT-PCR. Thus, the arpR mutation negatively affected arsA transcription.
Fig 2.
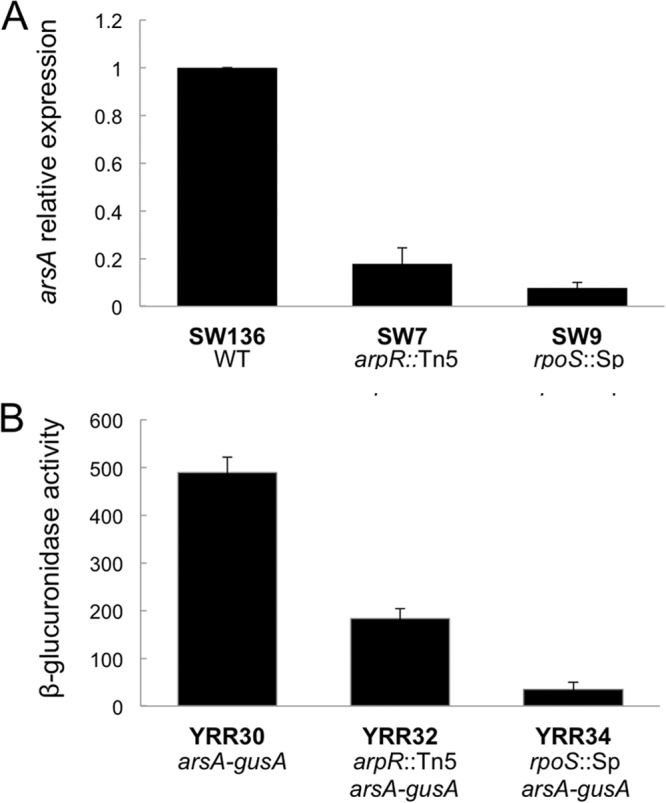
ArpR and RpoS positively regulate the expression of arsA. (A) Expression of arsA of strain SW136 (WT) and its isogenic mutants SW7 (arpR::Tn5) and SW9 (rpoS::Sp) as measured by qRT-PCR. The data are presented as fold changes of arsA mRNA levels of the SW7 and SW9 mutant strains relative to that of the parental strain (SW136). (B) Expression of arsA, quantified as β-glucuronidase activities, in strains YRR30 (WT derivative), YRR32 (SW7 derivative) and YRR34 (SW9 derivative), containing a chromosomal arsA::gusA transcriptional fusion. One unit of β-glucuronidase activity corresponds to 1 nmol of substrate (X-Gluc) hydrolyzed min−1 mg protein−1. Determinations for both panels were made from bacterial cultures grown for 36 h in liquid BBOH medium at 30°C. Error bars represent standard deviations.
To start characterizing the promoter region of arsA, the transcription start site of arsA was identified by primer extension analysis with total RNA isolated from cells induced to encyst for 36 h on BBOH medium, as described in Materials and Methods. A single cDNA product of arsA was identified (Fig. 3A), defining the transcription start site for this gene 253 nt upstream of the ATG start codon (Fig. 3B).
Fig 3.

Inactivation of arpR or rpoS negatively affects arsA expression from a single promoter. (A) Primer extension mapping of arsA transcription initiation 36 h after encystment induction in A. vinelandii SW136 (lane 1) and mutant derivatives carrying the arpR::Tn5 mutation (lane 2) or rpoS::Sp (lane 3). (B) Nucleotide sequence of the upstream regulatory region of arsA. The transcription start site mapped by primer extension is shown in bold and marked by a black triangle. The −35 and −10 consensus sequences for the P1 promoter are shown in bold and underlined. The arsA translation initiation codon is shown in bold and marked by an arrow. The analysis was performed using the primer 5′-GAGGATCGACGAACGGAC-3′ and 50 μg of RNA isolated from cells grown for 36 h in liquid BBOH medium.
By using the bacterial promoter recognition program BPROM (http://www.softberry.com/berry.phtml?topic=bprom&group=programs&subgroup=gfindb), −35 and −10 promoter elements showing similarity to RpoD-dependent promoters were identified in the arsA upstream region, in correspondence with the transcriptional start site identified (Fig. 3B). In the primer extension experiment, no cDNA product was observed for RNA from the arpR mutant strain SW7 (Fig. 3A), further confirming the regulation of arsA expression by ArpR.
ArpR is the activator of arsA transcription.
To test the possible direct regulation of arsA expression by ArpR, the arpR gene was cloned into the expression plasmid pBAD/His A (Invitrogen) under the control of an arabinose-inducible promoter and fused to an N-terminal histidine tag, resulting in plasmid pBAD-ArpR. Additionally, a plasmid compatible with pBAD-ArpR and containing the arsA regulatory region fused to the chloramphenicol acetyltransferase reporter gene (cat) was constructed (plasmid pACYC/PKK-ArsA). Both the pBAD-ArpR and pACYC/PKK-ArsA plasmids were transformed into Escherichia coli DH5α. In this strain, the induction of arpR expression produced chloramphenicol-resistant colonies, whereas no growth was obtained under uninduced conditions in the presence of this antibiotic (Fig. 4A), showing activation of arsA-cat transcription by ArpR. Also, the expression of arsA was measured as the CAT activity in liquid medium without chloramphenicol, under arpR-inducing and noninducing conditions. The CAT activity doubled with respect to that under uninduced conditions 3 h after the ArpR regulator was induced, and it reached a 20-fold higher activity after 12 h (Fig. 4B). The expression of ArpR after induction was confirmed by Western blot analysis (Fig. 4C).
