

Abstract
Integrating conjugative elements (ICEs) of the SXT/R391 family are major contributors to the spread of antibiotic resistance genes. These elements also catalyze their own diversity by promoting inter-ICE recombination through the action of the RecA-independent homologous recombination system that they encode. Here, we report that expression of this recombination system, which consists of the single-stranded DNA annealing protein Bet and the exonuclease Exo, is induced by DNA-damaging agents via ICE-encoded transcriptional regulators. We show that the bet and exo genes are part of a large polycistronic transcript that contains many conserved ICE genes that are not involved in the main integration/excision and conjugative transfer processes. We show that although the recombination genes are highly transcribed, their translation is subject to additional strong regulatory mechanisms. We also show that an ICE-encoded putative single-stranded DNA binding protein (Ssb) limits hybrid ICE formation. Finally, a thorough in silico analysis reveals that orthologues of Bet and Exo are widely distributed in bacterial strains belonging to very distantly related bacterial species and are carried by various mobile genetic elements. Phylogenetic analyses suggest that the annealing proteins and exonucleases that compose these systems sometimes have different evolutionary origins, underscoring the strong selective pressure to maintain the functionality of these unrelated cooperating proteins.
INTRODUCTION
Bacteria have the capacity to rapidly adapt to a changing environment due their ability to acquire and share genes, providing them with new selectable traits. One of the most striking examples of bacterial genome plasticity is the emergence and spread of antibiotic resistance genes. This exchange of genes occurs through horizontal gene transfer (HGT) events such as transformation, transduction, and conjugation. HGT events can be mediated by an array of mobile genetic elements (MGEs) such as conjugative plasmids, bacteriophages, and integrating conjugative elements (ICEs), which are also known as conjugative transposons (1–4). Stable integration of the features acquired by HGT, through site-specific recombination, transposition, or homologous recombination, allows their vertical transmission as well (5).
ICEs of the SXT/R391 family are mobile genetic elements that greatly contribute to the spread of antibiotic resistance genes in Vibrio cholerae and related gammaproteobacterial species (6, 7). These elements are found site-specifically integrated in the chromosome of their host. Under certain conditions, ICEs of the SXT/R391 family are excised from their host's chromosome as a circular, covalently closed molecule and transferred via conjugation to a new recipient cell in the form of a single-stranded DNA substrate (8). All members of this family share a conserved set of 52 core genes, of which 25 are required for their key functions of integration/excision, conjugative transfer and regulation (7). Newly identified ICEs are classified in this family based on the conservation of the scaffold of conserved genes, the presence of a conserved P4-like site-specific tyrosine recombinase and on their site-specific integration into the 5′ end of prfC, which encodes the peptide chain release factor RF3 (9). Interspersed in intergenic regions of this conserved backbone are variable DNA sequences that confer a wide selection of accessory functions, for instance, antibiotic or heavy metal resistances, and genes involved in modifying cell-signaling pathways (10–18). Similar to the induction of the lytic cycle of bacteriophage λ in lysogenic cells, the expression of the transfer genes of SXT/R391 ICEs is derepressed under conditions that trigger the host's SOS response. Under noninducing conditions, the master repressor SetR, which is encoded by these elements, prevents the expression of the transcriptional activators SetCD. DNA-damaging agents such as mitomycin C, UV light, and the antibiotic ciprofloxacin alleviate this repression, allowing SetCD-mediated activation of the conserved genes involved in excision and conjugative transfer (19).
SXT/R391 ICEs also encode a RecA-independent homologous recombination system similar to the bacteriophage λ Red system. In a previous study, we demonstrated that this recombination system promotes the formation of hybrid ICEs by catalyzing the recombination of two ICEs integrated in a tandem fashion (20). The recombination system carried by SXT/R391 ICEs comprises two genes: s065, encoding a single-stranded DNA annealing protein (21), and s066, encoding a 5′-3′ exonuclease (22). These proteins are, respectively, related to λBet and λExo, and we therefore refer to s065 as bet and to s066 as exo. Homologues of the SXT/R391 recombination genes are also found in conjugative plasmids of the A/C incompatibility group (IncA/C) (Fig. 1). These multidrug resistance plasmids are widespread in Salmonella and other enterobacteria from agricultural sources, and it was recently demonstrated that most of the conserved genes of SXT/R391 ICEs are also present in this family of conjugative plasmids (7).
Fig 1.
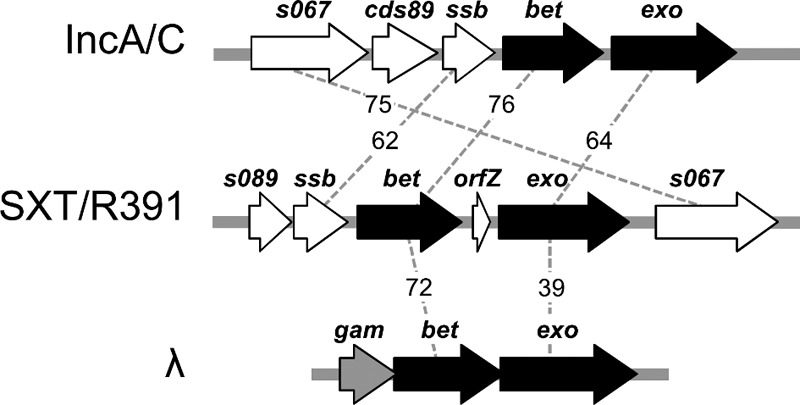
Schematic comparison of the recombination loci of SXT/R391 ICEs, IncA/C plasmids, and bacteriophage λ. SXT/R391 bet and exo and their orthologues are represented in black. Surrounding genes that have no known role in recombination are represented in white. λgam has no orthologue in SXT/R391 ICEs and IncA/C plasmids and is presented in gray. Dotted lines and associated numbers indicate the percent similarity between orthologous genes.
Although the λ Red and SXT/R391 recombination genes are functionally similar, they differ in their genetic organization. λbet and λexo are located in the λ pL operon with other genes involved in homologous and site-specific recombination and are expressed early in the lytic cycle (23, 24). λgam, a gene coding for an inhibitor of the host exonuclease RecBCD, is found directly upstream of λbet and is transcribed with the recombination genes (Fig. 1). The product of λgam protects the phage's linear double-stranded DNA (dsDNA) by inhibiting the ability of RecBCD to bind dsDNA ends (25). No homologue of λgam exists in SXT/R391 ICEs or IncA/C plasmids; instead, a gene encoding a putative single-stranded DNA-binding protein (ssb) is found upstream of bet. In SXT/R391 ICEs, bet and exo are separated by a 288-bp stretch that contains a small 141-bp open reading frame (ORF) of unknown function, termed orfZ (Fig. 1). Upstream of ssb is found a gene of unknown function, s089. No homologues of orfZ or s089 are found in the IncA/C plasmids.
Here, we report that the RecA-independent homologous recombination functions of SXT/R391 ICEs are induced by the DNA-damaging agent mitomycin C via the main transcriptional activators SetCD. Our results also indicate that the recombination functions are further regulated at the translational level. Furthermore, deletion of ssb significantly increased Bet-Exo-mediated hybrid ICE formation, suggesting that it could act as a modulator of the recombination activity. Our in silico analysis revealed that similar recombination systems are widely distributed in a large number of strains belonging to very diverse bacterial taxonomic orders and that they are present not only in ICEs and λ-like bacteriophages but also in conjugative plasmids from various incompatibility groups. Finally, our results also suggest different evolutionary origins for the Bet and Exo proteins that form the recombination system of SXT/R391 ICEs.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media.
The strains and plasmids used in this study are described in Table 1. These strains were routinely grown in Luria-Bertani (LB) broth at 37°C in an orbital shaker/incubator. Antibiotics were used at the following concentrations: ampicillin, 30 μg/ml (pGG2B only) or 100 μg/ml; tetracycline, 12 μg/ml; sulfamethoxazole, 32 μg/ml; trimethoprim, 4 μg/ml; spectinomycin, 50 μg/ml, kanamycin, 50 μg/ml. When required, bacterial cultures were supplemented with l-arabinose (0.2%, wt/vol) or mitomycin C (100 or 200 ng/ml).
Table 1.
