

Abstract
It has recently been suggested that bactericidal antibiotics, including aminoglycoside antibiotics (AGAs), and toxic small molecules, such as hydroxyurea (HU), kill bacteria the same way, namely, by generating reactive oxygen species (ROS) via a process requiring activation of the Cpx stress response. We suggest an opposite, protective role for Cpx. We have confirmed the initial finding that cpxA null mutations confer resistance to HU. However, the two-component sensor CpxA is both a kinase and a phosphatase, and previous work from our lab has shown that removing CpxA can activate the stress response owing to buildup of the phosphorylated response regulator (CpxR∼P) that occurs in the absence of the phosphatase activity. We show that a dominant cpxA* mutation that constitutively activates the Cpx stress response confers a high level of resistance to both HU and AGAs in a CpxR-dependent manner. In contrast, inactivating the CpxR response regulator by mutating the phosphorylation site (D51A) or the putative DNA-binding motif (M199A) does not increase resistance to HU or AGAs. Taken together, these results demonstrate that activation of the Cpx stress response can protect cells from HU and AGAs. However, the Cpx response does not increase resistance to all classes of bactericidal antibiotics, as the cpxA* mutants are not significantly more resistant to fluoroquinolones or β-lactams than wild-type cells. Thus, it seems unlikely that all bactericidal antibiotics kill by the same mechanism.
INTRODUCTION
The mechanism by which aminoglycoside antibiotics (AGAs) kill bacteria has been a subject of interest for decades, and classic studies link the mechanism with alterations in the cellular membrane (1). It is known that AGAs bind to the A-site of the ribosome, leading to mistranslation (2). Incorporation of these mistranslated proteins into the membrane could alter this essential structure to an extent that renders the cell inviable (2). Consistent with this idea, certain AGAs (such as gentamicin) trigger activation of the Cpx envelope stress response (3). Misfolded proteins, including inner membrane (IM) proteins, are also known inducers of the Cpx response (4). Recent work suggests that induction of the Cpx response contributes to the bactericidal nature of these antibiotics by stimulating the formation of reactive oxygen species (ROS) (5). Antibiotics like fluoroquinolones that inhibit DNA gyrase or small toxic molecules that inhibit DNA replication by targeting ribonucleotide reductases, such as hydroxyurea (HU), are also thought to generate misfolded envelope proteins and lead to killing by a similar mechanism (6, 7). Indeed, many bactericidal antibiotics, including the β-lactams, are thought to ultimately cause cell death by stimulating formation of ROS (5).
The Cpx two-component system is composed of the histidine sensor kinase/phosphatase CpxA and its cognate response regulator CpxR (8–10). Evidence suggests that this system senses extracytoplasmic stresses, such as misfolded envelope proteins, and transcriptionally mediates an adaptive response (10–13). Many of the Cpx-controlled genes upregulated by this adaptive response encode protein folding and degradation factors (14, 15). Signals are transduced from CpxA to CpxR via conserved phosphotransfer reactions that are characteristic of two-component systems (11). Inducing cues cause a conformational change in CpxA, stimulating the autophosphorylation of a conserved histidine residue. Once this residue is phosphorylated, CpxA acts as a kinase and phosphorylates a conserved aspartate residue on CpxR. Phosphorylated CpxR (CpxR∼P) acts as a transcriptional activator (8, 16). In the absence of induction, unphosphorylated CpxA acts as a phosphatase that removes phosphoryl groups from CpxR, thereby maintaining the response regulator in an inactive state even in the presence of heterologous kinases and small-molecule phospho donors such as acetyl-phosphate (8). In mutant strains exhibiting constitutive Cpx activity (cpxA*), the mutant CpxA* protein still functions as an autokinase and CpxR kinase but lacks phosphatase activity. This leads to high levels of CpxR∼P and hyperactivation of the Cpx response (8, 11).