Fig 4.
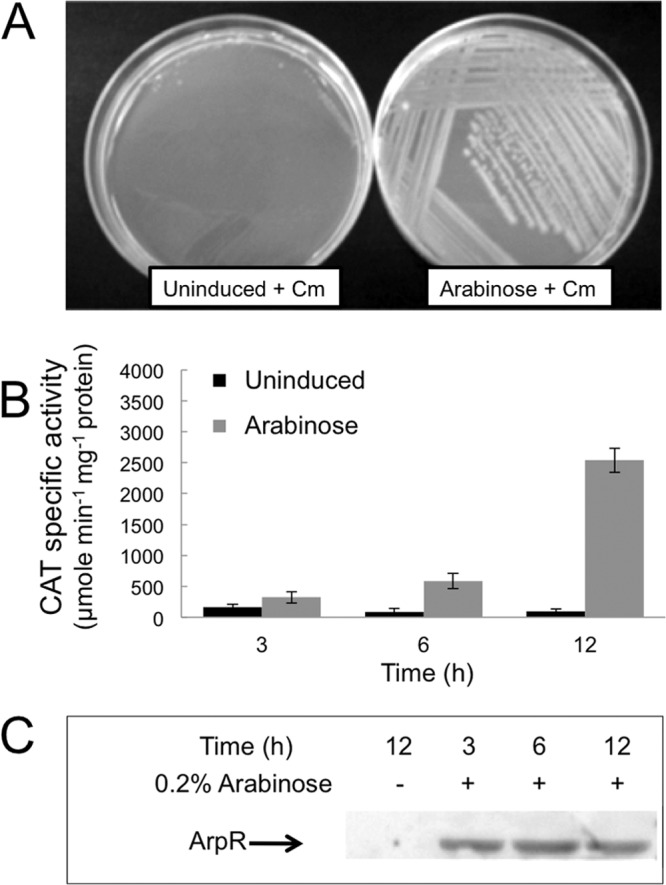
ArpR is the activator of arsA transcription. (A) Growth of E. coli DH5α carrying plasmids pBAD-ArpR, expressing ArpR from an arabinose-inducible promoter, and pACYC/PKK-ArsA, containing the arsA regulatory region fused to the chloramphenicol acetyltransferase reporter gene (cat). The experiment was done on LB medium plates supplemented with chloramphenicol (Cm) in the presence or absence of 0.2% arabinose. (B) Expression of the arsAp-cat transcriptional fusion in E. coli DH5α transformed with the same plasmids as those in panel A, measured as CAT activity. The experiment was done in liquid LB medium at 37°C. The activity was determined after 3, 6, and 12 h of growth in the presence or absence of arabinose. The CAT specific activity was quantified as μmol of substrate (chloramphenicol) hydrolyzed min−1 mg protein−1. Error bars represent the standard deviations. (C) Western blot analysis showing heterologous expression of His-tagged ArpR from plasmid pBAD-ArpR in the presence of arabinose, but not in its absence, even after 12 h of growth. Monoclonal anti-His antibodies were used, and the samples were taken from parallel cultures grown under the same conditions used for activity determinations.
To confirm the direct regulation of arsA by ArpR, we determined whether ArpR binds to the arsA promoter region by performing EMSA using a purified MBP-ArpR fusion protein (Fig. 5). A DNA fragment of 291 bp (positions −462 to −171 with respect to the ATG start codon) containing the upstream regulatory region of arsA was amplified by PCR and subjected to an EMSA as described in Materials and Methods. ArpR binds to the arsA regulatory region but not to a nonspecific 225-bp DNA fragment corresponding to the upstream regulatory region of the rpoS gene, which was used as a negative control. Competition assays using the unlabeled arsA regulatory region or the nonspecific rpoS fragment were carried out as controls to demonstrate specificity. Only the former competed with the labeled fragment (Fig. 5A). As an additional negative control, the assay was also carried out with MBP. MBP was not able to bind the arsA regulatory region (Fig. 5B). These results indicate that ArpR specifically binds the regulatory region of arsA.
Fig 5.

ArpR specifically binds the regulatory arsA region. Electrophoretic mobility shift assays were performed to analyze ArpR binding to the regulatory region of arsA. (A) Labeled DNA fragments (10 nM) containing the regulatory region of arsA were incubated with increasing concentrations of MBP-ArpR (0 to 1,000 nM). ArpR binding to arsA was further analyzed by competitive EMSA. As a negative control, a fragment containing the regulatory region of rpoS was included in the DNA binding reaction mix. The labeled DNA fragment containing the regulatory region of arsA was mixed with 1,000 nM MBP-ArpR in the presence or absence of a 100-fold excess of unlabeled specific (arsA) or nonspecific (rpoS) competitor. (B) EMSA with MBP as a negative control. The DNA-protein complexes were resolved in a nondenaturing 6% polyacrylamide gel.