E. coli strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotypea | Reference or source |
|---|---|---|
| E. coli strains | ||
| BW25113 | F− Δ(araD-araB)567 ΔlacZ4787(::rrnB3) λ− rph-1 Δ(rhaD-rhaB)568 hsdR514 | 26 |
| BW27784 | F− Δ(araD-araB)567 ΔlacZ4787(::rrnB-3) λ− Δ(araH-araF)570(::FRT) ΔaraEp-532::FRT ϕPcp18 araE533 Δ(rhaD-rhaB)568 hsdR514 | 58 |
| CAG18439 | MG1655 lacZU118 lacI42::Tn10 (Tcr) | 59 |
| VB112 | MG1655 Rfr | 60 |
| HW220 | CAG18439 prfC::SXT (Tcr Sur Tmr) | 61 |
| JO99 | CAG18439 prfC::R391 (Tcr Knr) | 62 |
| VB40 | CAG18439 ΔlacZ prfC::SXT::lacZ (Tcr Sur Tmr) | 20 |
| GG13 | CAG18439 ΔgalK prfC::R391::galK (Tcr Knr) | 20 |
| VB17 | BW25113 prfC::SXT (Sur Tmr) | V. Burrus |
| GG247 | BW25113 ΔrecA prfC::SXT (Sur Tmr) | This study |
| GG242 | BW25113 prfC::SXT ΔsetDC (Sur Tmr) | This study |
| GG55 | VB112 ΔrecA (Rfr) | 20 |
| GG66 | VB112 prfC::[SXT::lacZ]-[R391::galK] (Rfr Sur Tmr Knr) | 20 |
| GG67 | VB112 prfC::[SXT::lacZ Δs089]-[R391::galK Δs089] (Rfr Sur Tmr Knr) | This study |
| GG68 | VB112 prfC::[SXT::lacZ Δssb]-[R391::galK Δssb] (Rfr Sur Tmr Knr) | This study |
| VB47 | CAG18439 ΔgalK ΔrecA (Tcr) | 20 |
| GG209 | BW25113 prfC::SXT bet′-′lacZ (Sur Tmr) | This study |
| GG215 | BW25113 prfC::SXT exo′-′lacZ (Sur Tmr) | This study |
| Plasmids | ||
| pBAD30 | orip15A araC PBAD | 27 |
| pGG2B | pBAD30::setDC | This study |
| pGG7 | pBAD::orfZ | This study |
| pGG32 | pCR2.1::Igr(s063-s089)SXT | This study |
| pVI67 | pAH57 Δ(xisλ-intλ)::setDC(Ts) | 20 |
| pVI36 | Spr template for one-step chromosomal gene inactivation | 60 |
| pKD13 | Knr template for one-step chromosomal gene inactivation | 21 |
| pVI42B | pVI36 BamHI::Plac-lacZ | 20 |
| pDPL373 | pBAD::TA−10-bet′-′lacZ | This study |
| pDPL374 | pBAD::TA−24-bet′-′lacZ | This study |
| pDPL375 | pBAD::TA−53-bet′-′lacZ | This study |
| pDPL376 | pBAD::TA−11-exo′-′lacZ | This study |
| pDPL377 | pBAD::TA−34-exo′-′lacZ | This study |
| pDPL378 | pBAD::TA−41-exo′-′lacZ | This study |
Apr, ampicillin resistant; Cmr, chloramphenicol resistant; Knr, kanamycin resistant; Rfr, rifampin resistant; Sur, sulfamethoxazole resistant; Smr, streptomycin resistant; Spr, spectinomycin resistant; Tcr, tetracycline resistant; Tmr, trimethoprim resistant; Ts, thermosensitive.
Plasmid and strain construction.
The oligonucleotides used in this study are described in Table S1 in the supplemental material. Deletion and fusion mutants were constructed by using the one-step chromosomal gene inactivation technique of Datsenko and Wanner (26). All deletions were designed to be nonpolar. The ΔrecA mutation was introduced into Escherichia coli BW25113 using the primer pair recAWF/recAWR (20) and plasmid pVI36 as the template. The ΔsetDC mutation was introduced into E. coli BW25113 SXT using the primer pair setD2wF/setC2wR and plasmid pKD13 as the template. The Δs089 and Δssb mutations were introduced into SXT (in strain HW220) using the primer pairs s089WF/s089WR and ssb1WF/ssb1WR, respectively, and template plasmid pVI36. The corresponding mutations were introduced into R391 (in strain JO99) using the primer pairs R66WF/s089WR and ssb2WF/ssb2WR, respectively, and pVI36 as the template. Plac-lacZ and Plac-galK were introduced into strains containing SXT and R391 deletion mutants, using generalized P1vir transduction and E. coli VB40 and GG13 as donor strains. Strains containing tandem arrays were constructed by successively transferring SXT::lacZ and R391::galK (or their corresponding deletion derivatives) into GG55, yielding strains GG66 to GG68. Plasmid pVI67 was introduced into GG55 prior to the transfer of the ICEs. lacZ translational fusions of bet and exo were constructed in BW25113 SXT using the primer pairs 65lacZ42BF/65lacZ42BR and 66lacZ42BF/66lacZ42BR, respectively, and plasmid pVI42B as the template. For both bet and exo, lacZ was fused to the 8th codon of the gene. All deletions and fusions were verified by PCR amplification using primers flanking the deletion, cloning, and sequencing. Plasmid pGG2B was constructed by amplifying setDC from strain HW220 using the primer pairs setDF/setC2R, subcloning into pCR2.1TOPO (Invitrogen), and recloning into pBAD30 (27). Plasmid pGG7 was constructed by amplifying orfZ from strain HW220 using the primer pair orfZTOPOF/corfZR and cloning into pBAD-TOPO (Invitrogen). Plasmid pGG32 was constructed by amplifying the intergenic region located between s063 and s089 in SXT from E. coli HW220 using the primer pair 6389F/6389R and cloning into pCR2.1TOPO. Plasmids pDPL373, pDPL374, and pDPL375 were constructed by cloning into pBAD-TOPO the bet′-′lacZ translational fusion and various lengths of its 5′ untranslated region (UTR) that were amplified using the primers betTAlacZF1, betTAlacZF2, and betTAlacZF3, respectively, with primer lacZTAcommonR and genomic DNA of GG209 as the template. Plasmids pDPL376, pDPL377, and pDPL378 were similarly constructed for the exo′-′lacZ translational fusion and its 5′ UTR using primers exoTAlacZF1, exoTAlacZF2, and exoTAlacZF3, respectively, with primer lacZTAcommonR and genomic DNA of GG215 as the template. The resulting six plasmids were introduced into E. coli BW27784. All plasmid inserts were verified by sequencing by Centre d'Innovation Génome Québec (McGill University, Montreal, QC, Canada). E. coli was transformed by electroporation in 1-mm-gap cuvettes in a GenePulser Xcell apparatus (Bio-Rad) set at 2.5 μF, 200 Ω, and 1.8 kV.
RNA extraction and cDNA synthesis.
Briefly, RNA extractions were performed as follows. Cultures were grown for 14 to 16 h in LB broth containing the appropriate antibiotics, diluted 1:100 in fresh medium in the absence of antibiotics (except when ampicillin was added for the maintenance of the pGG2B vector), and grown to an optical density at 600 nm (OD600) of 0.2. They were then diluted 1:100 a second time and grown to an OD600 of 0.2. Each culture was then split between two flasks, of which one was induced with 100 ng/ml mitomycin C (MMC) or 0.02% l-arabinose (when using pGG2B), and both were incubated for 2 h at 37°C. A 1-ml sample of each culture was used for total RNA extraction using an RNeasy minikit and RNAprotect bacterial reagent (Qiagen) following the manufacturer's instructions. Once purified, the RNA samples were subjected to gDNA digestion using Turbo DNase (Ambion) and following the manufacturer's instructions. cDNA was synthesized using 1 μg of RNA, 50 ng of random hexamers, or 2 pmol of the gene-specific primer s073RT (Integrated DNA Technologies) (see Table S1 in the supplemental material), and the reverse transcriptase SuperScript II (Invitrogen), following the manufacturer's instructions. Control reactions in the absence of reverse transcriptase (no-RT reactions) were performed for each sample.
Real-time quantitative PCR analysis.
qRT-PCR was performed by measuring the increase of fluorescence using the Quantifast SYBR green mix (Qiagen) in an Eppendorf RealPlex (Eppendorf). Primers were designed to amplify an internal fragment of ca. 180 bp of the reference genes (rpoZ and gyrA) (28) or of each gene of interest (see Table S1 in the supplemental material). The expression ratios were calculated using the 2−ΔΔCT method, and the difference in the transcript levels of each gene of interest was calculated compared to the transcript level of rpoZ and gyrA using the 2−ΔCT method. The experiments were carried out with a minimum of three biological replicates, each with three technical replicates.
Primer extension analysis.
Primer extension analysis was carried out using 1 and 5 μg of total RNA from BW25113 SXT cultured in the absence (CTL) or presence of mitomycin C (MMC) at 100 ng/ml, with radiolabeled TSS89up4 primer ([γ-32P]ATP, 3000 Ci/mmol, 10 mCi/ml [PerkinElmer]) and a primer extension system with Primer Extension System-AMV Reverse Transcriptase (Promega) following the manufacturer's instructions. A control reaction in which water replaced the RNA was performed. A 3-μl portion of each primer extension reaction was directly mixed with an equal volume of loading dye, and the primer extension products were migrated in a 0.8% acrylamide–bis-acrylamide (19:1) gel in a SequiGenGT nucleic acid sequencing cell (Bio-Rad) alongside the sequencing reaction of the intergenic region located between s063 and s089 in SXT. Sanger sequencing of the 367-bp intergenic region located between s063 and s089 in SXT was performed using 5 μg of plasmid pGG32 as the template, [α-32P]dATP (800 Ci/mmol, 10 mCi/ml) (PerkinElmer), primer TSS89up4, and the Sequenase version 2.0 kit (Affymetrix). Signal strength was detected using a Storm 860 molecular imager (GMI).