A role for Cpx in the generation of ROS that would contribute to the cell death caused by various antibiotics was unexpected, given the proposed function of the Cpx system in combating envelope stress as described above. Indeed, there are several experimental observations that argue against a role for the Cpx response in promoting stress-induced cell death. First, the cpxA* mutations were initially identified as suppressors of the toxicity caused by misfolded or mistargeted proteins in the cell envelope (4, 10, 17, 18). Second, the cpxA* mutation confers resistance to amikacin, an AGA (19). Work presented in this study explains these seemingly paradoxical results and also suggests that all bactericidal drugs do not kill via the same mechanism.
MATERIALS AND METHODS
Strains, media, and culture conditions.
All strains used in this study are isogenic derivatives of Escherichia coli MC4100 constructed by generalized transduction with bacteriophage P1 and are listed in Table 1 (22). To construct the cpxA24 cpxR::kan double mutant, a cpxR::kan insertion/deletion construct was introduced into the cpxA24 strain by homologous recombination using previous described methods (23). This kan insertion does not confer resistance to amikacin or gentamicin.
Table 1.
E. coli strains and plasmids used in this study
| Strain or plasmid | Relevant characteristic(s) | Reference(s) and/or source |
|---|---|---|
| Strains | ||
| MC4100 | F− araD139 Δ(argF-lac)U169 rpsL150 relA1 flbB5301 deoC1 ptsF25 thi | 3 |
| PTM163 | MC4100 cpxA24 zii::Tn10 | This study; 8, 16 |
| PTM180 | MC4100 cpxA17 metF::kan | This study; 18; lab stock |
| PTM197 | MC4100 cpxR M199A zii::Tn10 | This study; 13 |
| PTM198 | MC4100 cpxR D51A zii::Tn10 | This study; lab stock |
| PTM202 | MC4100 carrying pLD404 | This study; 17 |
| PTM208 | MC4100(pLD404) cpxR D51A zii::Tn10 | This study |
| PTM329 | MC4100 cpxA::kan | This study; 20 |
| PTM436 | MC4100 cpxR::spc | This study; 15 |
| PTM441 | MC4100 cpxA24 cpxR::kan | This study |
| PTM467 | MC4100 baeS::Tn10-cam | This study; 21 |
| PTM472 | MC4100 rseA(Oc) nadB::Tn10 | Jaclyn Schwalm, unpublished data |
| Plasmid | ||
| pLD404 | NlpE overexpression plasmid | 17 |
Media and chemicals.
All strains were grown in Luria-Bertani (LB) broth or agar at 37°C unless otherwise indicated. Strains containing cpxA* alleles were maintained on LB agar containing amikacin (Sigma) at 3 μg/ml. Plasmid-containing strains were grown in LB agar supplemented with ampicillin (Sigma) at 100 μg/ml.
Sensitivity assays.
For drug sensitivity assays, strains were serially diluted and plated on LB agar containing increasing amounts of HU, gentamicin, or amikacin. CFU were enumerated after 24 h of growth at 37°C. Between 10 mM and 30 mM HU were required to observe significant cell death in plating assays. Sensitivity of strains was also determined by efficiency of plating (EOP). Strains were serially diluted and replica plated onto LB agar plates supplemented with either 20 mM HU, 3 μg/ml amikacin, or 5 μg/ml gentamicin.
RESULTS
Activation of the Cpx response confers resistance to AGAs and HU.
Recent work has shown that null mutations in cpxA confer resistance to AGAs and HU (6, 24), and we have confirmed this result: strains carrying cpxA::kan are resistant to HU and the AGAs amikacin and gentamicin (Fig. 1). Previous studies suggested that this increased level of resistance occurs because HUs and AGAs induce the Cpx response, and this in turn increases production of ROS that cause cellular damage leading to cell death (5, 7, 24). Like many two-component sensors, CpxA functions not only as an autokinase and a kinase to activate CpxR but also as a phosphatase that maintains CpxR in an unphosphorylated state in the absence of inducing stimuli (8). Importantly, null mutations of cpxA disrupt all three activities. The absence of phosphatase activity in particular raises the possibility that the Cpx response will actually be upregulated in cpxA null mutants owing to the unrestrained phosphorylation of CpxR by small-molecule phospho donors or heterologous kinases (15, 16). This alternative explanation suggests that cpxA null mutations confer resistance not by inactivating but by activating the Cpx stress response. According to this view, the Cpx response is activated in cpxA null mutant strains, and this activation does not contribute to cell death but rather prevents it.