Inactivation of the sigma factor RpoS negatively affects arsA expression.
It was recently published that RpoS is involved in control of the synthesis of ARs in A. vinelandii ATCC 9046, as an rpoS mutant was unable to synthesize these lipids (11). However, the mechanism of regulation was not established. In order to further investigate this regulation, we determined the effect of the inactivation of rpoS on the expression of arsA. We constructed an rpoS mutant derivative of SW136 (strain SW9) and tested the effect of the mutation on the production of ARs. As expected, strain SW9 cells induced to encyst were not stained with Fast Blue B (Fig. 1A), and the amount of ARs quantified showed a strong negative effect of rpoS inactivation on the synthesis of ARs (Fig. 1B), in accordance with the results reported by Cocótl-Yañez et al. (11) for A. vinelandii strain ATCC 9046. When the relative expression level of arsA in the forming cysts of the rpoS mutant SW9 was measured by qRT-PCR, we found that it was more than 10 times lower than that in the wild-type strain SW136 (Fig. 2A). This was confirmed by constructing strain YRR34, an SW9 derivative containing the same chromosomal arsA-gusA transcriptional fusion present in strains YRR30 and YRR32 (Fig. 2B). The expression of arsA in the rpoS mutant YRR34, measured as β-glucuronidase activity, was 12 times lower than that of the YRR30 strain, carrying the wild-type rpoS allele (Fig. 2B). A similar result was observed in the primer extension analysis in comparing the amounts of arsA cDNA obtained from the mRNAs from SW136 and SW9 cells induced to encyst (Fig. 3A). Thus, the previously reported negative effect of rpoS inactivation on the synthesis of ARs is exerted at the level of transcription of the ars biosynthetic operon.
RpoS regulates arsA expression indirectly through the control of arpR transcription.
To investigate whether the control of arsA expression by RpoS is exerted through the regulation of the ArpR activator, we determined the effect of the rpoS inactivation on arpR expression during encystment. The amount of arpR mRNA measured by qRT-PCR was more than 10 times lower in the rpoS mutant than in the wild-type strain. To further confirm the effect of the rpoS mutation on arpR transcription, an arpR-gusA transcriptional fusion was introduced into the chromosome of the wild-type strain SW136 and its rpoS mutant SW9, producing strains YRR50 and YRR52, respectively. The expression of arpR, measured as β-glucuronidase activity, was impaired in the strain containing the rpoS inactivation (only 2% of the activity found in YRR52), confirming the positive control of arpR expression by RpoS.
To establish if the control of RpoS on arsA expression is exerted solely through the regulation of arpR transcription, the ArpR protein was expressed in strain SW9 in trans from a constitutive RpoS-independent promoter by using plasmid pBBR-ArpR, the same plasmid previously used to complement the arpR mutant SW7. The expression of ArpR reestablished the production of ARs of the rpoS mutant to levels similar to those of the complemented arpR mutant (Fig. 6A and B).
Fig 6.
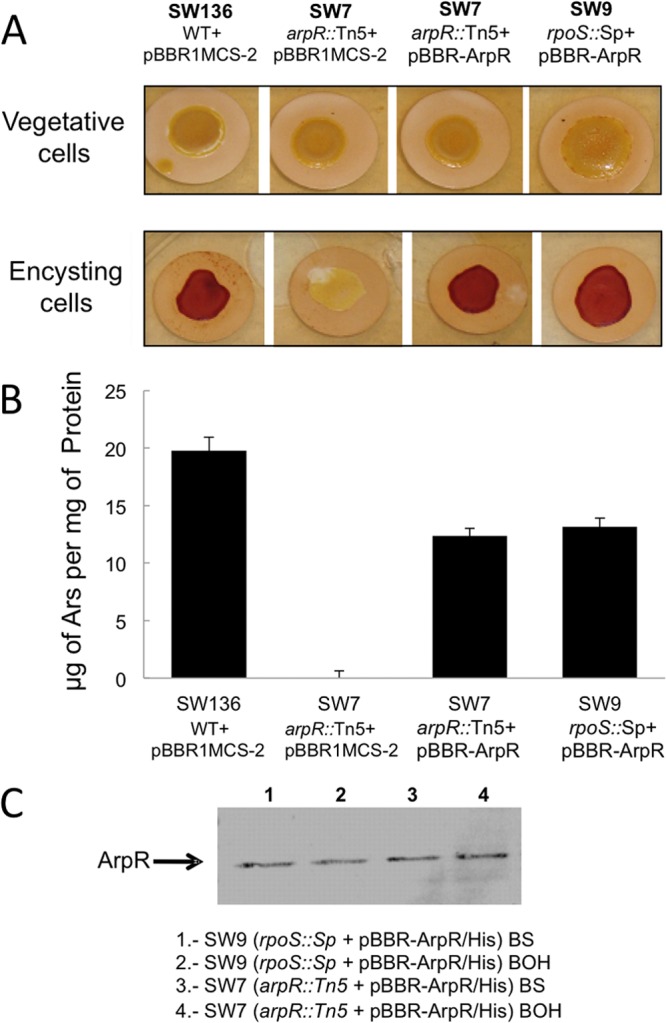
ArpR restores the synthesis of ARs in the rpoS mutant strain during encystment but not in vegetative cells. (A) Staining of ARs produced by the A. vinelandii SW7 and SW9 mutant strains grown on agar-fortified medium under vegetative growth or encystment-inducing conditions. Strains were transformed with plasmid pBBR-ArpR, carrying a constitutively expressed ArpR gene. As a negative control, the empty pBBRIMCS-2 plasmid was transformed into the SW136 and SW7 strains. (B) Quantification of ARs produced by the complemented strains under the encystment conditions shown in panel A. (C) Western blot analysis showing expression of His-tagged ArpR from plasmid pBAD-ArpR in strains SW7 and SW9. Samples were taken from vegetative and encysting cultures. Monoclonal anti-His antibodies were used.