Semiquantitative PCR.
PCRs aimed at amplifying s063, s989, and bet were carried out using cDNA prepared with the gene-specific primer s073RT (see Table S1 in the supplemental material). Each reaction mixture contained a 1 μM concentration of each primer, a 100 μM concentration of each deoxynucleoside triphosphate (dNTP), and 1 U Taq DNA polymerase (New England BioLabs); 1 μl of a 1:10 dilution of the cDNA synthesis reaction, 1 μl of a 1:10 dilution of the no-RT reactions (negative control), and 500 pg BW25113 SXT gDNA (positive control) were used as the templates for PCR amplification. PCR conditions were as follows: (i) 3 min at 95°C, (ii) 28 cycles of 30 s at 95°C, 30 s at the appropriate annealing temperature, and 30 s at 72°C, and (iii) 5 min at 72°C. After PCR, the samples were mixed with loading dye (orange G in 30% glycerol [vol/vol]) at a final concentration of 1×, and 15 μl of each reaction mixture was migrated in a 2% agarose gel in 1× TAE (Tris-acetate-EDTA) buffer, alongside 1 μg 2-log DNA ladder (New England BioLabs). Migrated gels were colored in an ethidium bromide bath (3 μg/ml) for 30 min and then soaked for 30 min in distilled water. Gels were visualized under UV light in a GelDocXR system (Bio-Rad) and analyzed using Quantity One v6.3 software (Bio-Rad).
β-Galactosidase assays.
β-Galactosidase assays were carried out using translational fusions of lacZ to bet or exo in BW25113 SXT or strain BW27784 containing pDPL plasmids, under induced or control conditions. Briefly, strains were grown for 14 to 16 h in LB with the appropriated antibiotics, then refreshed at 1:100, and grown to an OD600 of 0.2 in fresh LB broth in the absence of antibiotics, except when ampicillin was used to maintain pGG2B or pDPL plasmids. Cultures were split into 4-ml samples and grown in the absence or presence of MMC at 100 ng/ml or 200 ng/ml (for chromosomal fusions) for 2 or 16 h or in the presence or absence of l-arabinose 0.2% vol/vol (for assays performed with pDPL plasmids). β-Galactosidase activity was assessed using 100- or 500-μl samples as described elsewhere (29).
Bioinformatics analyses.
The predicted proteomes of 2,714 plasmids, 4,093 viruses, and 1,706 complete microbial genomes available in March 2012 were obtained from the RefSeq database (30) and analyzed to identify putative recombination systems similar to the SXT/R391 system. We screened this data set using profile hidden Markov models of the RecT, YqaJ, and Gam families with HMMsearch from the HMMER v3.0 software package (31). The HMM profiles of Bet, Exo, and λGam were recovered from the Pfam 25.0 database, as follows: for Bet, RecT and PF03837; for Exo, YqaJ and PF09588; for λGam, Gam and PF06064. The sequences corresponding to each identified protein were downloaded and aligned using MUSCLE multiple sequence alignment software (32). Maximum-likelihood phylogenies of proteins belonging to the RecT and YqaJ families were generated using the PhyML v3.0 program (33) with the HKY85 substitution. Tree topologies were optimized by PhyML using the NNI and SPR methods model, and the starting trees were estimated using BioNJ. Branch support of the phylogenies was estimated using nonparametric bootstrap (100 replicates). Phylogenetic analyses were computed using amino acid alignment generated by MUSCLE, and the poorly aligned regions were removed with trimAl v1.2 software using the automated heuristic approach (34) prior to phylogenetic analyses. Phylogenetic trees were viewed in iTOL v2 (35) and are available as a shared project on the iTOL website (http://itol.embl.de/shared/GGarriss). Sequence alignments were performed using ClustalW (36).
RESULTS
DNA-damaging agents induce the expression of ICE recombination genes.
In order to gain a better understanding of the conditions that favor hybrid ICE formation, we investigated the conditions that induce the transcription of bet and exo. Since all the conserved genes of SXT/R391 ICEs involved in integration/excision and conjugative transfer are induced under conditions that trigger their host's SOS response, we decided to verify whether the DNA-damaging agent mitomycin C (MMC) would also promote the transcription of the recombination genes. We measured by reverse transcription-quantitative PCR (RT-qPCR) the effect of the exposure to MMC on the transcript levels of bet and exo, using E. coli BW25113 harboring SXT (VB17). Upon exposure to MMC, the relative expression levels (2−ΔΔCT) of bet and exo were ∼50 times higher than under the control condition (Fig. 2A). However, when an isogenic recA-null mutant (GG247) was used, the induction of bet and exo expression was abolished (expression ratio of ≈1), indicating that the pathway by which these genes are induced requires recA. In order to determine if this increase relies on the ICE's main activators, SetC and SetD, and not on the alleviation of repression by the host-encoded SOS response repressor LexA, we carried out the same experiments using a strain harboring an SXT ΔsetCD mutant (GG242). Our results showed that in the absence of setCD, induction of bet and exo transcription with MMC was unachievable (expression ratio of ≈1). This deletion was more than successfully complemented using plasmid pGG2B, which carries setCD under the control of an arabinose-inducible promoter (expression ratio > 5,000). Furthermore, the absence of recA, which is required for derepression of setCD expression, could be bypassed by providing SetCD from pGG2B, which resulted in a >6,000-fold increase of bet and exo transcript levels. Our results show that agents that trigger the host's SOS response induce the transcription of ICE-encoded recombination functions, through the action of recA and the ICE-encoded transcriptional activators setCD.
Fig 2.
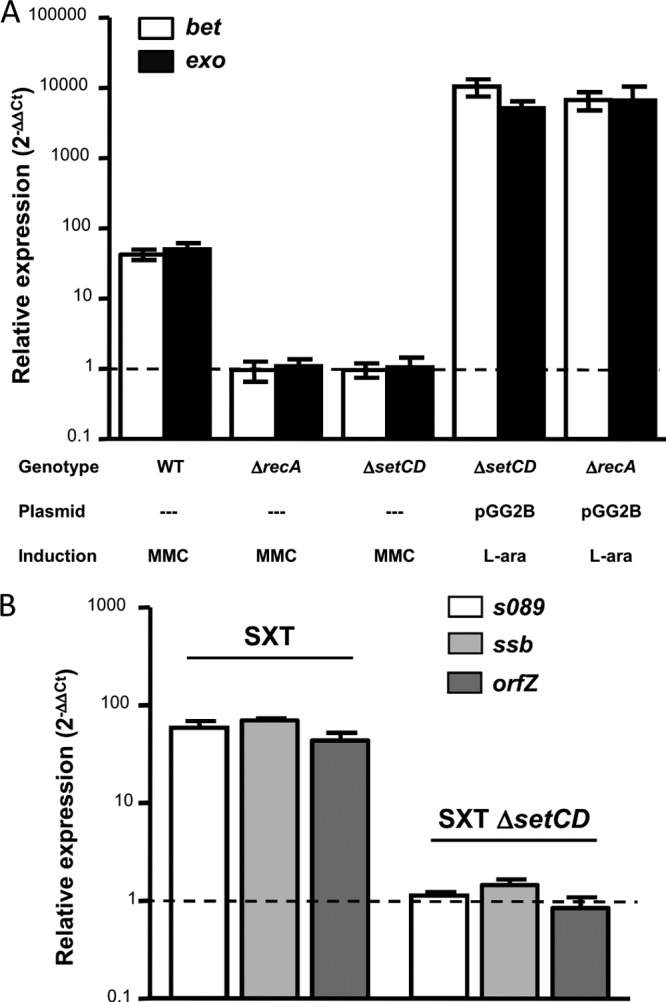
Expression of bet and exo is induced by mitomycin C. (A) Relative expression of bet and exo. The genotype of the ICE or strain, the presence of the setCD expression vector pGG2B, and induction with mitomycin C (MMC) or l-arabinose for pGG2B (L-ara) are indicated below the graph. (B) Relative expression of s089, ssb, and orfZ in an SXT or SXT ΔsetCD background upon induction with MMC. Bars represent the ratio (2−ΔΔCT) between an induced and the noninduced conditions, using rpoZ as an internal reference as determined by RT-qPCR, and are means from a least three independent biological replicates. Standard deviations are indicated. Dotted lines indicate an expression ratio of 1 (no difference between the control and induced conditions).
We decided to verify whether the transcription of the two genes located upstream the recombination system, s089 and ssb, as well orfZ, located between bet and exo, responded to the same induction conditions as bet and exo using the same RT-qPCR assays. Our results show that the exposure to MMC causes a SetCD-dependent increase of s089, ssb, and orfZ transcripts comparable to that obtained for bet and exo (Fig. 2B).
The recombination genes are part of a polycistronic transcript that spans 11 kb of conserved ICE DNA and is driven from a MMC-induced promoter located upstream s089.