Fig 1.

The cpxA::kan null and constitutive cpxA* mutations confer resistance to hydroxyurea (HU) and aminoglycoside antibiotics (AGAs), while null mutations in cpxR do not. (A) Survival of strains was measured by enumerating CFU on LB agar plates supplemented with increasing concentrations of HU following overnight growth at 37°C. The strains tested include E. coli MC4100, PTM163 (cpxA*), PTM197 (cpxR M199A [cpxR gene with an M-to-A change at position 199 that inactivates the putative DNA-binding motif of the CpxR response regulator]), PTM198 (cpxR D51A [cpxR gene with a D-to-A change at position 51 that mutates the phosphorylation site of the CpxR response regulator]). (B) Efficiency of plating (EOP) assay on LB agar plates supplemented with 20 mM HU, 3 µg/ml amikacin, or 5 μg/ml gentamicin grown at 37°C. Sensitivity was determined for the following strains: MC4100, PTM163 (cpxA*), PTM197 (cpxR M199A), PTM198 (cpxR D51A), and PTM436 (cpxR::spc).
To test the alternative explanation that Cpx activity actually protects cells from AGAs and HU, we took advantage of a constitutively active mutation that abolishes the phosphatase activity, but not the kinase activity, of CpxA (termed cpxA*). Compared with null mutations in cpxA, these cpxA* mutations confer resistance to HU and AGAs to comparable levels (Fig. 1B). Multiple cpxA* alleles have been isolated, with all identified mutations clustering in three regions of CpxA: the periplasmic domain, the second transmembrane helix, and the region adjacent to the predicted site of autophosphorylation (8). The cpxA24 allele, which encodes a variant of CpxA lacking 32 amino acids in a periplasmic loop, exhibits one of the highest levels of induction among all constitutive cpx mutations (8) and is the cpxA* allele used exclusively hereafter.
To confirm that the resistance to HU and AGAs conferred by a cpxA null mutation is due to the unrestrained phosphorylation of CpxR and the accumulation of CpxR∼P, we determined drug sensitivity in a mutant strain lacking both CpxA and CpxR. A spectinomycin resistance cassette insertion in cpxR has polar effects on cpxA and is effectively null for both genes (13). The cpxR::spc allele does not ameliorate sensitivity to HU or AGAs (Fig. 1B). This mutant, which is null for both cpxR and cpxA, suggests that the resistance of the cpxA null mutant strain to HU and AGAs is dependent on the presence of functional cpxR and is due to the accumulation of CpxR∼P. This demonstrates that the activation of the Cpx response increases survival in the presence of HU or AGAs.
To demonstrate that inactivating the Cpx response did not confer resistance to HU or AGAs, we used characterized point mutations in cpxR that abolish or reduce function of the response regulator. Mutating the phosphorylation site of CpxR (D51A) did not confer resistance to HU (Fig. 1). Similarly, mutating the putative DNA-binding site of CpxR (M199A) did not confer detectable resistance to HU (Fig. 1) (13). This further demonstrated that it is the activation of the Cpx response that confers resistance to HU and AGAs. Disrupting the response regulator effectively inactivates the Cpx response; survival in the presence of HU and AGAs in this context is similar to that observed for wild-type strains.
Resistance of cpxA* mutants is not due to pleiotropic effects.
The cpxA* mutation causes many pleiotropic effects, some of which could be the consequence of hyperphosphorylated CpxR and some of which could result from the phosphorylation of heterologous response regulators by CpxA (10). To ensure that the increased resistance of cpxA* to AGAs and HU was an on-pathway effect, mediated by physiologically normal levels of CpxR∼P, we examined the impact of activating the Cpx response by other means.
Overproducing the outer membrane lipoprotein NlpE induces the Cpx response, albeit to a lower level than is observed in the cpxA* mutant (13, 17). Consistent with our hypothesis, NlpE overproduction caused resistance to HU, but to a lesser degree than the cpxA* mutation did (Fig. 2). This intermediate level of resistance suggests that the level of Cpx activation determines the degree of resistance to HU.