ArpR is expressed only during encystment.
We recently demonstrated that arsABCD expression is induced in cells undergoing encystment (2). To investigate if the same expression pattern occurs for the regulator ArpR, we compared arpR expression in vegetative and encysting cells of the wild-type and rpoS mutant strains (grown on BS and BBOH medium, respectively) by RT-PCR. We observed that arpR in the wild-type strain was expressed only under encystment induction conditions (Fig. 7A). To confirm this result, we examined the transcription of arpR in vegetative and encysting cells by measuring β-glucuronidase activity in strain YRR50, carrying the chromosomal arpR-gusA transcriptional fusion. As shown in Fig. 7B, arpR transcription increased after encystment induction, reaching values 120-fold higher than those of the uninduced vegetative cells. The results described above showed that arpR has an expression pattern similar to that observed for arsA expression (2).
Fig 7.
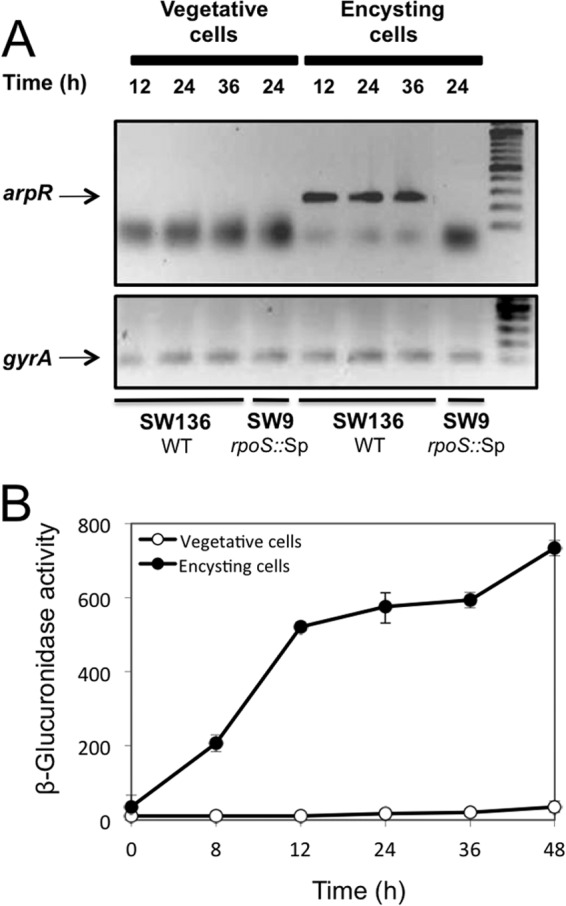
ArpR is expressed under encystment-inducing conditions. (A) RT-PCR assay to monitor arpR expression in vegetative and encysting cells of A. vinelandii SW136 and its rpoS mutant SW9. Total RNA was extracted from cells grown at 30°C for 12, 24, or 36 h under vegetative growth conditions (liquid BS medium) or with encystment induction (liquid BBOH medium). cDNA was synthesized as reported previously (25). The expression of gyrA was used as a control of constitutive expression. Both the arpR and gyrA fragments were amplified by PCR with Taq polymerase (Altaenzyme). The PCR products were resolved in a 1% agarose gel. (B) Expression of arpR during growth in liquid BS or BBOH medium, measured as β-glucuronidase activities of an arpR::gusA transcriptional fusion in strain YRR50. One unit of β-glucuronidase activity corresponds to 1 nmol of substrate (X-Gluc) hydrolyzed min−1 mg protein−1. Average values ± standard deviations are shown.
The expression of arpR during vegetative growth is insufficient to induce the synthesis of ARs.