The recombination genes are located within an ∼11-kb conserved region of the SXT/R391 ICEs positioned between two inversely transcribed genes, s063 (SXT/R391 conserved core gene) and s074 (nonconserved gene present in SXT) (Fig. 3A) (7). Eight nearly identical and perfectly syntenic genes are located downstream of exo in this region. These genes play no role in conjugative transfer, and their respective functions remains obscure (7). The similar response of the genes between s089 and exo to the exposure to MMC, combined with the fact that in λ the recombination genes are present in a single transcript (23), led us to hypothesize that a similar operon structure might also be found in SXT/R391 ICEs. The overall genetic organization of this locus led us to investigate whether all the genes between s063 and s074 are cotranscribed with bet-exo.
Fig 3.

Operon structure of the SXT/R391 homologous recombination system. (A) Schematic representation of the s089-s073 region of the SXT/R391 backbone. Genes are represented as follows: black, recombination genes; white, genes of other or unknown function; gray, immediate borders of the s089-s073 locus. The relative positions of the reverse transcription primer s073RT as well as PCR primers used to amplify bet, s063, and s089 are indicated. Dotted lines shows the reverse transcription product, and results of PCR are indicated as positive or negative. (B) A 2% agarose gel from an assay to amplify s063, s089, and bet on the s073RT product. VB17 genomic DNA (gDNA) and reverse transcription samples in the absence of reverse transcriptase (No RT) were used, respectively, as positive and negative PCR controls. MMC, MMC-induced samples; M, molecular weight marker.
We carried out a reverse transcription experiment using a primer located at the 3′ end of s073 (s073RT) and RNA obtained from MMC-treated VB17 cells (Fig. 3A). We then used PCR to amplify fragments located at the 5′ ends of s063, s089, and bet to determine if any of these genes are cotranscribed with s073. Our results show that s089 and bet are part of a transcript that encompasses ca. 11 kb of conserved ICE DNA (Fig. 3B) and that expression of all of the genes from s089 to s073 is induced by MMC. Moreover, the impossibility of amplifying s063 from the same cDNA sample indicates that this transcript originates within the intergenic region lying between divergently transcribed s063 and s089 (Fig. 3).
In order to determine the precise location of the promoter driving the transcription of the recombination genes, we analyzed the 367-bp intergenic region located between s063 and s089 by primer extension, using total RNA from E. coli VB17 prepared in noninduced and MMC-induced conditions. Our results show that the transcription start site is located 235 bp from the start codon of s089 and corresponds to a C in the nontranscribed DNA strand (Fig. 4). Despite the presence of a large 5′ UTR, we found no evidence of another ORF in the region located between the transcriptional and translation start sites of the recombination system. As expected, the intensity of the band is dependent on the presence of MMC and also on the concentration of RNA used for the primer extension assay. A second band of lesser intensity is observed at A+2, but this might be due to imprecise transcription initiation (Fig. 4A). Further analysis of the sequence upstream of C+1 revealed likely putative −35 (CTACCC) and −10 (TACACT) promoter elements (Fig. 4B). No other DNA elements that contribute to promoter recognition by E. coli σ70 (37) could be identified in this region.
Fig 4.

Promoter region of the SXT/R391 homologous recombination system. (A) Primer extension analysis of the transcribed strand of the s063-s089 intergenic region. No RNA, negative control; CTL, noninduced control samples; MMC, MMC-induced samples; G, A, T and C, Sanger sequencing reaction lanes. Concentrations of RNA used for the primer extension reaction are indicated. Each well was loaded with equal reaction volumes. A partial read of the sequencing reaction is located to the right of the gel, with arrows indicating the transcription start sites on the transcribed strand. (B) Schematic representation of the promoter region. Positions of the two transcription start sites are indicated by angled arrows. Deduced −35 (5′-TTGACA-3′) and −10 (5′-TATAAT-3′) promoter elements recognized by E. coli σ70 are shown in bold capital letters. s063 and s089 are represented by gray and white arrows, respectively. Lengths (in base pairs) of the spacers between represented regions are indicated.
A gene encoding a single-stranded DNA binding protein modulates hybrid ICE formation.
The presence of s089 and ssb in the same transcript as bet and exo led us to investigate their possible involvement in Bet-Exo-mediated hybrid ICE formation. We constructed recA donor strains harboring tandem arrays of Δs089 and Δssb mutants of SXT and R391 and used them in conjugation assays with a recA recipient strain (VB47), using a quantitative detection assay designed to study hybrid ICE formation (20). These experiments were carried out in a ΔrecA background to prevent RecA-mediated recombination and ensure that all hybrid ICEs are formed by Bet-Exo-mediated recombination (20). We bypassed the requirement for RecA for the activation of expression of the conjugative transfer genes (19, 20) as well as for expression of bet and exo by providing setCD in trans from plasmid pVI67. We found that although s089 does not appear to be involved in hybrid ICE formation, the deletion of ssb caused a 3-fold increase of the percentage of exconjugants containing a hybrid ICE (P < 0.001) (Fig. 5). These results show clearly that the absence of Ssb allows Bet-Exo to more efficiently catalyze inter-ICE recombination.
Fig 5.
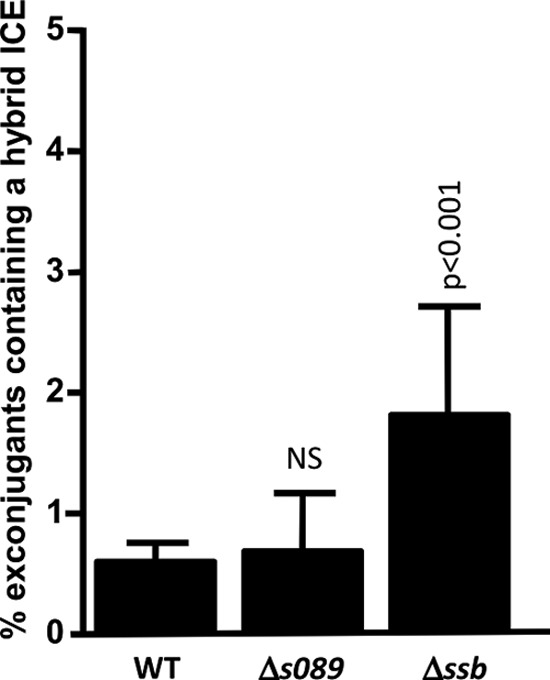
Impact of s089 and ssb on hybrid ICE formation. recA donor strains containing wild-type SXT-R391 tandem arrays or tandem arrays of Δs089 and Δssb mutants of SXT and R391 were used in mating experiments with a recA recipient strain. Values are means and standard deviations from at least three independent experiments. One-way ANOVA with Dunnett's multiple comparison test was used to compare the means for hybrid-ICE-containing exconjugant colonies of each deletion mutant with the WT. The confidence intervals (p) for the comparisons are indicated. NS, not statistically different from the WT.
Expression of bet and exo is subject to strong translational regulation.
To determine whether the translation of bet and exo is consistent with the strong accumulation of their mRNA transcript upon MMC exposure, we used single-copy chromosomal lacZ translational fusions of the two genes in E. coli BW25113 derivatives (GG209 and GG215) to perform β-galactosidase assays. Interestingly, detectable translation of bet′-′lacZ and exo′-′lacZ occurred only after extensive exposure to MMC. Indeed, no differences in β-galactosidase activity between the induced and control samples were observed when the cultures were induced under conditions similar to those used for our RT-qPCR analysis (2 h with 100 ng/ml MMC). We confirmed by RT-qPCR with primers targeting lacZ that the mRNA transcript level of lacZ for each fusion was comparable to that obtained for the corresponding wild-type genes (see Fig. S1 in the supplemental material). We found that a 16-h induction with 200 ng/ml MMC was required for the detection of a significant induction of β-galactosidase activity for the bet and exo fusions. As expected, the translation of bet and exo was increased by MMC in a recA+ strain: β-galactosidase activities were 3.56 ± 0.93 and 0.52 ± 0.23 Miller units (means ± standard deviations) for the bet and exo fusions, respectively, after MMC induction, compared with 0.00 ± 0.01 Miller units for both fusions without induction.
However, the comparison of RT-qPCR and β-galactosidase data revealed important differences in the transcriptional and translational profiles of bet and exo. Indeed, upon induction with MMC, bet and exo conserved the same relative mRNA amounts relative to the reference gene rpoZ (2−ΔCT) (Fig. 6A). However, β-galactosidase assay results demonstrate that exo is significantly less translated (nearly a 5-fold difference) than bet (P < 0.001) (Fig. 6B). Therefore, although bet and exo are part of the same transcript, the production of the Bet and Exo proteins is differentially regulated. As RT-qPCR assays ruled out the possibility that the difference observed between transcriptional and translational data was due to possible polar effects resulting from the lacZ fusions (see Fig. S1 in the supplemental material), our results suggest that expression of bet and exo is subject to an additional posttranscriptional or translational regulatory mechanism.
Fig 6.
