

Abstract
The major outer sheath protein (Msp) is a primary virulence determinant in Treponema denticola, as well as the parental ortholog for the Treponema pallidum repeat (Tpr) family in the syphilis spirochete. The Conserved Domain Database (CDD) server revealed that Msp contains two conserved domains, major outer sheath proteinN (MOSPN) and MOSPC, spanning residues 77 to 286 and 332 to 543, respectively, within the N- and C-terminal regions of the protein. Circular dichroism (CD) spectroscopy, Triton X-114 (TX-114) phase partitioning, and liposome incorporation demonstrated that full-length, recombinant Msp (MspFl) and a recombinant protein containing MOSPC, but not MOSPN, form amphiphilic, β-sheet-rich structures with channel-forming activity. Immunofluorescence analysis of intact T. denticola revealed that only MOSPC contains surface-exposed epitopes. Data obtained using proteinase K accessibility, TX-114 phase partitioning, and cell fractionation revealed that Msp exists as distinct OM-integrated and periplasmic trimers. MspFl folded in Tris buffer contained slightly less β-sheet structure than detergent-folded MspFl; both forms, however, partitioned into the TX-114 detergent-enriched phase. CDD analysis of the nine Tpr paralogs predicted to be outer membrane proteins (OMPs) revealed that seven have an Msp-like bipartite structure; phylogenetic analysis revealed that the MOSPN and MOSPC domains of Msp are most closely related to those of TprK. Based upon our collective results, we propose a model whereby a newly exported, partially folded intermediate can be either processed for OM insertion by the β-barrel assembly machinery (BAM) or remain periplasmic, ultimately forming a stable, water-soluble trimer. Extrapolated to T. pallidum, our model enables us to explain how individual Tprs can localize to either the periplasmic (e.g., TprK) or OM (e.g., TprC) compartments.
INTRODUCTION
The cultivatable oral spirochete Treponema denticola is the best characterized of the approximately 60 treponemal phylotypes within the complex polymicrobial consortium associated with periodontal disease (1–3), a chronic inflammatory condition of the gingiva that causes bone resorption and tooth loss (4, 5). Reports ascribing a plethora of biological activities to the major outer sheath protein (Msp/TDE0405) of T. denticola have established this prominent, outer membrane (OM)-associated polypeptide as one of the bacterium's principal virulence determinants (3). In addition to forming large, depolarizing channels in artificial and HeLa cell membranes (6, 7), Msp binds to extracellular matrix components and cell adhesion molecules (8–10, 64), induces cytopathic effects in epithelial cells (11), perturbs cytoskeletal actin dynamics and intracellular calcium flux in fibroblasts (12, 13), inhibits neutrophil chemotaxis (14, 15), and activates neutrophils, inducing them to release matrix metalloproteases and other tissue-damaging enzymes (16, 17). Msp also forms detergent-stable trimers that physically associate with dentilisin (11, 18, 19), a protease complex that degrades extracellular matrix and disrupts intercellular junctions (20, 21). Notably, despite the considerable attention devoted to Msp over the years, very little is known about its structure and membrane topology.
Treponema pallidum is the agent of venereal syphilis, a multistage, sexually transmitted disease renowned for its protean clinical manifestations and protracted natural history (22). An obligate human parasite (23, 24), T. pallidum is believed to have evolved by reductive evolution from commensal treponemes such as T. denticola (25, 26). Since the discovery of rare outer membrane proteins (OMPs) more than 2 decades ago (27, 28), efforts to identify these elusive molecules have been hampered by many factors, including their low abundance, their lack of sequence relatedness to prototypical OMPs in other diderms, the lability of the treponemal OM during experimental manipulation, and the syphilis spirochete's refractoriness to in vitro cultivation (24, 29–34). To circumvent these limitations, we devised a novel computational approach (35) based on the assumption that rare OMPs form β-barrels as in other diderms (36–38). Among the spirochete's predicted OMPs were 9 members of the T. pallidum repeat (Tpr) family (35), paralogs initially suspected of being OM-spanning proteins because of their sequence relatedness to Msp (34, 39). Recently, we reported that TprC/D (TP0117/TP0131; here referred to as TprC), the highest ranked Tpr candidate, meets criteria as a bona fide rare OMP: low copy number, extensive β-sheet secondary structure, amphiphilicity, and surface exposure (40). Additionally, TprC is trimeric, forms pores, and, as predicted by the Conserved Domain Database (CDD) server (54), possesses a two-domain architecture in which the C-terminal domain is responsible for β-barrel and pore formation (40). Also among the candidates, though in the lowest-ranked cluster, was TprK, the most extensively studied Tpr family member. A number of reports have contended that TprK is an OMP that undergoes sequence and antigenic variation and facilitates immune evasion by T. pallidum (24, 33, 39, 42, 43). In our hands, however, TprK from the Nichols strain is periplasmic, lacks amphiphilicity, and does not display sequence variability in the rabbit model of experimental syphilis (35, 44).
We reasoned that detailed structural, physicochemical, and topological analysis of Msp would enhance our understanding of its many virulence-related properties in T. denticola and, as the parental ortholog, yield information applicable to the entire Tpr family in T. pallidum. Herein, we show that Msp, like TprC, has a bipartite architecture; importantly, only the C-terminal domain is surface exposed in T. denticola and able to form a β-barrel that can insert into liposomes and form channels. Consistent with immunolabeling studies localizing Msp to both the OM and periplasm of T. denticola (19, 45), we show that the native protein exists as OM-integrated and periplasmic trimers with distinct physical properties. Based upon our collective results, we propose a model whereby a newly exported, partially folded intermediate can be either processed for OM insertion by the β-barrel assembly machinery (BAM) or remain periplasmic, ultimately forming a stable, water-soluble trimer. Extrapolated to T. pallidum, our model enables us to explain how individual Tprs can localize to either the periplasmic (e.g., TprK) or OM (e.g., TprC) compartments in T. pallidum.
MATERIALS AND METHODS
Ethics statement.
Animal protocols described in this work strictly follow the recommendations of the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the University of Connecticut Health Center Animal Care Committee under the auspices of Animal Welfare Assurance A347-01.
Propagation of Treponema denticola.
T. denticola ATCC 35405 was grown in new oral spirochete (NOS) broth (8) supplemented with 10% heat-inactivated normal rabbit serum using the GasPak Plus anaerobic system (Becton, Dickinson, Cockeysville, MD).
Bioinformatics analysis.
Multiple sequence alignments and phylogenetic analysis were performed in ClustalX (46) using sequences in the NCBI database. The NCBI conserved domain database (CDD) server (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (54) was used to identify conserved domains within Msp and the Tprs.
Cloning of msp and troA genes.
DNAs encoding full-length Msp/TDE0405 (without signal sequence) (MspFl; accession number NP_971019) and TroA/TDE1226 (accession number NP_971833) were PCR amplified from T. denticola 35405 genomic DNA using the primers listed in Table 1. The resulting amplicons were cloned, respectively, into the EcoRI/HindIII and BamHI/XhoI restriction sites of the expression vector pET23b (Novagen, San Diego, CA). DNAs encoding the major outer sheath proteinN (MOSPN) and MOSPC domains within the N- and C-terminal regions of Msp (MspN and MspC, respectively) were amplified from the pET23b plasmid harboring full-length msp by using the primers listed in Table 1 and cloned into the EcoRI and HindIII restriction sites of pET23b (Novagen, San Diego, CA). All constructs were confirmed by nucleotide sequencing.
Table 1.
Primers used in this study
| Primer designation | Sequence (5′–3′)a | T. denticola genome coordinates |
|---|---|---|
| MspFL (5′) | GATAGAATTCTATGGCAAAGGCTTCTGTAAACT | 455661–455684 |
| MspFL (3′) | GATCAAGCTTGTAGATAACTTTAACACCGA | 457238–457218 |
| MspN (5′) | GATAGAATTCTATGAACTTTGATCTTGGTGTAA | 455838–455860 |
| MspN (3′) | GATAAAGCTTTAAATCAAGTCCGAAA | 456468–456452 |
| MspC (5′) | GATAGAATTCTATGGGATTGAAACTCGGATCA | 456603–456624 |
| MspC (3′) | GATCAAGCTTGTAGATAACTTTAACACCGA | 457238–457218 |
| TroA (5′) | GATAGGATCCTATGACAAAAAAATTAATAGTCTTA | 1260573–1260549 |
| TroA (3′) | GATACTCGAGTTTTTTTAAGGCATCTATTATCG | 1259632–1259655 |
Underlined sequences indicate restriction sites.