To determine if the expression of ArpR is the only factor determining the expression of the ars structural genes, we expressed ArpR in vegetative cells of the arpR and rpoS mutants in trans from plasmid pBBR1-arpR. This plasmid complemented the arpR mutant and reestablished the synthesis of ARs in the rpoS mutant during encystment, but no ARs were synthesized in vegetative cells (Fig. 6A and B), although the ArpR protein was produced as shown by a Western blot analysis (Fig. 6C), showing that the expression of ArpR is not enough to determine production of ARs. This result suggested that some other factor is needed for the synthesis of ARs.
Coinducers are recognized as being important for the function of LTTRs and often appear to contribute to a feedback loop in which a product or intermediate of a given metabolic pathway activated by an LTTR acts as the coinducer necessary for the transcriptional activation or repression (29).
Considering both the possible requirement of a coinducer and the encystment-inducing properties of molecules such as β-hydroxybutyrate or n-butanol, which has to be metabolized through β-hydroxybutyrate to induce encystment (30), we tested the effect of β-hydroxybutyrate and the metabolically related molecules hydroxybutyryl-CoA and acetoacetyl-CoA on ArpR binding to the regulatory region of arsA by using EMSA. Because ARs are synthesized from malonyl-CoA in A. vinelandii (31), we also tested this molecule. The addition of acetoacetate, malonyl-CoA, or β-hydroxybutyrate at concentrations of up to 12 mM did not affect the binding reactions (data not shown); however, the presence of 1 to 12 mM acetoacetyl-CoA considerably increased the in vitro binding of ArpR to the arsA promoter region (Fig. 8).
Fig 8.
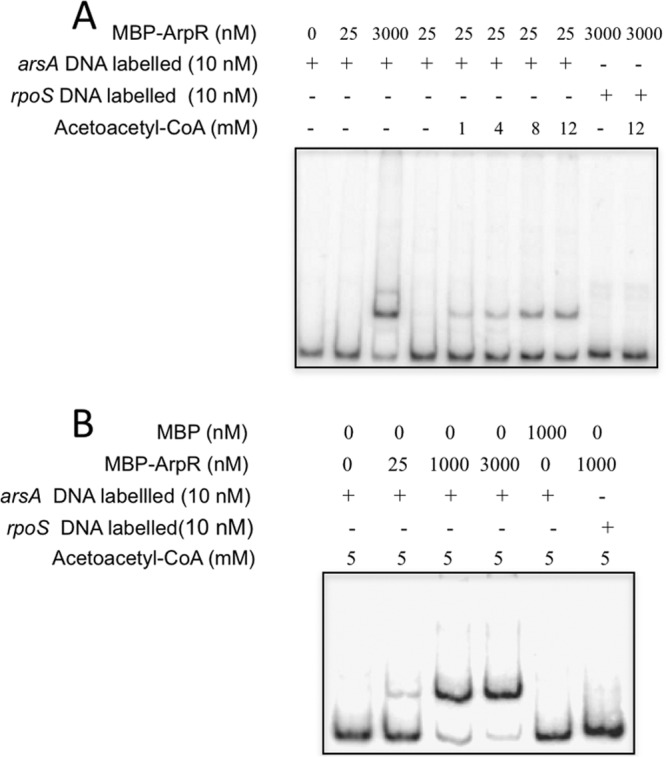
Acetoacetyl-CoA increases the affinity of ArpR for the regulatory region of arsA. EMSA was performed using an arsA DNA fragment in the absence or presence of different amounts of acetoacetyl-CoA. The labeled DNA fragment containing the regulatory region of arsA was incubated with increasing concentrations of MBP-ArpR (0, 0.1, and 8.0 μM) and 0, 1, 4, 8, or 12 mM acetoacetyl-CoA. As a negative control, a 225-bp fragment containing the regulatory region of rpoS was included in each DNA binding reaction mix. (B) EMSA with MBP as a negative control.
In order to estimate the increase in affinity in the presence of acetoacetyl-CoA, the apparent Kd was estimated in both the absence and presence of this putative coinducer. This constant was deduced from the concentration of ArpR required to bind 50% of the arsA DNA fragment in an EMSA (not shown). A value of 17.2 μM was determined for the protein in the absence of the inducer, with a value of 85.7 nM in its presence.
When the effect of in vivo addition of acetoacetyl-CoA was tested on Burk's sucrose medium (on which ARs are not produced), an induction of the synthesis of ARs was observed in the wild-type strain SW136 (Fig. 9A). This induction was dependent on ArpR, as the arpR mutant did not produce ARs under these conditions, unless the regulator was expressed independently from RpoS from plasmid pBBR-ArpR, and because the rpoS mutant, expressing ArpR from a promoter nondependent on this sigma factor, produced ARs.
Fig 9.