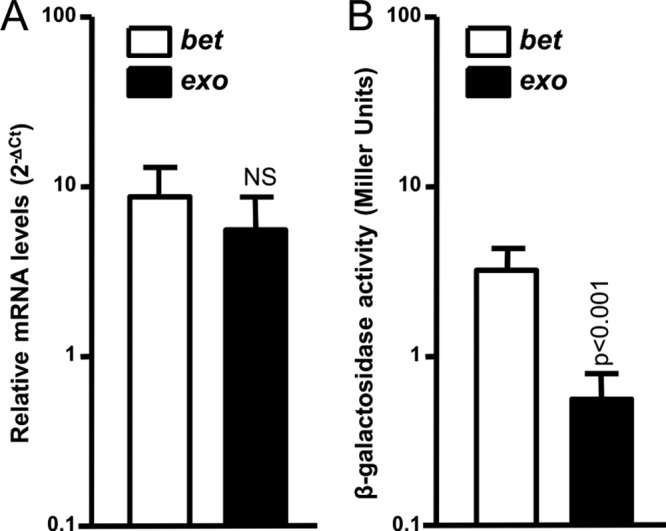
Relative quantification of the mRNA level and translation of bet and exo in the presence of MMC. (A) mRNA levels are expressed as the relative quantification by RT-qPCR (2−ΔCT) of each gene relative to rpoZ, in a WT background upon induction with MMC. (B) Translation results are expressed as Miller units obtained from β-galactosidase assays with bet′-′lacZ and exo′-′lacZ translational fusions (lacZ fused to the 8th codon of each gene) upon induction with MMC. Values are means and standard deviations from at least three independent experiments. One-way ANOVA with Tukey's multiple comparison test was used to compare the means between bet and exo transcription and translation. The mean number of Miller units obtained for exo was statistically significantly different from the values for bet, with a confidence interval of P < 0.001. NS, not statistically different.
The SXT/R391 recombination system differs from other recombination systems composed of proteins related to Bet and Exo by the presence of a gene of unknown function, termed orfZ, located between the bet and exo genes (Fig. 1). Although OrfZ does not have any known homologues, its small size suggests that it could act as a transcriptional or translational regulator. To test this hypothesis, we cloned orfZ under the control of an arabinose-inducible promoter (pGG7) and expressed it in trans in E. coli VB17. We used an RT-qPCR assay to measure the impact of its overexpression on the expression of bet and exo. Relative expression (2−ΔΔCT) of bet and exo remained unaffected by orfZ overexpression (expression ratio of ≈1). We also tested its impact on the translation of the single-copy chromosomal bet′-′lacZ and exo′-′lacZ fusions in the presence and absence of arabinose to induce the expression of orfZ from pGG7. No change in β-galactosidase activity was observed for these fusions upon overexpression of orfZ (expression ratio of ≈1). Taken together, these results suggest that orfZ does not participate in Bet-Exo-mediated recombination by regulating their expression. Furthermore, our analysis of the intergenic regions on each side of orfZ shows that they share some sequence similarity (see Fig. S4 in the supplemental material). The presence of orfZ only in SXT/R391 ICEs suggests that it results from an insertion event that led to the duplication of the bet-exo intergenic region and that it does not have a significant role in recombination (see below).
Conserved putative translational attenuators are found upstream of bet and exo.
We used the RibEx server (38) to assess the presence of putative regulatory elements that could affect bet and exo translation. We identified two putative translational attenuators located immediately upstream of bet (TAbet) and exo (TAexo) (Fig. 7A). Moreover, the attenuator located upstream of exo has a higher predicted stability (ΔG = −25.50 kcal mol−1) than the one found upstream of bet (ΔG = −21.60 kcal mol−1), which could also explain the stronger translational attenuation observed for the production of Exo. These structures are highly conserved in all SXT/R391 ICEs (see Fig. S2 in the supplemental material), and RibEx analyses place similar structures upstream of the bet and exo genes found in IncA/C plasmids (see Fig. S3 in the supplemental material). The presence of these putative translational attenuators is consistent with our experimental finding that the expression of bet and exo is subject to a strong translational regulation.
Fig 7.

The recombination locus contains two predicted translational attenuators. (A) The presence of attenuators was determined with the RibEx server (38) and is shown by black lollipops upstream of bet and exo. Dotted rectangles contain folding predictions made with Mfold v4.6 (63) for each attenuator, along with the sequence located between the attenuator and the translation start sites of bet or exo. Positions of the Shine-Dalgarno (SD) sequences (dashed line), translation start sites (underlining), and ΔG values are indicated for each attenuator. Positions of the first nucleotide included in the construction of the pDPL lacZ translational fusion plasmids are indicated as TA sites and are numbered from the AUG start codon for each structure. (B) β-Galactosidase activity of bet′-′lacZ and exo′-′lacZ translational fusions (lacZ fused to the 8th codon of each gene) expressed in Miller units in the presence and absence (CTL) of l-arabinose. Values are means and standard deviations from three independent experiments. The asterisk indicates that activity was below the detection level. One-way ANOVA with Tukey's multiple comparison test was used to compare the means for the bet′-′lacZ fusion under arabinose-inducing conditions.
In order to determine if these putative translational attenuators are indeed functional, we cloned bet′-′lacZ and exo′-′lacZ, conserving different lengths of the 5′ UTR upstream of each fusion, under the control of an arabinose-inducible promoter. This yielded three constructions for each fusion: one devoid of the putative attenuator sequence (TA−10 and TA−11), one containing a truncated attenuator sequence (TA−24 and TA−34), and one containing the full-length putative attenuator sequence (TA−53 and TA−41) (Fig. 7A). We then assayed the translation levels of these constructions using β-galactosidase assays. In agreement with our prediction, under arabinose-inducing conditions, the absence of TAbet (TA−10) allowed a higher translation level, which corresponds to an almost-8-fold increase compared to the level of translation obtained with the chromosomal fusion (Fig. 7B). In the presence of the full-length TAbet (TA−53), a translation level comparable to that obtained with the chromosomal fusion was obtained. The truncated construction (TA−24) gave intermediate results, consistent with the destabilization of the first stem-loop of the predicted structure (Fig. 7). Taken together, these results confirm that the predicted TAbet sequence acts as a translational attenuator. Results obtained with TAexo did not allow us to confirm that it acts as a translational attenuator, since similar translation levels were obtained regardless of the length of the 5′ UTR (Fig. 7B). However, the high levels of translation obtained from these constructions confirm that the chromosomal translational fusion is indeed functional and suggest that other regulatory mechanisms might be in play to limit translation of exo.
Similar recombination systems are widely distributed among various bacterial species and genetic elements.
The similarity of the SXT/R391 recombination genes with the λ Red system and with genes found in the IncA/C family plasmids led us to investigate the prevalence of similar recombination systems in all sequenced bacterial species and mobile genetic elements. We performed a large-scale in silico analysis using the data extracted from the NCBI RefSeq database of fully sequenced viral, plasmid, and microbial genomes. We screened the proteomes of the 8,513 genomes of the database with the HMM profiles associated with the respective functional domains of Bet (RecT, PF03837) and Exo (YqaJ, PF09588) using HMMER3 software (31). Based on this analysis, we found 412 proteins containing a RecT domain and 632 proteins containing a YqaJ domain. A total of 163 recombination systems were identified based on the presence of a putative recombinase of the RecT family and a predicted exonuclease of the YqaJ family in the same locus (see Table S2 in the supplemental material).
The vast majority (125 of 163) of the recombination systems we identified were found in sequenced microbial genomes. Each system was further analyzed to determine whether it was part of an unidentified prophage-like element, integrating conjugative element, or integrated plasmid. We reclassified 96 of these systems as belonging to prophage-like elements on the basis of the presence of genes encoding a phage-like integrase and other unmistakable phage features (e.g., tail fiber protein, major capsid protein, and tail tape measure protein) in the vicinity of the recombination system. Four of the 125 systems identified in microbial genomes belonged to the SXT/R391 ICEs ICESpuPO1 (CP000503), ICEPmiUSA1 (AM942759), ICEVchBan9 (CP001485) and to one previously unidentified putative SXT/R391 ICE in V. cholerae 2010EL-1786 (NC_016445). The remaining recombination systems found in microbial genomes that could not be reclassified were assumed to be located on genomic islands (GIs). Most of the plasmid-borne recombination systems (8 of 14) were found in IncA/C plasmids, the closest SXT/R391 relatives. Surprisingly, three systems were identified in conjugative plasmids belonging to the IncP-7 group (pCAR1, pCAR1.2, and pDK1), one in a plasmid of the IncT group (Rts1), as well as two in nonconjugative plasmids pMAQU02 and pALVIN02.
We analyzed the distribution of the identified recombination systems according to the taxonomic order of their host. As expected, more recombination systems (69 of 163) were identified in strains belonging to the order Enterobacteriales, the predominant hosts of lambdoid phages, than in strains of any other order. The remaining 94 systems were distributed among strains belonging to very diverse taxonomic orders, some of them very distant from SXT/R391 ICEs' hosts, such as the orders Rhizobiales and Thermonoanaerobacterales (Fig. 8; also, see Table S2 in the supplemental material).