Expression, purification, and folding of recombinant proteins.
Expression, purification, and folding of Escherichia coli OmpG (47) and OmpF (48) were described previously. MspFl, MspN, and MspC were expressed in the E. coli C41(DE3) strain (Agilent Technologies, Inc., Santa Clara, CA) and purified and folded essentially as described previously for full-length TprC and recombinants containing the TprC MOSPN and MOSPC domains (40). To produce water-soluble Msp, denatured, purified protein was dialyzed for 8 h at 4°C against 2 liters of 10 mM Tris buffer, pH 8.0, with one buffer change.
T. denticola TroA was expressed in the Rosetta-gami (DE3) strain (Agilent Technologies, Inc., Santa Clara, CA). For purification, the harvested cell pellet was resuspended with 20 ml of 50 mM Tris (pH 7.5), 300 mM NaCl, 10 mM imidazole, 10% glycerol, 100 μg of lysozyme, and 100 μl of PIC and stored at −20°C. After thawing, the bacterial suspension was lysed by sonication for three 30-s pulses interspersed with 30 s of rest on ice. The supernatants were cleared of cellular debris by centrifugation at 18,000 × g for 20 min at 4°C and applied to a Superflow Ni-nitrilotriacetic acid (NTA) (Qiagen, Valencia, CA) immobilized metal-affinity chromatography (IMAC) column, which had been equilibrated with buffer A (50 mM Tris [pH 7.5], 300 mM NaCl, 10% glycerol). The protein was eluted with buffer A supplemented with 250 mM imidazole. Fractions containing the protein were concentrated using an Amicon-Ultra concentrator (Millipore, Billerica, MA) with a nominal molecular mass cutoff 10 kDa and dialyzed into phosphate buffer saline (PBS).
Protein concentrations were determined by measuring A280 in 20 mM sodium phosphate (pH 6.5) and 6 M guanidine hydrochloride (49). Molar extinction coefficients (M−1 cm−1) were calculated using the ProtParam tool provided by the ExPASy proteomics server (50).
Far-UV circular dichroism spectroscopy.
Far-UV circular dichroism (CD) spectroscopy was performed using a JASCO J-715 spectral polarimeter as described previously (40).
Tryptophan fluorescence.
Spectra were obtained using a Hitachi F-2500 fluorescence spectrometer as described previously (40).
Fractionation of T. denticola and assessment of the oligomeric state of native Msp.
To generate membrane and soluble fractions from freshly harvested T. denticola, 2 × 108 organisms were resuspended in Tris buffer (20 mM Tris-HCl [pH 8.0], 100 mM NaCl) and disrupted by sonication for three 20-s pulses interspersed with 30 s of rest on ice. Total membrane and soluble fractions were obtained by centrifugation at 100,000 × g for 45 min. The fractions were subjected to TX-114 phase partitioning or SDS-PAGE with or without boiling to assess the oligomeric state of native Msp (see below). For the latter, portions of each fraction were solubilized in 1× Laemmli sample buffer (SB) (Bio-Rad, Hercules, CA); one of each was boiled for 10 min. Subsequently, the fractions were resolved by SDS-PAGE, and the gel, including the stack, was transferred to nitrocellulose membranes (0.45 μM pore size, GE Healthcare) at 25 V for 25 min using a semidry apparatus (Bio-Rad). Membranes were blocked for 1 h with PBS, 5% nonfat dry milk, 5% fetal bovine serum, and 0.1% Tween 20 and probed overnight at 4°C with primary antibodies directed against the Msp at a dilution of 1:10,000. After being washed with PBS and 0.05% Tween 20 (PBST), the membranes were incubated for 1 h at 4°C with a horseradish peroxidase (HRP)-conjugated goat anti-rat antibody (Southern Biotech, Birmingham, AL) at a dilution of 1:30,000. Following washes with PBST, the immunoblots were developed using the SuperSignal West Pico chemiluminescent substrate (Thermo Fisher Scientific).
Triton X-114 phase partitioning.
Triton X-114 (TX-114) phase partitioning was performed with 10 μg of recombinant protein or ∼2 × 106 T. denticola cells using standard protocols (35, 51). For protease-treated samples, treponemes were incubated with 100 μg of proteinase K (PK) for 1 h at 37°C prior to phase partitioning.
Blue-native PAGE.
Blue-native (BN) PAGE of MspFl was performed as described previously (40).
Preparation of liposomes.
Liposomes were prepared as previously described (40).
Liposome floatation assay.
Liposome floatation assays were performed as previously described (40).
Pore formation assay.
Pore formation assays were performed using large unilamellar vesicles (LUVs) loaded with the fluorophore Tb(DPA)33− as previously described (40, 48).
Immunologic reagents.
Rat polyclonal antisera directed against Msp and isolated T. denticola endoflagellar filaments were described previously (45). Rat antisera directed against MspN, MspC, and TroA were generated in female Sprague-Dawley rats by intraperitoneal injection with 30 μg of purified protein in a 1:1 mixture of PBS (pH 7.4) and complete Freund's adjuvant; 4 and 6 weeks later, animals received 15-μg booster doses in a 1:1 mixture of PBS and incomplete Freund's adjuvant. Hybridoma clones HIS-1, a murine monoclonal antibody specific for poly-histidine tags, was obtained from Sigma-Aldrich.
Quantitative immunoblot analysis.
The number of copies of Msp (molecular mass of 55,227 Da) per T. denticola cell was determined by quantitative immunoblotting as previously described (40, 47).
Accessibility of native Msp to surface proteolysis.
The accessibility of Msp to proteolysis in intact treponemes was assessed as recently described for TprC and TP0326 in T. pallidum (40, 47). Immunoblotting was performed using antisera against MspN and MspC.
Localization of MspN and MspC in T. denticola by gel microdroplet immunofluorescence assay.
Mid-logarithmic-phase T. denticola cells were encapsulated in microdroplets of low-melting-point agarose as previously described (52, 53). Encapsulated organisms were probed in a two-step process. In the first step, rat antiserum directed against MspN, MspC, or endoflagella (each diluted 1:200) was added to the bead suspensions (0.2 to 0.3 ml) in the presence or absence of 0.05% (vol/vol) Triton X-100. In the second step, encapsulated organisms were fixed with 4% (vol/vol) paraformaldehyde and extensively washed with PBS and NOS broth prior to the addition of primary antibody. Samples were incubated with gentle mixing in a 34°C water bath for 2 h. The beads then were washed three times by low-speed centrifugation (100 × g) and resuspended in NOS broth followed by incubation for 1 h at 34°C with 1 μg/ml of goat anti-rat Alexa Fluor 488 or Alexa Fluor 594 conjugates (Invitrogen). The beads then were washed three times with NOS broth and observed with a epifluorescent Olympus BX-41 microscope using a 100× (1.4-numerical-aperture [NA]) oil immersion objective equipped with a Retiga Exi charge-coupled-device (CCD) camera (Q Imaging, Tucson, AZ) and fluorescein isothiocyanate (FITC) and rhodamine omega filter sets. For each sample, three slides were prepared and approximately 100 organisms were scored for labeling.
RESULTS
Recombinant Msp has a bipartite architecture.