The presence of acetoacetyl-CoA under vegetative growth conditions induces the expression of the arsA and arpR genes and the synthesis of ARs. (A) Staining of ARs produced by A. vinelandii SW136 and the arpR and rpoS mutants SW7 and SW9, carrying plasmid pBBRIMCS-2 or pBBR-ArpR. (B) Production levels of ARs of A. vinelandii SW136 at different times. (C) arsA expression measured as the β-glucuronidase activity of strain YRR30, which contains an arsA::gusA reporter fusion. (D) arpR expression measured as the β-glucuronidase activity of strain YRR50, which contains an arpR::gusA reporter fusion. For the experiments in all panels, the strains were grown on agar-fortified BS medium in the absence (no inducer) or presence of 5 mM acetoacetyl-CoA. One unit of β-glucuronidase activity corresponds to 1 nmol of substrate (X-Gluc) hydrolyzed min−1 mg protein−1. Data are averages for two independent experiments. Error bars represent standard deviations.
A time course of the production of ARs by the wild-type strain in the presence or absence of acetoacetyl-CoA clearly showed an induced synthesis of these lipids in the presence of this compound (Fig. 9B). As expected, the expression of arpR and arsA, determined for the strains containing the chromosomal arpR-gusA and arsA-gusA transcriptional fusions, followed a similar induction pattern (Fig. 9C and D).
DISCUSSION
In A. vinelandii, the expression of the AR biosynthetic genes, arsABCD, is induced during encystment, in accordance with the synthesis of these lipids (2). We identified the regulator ArpR, whose inactivation impairs arsABCD expression and the synthesis of ARs. ArpR directly activates transcription of the biosynthetic genes from a single putative RpoD-dependent promoter under encystment-inducing conditions, transcribing the gene from 253 nt upstream of the start codon. The reason for the long leader in the arsA mRNA is not known, but it could be related to other regulatory phenomena. The expression of arpR is very low in vegetative cells and is highly induced (120-fold) during encystment. This expression pattern is in correspondence with arsABCD expression, the synthesis of ARs, and the percentage of mature cysts formed under each condition (2); thus, ArpR is the regulator controlling the synthesis of ARs during encystment.
ArpR is not the only regulator involved in the control of the synthesis of ARs. It was recently reported that the sigma factor RpoS is required for the production of ARs (11). Here we show that RpoS regulates the expression of the arsABCD biosynthetic genes through the control of arpR transcription. This was demonstrated by the suppression of the AR-negative phenotype of the rpoS mutant when ArpR was expressed from an RpoS-independent promoter. The analysis of the sequence of the arpR regulatory region showed a putative rpoS-dependent promoter (15), suggesting direct regulation of arpR by this sigma factor, although this control could be indirect.
In addition to the lack of ARs in the rpoS mutant, Cocótl-Yañez et al. (11) reported that other genes essential for cyst formation could be regulated by RpoS, because the cysts of this mutant were unable to resist desiccation and completely lacked the exine and intine layers when observed through electron microscopy. The lack of ARs in the rpoS mutant is not responsible for this phenotype, because these lipids play a structural role in the exine layer, but they are not essential for either cyst formation or desiccation resistance (2). The cyst morphology of the arpR mutant, observed through electron microscopy, and its desiccation resistance were very similar to the phenotype of the arsA mutant but different from that of the rpoS mutant (data not shown). These results suggest that ArpR has no other role in encystment and demonstrate that in addition to arpR, RpoS regulates the expression of other genes needed for mature cyst formation. The other roles in encystment played by this sigma factor remain to be investigated.
Usually, the LTTRs require a coinducer to activate expression of their target genes, and this inducer is typically a metabolic intermediate or substrate for the degradation pathway being regulated (32). The lack of AR synthesis in BS medium, even after ArpR expression in trans from plasmid pBBR1-arpR, suggested the need for either an additional regulator or an inducer. Some lines of evidence suggested the involvement of a metabolic signal in encystment induction. β-Hydroxybutyrate or related metabolites induce this differentiation process (33). Also, a mutant impaired in PHB biosynthesis showed a phenotype of increased encystment and production of ARs, even under vegetative growth conditions (23). Here we show that acetoacetyl-CoA increases the affinity of ArpR for the promoter region of arsA in vitro. This metabolite can be generated during PHB synthesis from the condensation of two molecules of acetyl-CoA or as a result of PHB mobilization or the catabolism of β-hydroxybutyrate, butanol, or crotonate (34). ArpR binds specifically to its target DNA, and acetoacetyl-CoA increases its apparent affinity for the arsA regulatory region. We also observed in vivo that the presence of acetoacetyl-CoA in the medium was able to induce the expression of the arsA and arpR genes, as well as the synthesis of ARs, even under vegetative growth conditions. These results suggest that this molecule could be a metabolic signal sensed by this regulatory system for the transcriptional activation of the synthesis of ARs during encystment.
For many LTTRs, coinducer binding is presumed to affect transcription through the induction of a conformational change of the promoter-regulator complex already formed, resulting in preferential binding to different locations in the promoter region, affecting DNA bending and interaction with RNA polymerase (29). However, there are some examples where coinducer binding has been shown to result in changes in the binding affinity for the target promoter. One interesting example is found in MvfR of Pseudomonas aeruginosa, as two different coinducers (3,4-dihydroxy-2-heptylquinoline and its precursor, 4-hydroxy-2-heptylquinoline) enhance MvfR binding to the target pqsA promoter (35). Another example is found in E. coli OxyR, where the sensing of H2O2 alters its conformation and its DNA affinity, and although this protein does not respond to a classical coinducer but to a redox modification, the induced changes in this protein influence its DNA-binding affinity (36, 37). In the case of ArpR, more work is needed to establish if the ligand acetoacetyl-CoA induces conformational changes, modifies the location for DNA binding, or changes the affinity of this regulator for the target promoters.