Fig 2.
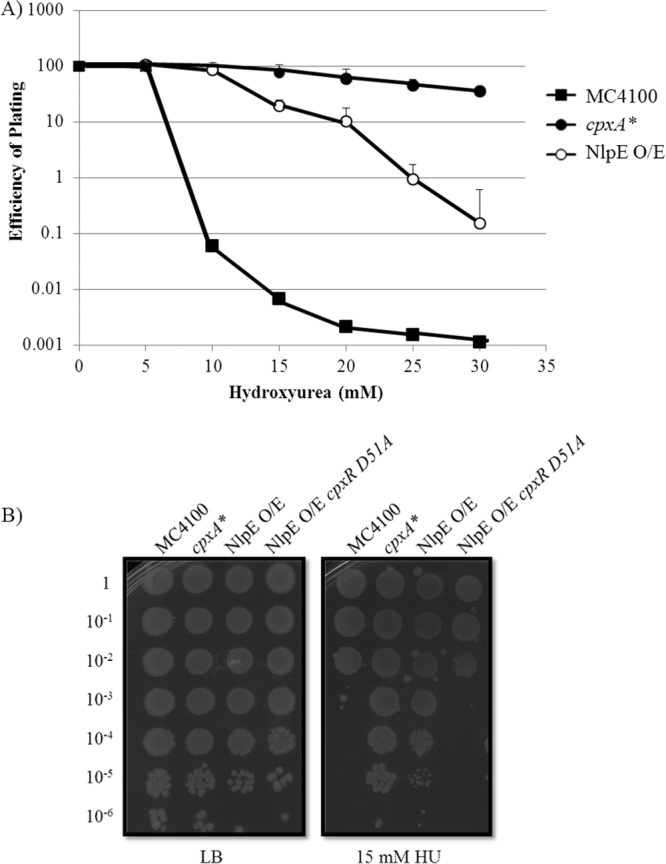
Overexpression of the outer membrane lipoprotein NlpE activates the Cpx response, which is sufficient to confer resistance to hydroxyurea (HU). (A) Survival of strains was measured by enumerating CFU on LB agar plates supplemented with increasing concentrations of HU. (B) EOP assay (as described in the legend to Fig. 1) of E. coli strains MC4100, PTM163 (cpxA*), PTM202 (NlpE overexpressed [NlpE O/E]), and PTM208 (NlpE O/E cpxR D51A) on 15 mM HU.
To show that resistance is conferred by downstream members of the Cpx regulon and is not the consequence of phosphorylation of a noncognate response regulator, we used tests of epistasis. In strains producing CpxR D51A, NlpE overproduction no longer conferred resistance to HU (Fig. 2). Similarly, the ameliorating effect of cpxA* is also dependent on functional cpxR; resistance to both HU and AGAs is abolished when a cpxR null mutation is introduced into a cpxA* background (Fig. 3), a fact noted previously for amikacin resistance (25). Hence, removing the cognate response regulator of the Cpx response abolished the resistance conferred by an activated CpxA kinase. These results demonstrate that resistance to HU and AGAs conferred by the activated Cpx response is due to phosphorylation of CpxR and concomitant changes in Cpx regulon expression.
Fig 3.

Deletion of cpxR in a cpxA* background abolishes resistance to HU and AGAs. EOPs of strains MC4100, PTM163 (cpxA*), and PTM441 (cpxA* cpxR::kan) on LB agar supplemented with 20 mM HU, 3 µg/ml amikacin, or 5 μg/ml gentamicin at 37°C.
Multiple Cpx regulon members may be responsible for resistance.