We began by using the NCBI's CDD server (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (54) to query the Msp sequence for annotated domains. This analysis revealed potential functional units spanning residues 77 to 286 (MOSPN) and 332 to 543 (MOSPC) within the protein's N- and C-terminal regions (Fig. 1A). Based on our recent studies with the T. pallidum paralog TprC (40), which also contains these domains, we conjectured that only MOSPC would be capable of folding into a β-barrel. To test this idea, we used far-UV CD spectroscopy to assess the secondary structures of folded, full-length Msp (MspFl) and recombinant proteins containing the two domains (designated MspN and MspC, respectively). For these experiments and unless otherwise stated, both MspFl and MspC were folded in buffer containing the detergent DDM (n-dodecyl β-d-maltoside) as described in Materials and Methods; folding was monitored by tryptophan fluorescence. The CD spectra and deconvoluted data are shown in Fig. 1B and Table 2, respectively. MspFl displayed broad minima centering on 218 nm, indicating a preponderance of β-structure, as did the E. coli OmpG control, a 14-stranded β-barrel (55, 56). MspN and MspC displayed clearly disparate CD spectra; the former was a mixture of α-helix and β-sheet, whereas the latter consisted predominantly of β-sheet with very little α-helix.
Fig 1.

(A) Domain structure of Msp predicted by the CDD server (54). (B) CD spectra of MspFl, E. coli OmpG, MspN, and MspC. MspFl (5 μM) and MspC (5 μM) were folded in DDM buffer; OmpG (5 μM) was folded in 50 mM NaCl, 10 mM Tris (pH 7.5), and 0.2% n-octyl-β-d-glucopyranoside, and MspN (5 μM) was folded in 50 mM Tris (pH 7.5) 50 mM NaCl.
Table 2.
Secondary structure of Msp constructs and E. coli OmpG determined by CD spectroscopy
| Protein | β-Sheet (%) | α-Helix (%) | Random coil (%) |
|---|---|---|---|
| MspFl-folded in DDM | 50.7 | 18.6 | 30.7 |
| MspN | 32.7 | 25.2 | 42.1 |
| MspC | 58.6 | 4.5 | 36.9 |
| OmpG | 54.5 | 4.5 | 36.9 |
| MspFl-folded in Tris | 40.8 | 20.6 | 38.6 |
MspFl and MspC, but not MspN, are amphiphilic and form channels in large unilamellar vessels.
We next used TX-114 phase partitioning to examine which, if any, of the three recombinant proteins possess the amphiphilic character typical of an OM-spanning protein. As shown in Fig. 2A, both MspFl and MspC partitioned exclusively into the detergent-enriched phase, whereas MspN was hydrophilic. To extend these results, we examined the ability of the three proteins to incorporate into liposomes. Following separation on discontinuous sucrose gradients, MspFl and MspC were recovered from the liposome-containing top fractions, whereas MspN sedimented in the bottom fraction containing unincorporated material (Fig. 2B). Msp has been noted to form large pores in both artificial and HeLa cell membranes (6, 7). Consistent with these results, we found that MspFl promoted significantly greater efflux of the small (10-Å, 650-Da) fluorophore Tb(DPA)33− from LUVs than did E. coli OmpF, an archetypal porin (57) (Fig. 2C). Importantly, MspC displayed even greater channel-forming activity than MspFl, while pore formation by MspN was negligible (Fig. 2C). β-Barrel-forming proteins characteristically retain a high degree of β-sheet content when solubilized in SDS at room temperature and, consequently, run with lower apparent molecular masses by SDS-PAGE than when denatured by boiling; this property is termed heat modifiability (58, 59). Surprisingly, none of the recombinant proteins exhibited this property (data not shown).
Fig 2.

MspFl and MspC, but not MspN, are amphiphilic and possess channel-forming activity. (A) A total of 10 μg each of MspFl, MspN, and MspC was phase partitioned in TX-114 and stained with GelCode Blue (Thermo Scientific) following SDS-PAGE. Molecular mass standards (kDa) are indicated on the right. (B) Liposomes were reconstituted with 10 μg each of MspFl, MspN, and MspC followed by sucrose density gradient ultracentrifugation. Following SDS-PAGE, fractions were subjected to immunoblotting with antisera directed against MspFl. Lanes: top fractions (TF) contain liposome-incorporated material, whereas middle and bottom fractions (MF and BF, respectively) contain unincorporated material. Molecular mass standards (kDa) are indicated on the right. (C) Quenching of Tb(DPA)33− encapsulated in LUVs following incubation (30 min) with 100 nM E. coli OmpF, MspFl, MspN, and MspC in 50 mM Tris (pH 7.5) and 100 mM NaCl supplemented with 5 mM EDTA. Each bar represents the mean ± standard error of the mean (SEM) from three independent experiments. P values of <0.05 (Student's t test) were considered significant.
Native Msp is present in high copy number in T. denticola but only the MOSPC domain is surface exposed.
Having established the bipartite domain structure of Msp using recombinant proteins, we next turned our attention to the native polypeptide in T. denticola. Although Msp is known to be prominent in T. denticola cell lysates (18), to the best of our knowledge, its expression on a per-cell basis has never been assessed. Quantitative immunoblotting, shown in Fig. 3A, revealed that, in mid-logarithmic phase, each T. denticola cell expresses an average (mean from three independent experiments) of 2 × 105 copies of the protein; of note, this value is more than 1,000-fold greater than the per-cell copy number of the T. pallidum ortholog TprC (40).
Fig 3.

Msp is expressed in extremely high copy number in T. denticola, but only MspC is surface exposed. (A) Quantitative immunoblot analysis of Msp expressed in T. denticola. T. denticola lysates (2.0 × 106 organisms) were immunoblotted with anti-MspFl antiserum; a standard curve generated from densitometric values obtained for graded amounts of MspFl was used to determine the copy number of Msp per cell. Molecular mass standards (kDa) are indicated on the left. (B) Intact T. denticola or organisms treated with TX-100 (0.05%) were encapsulated in gel microdroplets and probed with rat antisera against MspN, MspC, or anti-T. denticola flagella (45). Antibody binding was detected with goat anti-rat Alexa Fluor 488 (green) or Alexa Fluor 594 (red) conjugates.
The structural data derived from the recombinant proteins leads to the prediction that MOSPC, the β-barrel domain, has surface-exposed epitopes, while the hydrophilic MOSPN domain is periplasmic. To test this conjecture, we first generated rat antisera against MspN and MspC; preliminary immunoblots revealed that the two antisera had comparable sensitivities and were capable of detecting subnanogram quantities of MspFl (data not shown). To establish the cellular location of the two domains, we performed indirect immunofluorescence analysis of treponemes encapsulated in agarose beads (gel microdroplets) (35, 45, 52, 53). This method is advantageous because it not only preserves the integrity of fragile treponemal OMs during immunolabeling studies but also allows controlled removal of OMs by detergent solubilization in order to detect subsurface antigens. As shown in Fig. 3B, labeling of intact organisms was observed exclusively with anti-MspC antiserum, while labeling of detergent-treated organisms was seen with both anti-MspN and anti-MspC antisera. Also noteworthy is that 100% of the treponemal population showed surface labeling with anti-MspC antiserum. Antiflagellar antiserum, used to assess OM integrity, labeled only detergent-treated organisms, while normal rat serum failed to label either intact or detergent-treated organisms (data not shown).
Full-length forms of native Msp reside in both the outer membrane and periplasmic space.
One interpretation of the above-described immunolabeling results was that full-length forms of Msp reside in both the OM and periplasm, a possibility we considered in our previous study of Msp (45). Three complementary approaches were employed to verify this idea.
First, we tested the accessibility of Msp to proteinase K (PK) in intact organisms. The amounts of Msp detected by immunoblotting with antisera directed against either MspN or MspC (Fig. 4A and B, respectively) showed barely perceptible decreases in organisms exposed to increasing concentrations of PK. One difference in the results obtained with the two antisera was notable, however; degradation products, including an approximate 25-kDa band (very noticeable at the highest PK concentration, 50 μg/ml), were detected only with anti-MspN antibodies (compare Fig. 4A and B). Msp was fully degraded in organisms disrupted with TX-100 and lysozyme prior to treatment with PK (Fig. 4C), ruling out the possibility that the protein is intrinsically PK resistant. Furthermore, neither the flagellar sheath protein (FlaA) nor TroA (the periplasmic substrate-binding component for an ABC-type metal transporter) (60) was affected by PK treatment of intact organisms, while both were completely degraded in disrupted organisms (Fig. 4C). Organisms incubated with the highest concentration of PK retained full motility under dark-field microscopy, an additional indication that OM integrity was maintained during incubation with the enzyme.