Little is known about the genetic control of encystment in bacteria. In A. vinelandii, RpoS (11), the sigma factor AlgU (7, 8), and the response regulator AlgR (6) are known to be required for encystment. However, whereas these regulators also control genes during vegetative growth, ArpR is the first cyst-specific regulator to be described, and its role in encystment is now clear.
ACKNOWLEDGMENTS
This work was supported by grants 127979 from CONACyT and IN221809 and IT200612 from DGAPA-PAPIIT-UNAM. Yanet Romero thanks CONACyT for M.Sc. and Ph.D. scholarships.
We are grateful to Gloria Soberón-Chávez and Victor Bustamante for critical discussions on this work. We acknowledge Leticia Olvera for her technical support in the analysis of transcription of arpR.
Footnotes
Published ahead of print 1 February 2013
REFERENCES
- 1. Sadoff HL. 1975. Encystment and germination in Azotobacter vinelandii. Bacteriol. Rev. 39:516–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Segura D, Vite O, Romero Y, Moreno S, Castañeda M, Espín G. 2009. Isolation and characterization of Azotobacter vinelandii mutants impaired in alkylresorcinol synthesis: alkylresorcinols are not essential for cyst desiccation resistance. J. Bacteriol. 191:3142–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lin LP, Sadoff HL. 1968. Encystment and polymer production by Azotobacter vinelandii in the presence of beta-hydroxybutyrate. J. Bacteriol. 95:2336–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stasiuk M, Kozubek A. 2010. Biological activity of phenolic lipids. Cell. Mol. Life Sci. 67:841–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reusch RN, Sadoff HL. 1983. Novel lipid components of the Azotobacter vinelandii cyst membrane. Nature 302:268–270 [DOI] [PubMed] [Google Scholar]
- 6. Nuñez C, Moreno S, Soberón-Chavez G, Espín G. 1999. The Azotobacter vinelandii response regulator AlgR is essential for cyst formation. J. Bacteriol. 181:141–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moreno S, Najera R, Guzman J, Soberon-Chavez G, Espin G. 1998. Role of alternative sigma factor algU in encystment of Azotobacter vinelandii. J. Bacteriol. 180:2766–2769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. León R, Espín G. 2008. FlhCD but not FleQ regulates flagella biogenesis in Azotobacter vinelandii and is under AlgU and CydR negative control. Microbiology 154:1719–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castañeda M, Sanchez J, Moreno S, Nuñez C, Espín G. 2001. The global regulators GacA and σs form part of a cascade that controls alginate production in Azotobacter vinelandii. J. Bacteriol. 183:6787–6793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hernández-Eligio A, Castellanos M, Moreno S, Espín G. 2011. Transcriptional activation of the Azotobacter vinelandii polyhydroxybutyrate biosynthetic genes phbBAC by PhbR and RpoS. Microbiology 157:3014–3023 [DOI] [PubMed] [Google Scholar]
- 11. Cocótl-Yañez M, Sampieri A, Moreno S, Nuñez C, Castañeda M, Segura D, Espín G. 2011. Roles of RpoS and PsrA in cyst formation and alkylresorcinol synthesis in Azotobacter vinelandii. Microbiology 157:1685–1693 [DOI] [PubMed] [Google Scholar]
- 12. Funa N, Ozawa H, Hirata A, Horinouchi S. 2006. Phenolic lipid synthesis by type III polyketide synthases is essential for cyst formation in Azotobacter vinelandii. Proc. Natl. Acad. Sci. U. S. A. 103:6356–6361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kovach ME, Elzer PH, Hill DS, Robertson GT, Farris MA, Roop RM, II, Peterson KM. 1995. Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes. Gene 166:175–176 [DOI] [PubMed] [Google Scholar]
- 14. Brosius J. 1984. Plasmid vector for the selection of promoters. Gene 27:151–160 [DOI] [PubMed] [Google Scholar]
- 15. Peralta-Gíl M, Segura D, Guzmán J, Servín-Gonzalez L, Espín G. 2002. Expression of the Azotobacter vinelandii poly-β-hydroxybutyrate biosynthetic gene phbB is driven by two overlapping promoters and is dependent on the transcriptional activator PhbR. J. Bacteriol. 184:5672–5677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kennedy C, Gamal R, Humphrey R, Ramos J, Brigle K, Dean D. 1986. The nifH, nifM and nifN genes of Azotobacter vinelandii: characterization by Tn5 mutagenesis and isolation from pLARF1 gene banks. Mol. Gen. Genet. 205:318–325 [Google Scholar]