The Cpx response is one of the primary envelope stress response systems in E. coli, which play overlapping but distinct roles in monitoring the integrity of the cell envelope (11). Some important Cpx regulon members, such as spy and degP, are coregulated by other envelope stress responses. If any of these coregulated Cpx regulon members were involved in conferring resistance to AGAs or HU, then activation of the other stress response involved in their regulation should also cause resistance. The two envelope stress responses that coregulate Cpx regulon members are σE and BaeRS. As with cpxA*, mutations that cause constitutive activation of the σE and BaeRS stress responses have been isolated. The activity of σE is controlled by the anti-σ factor RseA. Degradation of RseA frees σE to become active, so null mutations in rseA cause constitutive activity of σE (26, 27). The BaeRS response can be activated by a gain-of-function mutation baeS::Tn10-cam (21). However, upregulation of neither σE nor BaeRS led to an increase in resistance to HU or AGAs (Fig. 4). This demonstrates that the increased resistance to HU and AGAs conferred by cpxA* is specific to the Cpx response. It also suggests that the Cpx regulon member responsible for conferring resistance is not coregulated by the σE or Bae stress response systems.
Fig 4.

Activation of the σE or BaeRS stress responses does not confer resistance to HU or AGAs. EOP assays were performed on strains MC4100, PTM163 (cpxA*), PTM472 (rseA with ochre stop codon [rseA(Oc)]), and PTM467 (baeS::Tn10-cam). rseA null mutations such as rseA(Oc) activate the σE stress response to its highest level. Similarly, a Tn10-cam insertion in baeS activates the Bae response. In neither case, does activation of these envelope stress response systems increase resistance to HU or AGAs.
Having clarified the relationship between Cpx activation and antibiotic resistance, we sought to identify the Cpx regulon member(s) responsible for resistance to HU and AGAs. Using a candidate approach, we individually constructed null mutations in published Cpx regulon members in both wild-type and cpxA* strain backgrounds (Table 2). None of the mutations in the regulon members upregulated by the Cpx response decreased the resistance conferred by cpxA*. Similarly, disrupting genes that are known to be downregulated by the Cpx response in a wild-type background did not increase resistance to HU or AGAs. This seems to suggest that no single known regulon member is necessary to confer resistance to HU and AGAs, although it is possible that one regulon member may be sufficient.
Table 2.
Cpx regulon members tested for a role in resistance to AGAs and HUa
| Cpx regulon members tested |
|---|
| Upregulated genes |
| cpxP |
| rdoA-dsbA |
| psd |
| ydeH |
| ppiD |
| ycfS |
| yccA |
| spy |
| ybaJ |
| ung |
| yebE |
| yqjA |
| ppiA |
| ydeH |
| htpX |
| ftnB |
| degP |
| ompC |
| acrD |
| secA |
| Downregulated genes |
| efeU |
| mdtABCD |
| ompF |
| tsr |
| aroK |
| aer |
| csgDEFG |
| glpABC |
The candidate list was derived from a previously published report (14).
The Cpx response confers resistance to some, but not all, bactericidal antibiotics.
It has been suggested that all bactericidal antibiotics kill via the same mechanism (namely, the generation of ROS) to which the activation of the Cpx response contributes. Accordingly, we sought to determine whether activation of Cpx confers resistance to other antimicrobial compounds. Although cpxA* confers resistance to HU and AGAs, this mutation does not increase resistance to fluoroquinolones, such as norfloxacin, or β-lactams, such as ampicillin (Fig. 5). Thus, the Cpx response confers resistance to a subset of toxic molecules and antibiotics, not to all bactericidal antibiotics. The fact that activation of the Cpx response causes resistance only to a subset of bactericidal antibiotics suggests that this class of antibiotic may not kill bacteria via a common mechanism.
Fig 5.