Fig 4.
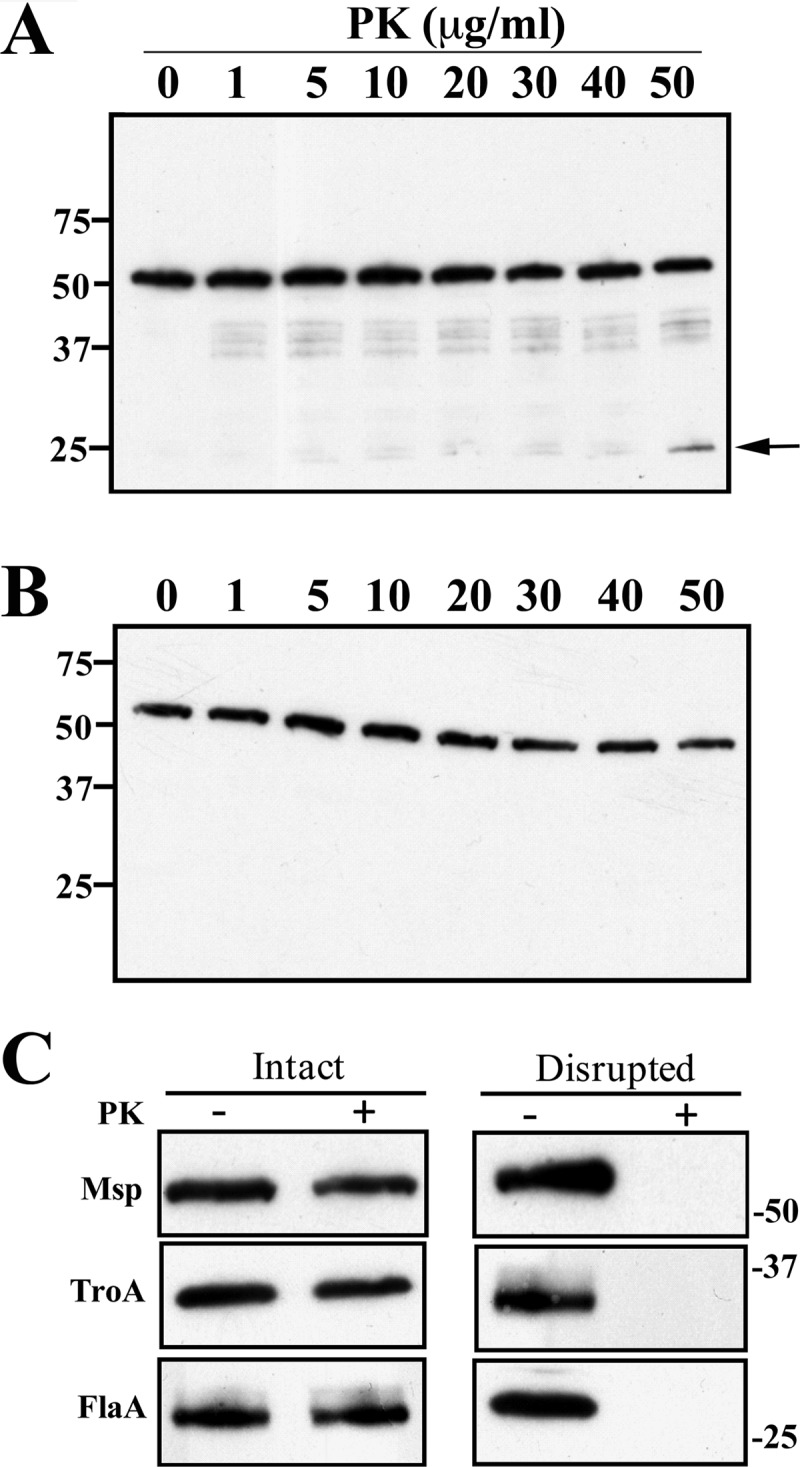
Limited accessibility of Msp to surface proteolysis in T. denticola. Immunoblot analysis of Msp, detected using anti-MspN (A) or anti-MspC (B) antiserum in motile treponemes (1.0 × 108 organisms/lane) treated for 1 h with graded concentrations of proteinase K (PK). Molecular mass standards (kDa) are shown on the left. The arrow in panel A designates the ∼25-kDa degradation product reactive with anti-MspN antiserum. (C) PK accessibility of Msp and two periplasmic controls (TroA and FlaA) in intact and detergent lysozyme-treated organisms incubated with (+) or without (−) 50 μg/ml of PK. Each lane represents 1.0 × 106 T. denticola immunoblotted with rat antisera directed against MspFl, TroA, or flagella. Molecular mass standards (kDa) are indicated on the left of panels A and B.
As a second approach, we performed TX-114 phase partitioning; here, we reasoned that full-length Msp would be detected in both aqueous and detergent-enriched phases if T. denticola expresses OM-spanning and periplasmic forms. Figure 5A shows that this, in fact, was the case. Moreover, when PK-treated organisms were phase partitioned, full-length Msp was detected only in the aqueous phase (Fig. 5B), indicating that the amphiphilic (i.e., OM-associated) form was degraded. Immunoblotting of PK-treated organisms with antibodies to the two domains yielded highly similar results but with one important difference: an approximate 25-kDa polypeptide was detected only when the aqueous phase from PK-treated organisms was reacted with anti-MspN antiserum (compare Fig. 5B and C). This fragment, therefore, has to be derived from the periplasmic (i.e., PK-inaccessible), MOSPN-containing portion of Msp.
Fig 5.

T. denticola contains both hydrophilic and amphiphilic forms of Msp, but only the latter is PK accessible in intact organisms. Motile T. denticola without (A) or with (B and C) PK treatment (50 μg/ml, 1 h) was subjected to TX-114 phase partitioning followed by SDS-PAGE and immunoblotting with antibodies to MspFl, MspN, and MspC (panels A, B, and C, respectively). The arrowhead in panel B indicates an ∼25-kDa degradation product detected only with anti-MspN antiserum. Lanes: whole cells (WC), TX-114 insoluble material (Ins), aqueous (Aq), and detergent-enriched (Det) phases. Molecular mass standards (kDa) are indicated on the left of each panel.
Third, we separated T. denticola into membrane and soluble fractions by ultracentrifugation, which then were separately phase partitioned in TX-114. Figure 6A shows that native, full-length Msp in the membrane fraction partitioned exclusively into the detergent phase, while Msp in the soluble fraction was recovered only in the aqueous phase. Native Msp forms SDS-stable trimers that dissociate upon boiling (18, 19). The SDS-PAGE gels in Fig. 6B show that Msp in both the membrane and soluble fractions is trimeric (Fig. 6B). BN-PAGE showed that MspFl also is trimeric (after subtracting ∼50-kDa DDM micelles) (Fig. 6C).
Fig 6.

T. denticola membrane and soluble fractions contain trimeric forms of Msp. (A) Membrane and soluble fractions of T. denticola (MF and SF, respectively) were phase partitioned with TX-114 followed by SDS-PAGE and immunoblot analysis using anti-MspFl antiserum. Molecular mass standards (kDa) are indicated on the right. (B) Membrane and soluble fractions were separated by SDS-PAGE without (−) and with (+) boiling followed by immunoblot analysis with anti-MspFl antiserum. Molecular mass standards (kDa) are indicated on the left. (C) BN-PAGE showing trimer formation by MspFl. Molecular mass standards (kDa) are indicated on the left.
Recombinant MspFl can fold into water- and detergent-soluble forms.