- 17. Bali A, Blanco G, Hill S, Kennedy C. 1992. Excretion of ammonium by a nifL mutant of Azotobacter vinelandii fixing nitrogen. Appl. Environ. Microbiol. 58:1711–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. 1951. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193:265–275 [PubMed] [Google Scholar]
- 19. Puente JL, Bieber D, Ramer SW, Murray W, Schoolnik GK. 1996. The bundle-forming pili of enteropathogenic Escherichia coli: transcriptional regulation by environmental signals. Mol. Microbiol. 20:87–100 [DOI] [PubMed] [Google Scholar]
- 20. Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 21. Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74:5463–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson KJ, Sessitsch A, Corbo JC, Giller KE, Akkermans AD, Jefferson RA. 1995. Beta-glucuronidase (GUS) transposons for ecological and genetic studies of rhizobia and other gram-negative bacteria. Microbiology 141:1691–1705 [DOI] [PubMed] [Google Scholar]
- 23. Segura D, Cruz T, Espín G. 2003. Encystment and alkylresorcinol production by Azotobacter vinelandii strains impaired in poly-β-hydroxybutyrate synthesis. Arch. Microbiol. 179:437–443 [DOI] [PubMed] [Google Scholar]
- 24. Tluscik F, Kozubek A, Mejbaum-Katzenellenbogen W. 1981. Alkylresorcinols in rye (Secale cereale L.) grains. VI. Colorimetric micromethod for the determination of alkylresorcinols with the use of diazonium salt, Fast Blue B. Acta Soc. Bot. Pol. 50:645–651 [Google Scholar]
- 25. Noguez R, Segura D, Moreno S, Hernández A, Juárez K, Espín G. 2008. Enzyme INtr, Npr and IIANtr are involved in regulation of the poly-β-hydroxybutyrate biosynthetic genes in Azotobacter vinelandii. J. Mol. Microbiol. Biotechnol. 15:244–254 [DOI] [PubMed] [Google Scholar]
- 26. Barry T, Geary S, Hannify S, Mac Gearailt C, Shalloo M, Heery D, Gannon F, Powell R. 1992. Rapid mini-preparations of total RNA from bacteria. Nucleic Acids Res. 20:4940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 28. Setubal JC, dos Santos P, Goldman BS, Ertesvåg H, Espin G, Rubio LM, Valla S, Almeida NF, Balasubramanian D, Cromes L, Curatti L, Du Z, Godsy E, Goodner B, Hellner-Burris K, Hernandez JA, Houmiel K, Imperial J, Kennedy C, Larson TJ, Latreille P, Ligon LS, Lu J, Mærk M, Miller NM, Norton S, O'Carroll IP, Paulsen I, Raulfs EC, Roemer R, Rosser J, Segura D, Slater S, Stricklin SL, Studholme DJ, Sun J, Viana CJ, Wallin E, Wang B, Wheeler C, Zhu H, Dean DR, Dixon R, Wood D. 2009. The genome sequence of Azotobacter vinelandii, an obligate aerobe specialized to support diverse anaerobic metabolic processes. J. Bacteriol. 191:4534–4545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maddocks SE, Oyston PCF. 2008. Structure and function of the LysR-type transcriptional regulator (LTTR) family proteins. Microbiology 15:3609–3623 [DOI] [PubMed] [Google Scholar]
- 30. Gama-Castro S, Núñez C, Segura D, Moreno S, Guzmán J, Espín G. 2001. Azotobacter vinelandii aldehyde dehydrogenase regulated by sigma(54): role in alcohol catabolism and encystment. J. Bacteriol. 183:6169–6174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyanaga A, Funa N, Awakawa T, Horinouchi S. 2008. Direct transfer of starter substrates from type I fatty acid synthase to type III polyketide synthases in phenolic lipid synthesis. Proc. Natl. Acad. Sci. U. S. A. 105:871–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schell MA. 1993. Molecular biology of the LysR family of transcriptional regulators. Annu. Rev. Microbiol. 47:597–626 [DOI] [PubMed] [Google Scholar]
- 33. Reusch RN, Sadoff HL. 1981. Lipid metabolism during encystment of Azotobacter vinelandii. J. Bacteriol. 145:889–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sudesh K, Fukui T, Iwata T, Doi Y. 2000. Factors affecting the freeze-fracture morphology of in vivo polyhydroxyalkanoate granules. Can. J. Microbiol. 46:304–311 [PubMed] [Google Scholar]
- 35. Xiao G, Déziel E, He J, Lépine F, Lesic B, Castonguay M-H, Milot S, Tampakaki AP, Stachel SE, Rahme LG. 2006. MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol. Microbiol. 62:1689–1699 [DOI] [PubMed] [Google Scholar]
- 36. Kullik I, Toledano MB, Tartaglia LA, Storz G. 1995. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for oxidation and transcriptional activation. J. Bacteriol. 177:1275–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kullik I, Stevens J, Toledano MB, Storz G. 1995. Mutational analysis of the redox-sensitive transcriptional regulator OxyR: regions important for DNA binding and multimerization. J. Bacteriol. 177:1285–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]