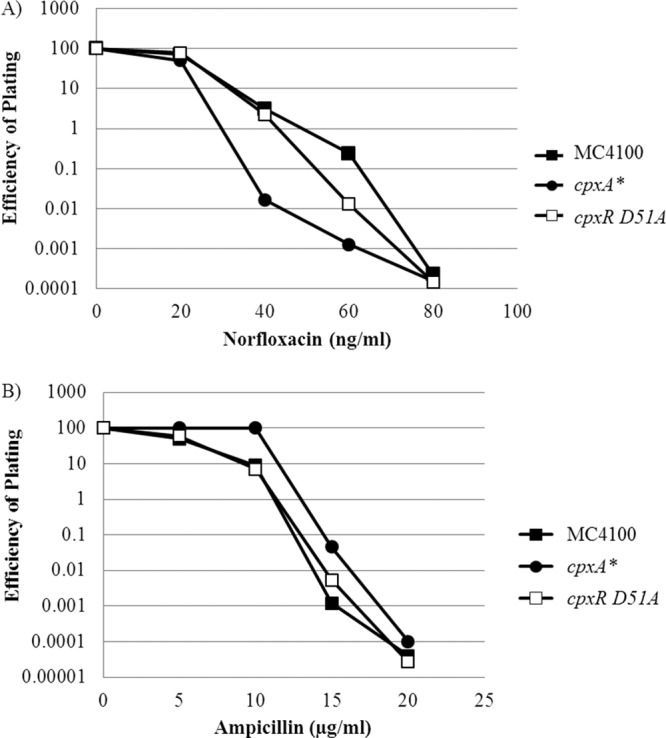
cpxA* causes resistance to only certain classes of bactericidal antibiotics. (A and B) Sensitivity of strains MC4100, PTM163 (cpxA*), and PTM198 (cpxR D51A) to fluoroquinolones such as norfloxacin (A) or β-lactams such as ampicillin (B) was performed as described in the legend to Fig. 1.
DISCUSSION
We describe a novel role for the Cpx two-component regulatory system in combating the stress caused by AGAs and the small toxic molecule HU. Although it has been known for years that cpxA* mutations confer resistance to amikacin (19), this phenotype was regarded as a consequence of the pleiotropic effects caused by hyperphosphorylation of the response regulator CpxR. Our results suggest that resistance to these toxic small molecules is directly mediated by the Cpx response. Our results also indicate that the combined actions of multiple Cpx regulon members are required to confer resistance to these toxic agents, as no single documented regulon member is epistatic to cpxA*.
The Cpx response was first characterized as an envelope stress response triggered by misfolded or mistargeted outer membrane proteins. A processing-defective mutant of the maltoporin, LamBA23D (18) and a toxic “tribrid” fusion of LamB-LacZ-PhoA (18, 28) both cause extracytoplasmic toxicity that is suppressed by cpxA*. The nature of the toxicity caused by LamB-LacZ-PhoA is unclear. However, LamBA23D is tethered to the inner membrane (IM) by its uncleaved signal sequence, and it is likely that it alters the IM in some deleterious way. Recent work has reinforced the likelihood that the Cpx response both senses and alleviates problems at the IM (4, 29, 30), and we suggest that this may be the primary role of this envelope stress response. In support of this idea, several known Cpx regulon members involved in protein quality control are located at the IM (4, 14, 29, 30).
A role for the Cpx two-component system in AGA resistance is consistent both with a role for this stress response in combating IM toxicity and with the current view that AGAs kill by promoting incorporation of mistranslated proteins into (and the consequent perturbation of) this compartment (2). It seems likely that the Cpx response combats the problem by promoting the degradation of mistranslated IM proteins. However, we do not yet understand how these aberrant proteins are selectively recognized, and we have not identified the protease that degrades them.
A role for Cpx in conferring resistance to HU is much less clear. The target of HU is class I ribonucleotide reductase (31, 32). Inhibition of this enzyme causes a fatal stalling of the replication fork owing to a lack of available deoxyribonucleotide triphosphate substrates (31, 32). It is not immediately obvious how a block to DNA replication could cause fatal problems at the IM. One theory posits that all bactericidal antibiotics, including AGAs, and toxic molecules like HU lead to IM perturbations that result in the generation of reactive oxygen species (ROS), which in turn cause cell death by damaging cellular macromolecules (5, 7). This theory predicts that a stress response that effectively combats these IM perturbations would confer some level of resistance to all bactericidal antibiotics and relevant toxic small molecules. However, we find that this is not the case. The Cpx response specifically confers resistance to AGAs and HU, but not to all bactericidal antibiotics. For example, the cpxA* mutation does not confer detectable resistance to either fluoroquinolones or β-lactams. This is true even though the fluoroquinolones, like HU, block DNA replication. Accordingly, we think it unlikely that all bactericidal antibiotics cause cell death by a similar, ROS-related mechanism.