The finding that native Msp exists as both amphiphilic and hydrophilic proteins raised the question of whether it is possible to generate a water-soluble form of recombinant MspFl. As described in Materials and Methods, MspFl was folded in Tris buffer alone or in the buffer containing DDM; we found that a water-soluble form of MspFl could, indeed, be generated. Moreover, the far-UV CD spectra of the two forms of MspFl revealed that both contain a preponderance of β-sheet structure (Fig. 7A). However, MspFl folded in Tris buffer contained slightly less β-sheet and correspondingly more random coil, while the α-helical contents of the two forms were almost identical (Table 2). Strikingly, in contrast to periplasmic, native Msp, MspFl partitioned exclusively into the TX-114 detergent-enriched phase (Fig. 7B).
Fig 7.

Characterization of a water-soluble form of MspFl. (A) Far-UV CD spectra of MspFl folded in DDM (5 μM) and Tris buffer (3 μM). (B) TX-114 phase partitioning of water- and detergent-soluble forms (WS and DS, respectively) of MspFl. Molecular mass standards (kDa) are indicated on the right.
Phylogenetic relationships between Msp and Tpr proteins predicted to be T. pallidum OMPs.
Our ranked clusters of predicted T. pallidum OMPs contain nine members of the Tpr family (35). Lastly, we examined the structural and evolutionary relationships between these paralogs and Msp, which is believed to be representative of the parental ortholog (34, 39). Notably, the CDD server revealed that seven of the nine contain MOSPN and MOSPC domains, while two, TprA and TprF, contain only MOSPN domains (Fig. 8A). Phylogenetic analysis revealed that Msp is most closely related to TprK and relatively distantly related to TprC (Fig. 8B). These relationships were preserved in separate comparisons between MOSPN and MOSPC domains (data not shown).
Fig 8.

Predicted architectural and phylogenetic relationships between Msp and Tpr proteins within the OM. (A) CDD analysis of the nine Tpr proteins predicted to be OMPs (35). (B) Phylogenetic relationships between Msp and the eight Tprs. The phylogenetic analysis was carried out using the ClustalX (46) program using 10,000 bootstrap trials. The phylogenetic tree was viewed with the help of TreeView32 software. Bootstrap values (%) are indicated for the major branch points in the tree.
DISCUSSION
Theoretical considerations derived from innumerable investigations on the topology and export of integral membrane proteins (38, 57, 61–63), as well as the practical need for water-filled channels to enhance uptake of nutrients, led to the supposition that OMPs in spirochetes employ the β-barrel scaffold characteristic of OM-spanning proteins in other proteobacteria (32, 64, 65). Our recent studies with T. pallidum (35, 40) and, in this report, with T. denticola have garnered substantial evidence to validate this premise. Additionally, identification of spirochetal orthologs for BamA (47, 66), the central component of the molecular machine that catalyzes insertion of newly exported proteins into the OM bilayers, implies that β-barrel formation is essential for OM biogenesis in spirochetes as in all other diderms (36, 67). As a high-copy-number protein in a genetically manipulable spirochete, Msp has the potential to become a model system for elucidating the structural features and export pathways of a novel class of OMPs that appears to have evolved to meet the demands of the myriad microenvironments inhabited by pathogenic and commensal treponemes (2, 24, 68–70).
When the Msp sequence was published in 1996 (64), it was assumed that the entire 57-kDa polypeptide encompasses the β-barrel. Our recent examination of the T. pallidum Msp paralog TprC (40) prompted us to revisit this topologic conception. CDD analysis (54) predicted that TprC contains two conserved domains (MOSPN and MOSPC) derived from the Msp sequence, though providing no inkling as to the cellular location or function of either. With E. coli OmpA as a precedent (71–73), we expected that MOSPN would contain the integral membrane portion of TprC and that MOSPC would be periplasmic. Studies with recombinant proteins revealed, much to our surprise, that the MOSPC domain is responsible for β-barrel formation, pore-forming activity, and trimerization. We began the present study using a similar physicochemical approach to show that recombinant Msp possesses bipartite topology and that the MOSPC domain comprises the amphiphilic, pore-forming β-barrel. We then confirmed by immunofluorescence analysis and proteolysis of motile T. denticola that MOSPC is surface exposed, while MOSPN is periplasmic. These results establish unequivocally that Msp and TprC adhere to the same architectural blueprint in their native OM settings.
Over the years, numerous activities have been described for Msp (3, 6–19); our studies provide a topologic framework for assessing these claims. As one example, binding and biological assays of Msp often have employed isolated, native protein (7, 8, 10, 11, 13, 14, 64). Inasmuch as β-barrels are stably integrated into OM bilayers via a minimum of eight transmembrane domains (38, 74), thermodynamic considerations alone lead one to question the biological relevance of such experimental strategies. Interpretation of these results is complicated further by the unknown conformational state of the protein reagents used and the lack of data as to the regions of the polypeptide interacting with target cells and molecules. Edwards et al. (9) attempted to address these uncertainties by mapping the regions of Msp that mediate binding to extracellular matrix components. They reported that binding activity and surface-exposed epitopes are confined to the N-terminal half of the protein, findings that are irreconcilable with a periplasmic MOSPN domain. Topological questions, though not insurmountable ones, also arise in considering how Msp interacts with dentilisin in light of recent data showing that individual polypeptides within the protease complex reside on different sides of the OM. One function, pore formation, is readily compatible with the bipartite architecture of Msp. Based upon conductance measurements in black lipid membranes, Egli et al. (6) concluded that isolated, native Msp forms extremely large channels, estimated at 3.4 nm. While our efflux assay does not permit estimation of the size of the channel generated by MspFl or MspC, both recombinant proteins promoted significantly greater efflux of Tb(DPA)33− than did E. coli OmpF (known exclusion limit of 600 Da). Although MspC is somewhat smaller than classical porins, it is important to note that structural features, in addition to number of transmembrane segments, including the size and position of external loops, the residues lining the channel, their spatial arrangements, and the contour of the pore, greatly influence substrate specificity and conductance characteristics (73, 75–77).
In our prior examination of Msp (45), we challenged two prevailing views about the molecule. One is that it forms hexagonal arrays (6, 18). Using several electron microscopy techniques, including freeze fracture to expose the interior of the OM bilayer (78) and biochemical analysis of T. denticola detergent extracts, we concluded that the lattice-like structure thought to be Msp is actually the peptidoglycan sacculus. The other is that Msp is exclusively an OMP. Although we observed surface exposure and OM association by immunolabeling at the light and electron microscopy levels, we also detected substantial amounts of Msp within the periplasmic space (45). Recently, Godovikova et al. (19) disputed this contention; their immunofluorescence images, however, clearly show labeling of treponemes without OMs as well as intact organisms. Data obtained herein using combinations of PK accessibility, TX-114 phase partitioning, and cell fractionation collectively make a compelling argument for the existence of OM-integrated (i.e., amphiphilic) and periplasmic (i.e., hydrophilic) conformers. The combined PK/TX-114 experiments are particularly informative with respect to the bipartite architecture of the OM-integrated form of Msp and the full-length native protein's dual compartment location. If MOSPC alone comprises the β-barrel of the OM-integrated form and Msp resides in both the OM and periplasmic compartments, PK treatment of intact organisms should (i) eliminate the amphiphilic, full-length protein, generating an ∼25-kDa hydrophilic remnant consisting largely of MOSPN, and (ii) yield a full-length, hydrophilic protein, the PK-shielded periplasmic conformer. This prediction is explicitly fulfilled. Although we have not yet rigorously determined the relative proportions of the two forms, the small decrement in full-length protein and the correspondingly small amount of the 25-kDa degradation product detected following PK treatment of intact treponemes suggest that the OM conformer is the minority species.