Fig 8.

Phylogenetic trees of single-stranded DNA annealing proteins and cognate exonucleases encoded by gene pairs found in sequenced microbial, plasmid, and bacteriophage genomes. (A) Recombination proteins containing a RecT domain. (B) Exonucleases containing a YqaJ domain. Label colors indicate orders of bacterial strains in which each protein was found (inner wheel). Outer color strips indicate the element carrying each protein (outer wheel). Filled and open circles indicate the presence and absence, respectively, of Gam. Relevant elements are indicated by arrows. Bootstrap values of >80 are indicated.
SXT/R391 Bet and Exo have different evolutionary origins.
We carried out a phylogenetic analysis to determine the evolutionary relationships between the 163 pairs of RecT domain- and YqaJ domain-containing proteins identified (Fig. 8). As SXT and R391 are found in genomes that are not fully assembled and are thus absent from the RefSeq database, we manually added their respective Bet and Exo sequences to our data set. Our analysis shows that the predicted recombinases and exonucleases identified in phages or GIs seem to cluster relative to their host's taxonomic order. However, proteins encoded by ICEs and plasmids tend to cluster relative to the family of mobile genetic elements to which they belong (Fig. 8). For instance, the Bet and Exo homologues of IncA/C plasmids form monophyletic groups, closely related to the corresponding ones of SXT/R391 ICEs. Similarly, plasmids belonging to the IncP-7 and IncT (Rts1) families also cluster together (Fig. 8).
Our analysis shows that partner recombinases and exonucleases do not always share a common origin. Indeed, the Bet proteins of SXT/R391 ICEs and IncA/C plasmids seem to have evolved from the ancestor of Bet proteins of λ-like phages (Fig. 8A). However, their cognate Exo proteins are very distantly related to those encoded by lambdoid phages (Fig. 8B). Moreover, while the YqaJ domain proteins encoded by all the plasmid- and ICE-borne recombination systems derive from the same common ancestor (Fig. 8B), their cognate RecT domain proteins have different phylogenetic relationships: RecT domain proteins from SXT/R391 ICEs and IncA/C plasmids form a monophyletic group, while those from IncP-7 plasmids (pCAR1, pCAR1.2, and pDK1), the IncT plasmid Rts1, and plasmid pMAQU02 form another distantly related group, more closely related to those derived from phages and prophage-like elements from Enterobacteriales and Burkholderiales (Fig. 8A). We also found that all plasmids but pALVIN02 harbor a gene encoding an Ssb in the vicinity of the predicted recombinase gene (see Table S2 in the supplemental material). Interestingly, pALVIN02 is the only plasmid that harbors a RecT orthologue unrelated to other plasmid-borne putative recombinase proteins. Although most bacteriophages from the order Enterobacteriales encode a homologue of λGam, none was found in the recombination systems carried by ICEs or plasmids (Fig. 8A). Finally, the only six occurrences of an ORF related to orfZ located between the recombinase- and exonuclease-encoding genes were found in SXT/R391 ICEs.
DISCUSSION
In previous studies, we demonstrated that ICEs of the SXT/R391 family generate their own genetic diversity by mediating recombination events between two ICEs arranged in a tandem array (20, 39). Two ICE-encoded proteins, Bet and Exo, which together function as a RecA-independent homologous recombination system, are able to catalyze a significant part of these events, generating functional ICEs with new features (20). Here, we investigated the conditions that regulate the expression of bet and exo to better understand which signals trigger the formation of hybrid ICEs. Like the host's SOS response, the expression of both genes is induced by DNA-damaging agents. RecA, as well as the ICE-encoded transcriptional regulators SetC and SetD, was found to be required for mitomycin C-induced expression of bet and exo. These results show that although they are induced during the SOS response, the recombination genes are not under the control of their host's SOS repressor, LexA. Instead, like the tra genes, bet and exo are under the control of the ICE-encoded main repressor SetR, which represses the expression of the SetCD transcriptional activator complex (11, 19, 40). SXT/R391 recombination functions are induced in response to DNA damages, the same conditions that trigger the conjugative transfer of these mobile elements to a new host (19), consequently providing the immediate possibility to segregate functional recombinants. Moreover, as we have previously shown that Bet-Exo and RecA homologous recombination pathways can cooperate to enhance hybrid ICE formation, their coinduction during SOS response highlights the extent to which these elements take advantage of their host's cellular function to enhance their own diversity (20). Additionally, in the assay used to study hybrid ICE formation (20), derepression of SetCD in a recA+ context probably depends upon the transient expression of the SOS response in a subpopulation of donor cells (41), since we did not provide the direct action of a DNA-damaging agent. Under those conditions, the total levels of RecA and Bet-Exo are expected to be much lower. Combined with the results we present here, this suggests that the percentage of hybrid ICEs obtained during those assays is possibly an ample underestimation of the real amount of hybrids that can be formed in the presence of DNA-damaging agents. Baharoglu and colleagues (42, 43) have recently demonstrated that, besides exposure to most antibiotics, conjugation itself induces the SOS response in V. cholerae, the natural host of SXT/R391 ICEs. Together with our results, this observation sheds new light on the major impact that these elements can have on their host's genome plasticity. Therefore, not only are bet and exo expressed in the induced donor cells, but they are also likely expressed in the fresh exconjugants, as the pool of SetR in the recipient cells has to build up to repress setCD expression. In addition, we anticipate that the accumulation of SetR could be slowed down by its simultaneous autocleavage, which would be promoted by the coprotease activity of RecA bound to the incoming single-stranded DNA.
In bacteriophage λ, λbet and λexo are part of the pL operon, which is also induced by DNA-damaging agents (23, 44). We, as well as others, have discussed in the past the possibility that these homologous recombination systems could act as a repair pathway for salvaging ICE or phage DNA broken by exposure to DNA-damaging agents (20, 24). Our finding that the SXT/R391 bet and exo genes are specifically expressed in the presence of DNA-damaging agents strengthens this hypothesis. We have found that the bet and exo genes are part of a polycistronic transcript that is driven from an SOS-inducible promoter located in the intergenic region between s063 and s089. This transcript includes 12 other highly conserved and syntenic genes of the SXT/R391 backbone that have no apparent role in their transfer and is by far the largest set of contiguous genes of unknown function found in these elements (7). The conservation of these genes as well as their coexpression with the recombination and conjugative transfer genes suggests that at least some of them may have a role in recombination or in the fitness of these elements. Interestingly, 7 of the 14 genes of this locus, including ssb, bet, and exo, are also conserved in all the IncA/C conjugative plasmids, although their order differs slightly (7). To the best of our knowledge, the stimuli that trigger the conjugative transfer of IncA/C plasmids have not been identified. However, they lack a homologue of the ICE SetR repressor, suggesting that unless they rely on the host's LexA repressor, DNA damages are unlikely to activate their transfer. As a consequence, we are unable to predict which conditions trigger the expression of bet and exo orthologues in IncA/C plasmids.
The coexpression of bet and exo with genes located upstream of the recombination system (s089 and ssb) led us to investigate their role in hybrid ICE formation. We found that the deletion of s089 did not impact hybrid ICE formation, thus ruling out a role for the protein it encodes in recombination. However, we found that the deletion of ssb, a gene encoding a single-stranded DNA binding protein located immediately upstream bet, caused a significant (nearly 3-fold) increase in Bet-Exo-mediated hybrid ICE formation. Ssb proteins are known to be involved in a number of recombination, replication, and repair mechanisms, acting both cooperatively and competitively with a myriad of other proteins for binding to single-stranded DNA (ssDNA) (45, 46). Although we have not investigated its precise role in the recombination mechanism, it is possible that SXT Ssb limits the access of Bet to its substrate by competing for binding to ssDNA. This hypothesis is strengthened by recent findings that in vivo coexpression of SXT's Ssb-Bet-Exo diminishes by more than 2-fold the recombination frequency of a linear dsDNA substrate with the E. coli chromosome, compared with the coexpression of only Bet-Exo (22). Additionally, like λBet, SXT Bet is able to recombine ssDNA sequences that possess very short homologous sequences (36 bp) (21), and it has been demonstrated that λBet is able to promote strand exchange and displacement using short oligonucleotides containing several mismatches as substrates (47). In a context where the ICE recombination functions are induced by DNA-damaging agents, when large quantities of ssDNA would be available, it is possible that controlling the efficiency of recombination of sequences bearing only short homologous regions (36 bp [21]) would limit illegitimate recombination events, as they would lead to the formation of nonfunctional elements unable of transferring to a new host.