While activation of the Cpx response confers resistance to AGAs and HU, wild-type cells with an intact Cpx response remain sensitive to both. Why are wild-type cells not protected by this stress response? In particular, AGAs like gentamicin induce the Cpx response (3), yet this induction does not render wild-type cells resistant. We suggest that this disparity reflects a race between Cpx induction and cell killing, both of which are caused by these lethal agents. The harmful effects of these compounds reach lethal levels before Cpx induction reaches a sufficient level to confer resistance. In cpxA* mutants, the continuous presence of high levels of the stress combative proteins prevent the damage caused by these toxic compounds from reaching lethal levels.
Both null mutations in cpxA and constitutively active mutations of cpxA are sufficient to confer resistance to AGAs and HU. The phenotypes of the cpxA null mutant, the cpxA* constitutively active mutant, and the cpxR null mutants underscore the delicacy required when analyzing two-component signaling systems. Archetypical two-component signal transduction systems are made up of a sensor histidine kinase and a cognate response regulator. Most of these histidine kinases are multifunctional, acting not only as kinases and autokinases but also as phosphatases. In the absence of an activating signal that stimulates autophosphorylation, sensor kinases act as phosphatases to maintain the proper balance of phosphorylated and unphosphorylated response regulator species (33, 34). Given the dual function of many sensor kinases as phosphatases, analyzing only the phenotypes of null mutations in the gene for the histidine kinase may not provide an accurate picture of the impact of inactivating a given two-component system. In order to understand the full effect of disrupting a two-component signal transduction system, null mutations of both the histidine kinase and response regulator should be analyzed in concert.
ACKNOWLEDGMENTS
We thank members of the Silhavy lab for helpful comments and discussions.
Footnotes
Published ahead of print 18 January 2013
REFERENCES
- 1. Anand N, Davis BD, Armitage AK. 1960. Uptake of streptomycin by Escherichia coli. Nature 185:23–24 [DOI] [PubMed] [Google Scholar]
- 2. Davis BD, Chen LL, Tai PC. 1986. Misread protein creates membrane channels: an essential step in the bacterial action of aminoglycosides. Proc. Natl. Acad. Sci. U. S. A. 83:6164–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kashyap DR, Wang M, Liu L-H, Boons G-J, Gupta D, Dziarski R. 2011. Peptidoglycan recognition proteins kill bacteria by activating protein-sensing two-component systems. Nat. Med. 17:676–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shimohata N, Nagamori S, Akiyama Y, Kaback HR, Ito K. 2007. SecY alterations that impair membrane protein folding and generate a membrane stress. J. Cell Biol. 176:307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kohanski MA, Dwyer DJ, Collins JJ. 2010. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 8:423–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Davies BW, Kohanski MA, Simmons LA, Winkler JA, Collins JJ, Walker GC. 2009. Hydroxyurea induces hydroxyl radical-mediated cell death in Escherichia coli. Mol. Cell 36:845–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. 2007. A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130:797–810 [DOI] [PubMed] [Google Scholar]
- 8. Raivio TL, Silhavy TJ. 1997. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J. Bacteriol. 179:7724–7733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pogliano J, Lynch AS, Belin D, Lin ECC, Beckwith J. 1997. Regulation of Escherichia coli cell envelope proteins involved in protein folding and degradation by the Cpx two-component system. Genes Dev. 11:1169–1182 [DOI] [PubMed] [Google Scholar]
- 10. Vogt SL, Raivio TL. 2012. Just scratching the surface: an expanding view of the Cpx envelope stress response. FEMS Microbiol. Lett. 326:2–11 [DOI] [PubMed] [Google Scholar]
- 11. Raivio TL, Silhavy TJ. 2001. Periplasmic stress and ECF sigma factors. Annu. Rev. Microbiol. 55:591–624 [DOI] [PubMed] [Google Scholar]