At the time of our earlier report (45), it would not have been possible to generate a mechanistic hypothesis for the unprecedented finding that stably folded, trimeric forms of an exported protein can be integrated into the OM as a β-barrel and soluble within the periplasm. The critical implication of our results is that a newly exported precursor must enter divergent folding/localization pathways following emergence from the Sec translocon. The findings reported herein that recombinant, water-soluble MspFl (i) contains considerable secondary structure but modestly less β-sheet than its detergent-folded counterpart and (ii) becomes amphiphilic in the presence of TX-114 enable us to tentatively propose a two-pathway working model (Fig. 9) in accord with current thinking about OMP folding, protein export, and OM biogenesis in E. coli (36, 37, 67, 73, 79). We postulate that water-soluble MspFl resembles a partially folded intermediate that can either be chaperoned to the OMP assembly pathway for catalytic threading into the OM via the process known as β-augmentation (79, 80) or form a water-soluble trimeric conformer within the periplasm. What determines which of these two mutually exclusive pathways a newly exported Msp precursor takes? In E. coli, the terminal β-strand acts as a signature sequence that facilitates recognition of OMP precursors by the POTRA arm of BamA (81, 82). One possibility (depicted in Fig. 9) is that the diminished β-structure of water-soluble MspFl involves the region of the polypeptide containing the signature sequence. The folded β-barrel's lack of heat modifiability, indicating that the hydrogen bonds between N- and C-terminal β-strands that close the barrel are not as tight as in classical OMPs (38, 73, 83), provides some support for the idea. As a consequence of conformational flexibility in the region containing the signature sequence, the chaperone (presumably Skp, as T. denticola does not contain SurA) would have difficulty unloading its cargo to BamA (37, 81, 82). The resultant bottleneck would divert intermediates away from the OMP assembly pathway, creating the opportunity for stabilizing interactions among monomers that promote folding and trimerization within the periplasm. Competition for comparatively limited numbers of chaperone and POTRA binding sites also could drive intermediates toward this alternative, periplasmic pathway. Admittedly, much more experimentation, including structural comparison of the native OM-integrated and periplasmic conformers, will be needed to refine this hypothetical scenario.
Fig 9.

Proposed two-pathway model for generation of amphiphilic, OM-inserted and soluble, periplasmic Msp conformers. Msp precursor is exported across the cytoplasmic membrane (CM) by the Sec translocon. Once within the periplasm, the unfolded precursor acquires some secondary structure, becoming the “intermediate form.” The intermediate form can either be chaperoned via Skp (Tde2602) to the POTRA arm of BamA (Tde2601) for OM assembly (25) and trimerization (OMP conformer) or fold into a soluble trimeric periplasmic conformer. We hypothesize that in the intermediate form, a stretch of amino acids at the extreme C terminus of MOSPC (i.e., the signature sequence, shown in orange) containing the BamA recognition signal is unfolded but can become a β-strand once in contact with the POTRA1 domain of BamA. The resultant block to entry into the OMP assembly pathway drives the intermediate form toward the alternative, periplasmic pathway. The OMP conformer is depicted as trimerizing via the MOSPC domains based on our prior analysis of TprC (40). Presently, we have no data as to the regions of Msp that mediate trimerization of the periplasmic conformer. The components of the Bam complex in T. denticola, other than BamA, are unidentified (25) and, therefore, not shown.
An important outcome of our studies is that they validate Msp as a surrogate for conceptualizing topological, functional, and compartmental diversity within the paralogous Tpr family. The truncated Tprs that lack MOSPC domains are one example. TprA does not appear to contain a good N-terminal signal sequence (35) and, so, will not be exported. TprF, on the other hand, has a signal sequence (35) but lacks a MOSPC domain; without a MOSPC domain, we can safely predict that it resides within the periplasmic space and, thus, can be eliminated from further consideration as a candidate T. pallidum rare OMP. The bipartite TprC and TprK paralogs represent more complex cases. As noted earlier, all of the evidence we have generated to date indicates that TprC is an authentic rare OMP, while TprK is periplasmic (35, 40, 44). Msp is instructive in understanding this dichotomy. The fact that Msp exists as both OM-integrated and periplasmic forms strongly supports the contention that individual Tprs can segregate to either compartment. However, whereas Msp traverses both arms of the bifurcation pathway following export across the cytoplasmic membrane, TprC and TprK commit to one or the other. Our analysis of Msp points to the MOSPC domain as the key to this divergence in trafficking. While the MOSPC domain of TprC evidently possesses all of the requisites for recognition by the OM assembly machinery and insertion into the OM as a β-barrel, the corresponding domain in TprK must not; as a result, TprK becomes “trapped” within the periplasmic space. In the past, we have pointed out the extensive sequence differences between TprK and TprC, which include numerous residues within their MOSPC domains (35). One possible explanation for the inability of TprK to access the OM is that its MOSPC domain lacks the overall capacity to form a β-barrel. However, the relatively close phylogenetic relationship between Msp and TprK suggests an alternative explanation. As per our model for Msp trafficking, TprK might form an intermediate with considerable overall β-sheet structure (84) yet lack a signature sequence that can adopt the proper conformation for recognition by the OMP assembly machinery. As a result, it follows the alternative pathway, becoming a periplasmic constituent.
ACKNOWLEDGMENTS
This work was supported by NIH grants AI-26756 (J.D.R.) and AI-29735 (J.D.R. and M.J.C.).
Footnotes
Published ahead of print 1 March 2013
REFERENCES
- 1. Paster BJ, Boches SK, Galvin JL, Ericson RE, Lau CN, Levanos VA, Sahasrabudhe A, Dewhirst FE. 2001. Bacterial diversity in human subgingival plaque. J. Bacteriol. 183:3770–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Holt SC, Ebersole JL. 2006. The oral spirochetes: their ecology and role in the pathogenesis of periodontal disease, p 323–356 In Radolf JD, Lukehart SA. (ed), Pathogenic Treponema. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 3. Ellen RP. 2006. Virulence determinants of oral treponemes, p 357–386 In Radolf JD, Lukehart SA. (ed), Pathogenic Treponema. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 4. Amano A. 2010. Host-parasite interactions in periodontitis: microbial pathogenicity and innate immunity. Periodontol 2000 54:9–14 [DOI] [PubMed] [Google Scholar]
- 5. Hernandez M, Dutzan N, Garcia-Sesnich J, Abusleme L, Dezerega A, Silva N, Gonzalez FE, Vernal R, Sorsa T, Gamonal J. 2011. Host-pathogen interactions in progressive chronic periodontitis. J. Dent. Res. 90:1164–1170 [DOI] [PubMed] [Google Scholar]
- 6. Egli C, Leung WK, Muller KH, Hancock RE, McBride BC. 1993. Pore-forming properties of the major 53-kilodalton surface antigen from the outer sheath of Treponema denticola. Infect. Immun. 61:1694–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mathers DA, Leung WK, Fenno JC, Hong Y, McBride BC. 1996. The major surface protein complex of Treponema denticola depolarizes and induces ion channels in HeLa cell membranes. Infect. Immun. 64:2904–2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haapasalo M, Muller KH, Uitto VJ, Leung WK, McBride BC. 1992. Characterization, cloning, and binding properties of the major 53-kilodalton Treponema denticola surface antigen. Infect. Immun. 60:2058–2065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Edwards AM, Jenkinson HF, Woodward MJ, Dymock D. 2005. Binding properties and adhesion-mediating regions of the major sheath protein of Treponema denticola ATCC 35405. Infect. Immun. 73:2891–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Batista da Silva AP, Lee W, Bajenova E, McCulloch CA, Ellen RP. 2004. The major outer sheath protein of Treponema denticola inhibits the binding step of collagen phagocytosis in fibroblasts. Cell. Microbiol. 6:485–498 [DOI] [PubMed] [Google Scholar]