Our in silico analysis of the structure of several recombination systems shows that while a homologue of λGam is encoded by numerous phages, none is found in the plasmid or ICE-borne systems. λGam protects the phage linear dsDNA from degradation by inhibiting the host's RecBCD endonuclease (25, 48). Although we have not assessed the presence of other RecBCD inhibitors (46), the absence of λGam from plasmids and ICEs could be due to the fact that no step of these elements' normal “life cycle” involves a linear dsDNA molecule. Instead, all of these systems, with the exception of the one carried by the nonconjugative plasmid pALVIN02, contain a gene encoding an Ssb upstream of the gene encoding the Bet-like protein. Bacteriophage λ also carries a putative single-stranded DNA-binding protein, encoded by the gene ea10 (49). Although the role of this precise putative Ssb in λ recombination has not been evaluated, other phage-encoded Ssb proteins have been recognized for their positive effects on phage homologous recombination and replication (for a review, see reference 46 and references therein). However, the specific role of Ssb proteins in the recombination catalyzed by systems found in conjugative elements has not been investigated yet. One of the possible roles for the ICE-encoded Ssb could be to protect the ssDNA substrate of conjugative transfer from degradation, aberrant recombination prior to its circularization and synthesis of its complementary strand, or even to limit RecA binding to prevent overactivation of the host's SOS response.
The role of orfZ located between bet and exo remains to be determined. However, our results suggest that it does not act as a transcriptional or translational regulator of bet and exo expression. In addition, no homologues of orfZ were found in any of the recombination systems we have analyzed in silico. The presence of long and imperfect repeats (66% identity) flanking this gene in SXT/R391 ICEs (see Fig. S4 in the supplemental material) suggests that it results from an insertion event and supports the idea that it is not relevant to the recombination mechanism of systems related to Bet-Exo.
The expression profiles and genetic organization of bet and exo revealed that although mRNA transcripts of these genes accumulate at a high level in the presence of MMC, their translation remains weak. Our results are in accordance with other published data on the RecT-RecE recombination system of the rac prophage and on the λ Red system that show that overexpression of the recombinase with respect to its partner exonuclease favors recombination (50). We identified two putative translational attenuators, one located upstream of bet (TAbet) and the other upstream of exo (TAexo), that could account for the weak translation of the Bet- and Exo-LacZ fusion proteins. Our results suggest that TAbet is a genuine translational attenuator and when present causes a 10-fold reduction in the translation of the bet′-′lacZ fusion compared to when it is absent. On the other hand, the predicted translational attenuator located upstream of exo did not prove to effectively reduce translation. The high translation level of TA−11-exo′-′lacZ compared to TA−10-bet′-′lacZ (Fig. 7B) is in agreement with the predicted strength of the Shine-Dalgarno sequence upstream of exo (AAGGAG), which has a slightly better complementarity to the 3′ end of 16S RNA than the one found upstream of bet (CGGAGU) (Fig. 7A) (51). However, in its natural context, exo is much less translated than bet (Fig. 6B). Since TAexo does not seem to play a role as a translational attenuator in our artificial construction, another regulatory mechanism must be in play to reduce the efficient translation of Exo when it is part of the mRNA transcript initiated upstream of s089. Such translational regulation could depend upon the presence of an ICE-encoded small RNA, which would be present only in the chromosomal context of our translational fusions, and act by reducing the translation of Exo by binding to its 5′ UTR (for a review, see reference 52). Interestingly, while IncA/C plasmids lack an orfZ gene, an attenuator similar to TAbet is also found upstream the IncA/C bet gene, providing additional evidence for a common mode of translational regulation between these two recombination systems. Combined with our previously published results (20), our observations indicate that only very low levels of both Bet and Exo are sufficient to catalyze the formation of hybrid ICEs at high frequency.
The recombination proteins encoded by SXT and λ belong to the same families: λBet and SXT Bet belong to the RecT family (PF03837), and λExo and SXT Exo belong to the YqaJ family (PF0955), also sometimes referred to as the lambda exonuclease (LE) superfamily (53). Recombination systems composed of a putative recombinase of the RecT family and a putative exonuclease of the YqaJ family were identified in a variety of prophages, ICEs, and plasmids of different incompatibility groups as well as remnants of prophages or other genomic islands. Our analysis of the phylogenetic relationships between the predicted recombinase and exonuclease proteins from different origins suggests that those derived from phages cluster relative to the taxonomic order of their host, while those encoded by plasmids and ICEs cluster relative to the incompatibility group or family. This trend probably results from the narrow host range of phages, which would lead to fewer opportunities for interspecies gene exchange compared to plasmids and ICEs. Most mobile genetic elements are usually considered combinations of interchangeable functional modules that were originally defined as belonging to bacteriophages, conjugative plasmids, or transposons (6, 54–56). The widespread presence of recombination systems related to Bet-Exo in these mobile genetic elements might provide a suitable explanation for the exchange of such modules between different types of mobile elements, with a requirement for only very short homology regions on each side the different modules. The differences in the evolutionary relationships between the proteins of the RecT and YqaJ families suggest that many of the recombination systems identified have arisen by successive acquisition of the genes encoding these proteins rather than by the gain of a recombinase-exonuclease gene pair. While it is surprising that functionally cooperating proteins have been acquired from different origins, it is known that recombinases belonging to the RecT family are sometimes found associated with other types of exonucleases, such as RecE in the cryptic rac prophage (57), which has led to the assumption that the similar operon structures of different systems could arise independently due the selective pressure for the coexpression of cooperating proteins (57).
The prevalence of similar recombination genes in many mobile genetic elements found in nearly all bacterial orders suggests that mobile DNA may impact the plasticity and evolution of bacterial genomes far beyond the immediate benefit provided by selectable markers such as antibiotic resistance genes. Indeed, they could facilitate genome-wide rearrangement as well as incorporation of exogenous DNA into bacterial genomes by means of homologous recombination systems requiring much shorter identity regions than RecA-dependent pathways.
Supplementary Material
ACKNOWLEDGMENTS
We thank Eric Bordeleau for technical assistance with the HMM analysis. We are grateful to Nicolas Carraro, Eric Bordeleau, and Alain Lavigueur for insightful comments on the manuscript.
G.G. was the recipient a doctoral fellowship (Fonds de Recherche du Québec, Nature et Technologies, QC, Canada), and V.B. holds a Canada Research Chair in bacterial molecular genetics and is a member of the FRSQ-funded Centre de Recherche Clinique Étienne-Le Bel.
Footnotes
Published ahead of print 22 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.02090-12.
REFERENCES
- 1. Frost LS, Leplae R, Summers AO, Toussaint A. 2005. Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3:722–732 [DOI] [PubMed] [Google Scholar]
- 2. Juhas M, van der Meer JR, Gaillard M, Harding RM, Hood DW, Crook DW. 2009. Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33:376–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ochman H, Lawrence JG, Groisman EA. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299–304 [DOI] [PubMed] [Google Scholar]
- 4. Wiedenbeck J, Cohan FM. 2011. Origins of bacterial diversity through horizontal genetic transfer and adaptation to new ecological niches. FEMS Microbiol. Rev. 35:957–976 [DOI] [PubMed] [Google Scholar]
- 5. Didelot X, Maiden MC. 2010. Impact of recombination on bacterial evolution. Trends Microbiol. 18:315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wozniak RA, Waldor MK. 2010. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat. Rev. Microbiol. 8:552–563 [DOI] [PubMed] [Google Scholar]
- 7. Wozniak RA, Fouts DE, Spagnoletti M, Colombo MM, Ceccarelli D, Garriss G, Dery C, Burrus V, Waldor MK. 2009. Comparative ICE genomics: insights into the evolution of the SXT/R391 family of ICEs. PLoS Genet. 5:e1000786 doi:10.1371/journal.pgen.1000786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burrus V, Waldor MK. 2003. Control of SXT integration and excision. J. Bacteriol. 185:5045–5054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Burrus V, Marrero J, Waldor MK. 2006. The current ICE age: biology and evolution of SXT-related integrating conjugative elements. Plasmid 55:173–183 [DOI] [PubMed] [Google Scholar]
- 10. Beaber JW, Burrus V, Hochhut B, Waldor MK. 2002. Comparison of SXT and R391, two conjugative integrating elements: definition of a genetic backbone for the mobilization of resistance determinants. Cell. Mol. Life Sci. 59:2065–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Beaber JW, Hochhut B, Waldor MK. 2002. Genomic and functional analyses of SXT, an integrating antibiotic resistance gene transfer element derived from Vibrio cholerae. J. Bacteriol. 184:4259–4269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bordeleau E, Brouillette E, Robichaud N, Burrus V. 2010. Beyond antibiotic resistance: integrating conjugative elements of the SXT/R391 family that encode novel diguanylate cyclases participate to c-di-GMP signalling in Vibrio cholerae. Environ. Microbiol. 12:510–523 [DOI] [PubMed] [Google Scholar]