- 12. Yamamoto K, Ishihama A. 2005. Transcriptional response of Escherichia coli to external copper. Mol. Microbiol. 56:215–227 [DOI] [PubMed] [Google Scholar]
- 13. DiGiuseppe PA, Silhavy TJ. 2003. Signal detection and target gene induction by the CpxRA two-component system. J. Bacteriol. 185:2432–2440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J. Bacteriol. 191:1798–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Danese PN, Snyder WB, Cosma CL, Davis LJB, Silhavy TJ. 1995. The Cpx 2-component signal-transduction pathway of Escherichia coli regulates transcription of the gene specifying the stress-inducible periplasmic protease, DegP. Genes Dev. 9:387–398 [DOI] [PubMed] [Google Scholar]
- 16. Raivio TL, Popkin DL, Silhavy TJ. 1999. The Cpx envelope stress response is controlled by amplification and feedback inhibition. J. Bacteriol. 181:5263–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snyder WB, Davis LJB, Danese PN, Cosma CL, Silhavy TJ. 1995. Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J. Bacteriol. 177:4216–4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cosma CL, Danese PN, Carlson JH, Silhavy TJ, Snyder WB. 1995. Mutational activation of the Cpx signal transduction pathway of Escherichia coli suppresses the toxicity conferred by certain envelope associated stresses. Mol. Microbiol. 18:491–505 [DOI] [PubMed] [Google Scholar]
- 19. Rainwater S, Silverman PM. 1990. The Cpx proteins of Escherichia coli K-12: evidence that cpxA, ecfB, ssd, and eup mutations all identify the same gene. J. Bacteriol. 172:2456–2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2:2006.0008. doi:10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raffa RG, Raivio TL. 2002. A third envelope stress signal transduction pathway in Escherichia coli. Mol. Microbiol. 45:1599–1611 [DOI] [PubMed] [Google Scholar]
- 22. Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 23. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kohanski MA, Dwyer DJ, Wierzbowski J, Cottarel G, Collins JJ. 2008. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135:679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Wulf P, Lin ECC. 2000. Cpx two-component signal transduction in Escherichia coli: excessive CpxR-P levels underlie CpxA* phenotypes. J. Bacteriol. 182:1423–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dartigalongue C, Missiakas D, Raina S. 2001. Characterization of the Escherichia coli sigma(E) regulon. J. Biol. Chem. 276:20866–20875 [DOI] [PubMed] [Google Scholar]
- 27. Ruiz N, Silhavy TJ. 2005. Sensing external stress: watchdogs of the Escherichia coli cell envelope. Curr. Opin. Microbiol. 8:122–126 [DOI] [PubMed] [Google Scholar]
- 28. Snyder WB, Silhavy TJ. 1995. Beta-galactosidase is inactivated by intermolecular disulfide bonds and is toxic when secreted to the periplasm of Escherichia coli. J. Bacteriol. 177:953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shimohata N, Chiba S, Saikawa N, Ito K, Akiyama Y. 2002. The Cpx stress response system of Escherichia coli senses plasma membrane proteins and controls HtpX, a membrane protease with a cytosolic active site. Genes Cells 7:653–662 [DOI] [PubMed] [Google Scholar]
- 30. van Stelten J, Silva F, Belin D, Silhavy TJ. 2009. Effects of antibiotics and a proto-oncogene homolog on destruction of protein translocator SecY. Science 325:753–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rosenkranz HS, Winshell EB, Mednis A, Carr HS, Ellner CJ. 1967. Studies with hydroxyurea. VII. Hydroxyurea and synthesis of functional proteins. J. Bacteriol. 94:1025–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sinha NK, Snustad DP. 1972. Mechanism of inhibition of deoxyribonucleic acid synthesis in Escherichia coli by hydroxyurea. J. Bacteriol. 112:1321–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russo FD, Silhavy TJ. 1993. The essential tension: opposed reactions in bacterial two-component regulatory systems. Trends Microbiol. 1:306–310 [DOI] [PubMed] [Google Scholar]
- 34. Capra EJ, Laub MT. 2012. Evolution of two-component signal transduction systems. Annu. Rev. Microbiol. 66:325–347 [DOI] [PMC free article] [PubMed] [Google Scholar]