- 11. Fenno JC, Hannam PM, Leung WK, Tamura M, Uitto VJ, McBride BC. 1998. Cytopathic effects of the major surface protein and the chymotrypsinlike protease of Treponema denticola. Infect. Immun. 66:1869–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Visser MB, Koh A, Glogauer M, Ellen RP. 2011. Treponema denticola major outer sheath protein induces actin assembly at free barbed ends by a PIP2-dependent uncapping mechanism in fibroblasts. PLoS One 6:e23736 doi:10.1371/journal.pone.0023736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang Q, Ko KS, Kapus A, McCulloch CA, Ellen RP. 2001. A spirochete surface protein uncouples store-operated calcium channels in fibroblasts: a novel cytotoxic mechanism. J. Biol. Chem. 276:23056–23064 [DOI] [PubMed] [Google Scholar]
- 14. Puthengady Thomas B, Sun CX, Bajenova E, Ellen RP, Glogauer M. 2006. Modulation of human neutrophil functions in vitro by Treponema denticola major outer sheath protein. Infect. Immun. 74:1954–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magalhaes MA, Sun CX, Glogauer M, Ellen RP. 2008. The major outer sheath protein of Treponema denticola selectively inhibits Rac1 activation in murine neutrophils. Cell. Microbiol. 10:344–354 [DOI] [PubMed] [Google Scholar]
- 16. Ding Y, Haapasalo M, Kerosuo E, Lounatmaa K, Kotiranta A, Sorsa T. 1997. Release and activation of human neutrophil matrix metallo- and serine proteinases during phagocytosis of Fusobacterium nucleatum, Porphyromonas gingivalis and Treponema denticola. J. Clin. Periodontol. 24:237–248 [DOI] [PubMed] [Google Scholar]
- 17. Ding Y, Uitto VJ, Haapasalo M, Lounatmaa K, Konttinen YT, Salo T, Grenier D, Sorsa T. 1996. Membrane components of Treponema denticola trigger proteinase release from human polymorphonuclear leukocytes. J. Dent. Res. 75:1986–1993 [DOI] [PubMed] [Google Scholar]
- 18. Fenno JC, Wong GW, Hannam PM, Muller KH, Leung WK, McBride BC. 1997. Conservation of msp, the gene encoding the major outer membrane protein of oral Treponema spp. J. Bacteriol. 179:1082–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Godovikova V, Goetting-Minesky MP, Fenno JC. 2011. Composition and localization of Treponema denticola outer membrane complexes. Infect. Immun. 79:4868–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ishihara K, Kuramitsu HK, Miura T, Okuda K. 1998. Dentilisin activity affects the organization of the outer sheath of Treponema denticola. J. Bacteriol. 180:3837–3844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chi B, Qi M, Kuramitsu HK. 2003. Role of dentilisin in Treponema denticola epithelial cell layer penetration. Res. Microbiol. 154:637–643 [DOI] [PubMed] [Google Scholar]
- 22. Tramont EC. 2010. Treponema pallidum (syphilis), p 3035–3053 In Mandell GL, Bennett JE, Dolin R. (ed), Mandell, Douglas and Bennett's principles and practice of infectious diseases, vol 7 Churchill Livingtone Elsevier, Philadelphia, PA [Google Scholar]
- 23. Cox DL, Radolf JD. 2006. Metabolism of the Treponema, p 61–100 In Lukehart SA. (ed), Pathogenic Treponema. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 24. Lafond RE, Lukehart SA. 2006. Biological basis for syphilis. Clin. Microbiol. Rev. 19:29–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seshadri R, Myers GS, Tettelin H, Eisen JA, Heidelberg JF, Dodson RJ, Davidsen TM, DeBoy RT, Fouts DE, Haft DH, Selengut J, Ren Q, Brinkac LM, Madupu R, Kolonay J, Durkin SA, Daugherty SC, Shetty J, Shvartsbeyn A, Gebregeorgis E, Geer K, Tsegaye G, Malek J, Ayodeji B, Shatsman S, McLeod MP, Smajs D, Howell JK, Pal S, Amin A, Vashisth P, McNeill TZ, Xiang Q, Sodergren E, Baca E, Weinstock GM, Norris SJ, Fraser CM, Paulsen IT. 2004. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc. Natl. Acad. Sci. U. S. A. 101:5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Norris SJ, Weinstock GM. 2006. Comparative genomics of spirochetes, p 19–38 In Radolf JD, Lukehart SA. (ed), Pathogenic Treponema. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 27. Walker EM, Zampighi GA, Blanco DR, Miller JN, Lovett MA. 1989. Demonstration of rare protein in the outer membrane of Treponema pallidum subsp. pallidum by freeze-fracture analysis. J. Bacteriol. 171:5005–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Radolf JD, Norgard MV, Schulz WW. 1989. Outer membrane ultrastructure explains the limited antigenicity of virulent Treponema pallidum. Proc. Natl. Acad. Sci. U. S. A. 86:2051–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cox DL, Chang P, McDowall AW, Radolf JD. 1992. The outer membrane, not a coat of host proteins, limits antigenicity of virulent Treponema pallidum. Infect. Immun. 60:1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shevchenko DV, Akins DR, Robinson EJ, Li M, Shevchenko OV, Radolf JD. 1997. Identification of homologs for thioredoxin, peptidyl prolyl cis-trans isomerase, and glycerophosphodiester phosphodiesterase in outer membrane fractions from Treponema pallidum, the syphilis spirochete. Infect. Immun. 65:4179–4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radolf JD, Robinson EJ, Bourell KW, Akins DR, Porcella SF, Weigel LM, Jones JD, Norgard MV. 1995. Characterization of outer membranes isolated from Treponema pallidum, the syphilis spirochete. Infect. Immun. 63:4244–4252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Radolf JD. 1995. Treponema pallidum and the quest for outer membrane proteins. Mol. Microbiol. 16:1067–1073 [DOI] [PubMed] [Google Scholar]
- 33. Cameron CE. 2006. The T. pallidum outer membrane and outer membrane proteins, p 237–266 In Radolf JD, Lukehart SA. (ed), Pathogenic Treponema. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 34. Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, Sodergren E, Hardham JM, McLeod MP, Salzberg S, Peterson J, Khalak H, Richardson D, Howell JK, Chidambaram M, Utterback T, McDonald L, Artiach P, Bowman C, Cotton MD, Fujii C, Garland S, Hatch B, Horst K, Roberts K, Sandusky M, Weidman J, Smith HO, Venter JC. 1998. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281:375–388 [DOI] [PubMed] [Google Scholar]
- 35. Cox DL, Luthra A, Dunham-Ems S, Desrosiers DC, Salazar JC, Caimano MJ, Radolf JD. 2010. Surface immunolabeling and consensus computational framework to identify candidate rare outer membrane proteins of Treponema pallidum. Infect. Immun. 78:5178–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tommassen J. 2010. Assembly of outer-membrane proteins in bacteria and mitochondria. Microbiology 156:2587–2596 [DOI] [PubMed] [Google Scholar]
- 37. Hagan CL, Silhavy TJ, Kahne D. 2011. β-Barrel membrane protein assembly by the Bam complex. Annu. Rev. Biochem. 80:189–210 [DOI] [PubMed] [Google Scholar]
- 38. Wimley WC. 2003. The versatile Β-barrel membrane protein. Curr. Opin. Struct. Biol. 13:404–411 [DOI] [PubMed] [Google Scholar]
- 39. Centurion-Lara A, Castro C, Barrett L, Cameron C, Mostowfi M, Van Voorhis WC, Lukehart SA. 1999. Treponema pallidum major sheath protein homologue TprK is a target of opsonic antibody and the protective immune response. J. Exp. Med. 189:647–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anand A, Luthra A, Dunham-Ems S, Caimano MJ, Karanian C, LeDoyt M, Cruz AR, Salazar JC, Radolf JD. 2012. TprC/D (Tp0117/131), a trimeric, pore-forming rare outer membrane protein of Treponema pallidum, has a bipartite domain structure. J. Bacteriol. 194:2321–2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reference deleted.