- 13. Harada S, Ishii Y, Saga T, Tateda K, Yamaguchi K. 2010. Chromosomally encoded blaCMY-2 located on a novel SXT/R391-related integrating conjugative element in a Proteus mirabilis clinical isolate. Antimicrob. Agents Chemother. 54:3545–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwanaga M, Toma C, Miyazato T, Insisiengmay S, Nakasone N, Ehara M. 2004. Antibiotic resistance conferred by a class I integron and SXT constin in Vibrio cholerae O1 strains isolated in Laos. Antimicrob. Agents Chemother. 48:2364–2369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Juiz-Rio S, Osorio CR, de Lorenzo V, Lemos ML. 2005. Subtractive hybridization reveals a high genetic diversity in the fish pathogen Photobacterium damselae subsp. piscicida: evidence of a SXT-like element. Microbiology 151:2659–2669 [DOI] [PubMed] [Google Scholar]
- 16. Ahmed AM, Shinoda S, Shimamoto T. 2005. A variant type of Vibrio cholerae SXT element in a multidrug-resistant strain of Vibrio fluvialis. FEMS Microbiol. Lett. 242:241–247 [DOI] [PubMed] [Google Scholar]
- 17. Bani S, Mastromarino PN, Ceccarelli D, Le Van A, Salvia AM, Ngo Viet QT, Hai DH, Bacciu D, Cappuccinelli P, Colombo MM. 2007. Molecular characterization of ICEVchVie0 and its disappearance in Vibrio cholerae O1 strains isolated in 2003 in Vietnam. FEMS Microbiol. Lett. 266:42–48 [DOI] [PubMed] [Google Scholar]
- 18. Osorio CR, Marrero J, Wozniak RA, Lemos ML, Burrus V, Waldor MK. 2008. Genomic and functional analysis of ICEPdaSpaI, a fish-pathogen-derived SXT-related integrating conjugative element that can mobilize a virulence plasmid. J. Bacteriol. 190:3353–3361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beaber JW, Hochhut B, Waldor MK. 2004. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature 427:72–74 [DOI] [PubMed] [Google Scholar]
- 20. Garriss G, Waldor MK, Burrus V. 2009. Mobile antibiotic resistance encoding elements promote their own diversity. PLoS Genet. 5:e1000775 doi:10.1371/journal.pgen.1000775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Datta S, Costantino N, Zhou X, Court DL. 2008. Identification and analysis of recombineering functions from Gram-negative and Gram-positive bacteria and their phages. Proc. Natl. Acad. Sci. U. S. A. 105:1626–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen WY, Ho JW, Huang JD, Watt RM. 2011. Functional characterization of an alkaline exonuclease and single strand annealing protein from the SXT genetic element of Vibrio cholerae. BMC Mol. Biol. 12:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Court DL, Oppenheim AB, Adhya SL. 2007. A new look at bacteriophage lambda genetic networks. J. Bacteriol. 189:298–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poteete AR. 2001. What makes the bacteriophage lambda Red system useful for genetic engineering: molecular mechanism and biological function. FEMS Microbiol. Lett. 201:9–14 [DOI] [PubMed] [Google Scholar]
- 25. Murphy KC. 2007. The lambda Gam protein inhibits RecBCD binding to dsDNA ends. J. Mol. Biol. 371:19–24 [DOI] [PubMed] [Google Scholar]
- 26. Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Daccord A, Mursell M, Poulin-Laprade D, Burrus V. 2012. Dynamics of the SetCD-regulated integration and excision of genomic islands mobilized by integrating conjugative elements of the SXT/R391 family. J. Bacteriol. 194:5794–5802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miller JF. 1992. A short course in bacterial genetics. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 30. Pruitt KD, Tatusova T, Klimke W, Maglott DR. 2009. NCBI reference sequences: current status, policy and new initiatives. Nucleic Acids Res. 37:D32–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Eddy SR. 2009. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23:205–211 [PubMed] [Google Scholar]
- 32. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59:307–321 [DOI] [PubMed] [Google Scholar]
- 34. Capella-Gutierrez S, Silla-Martinez JM, Gabaldon T. 2009. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25:1972–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Letunic I, Bork P. 2011. Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 39:W475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948 [DOI] [PubMed] [Google Scholar]
- 37. Haugen SP, Ross W, Gourse RL. 2008. Advances in bacterial promoter recognition and its control by factors that do not bind DNA. Nat. Rev. Microbiol. 6:507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Abreu-Goodger C, Merino E. 2005. RibEx: a web server for locating riboswitches and other conserved bacterial regulatory elements. Nucleic Acids Res. 33:W690–W692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burrus V, Waldor MK. 2004. Formation of SXT tandem arrays and SXT-R391 hybrids. J. Bacteriol. 186:2636–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beaber JW, Waldor MK. 2004. Identification of operators and promoters that control SXT conjugative transfer. J. Bacteriol. 186:5945–5949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. McCool JD, Long E, Petrosino JF, Sandler HA, Rosenberg SM, Sandler SJ. 2004. Measurement of SOS expression in individual Escherichia coli K-12 cells using fluorescence microscopy. Mol. Microbiol. 53:1343–1357 [DOI] [PubMed] [Google Scholar]
- 42. Baharoglu Z, Bikard D, Mazel D. 2010. Conjugative DNA transfer induces the bacterial SOS response and promotes antibiotic resistance development through integron activation. PLoS Genet. 6:e1001165 doi:10.1371/journal.pgen.1001165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baharoglu Z, Mazel D. 2011. Vibrio cholerae triggers SOS and mutagenesis in response to a wide range of antibiotics: a route towards multiresistance. Antimicrob. Agents Chemother. 55:2438–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Oppenheim AB, Kobiler O, Stavans J, Court DL, Adhya S. 2005. Switches in bacteriophage lambda development. Annu. Rev. Genet. 39:409–429 [DOI] [PubMed] [Google Scholar]
- 45. Kuzminov A. 1999. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 63:751–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szczepanska AK. 2009. Bacteriophage-encoded functions engaged in initiation of homologous recombination events. Crit. Rev. Microbiol. 35:197–220 [DOI] [PubMed] [Google Scholar]
- 47. Li Z, Karakousis G, Chiu SK, Reddy G, Radding CM. 1998. The beta protein of phage lambda promotes strand exchange. J. Mol. Biol. 276:733–744 [DOI] [PubMed] [Google Scholar]
- 48. Marsic N, Roje S, Stojiljkovic I, Salaj-Smic E, Trgovcevic Z. 1993. In vivo studies on the interaction of RecBCD enzyme and lambda Gam protein. J. Bacteriol. 175:4738–4743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ineichen K, Shepherd JC, Bickle TA. 1981. The DNA sequence of the phage lambda genome between PL and the gene bet. Nucleic Acids Res. 9:4639–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Muyrers JP, Zhang Y, Buchholz F, Stewart AF. 2000. RecE/RecT and Redalpha/Redbeta initiate double-stranded break repair by specifically interacting with their respective partners. Genes Dev. 14:1971–1982 [PMC free article] [PubMed] [Google Scholar]
- 51. Komarova AV, Tchufistova LS, Supina EV, Boni IV. 2002. Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA 8:1137–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Waters LS, Storz G. 2009. Regulatory RNAs in bacteria. Cell 136:615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aravind L, Makarova KS, Koonin EV. 2000. Survey and summary. Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 28:3417–3432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Toleman MA, Walsh TR. 2011. Combinatorial events of insertion sequences and ICE in Gram-negative bacteria. FEMS Microbiol. Rev. 35:912–935 [DOI] [PubMed] [Google Scholar]
- 55. Toussaint A, Merlin C. 2002. Mobile elements as a combination of functional modules. Plasmid 47:26–35 [DOI] [PubMed] [Google Scholar]
- 56. Burrus V, Pavlovic G, Decaris B, Guedon G. 2002. The ICESt1 element of Streptococcus thermophilus belongs to a large family of integrative and conjugative elements that exchange modules and change their specificity of integration. Plasmid 48:77–97 [DOI] [PubMed] [Google Scholar]
- 57. Iyer LM, Koonin EV, Aravind L. 2002. Classification and evolutionary history of the single-strand annealing proteins, RecT, Redbeta, ERF and RAD52. BMC Genomics 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Khlebnikov A, Datsenko KA, Skaug T, Wanner BL, Keasling JD. 2001. Homogeneous expression of the P(BAD) promoter in Escherichia coli by constitutive expression of the low-affinity high-capacity AraE transporter. Microbiology 147:3241–3247 [DOI] [PubMed] [Google Scholar]
- 59. Singer M, Baker TA, Schnitzler G, Deischel SM, Goel M, Dove W, Jaacks KJ, Grossman AD, Erickson JW, Gross CA. 1989. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol. Rev. 53:1–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ceccarelli D, Daccord A, Rene M, Burrus V. 2008. Identification of the origin of transfer (oriT) and a new gene required for mobilization of the SXT/R391 family of ICEs. J. Bacteriol. 190:5328–5338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hochhut B, Waldor MK. 1999. Site-specific integration of the conjugal Vibrio cholerae SXT element into prfC. Mol. Microbiol. 32:99–110 [DOI] [PubMed] [Google Scholar]
- 62. Hochhut B, Beaber JW, Woodgate R, Waldor MK. 2001. Formation of chromosomal tandem arrays of the SXT element and R391, two conjugative chromosomally integrating elements that share an attachment site. J. Bacteriol. 183:1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406–3415 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.