- 42. Palmer GH, Bankhead T, Lukehart SA. 2009. ‘Nothing is permanent but change’—antigenic variation in persistent bacterial pathogens. Cell. Microbiol. 11:1697–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Giacani L, Molini BJ, Kim EY, Godornes BC, Leader BT, Tantalo LC, Centurion-Lara A, Lukehart SA. 2010. Antigenic variation in Treponema pallidum: TprK sequence diversity accumulates in response to immune pressure during experimental syphilis. J. Immunol. 184:3822–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hazlett KR, Sellati TJ, Nguyen TT, Cox DL, Clawson ML, Caimano MJ, Radolf JD. 2001. The TprK protein of Treponema pallidum is periplasmic and is not a target of opsonic antibody or protective immunity. J. Exp. Med. 193:1015–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Caimano MJ, Bourell KW, Bannister TD, Cox DL, Radolf JD. 1999. The Treponema denticola major sheath protein is predominantly periplasmic and has only limited surface exposure. Infect. Immun. 67:4072–4083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 47. Desrosiers DC, Anand A, Luthra A, Dunham-Ems SM, LeDoyt M, Cummings MA, Eshghi A, Cameron CE, Cruz AR, Salazar JC, Caimano MJ, Radolf JD. 2011. TP0326, a Treponema pallidum β-barrel assembly machinery A (BamA) orthologue and rare outer membrane protein. Mol. Microbiol. 80:1496–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Luthra A, Zhu G, Desrosiers DC, Eggers CH, Mulay V, Anand A, McArthur FA, Romano FB, Caimano MJ, Heuck AP, Malkowski MG, Radolf JD. 2011. The transition from closed to open conformation of Treponema pallidum outer membrane-associated lipoprotein TP0453 involves membrane sensing and integration by two amphipathic helices. J. Biol. Chem. 286:41656–41668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Edelhoch H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry 6:1948–1954 [DOI] [PubMed] [Google Scholar]
- 50. Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31:3784–3788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brusca JS, Radolf JD. 1994. Isolation of integral membrane proteins by phase partitioning with Triton X-114. Methods Enzymol. 228:182–193 [DOI] [PubMed] [Google Scholar]
- 52. Cox DL, Akins DR, Porcella SF, Norgard MV, Radolf JD. 1995. Treponema pallidum in gel microdroplets: a novel strategy for investigation of treponemal molecular architecture. Mol. Microbiol. 15:1151–1164 [DOI] [PubMed] [Google Scholar]
- 53. Cox DL, Akins DR, Bourell KW, Lahdenne P, Norgard MV, Radolf JD. 1996. Limited surface exposure of Borrelia burgdorferi outer surface lipoproteins. Proc. Natl. Acad. Sci. U. S. A. 93:7973–7978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, Zhang D, Bryant SH. 2013. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41:D348–D352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Subbarao GV, van den Berg B. 2006. Crystal structure of the monomeric porin OmpG. J. Mol. Biol. 360:750–759 [DOI] [PubMed] [Google Scholar]
- 56. Yildiz O, Vinothkumar KR, Goswami P, Kuhlbrandt W. 2006. Structure of the monomeric outer-membrane porin OmpG in the open and closed conformation. EMBO J. 25:3702–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67:593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beher MG, Schnaitman CA, Pugsley AP. 1980. Major heat-modifiable outer membrane protein in Gram-negative bacteria: comparison with the ompA protein of Escherichia coli. J. Bacteriol. 143:906–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Conlan S, Bayley H. 2003. Folding of a monomeric porin, OmpG, in detergent solution. Biochemistry 42:9453–9465 [DOI] [PubMed] [Google Scholar]
- 60. Brett PJ, Burtnick MN, Fenno JC, Gherardini FC. 2008. Treponema denticola TroR is a manganese- and iron-dependent transcriptional repressor. Mol. Microbiol. 70:396–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pugsley AP. 1993. The complete general secretory pathway in Gram-negative bacteria. Microbiol. Rev. 57:50–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Niederweis M, Danilchanka O, Huff J, Hoffmann C, Engelhardt H. 2010. Mycobacterial outer membranes: in search of proteins. Trends Microbiol. 18:109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb. Perspect. Biol. 2:a000414 doi:10.1101/cshperspect.a000414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fenno JC, Muller KH, McBride BC. 1996. Sequence analysis, expression, and binding activity of recombinant major outer sheath protein (Msp) of Treponema denticola. J. Bacteriol. 178:2489–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cullen PA, Cameron CE. 2006. Progress towards an effective syphilis vaccine: the past, present and future. Expert Rev. Vaccines 5:67–80 [DOI] [PubMed] [Google Scholar]
- 66. Lenhart TR, Akins DR. 2010. Borrelia burgdorferi locus BB0795 encodes a BamA orthologue required for growth and efficient localization of outer membrane proteins. Mol. Microbiol. 75:692–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ricci DP, Silhavy TJ. 2012. The Bam machine: a molecular cooper. Biochim. Biophys. Acta 1818:1067–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Antal GM, Lukehart SA, Meheus AZ. 2002. The endemic treponematoses. Microbes Infect. 4:83–94 [DOI] [PubMed] [Google Scholar]
- 69. Darveau RP. 2010. Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 8:481–490 [DOI] [PubMed] [Google Scholar]
- 70. Breznak JA. 2006. Termite gut spirochetes, p 421–445 In Radolf JD, Lukehart SA. (ed), Pathogenic Treponema. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 71. Smith SG, Mahon V, Lambert MA, Fagan RP. 2007. A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol. Lett. 273:1–11 [DOI] [PubMed] [Google Scholar]
- 72. Park JS, Lee WC, Yeo KJ, Ryu KS, Kumarasiri M, Hesek D, Lee M, Mobashery S, Song JH, Kim SI, Lee JC, Cheong C, Jeon YH, Kim HY. 2012. Mechanism of anchoring of OmpA protein to the cell wall peptidoglycan of the Gram-negative bacterial outer membrane. FASEB J. 26:219–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tamm LK, Hong H, Liang B. 2004. Folding and assembly of β-barrel membrane proteins. Biochim. Biophys. Acta 1666:250–263 [DOI] [PubMed] [Google Scholar]
- 74. Schulz GE. 2002. The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta 1565:308–317 [DOI] [PubMed] [Google Scholar]
- 75. Fairman JW, Noinaj N, Buchanan SK. 2011. The structural biology of Β-barrel membrane proteins: a summary of recent reports. Curr. Opin. Struct. Biol. 21:523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Van Gelder P, Dumas F, Bartoldus I, Saint N, Prilipov A, Winterhalter M, Wang Y, Philippsen A, Rosenbusch JP, Schirmer T. 2002. Sugar transport through maltoporin of Escherichia coli: role of the greasy slide. J. Bacteriol. 184:2994–2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ye J, van den Berg B. 2004. Crystal structure of the bacterial nucleoside transporter Tsx. EMBO J. 23:3187–3195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Satir BH, Satir P. 1979. Partitioning of intramembrane particles during the freeze-fracture procedure, p 43–49 In Rash JE, Hudson CS. (ed), Freeze fracture: methods, artifacts, and interpretations. Raven Press, New York, NY [Google Scholar]
- 79. Heuck A, Schleiffer A, Clausen T. 2011. Augmenting β-augmentation: structural basis of how BamB binds BamA and may support folding of outer membrane proteins. J. Mol. Biol. 406:659–666 [DOI] [PubMed] [Google Scholar]
- 80. Kim S, Malinverni JC, Sliz P, Silhavy TJ, Harrison SC, Kahne D. 2007. Structure and function of an essential component of the outer membrane protein assembly machine. Science 317:961–964 [DOI] [PubMed] [Google Scholar]
- 81. Knowles TJ, Jeeves M, Bobat S, Dancea F, McClelland D, Palmer T, Overduin M, Henderson IR. 2008. Fold and function of polypeptide transport-associated domains responsible for delivering unfolded proteins to membranes. Mol. Microbiol. 68:1216–1227 [DOI] [PubMed] [Google Scholar]
- 82. Bennion D, Charlson ES, Coon E, Misra R. 2010. Dissection of β-barrel outer membrane protein assembly pathways through characterizing BamA POTRA 1 mutants of Escherichia coli. Mol. Microbiol. 77:1153–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schulz GE. 2003. Transmembrane β-barrel proteins. Adv. Protein Chem. 63:47–70 [DOI] [PubMed] [Google Scholar]
- 84. Giacani L, Brandt SL, Puray-Chavez M, Reid TB, Godornes C, Molini BJ, Benzler M, Hartig JS, Lukehart SA, Centurion-Lara A. 2012. Comparative investigation of the genomic regions involved in antigenic variation of the TprK antigen among treponemal species, subspecies, and strains. J. Bacteriol. 194:4208–4225 [DOI] [PMC free article] [PubMed] [Google Scholar]